

Abstract
Aggregation of protein-based therapeutics is a challenging problem in the biopharmaceutical industry. Of particular concern are implications for product efficacy and clinical safety due to potentially increased immunogenicity of the aggregates. We used transmission electron microscopy (TEM) to characterize biophysical and morphological features of antibody aggregates formed upon controlled environmental stresses. TEM results were contrasted with results obtained in parallel by independent methods, including size exclusion chromatography, dynamic light scattering, microflow imaging and nanoparticle tracking. For TEM, stressed samples were imaged by negative staining and in the frozen-hydrated state. In both cases, aggregates appeared amorphous but differed in fine structural detail. Specifically, negatively stained aggregates were compact and consisted of smaller globular structures that had a notable three dimensional character. Elements of the native IgG structure were retained, suggesting that the aggregates were not assembled from denatured protein. In contrast, aggregates in frozen-hydrated samples appeared as extended, branched protein networks with large surface area. Using multiple scales of magnification, a wide range of particle sizes was observed and semi-quantitatively characterized. The detailed information provided by TEM extended observations obtained with the independent methods, demonstrating the suitability of TEM as a complementary approach to submicron particle analysis.
Keywords: protein aggregation, IgG antibody, imaging methods, image analysis, particle sizing
Introduction
Protein aggregation represents a major challenge in biopharmaceutical manufacturing. 1,2 It may occur during purification, formulation, shipment or storage and can lead to significant reduction in yield, bioavailability and potency of the final product. 3,4 Significant efforts are therefore employed to monitor and minimize aggregate formation.2-7 There is additional concern that aggregates may affect clinical safety given their potential for stimulating adverse immunogenicity in patients. 1,8-12 The molecular mechanisms by which protein aggregates induce unwanted immune responses are poorly understood, but it is likely that their physical and chemical structure play an important role. Particle structure can also be expected to have an impact on how aggregates are processed and eliminated in vivo, but lack of sufficient information on their morphological features have hampered a systematic investigation of this issue. Finally, aggregate morphology and specific attributes, such as inherent packing density, are likely to affect the response of analytical instruments that are commonly used in particle counting and sizing. Thus, there is a critical need for better morphological characterization of protein aggregates, including direct visual examination.
The wide range of aggregate sizes (nm to mm) and their generally unknown molecular features require the use of a diverse set of analytical tools for detection and characterization.13-15 Current technologies for aggregate characterization and quantification are well developed for particulates that exceed 10 μm or that are less than 0.1 μm in size, but there are virtually no technologies for both quantitative and morphological characterization of aggregates that fall in the 0.1-1 μm range. 13,16 Notably, it is well recognized that with size exclusion chromatography (SEC) sample dilution, exposure to high ionic strength mobile phases and/or adsorption of aggregates to column material can greatly alter the aggregate content and size distribution.17 Analytical ultracentrifugation and field flow fractionation are important alternatives for aggregation quantitation, though the low-throughput nature and difficulty of use associated with these technologies have limited their application mostly to corroboration of SEC results during method development.7
Furthermore, the fraction of protein that can be present as subvisible particles is often below the detection limit for loss of native protein mass by methods such as SEC 18. Direct counting and sizing of the particles by approaches such as microflow imaging, nanoparticle tracking analysis and Coulter counting have been shown to be valuable for analyses of these aggregates and with micron-sized and larger particles digital images are helpful for aggregate characterization. But there is still an unmet need for characterizing the morphologies of submicron particles found in therapeutic protein formulations.
Electron microscopy with its unique capability for providing direct visual information of size, shape and aggregation extent of a sample is a powerful tool in the arsenal of characterization techniques applied to protein therapeutics.19-21 Molecular electron microscopy uses advanced specimen preparation and imaging methods designed specifically to visualize complex biological samples under conditions close to their native hydration state. Automated data collection and processing software, linked to a relational database, provide the means to image and analyze samples in an efficient and reproducible manner, and sample throughputs are capable of addressing biopharmaceutical characterization needs in a statistically significant manner.22 Samples are preserved in solution by vitrification or by negative staining, and then imaged using a transmission electron microscope (TEM) controlled by automated software that enables imaging a significant portion of the specimen. 23-29
In this study, we used TEM to explore the morphology, size and distribution of antibody aggregates formed upon agitation and freeze-thaw stress in the presence or absence of the excipient polysorbate 20. The results were evaluated in the context of data obtained in parallel by more traditional aggregate and particle measurement technologies, including SEC, dynamic light scattering (DLS), microflow imaging (MFI) and nanoparticle tracking (NTA). TEM proved to be particularly useful in detecting and characterizing aggregates in the sub-micrometer range and providing specific information on aggregate microstructure. Moreover, automated data collection and image analysis allowed examination of a sufficiently large number of particles to permit semi-quantitation of their distribution in the sub-micrometer range. Thus, although a combination of analytical techniques is necessary to describe the full spectrum of aggregate characteristics, TEM is well suited as a complementary approach to submicron particle analysis.
Materials and Mehods
Materials
Intravenous Immunoglobulin G (IVIg), 100 mg/ml, (Baxter Healthcare Corporation, Westlake Village CA.) (Lot# LE12N107AB, Expiry: May 2016) was purchased from the University of Colorado-Boulder Apothecary. Glycine, sodium phosphate and sodium chloride were purchased from Fisher Scientific (Fair Lawn, NJ).
Methods
IVIg was diluted to 1 mg/ml using 0.2 M glycine, pH 4.2, the buffer in which the commercial product is prepared or 0.2 M glycine, pH 4.2 containing 0.01% (w/v) Tween 20. The IVIg solution in commercial buffer was agitated end-over-end for 48 hours at room temperature in a 50 ml conical centrifuge tube, frozen in liquid nitrogen and stored in a -80 °C freezer until analysis. IVIg with Tween 20 was agitated end-over-end for 10 days at room temperature in a 50 ml conical centrifuge tube, frozen in liquid nitrogen and stored in a -80 °C freezer until analysis. Before analysis the samples were thawed in a 25 °C water bath. After the sample had equilibrated to room temperature, characterization was conducted using SEC, MFI, NTA and DLS. Five individual samples were used for collecting the data.
Size-Exclusion Chromatography
An Agilent 1100 (Santa Clara, CA) chromatography system was used with a Tosoh G3000 SWXL 7.8 × 30 cm column. An in-line 0.22 μm filter was used with the column. The mobile phase was 200 mM sodium phosphate with 50 mM sodium chloride, pH 7.0. A flow rate of 0.7 ml/min was used with a run time of 20 min for each sample. IVIg was centrifuged at 14,000 × g for 10 minutes and the supernatant was loaded onto the column. Injection load was 25 μg IVIg and elution was monitored by absorbance at 280 nm. Monomer and dimer amounts were calculated as percentages of the total protein peak areas. Insoluble aggregate content was determined by comparing the loss of area under the curve for IVIg that was agitated and freeze-thawed to that of a control sample that was not stressed.
Micro-Flow Imaging
A Protein Simple (Ottawa, ON, Canada) 4200 MFI system was used for counting and sizing particles greater than 1 μm. The instrument was configured in ‘set point 3’ mode using a 100 μm flow cell. A sample volume of 0.5 ml was used of which 0.1 ml was purge volume and 0.35 ml was used for data acquisition.
Nanoparticle Tracking Analysis
A Nanosight (Amesbury, United Kingdom) LM20 system was used for counting and sizing submicron particles. The system was equipped with a laser sample chamber LM12B. Nanosight NS300 with a LM14 laser sample chamber was used for analyzing IVIg sample containing Tween20. A sample volume of 0.5 ml was delivered for analysis with a 1 ml all plastic silicone oil free syringe (National Scientific Company, TN). Sample videos were recorded for 60 seconds and processed using NTA 2.3 Build 127 software. For analysis a screen gain of 1 and a detection threshold of 10 were used.
Dynamic Light Scattering
A Zeta Sizer Nano ZS (Malvern, United Kingdom) was used to measure the hydrodynamic diameter of the aggregates. Sample volume of 0.2 ml was analyzed in a 0.45 ml quartz cuvette.
Sample preparation for electron microscopy
For negative stain microscopy, samples were prepared on continuous carbon films supported on nitrocellulose-coated 400 mesh copper grids (Ted Pella). A 3 μl drop of IVIg solution was applied to a freshly plasma-cleaned grid for 1 min and blotted to a thin film using filter paper. The sample was washed six times by floating the grid on a droplet of H2O for 1 min followed by staining on a droplet of 2 % (w/v) uranylformate for 1 min. The grid was blotted after each incubation and finally air-dried. For cryo-electron microscopy, frozen-hydrated samples of IVIg were prepared on C-flat grids (Protochips) coated with a thin layer of carbon. A 3 μl sample was applied to a freshly plasma-cleaned grid, blotted with filter paper and immediately vitrified in liquid ethane. Grids were handled and stored under liquid nitrogen until transfer to the electron microscope.
TEM image acquisition and analysis
Transmission electron microscopy was performed using an FEI Tecnai T12 electron microscope equipped with an FEI Eagle 4k × 4k CCD camera and operating at 120 kV. Images were collected at nominal magnifications of 6,500× (6.6 nm/pixel), 21,000× (0.5 nm/pixel) and 52,000× (0.21 nm/pixel) using the automated image acquisition software package Leginon. 25 The pixel size of the CCD camera was calculated using the diffraction pattern from a 2D catalase crystal with known cell parameters. Images were acquired at a nominal underfocus of -150 μm (6,500×), -5 μm (21,000×) and -4 to -2 μm (52,000) and electron doses of approximately 0.5 – 45 e/Å2. Images were analyzed using the Appion image processing software.30 After manual screening to ensure acceptable quality, images were randomly ordered for further analysis. Magnifications of 52,000×, 21,000× and 6,500× had a field view of 0.8 μm × 0.8 μm, 2.0 μm × 2.0 μm and 6.7 μm × 6.7 μm, respectively. For sizing of particles, contours were manually traced around each particle in the field of view of the image. Particle selection was based on the following criteria: (1) Particles had to be fully contained in the field of view; (2) Particles were selected only if they were at least partially contained in the holes (cryoEM); (3) Particle boundaries had to be clearly defined. The contours were then analyzed to extract several analytical metrics for the set including the maximum and minimum Feret diameter, area, perimeter, area equivalent diameter and circularity.
Results and Discussion
Generation of antibody aggregates
For the initial study, to induce formation of aggregates a stock solution of IVIg was diluted to 1 mg/ml with 0.2 M glycine buffer (pH 4.2) and agitated for 48 hours at room temperature. Aliquots were then flash-frozen in liquid nitrogen and stored at -80 °C. All measurements were performed immediately after thawing an aliquot at 25 °C. As described below, this protocol was able to convert a substantial fraction of IVIg into aggregates and provided sample that was well suited for comparison of results obtained by the various methods.
Imaging of antibody aggregates by TEM
For electron microscopy, antibody aggregates were imaged in either negative stain or the frozen-hydrated state. For negative staining, a thin layer of sample was deposited on a continuous carbon film, briefly incubated with a heavy metal ion-containing solution and subsequently air-dried. Surrounding the specimen with a heavy metal imparts higher contrast and preservation of material in the electron microscope. This method provides significant structural detail but can lead to alterations in sample appearance partly as a result of the drying process. Drying removes the native layer of hydration and thereby leads to some shrinkage of the specimen. It can also lead to collapse of large protein assemblies.31 Initial investigations indicated that the 1 mg/ml stressed antibody sample formed a densely packed mat of protein that prevented identification of individual components. Further dilution to 10 μg/ml was necessary to yield a satisfactory distribution of soluble and aggregated IVIg. A representative image of an aggregate surrounded by monomeric IgG as well as small oligomeric complexes is shown in figure 1A. Individual IgG molecules had the expected Y-shaped morphology with the Fab and Fc arms visible in the form of small rings. These rings were also apparent in the aggregate, suggesting that it was not formed from fully unfolded protein but molecules that had retained a significant degree of structural integrity. The aggregate measured approximately 110 nm × 70 nm and thus fell into the submicron size range, for which characterization is lacking with other technologies. A wide distribution of aggregates in the submicron range was observed (figure 1B), from small particles containing no more than a few antibody monomers to increasingly larger structures. The aggregates were generally amorphous, lacking any type of directionality or long-range order except for the ring-like domains of the antibody molecules themselves. Inspection of individual aggregates at higher magnification (figure 1C-E) revealed that they consisted of numerous smaller globules, suggesting that they may have arisen from assemblies of smaller nanoparticles.
Figure 1.
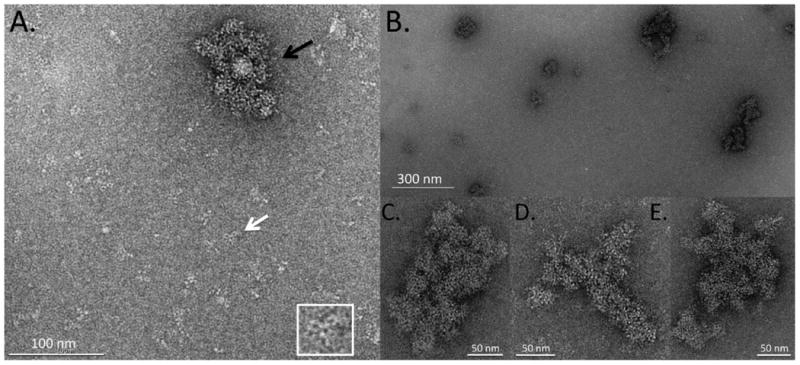
TEM images of negatively stained, stressed IVIg. (A) Representative image showing sub-micron IVIg aggregate (black arrow) surrounded by small oligomeric antibody complexes and monomeric IVIg molecules. A view of the monomeric IVIg molecule identified by the white arrow is shown at increased scale in the lower right corner. This antibody is oriented in a planar fashion on the grid, giving it a Y-shaped appearance. The three arms of the molecule are visible in the form of small rings. (B) Field of view at lower magnification showing distribution and size range of IVIg aggregates. (C to E) Individual IVIg aggregates at high magnification. The aggregates are compact and appear to be formed from smaller globules.
Because negative staining produces images under non-physiological conditions we did not characterize the aggregates in terms of size or size distribution by this method. For more quantitative characterization the sample was prepared by vitrification for cryo-electron microscopy (cryoEM). In cryoEM, the specimen is plunge-frozen in liquid ethane and imaged in the frozen-hydrated state. This method preserves the native structure of macromolecules including large protein assemblies but has the disadvantage of fairly low contrast.32 Thus, individual antibody molecules are not easily observed.
Aggregates in the frozen-hydrated state appeared as extended, branched protein networks with large surface areas (figure 2A and B). As already observed in the negatively stained sample, they were amorphous and highly irregular in shape. Also consistent with prior observations was their apparent make-up of smaller fragments, which were loosely packed into larger, highly porous structures. Numerous aggregates in the submicron range were detectable in addition to a wide range of larger structures, some of which extended over several micrometers (figure 2C). These larger structures had a mottled appearance indicating varying degrees of thickness, possibly as a result of the aggregates folding upon themselves.
Figure 2.
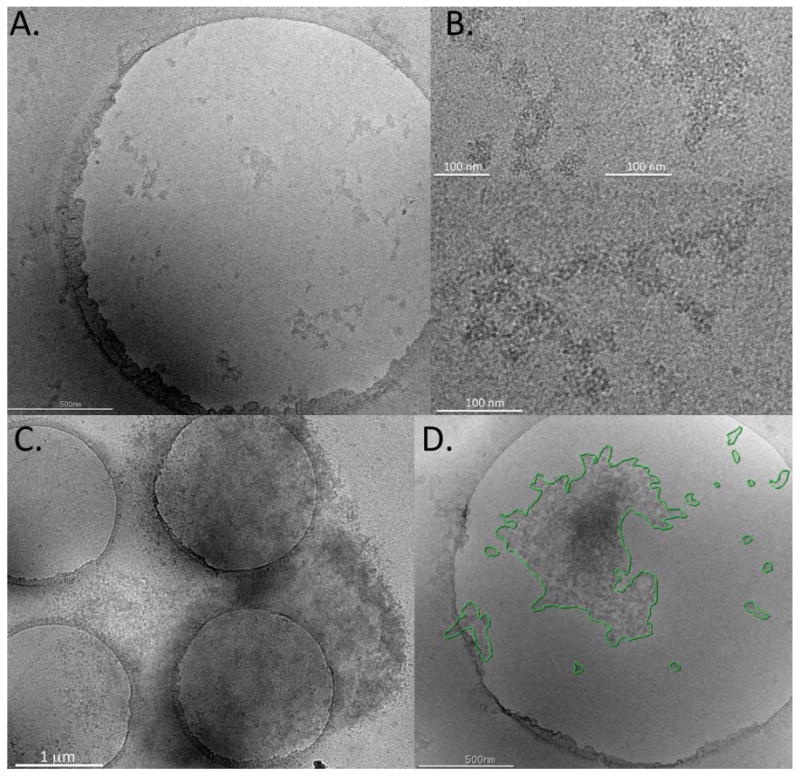
Images of stressed IVIg produced by cryo-electron microscopy. (A) Representative image showing sub-micron IVIg aggregates in the frozen-hydrated state. The sample is suspended in a thin layer of vitreous ice formed over holes (d=2 μm) in a carbon support film. (B) Higher magnification of sub-micron aggregates shown in (A). (C) Representative image of a larger aggregate extending over several holes and the carbon support film. (D) For semi-quantitative analysis, contours were traced manually around individual aggregates and automatically converted into area equivalent diameter (AED).
Taken together, the two different TEM methods provided valuable and unique information on IVIg aggregate characteristics. Negative staining revealed more detail on the microstructure of the component proteins, whereas cryo-electron microscopy was a more reliable indicator of aggregate shape and dimension.
Semi-quantitative assessment of aggregate size
Given the native structure of antibody aggregates in the frozen-hydrated state and availability of hundreds to thousands of aggregate images in a wide range of sizes, we developed a semi-automated particle-measuring tool to further explore distribution of aggregate sizes. To this end, an aggregate-contouring program was developed in which a human operator traces the outline of an aggregate and a computer algorithm calculates a variety of metrics including perimeter, area, maximum and minimum Feret diameter, circularity and area equivalent diameter (AED), i.e. the diameter of a circle with equivalent area to the tracing of the aggregate,.
The procedure was initially tested in a proof of concept experiment that employed 100 nm latex beads. The AED metric was used to monitor the size distribution and reproducibility of the method using images of the beads taken at 21,000× and 6,500× magnifications. Size distribution of the latex beads was highly reproducible (p ≫ 0.05) for three independent samplings with a mean particle diameter of 95.4 ± 6.9 nm (data not shown). This result was close to the mean particle diameter of 99 ± 19 nm determined by nanoparticle tracking analysis of the same sample (data not shown). As expected, the circularity of the beads was close to 1.0 at 0.957± 0.003.
The method was then applied to antibody aggregates imaged for the 1 mg/ml solution in the frozen-hydrated state. Aggregate selection was based on three criteria: (1) aggregates had to be fully contained in the field of view, (2) they had to be at least partially contained in the holes and (3) particle boundaries had to be clearly defined. An example of aggregates meeting these criteria is shown in figure 2D. In keeping with criterion (1), the stressed IVIg sample was imaged at three different magnifications (6,500×, 21,000× and 52,000×) to capture a wide range of aggregate sizes. As shown in figure 3, the aggregates formed three clusters with mean particle AED values of 64.3 nm (52,000×), 96.1 nm (21,000×) and 386 nm (6,500) (figure 3A-C). More particles were counted at the lowest magnification, but this was largely a consequence of the fact that the grid area sampled at 6,500× was more than 90 times larger than the area sampled at 52,000× and 16 times larger than that at 21,000x. Assuming that particle numbers scale linearly with area screened, the total number of particles at 21,000× and 52,000× would be ∼5,000 and ∼18,000, respectively, relative to ∼500 measured at 6,500. Although these numbers are likely to be imprecise, it is reasonable to conclude that particles in the lower nanometer range were significantly more abundant than those measuring several hundred nanometers in diameter.
Figure 3.
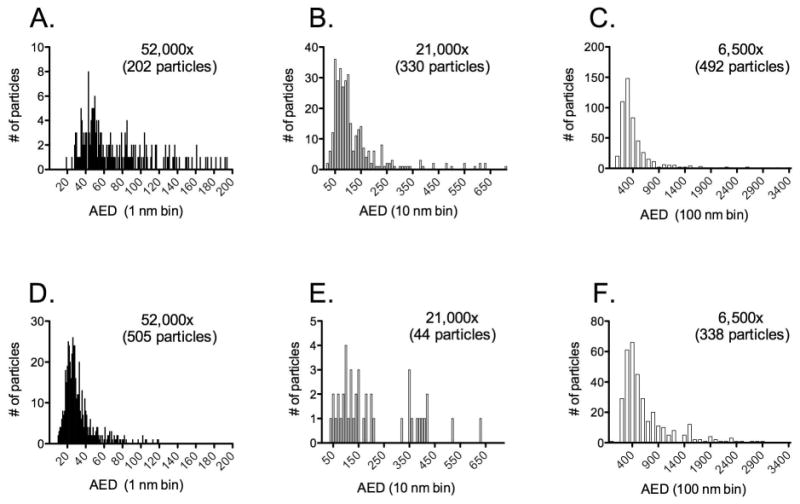
Size distribution of particles formed in the absence (panels A-C) or presence (panels D-F) of 0.01 % (w/v) Tween 20. Particles were imaged in the frozen-hydrated state and sized at three different magnifications: 6,500×, 21,000× and 52,000×. The number of particles measured at each magnification is indicated in the respective panel. Mean particle AED values in the absence of Tween 20 were of 64.3 nm (52,000×), 96.1 nm (21,000×) and 386 nm (6,500). The mean AED values in the presence of Tween 20 were 84.3 nm (52,000×), 131 nm (21,000×) and 502 nm (6,500×).
To further quantify morphological characteristics of the particles, they were binned more coarsely by size and additional shape parameters were determined for the various groups (Table 1). The resulting data provide useful information on the overall form of the particles but do not reflect their complex morphological features as observed by TEM.
Table 1. Morphological characteristics of IVIg particles.
| Magnification | Size bin(nm) | AEDa) (Mean ± STDc)) |
Particle perimeter/AED (Mean ± STD) |
Aspect ratiob) (Mean ± STD) |
|||
|---|---|---|---|---|---|---|---|
| -T20d) | +T20e) | -T20 | +T20 | -T20 | +T20 | ||
| 52,000× | 1 - 20 | 19.3 ± 0.0f) | 16.7 ± 2.6 | 3.75 ± 0.0f) | 3.38 ± 0.74 | 0.47 ± 0.0f) | 0.68 ± 0.17 |
| 20 - 40 | 34.1 ± 3.9 | 28.0 ± 5.3 | 3.51 ± 0.61 | 3.52 ± 1.05 | 0.63 ± 0.16 | 0.64 ± 0.22 | |
| 40 - 60 | 49.3 ± 5.1 | 42.2 ± 5.4 | 3.61 ± 0.62 | 4.34 ± 0.89 | 0.62 ± 0.14 | 0.57 ± 0.14 | |
| 60 - 80 | 70.1 ± 5.8 | 68.5 ± 5.5 | 4.04 ± 0.79 | 4.45 ± 0.97 | 0.55 ± 0.14 | 0.60 ± 0.14 | |
| 80 - 100 | 89.1 ± 5.5 | 85.8 ± 6.2 | 4.13 ± 0.54 | 4.34 ± 0.84 | 0.59 ± 0.11 | 0.56 ± 0.13 | |
| 100 - 150 | 121.5 ± 14.1 | 117.7 ± 17.2 | 4.57 ± 1.02 | 5.01 ± 1.37 | 0.60 ± 0.17 | 0.57 ± 0.15 | |
| 150 - 200 | 171.5 ± 14.9 | 165.9 ± 4.5 | 5.16 ± 1.06 | 5.25 ± 0.89 | 0.58 ± 0.13 | 0.58 ± 0.10 | |
| 21,000× | 1 - 50 | 39.8 ± 6.4 | 49.9 ± 0.0f) | 3.36 ± 0.78 | 3.92 ± 0.0f) | 0.69 ± 0.18 | 0.60 ± 0.0f) |
| 50 - 100 | 74.0 ± 14.2 | 74.8 ± 15.4 | 3.72 ± 1.17 | 3.88 ± 1.04 | 0.58 ± 0.20 | 0.58 ± 0.17 | |
| 100 - 150 | 120.4 ± 16.1 | 120.2 ± 15.2 | 4.13 ± 1.04 | 4.77 ± 1.52 | 0.60 ± 0.18 | 0.50 ± 0.17 | |
| 150 - 200 | 167.3 ± 12.7 | 167.4 ± 15.8 | 4.82 ± 0.94 | 5.14 ± 1.71 | 0.53 ± 0.12 | 0.72 ± 0.18 | |
| 200 - 250 | 223.9 ± 15.2 | 209.2 ± 6.8 | 5.31 ± 0.88 | 5.54 ± 1.96 | 0.56 ± 0.13 | 0.48 ± 0.08 | |
| 250 - 450 | 325.2 ± 60.7 | 381.4 ± 33.3 | 5.71 ± 2.04 | 5.49 ± 0.81 | 0.55 ± 0.19 | 0.55 ± 0.13 | |
| 450 - 650 | 567.2 ± 59.1 | 581.4 ± 73.0 | 5.49 ± 1.62 | 6.35 ± 1.83 | 0.62 ± 0.18 | 0.64 ± 0.12 | |
| 6,500× | 1 - 400 | 305.0 ± 58.6 | 321.1 ± 53.9 | 3.88 ± 1.29 | 3.65 ± 0.96 | 0.59 ± 0.21 | 0.61 ± 0.19 |
| 400 - 800 | 530.4 ± 100.1 | 544.8 ± 101.8 | 4.62 ± 1.60 | 3.92 ± 1.31 | 0.57 ± 0.21 | 0.62 ± 0.22 | |
| 800 - 1200 | 963.2 ± 138.1 | 946.1 ± 109.4 | 5.55 ± 1.64 | 4.57 ± 1.17 | 0.54 ± 0.19 | 0.59 ± 0.17 | |
| 1200 - 1600 | 1383.2 ± 129.8 | 1417.2 ± 138.3 | 5.03 ± 1.54 | 5.34 ± 1.22 | 0.62 ± 0.16 | 0.60 ± 0.13 | |
| 1600 - 2000 | 1802.4 ± 106.8 | 1849.5 ± 121.0 | 5.95 ± 1.70 | 5.46 ± 1.32 | 0.50 ± 0.08 | 0.59 ± 0.15 | |
| 2000 - 2700 | 2399.9 ± 221.0 | 2295.3 ± 180.6 | 5.05 ± 2.77 | 7.15 ± 1.95 | 0.68 ± 0.27 | 0.65 ± 0.14 | |
| 2700 - 3400 | 3165.0 ± 111.3 | 2851.7 ± 113.4 | 3.88 ± 0.14 | 6.45 ± 1.11 | 0.64 ± 0.01 | 0.59 ± 0.08 | |
AED, area equivalent diameter
Ratio of minimum to maximum Feret diameter
STD, standard deviation
-T20, Tween 20 absent during agitation and freeze/thaw stress
+T20, Tween 20 present during agitation and freeze/thaw stress
Standard deviation is zero because only one particle fell into this size bin.
Analysis of IVIg aggregates by alternate technologies
To place information obtained by TEM in the context of data obtained with more commonly employed technologies, the stressed IVIg sample was also analyzed by SEC, DLS, NTA and MFI. SEC indicated that 10% of the protein was lost to insoluble aggregates (data not shown) and of the remaining soluble fraction 10% were present in form of higher molecular weight species (figure 4A), presumably representing dimers and nanometer-sized soluble aggregates, which were also readily detected by TEM (figure 1A). DLS covered a wider size range and revealed the presence of nanometer to micrometer-sized particles (figure 4B). Specifically, three clusters were detected ranging from 6-20 nm, 40-100 nm and 500-1050 nm in size. While this was consistent with TEM observations, it is important to keep in mind that the DLS analysis software tries to fit a broad, polydisperse sample to a series of approximately monodisperse sizes. Therefore, the presence of several distinct peaks has to be interpreted with caution. This issue reinforces the value of TEM as a tool that directly visualizes the size distribution of particles in a given sample.
Figure 4.
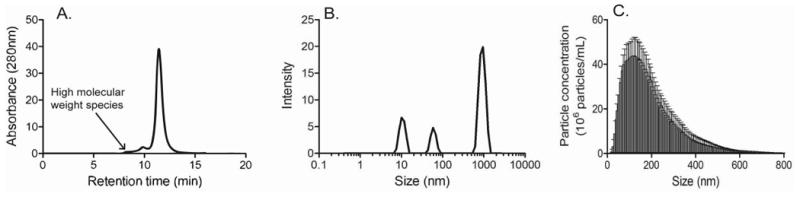
Characterization of IVIg aggregates by (A) Size exclusion chromatography, (B) Dynamic Light Scattering and (C) Nanoparticle Tracking Analysis. Five independent measurements were performed on each instrument. A representative result is shown for SEC and DLS in panels (A) and (B), respectively. Error bars in panel (C) indicate standard deviation of the mean.
NTA identified a broad particle size range from 30 nm, the apparent lower size detection limit of the instrument, to more than 600 nm with the greatest number of particles in the 50-230 nm range (figure 4C), in agreement with TEM data (figure 3A and B). This method also provided particle concentrations, which were on the order of 106 -107/ml for the various size bins.
Finally, MFI provided data on the concentration and appearance of particles in the micron range. The vast majority of the particles fell into the 1-2 micrometer size groups with significantly fewer particles detected above that range (figure 5A and B). Images of the particles captured by the instrument allowed inspection of their morphology and comparison with the attributes identified by TEM. As observed previously, the particles were highly heterogeneous and had an extended shape with large surface area (figure 5C). They appeared sheet-like and occasionally folded upon themselves, presumably as a result of being formed by rupture of a film of protein that arose at interfaces during agitation and freeze-thawing. Overall, these observations were consistent with those made by TEM, although TEM provided significantly more information on the microstructure of the aggregates owing to the much higher resolution and level of detail.
Figure 5.
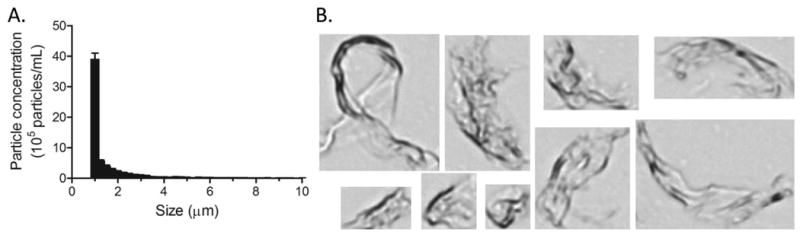
Micro Flow Imaging analysis of IVIg aggregates. (A) Particle concentration as a function of size for five independent measurements of the sample. Error bars indicate standard deviation of the mean. (B) Representative images of IVIg particles illustrating differences in shape and size. Size range is from 20-100 μm.
Analysis of IVIg aggregation in the presence of Tween 20
We next evaluated the effect of the nonionic surfactant Tween 20 on the formation of IVIg aggregates. Nonionic surfactants are often added to pharmaceutical protein formulations to minimize aggregation based on a variety of mechanisms by which these compounds confer protein stability. In the presence of 0.01 % (w/v) Tween 20, the IVIg sample had to be agitated for 10 days, compared to 2 days in its absence, to induce measurable amounts of aggregates. TEM analysis of negatively stained samples indicated that the morphology of the aggregates was largely unchanged (figure 6A). When viewed by cryoEM, however, some of the larger aggregates showed a more open, fenestrated structure that had not been as apparent in the absence of Tween 20 (figure 6B). Qualitatively, in the presence of the surfactant, the stressed sample appeared to contain a higher number of smaller-sized particles and this was confirmed by semi-quantitative sizing analysis (figure 3D-F). The mean AED values at the three magnifications were 84.3nm (52,000×), 131nm (21,000×) and 502nm (6,500×), similar to previous results. However, there was a clear shift towards smaller sized particles, which were approximately 12 times more abundant than in the absence of Tween 20.
Figure 6.
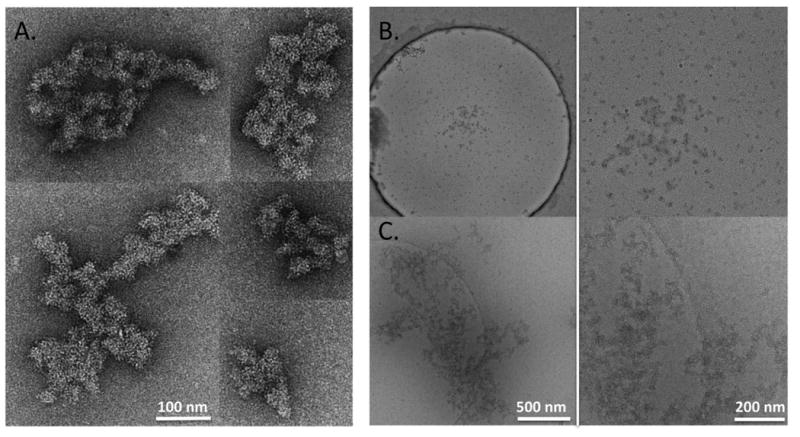
(A) Gallery of negatively stained IVIg aggregates formed in the presence of 0.01 % (w/v) Tween 20. (B and C) Cryo-electron microscopy of IVIg aggregates formed in the presence of 0.01 % (w/v) Tween 20. (B) shows abundance of small particles detected in the presence of the surfactant; a representative large aggregate in the same sample is shown in (C). The right panels of (B) and (C) represent higher magnification views of the central portion of the images shown to the left.
Analysis of the sample by SEC, DLS, NTA and MFI generally confirmed the trend towards formation of smaller particles in the presence of the surfactant, but there were some inconsistencies in the details between the data generated by these orthogonal technologies and those obtained by TEM. For example, SEC analysis suggested absence of soluble aggregates in the presence of Tween 20 (figure 7A) even though TEM analysis revealed the presence of a large number of particles in the low nanometer range (figure 3D). These soluble aggregates were readily detected by DLS, but DLS did not identify the larger aggregates also present in the sample (figure 7B). This was surprising as DLS results are usually biased towards the presence of large particles. NTA (figure 7C) and MFI (figure 8) were consistent with TEM analysis in that both technologies detected fewer particles in their respective size ranges. NTA did not register the spike in particles in the 10-30 nm range detected by TEM. This discrepancy was due to the apparent lower size detection limit of the NTA instrument, which was ca. 30 nm with the samples used in the current study. Surprisingly, MFI data showed a broader distribution of particle sizes than what had been observed in the absence of Tween 20 (figure 8). In addition, images of the aggregates showed the presence of long, thin fibers with considerable branching (figure 8), in contrast to their more globular appearance in the absence of the surfactant (figure 5).
Figure 7.
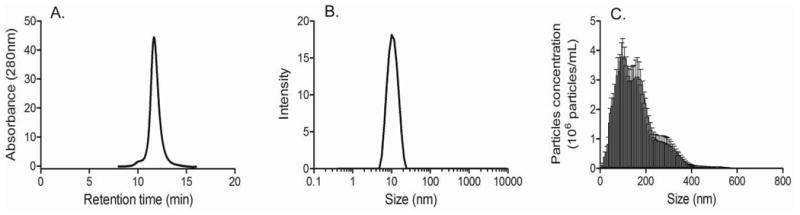
Characterization of IVIg aggregates formed in the presence of 0.01 % (w/v) Tween 20 by (A) Size exclusion chromatography, (B) Dynamic Light Scattering and (C) Nanoparticle Tracking Analysis. Five independent measurements were performed on each instrument. A representative result is shown for SEC and DLS in panels (A) and (B), respectively. Error bars in panel (C) indicate standard deviation of the mean.
Figure 8.
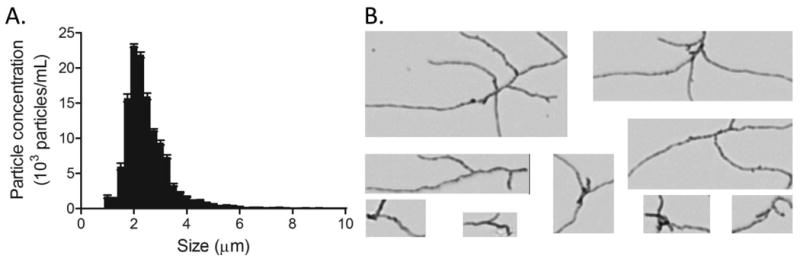
Micro Flow Imaging analysis of IVIg aggregates formed in the presence of 0.01 % (w/v) Tween 20. (A) Particle concentration as a function of size for five independent measurements of the sample. Error bars indicate standard deviation of the mean. (B) Representative images of IVIg particles illustrating differences in shape and size. Size range is from 20-100 μm.
To determine whether the difference in aggregate morphology was a result of the presence of Tween 20 or, alternatively, the longer agitation time, IVIg was agitated for 10 days in the absence of Tween 20. Analysis of the sample by MFI revealed mostly globular particles and only a few elongated, fibrillar structures (data not shown). Therefore, agitating an IVIg sample in the presence of Tween 20 for long enough to obtain reasonable levels of particles appears to result in more fibrillar particles than in the absence of the surfactant. The mechanism underlying this effect is currently unknown. It is also unclear why the fibrillar particles were not detected by TEM.
In summary, although the formation of large aggregates in the presence of Tween20 was significantly reduced, particles in the nanometer size range still formed, albeit much more slowly. These small particles can still present a significant problem with protein drug products.
Conclusions
Our results show that TEM can serve as an effective orthogonal method to characterize protein aggregates. In contrast to other currently employed technologies, TEM is capable of visualizing macromolecular specimen in a wide size range, from individual nanometer-sized molecules to large protein assemblies with dimensions of several micrometers. More importantly, TEM provides detailed information on the microstructure of the aggregates and can offer insights into the conformational integrity of the component proteins.
Our automated data collection methods provided a large enough number of images to justify semi-quantitative analysis of aggregate size and distribution. Tools developed toward that end will have to be improved as the current workflow continues to rely on manual selection of images to be analyzed and contouring of the aggregates. Automation of these two steps will further increase throughput and reduce potential bias. Despite these current limitations, semi-quantitative sizing and distribution results were in general agreement with those obtained by currently accepted technologies,
TEM revealed that aggregates were highly irregular in shape and porous in nature suggesting that their water content is substantial. These results were not unique to the IVIg aggregates investigated here but have been our general experience with antibodies stressed by freezing and thawing. Such attributes must be taken into account when converting particle concentrations into protein mass. A standard procedure is to determine the volume of a sphere from a given particle diameter and multiplying it with the density of a typical protein. Thus, the resulting protein mass estimates are likely to be associated with large errors and require correction by algorithms that have yet to be developed. Development of standards that better reflect the properties of protein aggregates for calibration of analytical instruments might be a more realistic solution to address this problem. TEM provides valuable information on aggregate characteristics to that end.
Acknowledgments
This work was supported by the National Institutes of Health grant NCATS 9R44TR000182.
References
- 1.Carpenter JF, Randolph TW, Jiskoot W, Crommelin DJ, Middaugh CR, Winter G, Fan YX, Kirshner S, Verthelyi D, Kozlowski S, Clouse KA, Swann PG, Rosenberg A, Cherney B. Overlooking subvisible particles in therapeutic protein products: gaps that may compromise product quality. J Pharm Sci. 2009;98(4):1201–1205. doi: 10.1002/jps.21530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang W. Protein aggregation and its inhibition in biopharmaceutics. Int J Pharm. 2005;289(1-2):1–30. doi: 10.1016/j.ijpharm.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 3.Cromwell ME, Hilario E, Jacobson F. Protein aggregation and bioprocessing. Aaps J. 2006;8(3):E572–579. doi: 10.1208/aapsj080366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mahler HC, Friess W, Grauschopf U, Kiese S. Protein aggregation: pathways, induction factors and analysis. J Pharm Sci. 2009;98(9):2909–2934. doi: 10.1002/jps.21566. [DOI] [PubMed] [Google Scholar]
- 5.Carpenter JF, Kendrick BS, Chang BS, Manning MC, Randolph TW. Inhibition of stress-induced aggregation of protein therapeutics. Methods Enzymol. 1999;309:236–255. doi: 10.1016/s0076-6879(99)09018-7. [DOI] [PubMed] [Google Scholar]
- 6.Narhi LO, Schmit J, Bechtold-Peters K, Sharma D. Classification of protein aggregates. J Pharm Sci. 2012;101(2):493–498. doi: 10.1002/jps.22790. [DOI] [PubMed] [Google Scholar]
- 7.Singh SK, Afonina N, Awwad M, Bechtold-Peters K, Blue JT, Chou D, Cromwell M, Krause HJ, Mahler HC, Meyer BK, Narhi L, Nesta DP, Spitznagel T. An industry perspective on the monitoring of subvisible particles as a quality attribute for protein therapeutics. J Pharm Sci. 2010;99(8):3302–3321. doi: 10.1002/jps.22097. [DOI] [PubMed] [Google Scholar]
- 8.Fradkin AH, Carpenter JF, Randolph TW. Immunogenicity of aggregates of recombinant human growth hormone in mouse models. J Pharm Sci. 2009;98(9):3247–3264. doi: 10.1002/jps.21834. [DOI] [PubMed] [Google Scholar]
- 9.Hermeling S, Crommelin DJ, Schellekens H, Jiskoot W. Structure-immunogenicity relationships of therapeutic proteins. Pharm Res. 2004;21(6):897–903. doi: 10.1023/b:pham.0000029275.41323.a6. [DOI] [PubMed] [Google Scholar]
- 10.Rosenberg AS. Effects of protein aggregates: an immunologic perspective. Aaps J. 2006;8(3):E501–507. doi: 10.1208/aapsj080359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schellekens H. Bioequivalence and the immunogenicity of biopharmaceuticals. Nat Rev Drug Discov. 2002;1(6):457–462. doi: 10.1038/nrd818. [DOI] [PubMed] [Google Scholar]
- 12.Schellekens H. Immunogenicity of therapeutic proteins. Nephrol Dial Transplant. 2003;18(7):1257–1259. doi: 10.1093/ndt/gfg164. [DOI] [PubMed] [Google Scholar]
- 13.den Engelsman J, Garidel P, Smulders R, Koll H, Smith B, Bassarab S, Seidl A, Hainzl O, Jiskoot W. Strategies for the assessment of protein aggregates in pharmaceutical biotech product development. Pharm Res. 2011;28(4):920–933. doi: 10.1007/s11095-010-0297-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Narhi LO, Jiang Y, Cao S, Benedek K, Shnek D. A critical review of analytical methods for subvisible and visible particles. Curr Pharm Biotechnol. 2009;10(4):373–381. doi: 10.2174/138920109788488905. [DOI] [PubMed] [Google Scholar]
- 15.Philo JS. A critical review of methods for size characterization of non-particulate protein aggregates. Curr Pharm Biotechnol. 2009;10(4):359–372. doi: 10.2174/138920109788488815. [DOI] [PubMed] [Google Scholar]
- 16.Ripple DC, Dimitrova MN. Protein particles: what we know and what we do not know. J Pharm Sci. 2012;101(10):3568–3579. doi: 10.1002/jps.23242. [DOI] [PubMed] [Google Scholar]
- 17.Carpenter JF, Randolph TW, Jiskoot W, Crommelin DJ, Middaugh CR, Winter G. Potential inaccurate quantitation and sizing of protein aggregates by size exclusion chromatography: essential need to use orthogonal methods to assure the quality of therapeutic protein products. J Pharm Sci. 2010;99(5):2200–2208. doi: 10.1002/jps.21989. [DOI] [PubMed] [Google Scholar]
- 18.Loyd R, Snyder JJK, Dolan JW. Introduction to modern chromatography. Hoboken NJ: John Wiley & Sons; 2010. [Google Scholar]
- 19.Correia I, Sung J, Burton R, Jakob CG, Carragher B, Ghayur T, Radziejewski C. The structure of dual-variable-domain immunoglobulin molecules alone and bound to antigen. MAbs. 2013;5(3):364–372. doi: 10.4161/mabs.24258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mulder AM, Carragher B, Towne V, Meng Y, Wang Y, Dieter L, Potter CS, Washabaugh MW, Sitrin RD, Zhao Q. Toolbox for non-intrusive structural and functional analysis of recombinant VLP based vaccines: a case study with hepatitis B vaccine. PLoS One. 2012;7(4):e33235. doi: 10.1371/journal.pone.0033235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao Q, Potter CS, Carragher B, Lander G, Sworen J, Towne V, Abraham D, Duncan P, Washabaugh MW, Sitrin RD. Characterization of virus-like particles in GARDASIL(R) by cryo transmission electron microscopy. Hum Vaccin Immunother. 2013;10(3) doi: 10.4161/hv.27316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lyumkis D, Moeller A, Cheng A, Herold A, Hou E, Irving C, Jacovetty EL, Lau PW, Mulder AM, Pulokas J, Quispe JD, Voss NR, Potter CS, Carragher B. Automation in single-particle electron microscopy connecting the pieces. Methods Enzymol. 2010;483:291–338. doi: 10.1016/S0076-6879(10)83015-0. [DOI] [PubMed] [Google Scholar]
- 23.Carragher B, Fellmann D, Guerra F, Milligan RA, Mouche F, Pulokas J, Sheehan B, Quispe J, Suloway C, Zhu Y, Potter CS. Rapid routine structure determination of macromolecular assemblies using electron microscopy: current progress and further challenges. J Synchrotron Radiat. 2004;11(Pt 1):83–85. doi: 10.1107/s0909049503023987. [DOI] [PubMed] [Google Scholar]
- 24.Cheng A, Leung A, Fellmann D, Quispe J, Suloway C, Pulokas J, Abeyrathne PD, Lam JS, Carragher B, Potter CS. Towards automated screening of two-dimensional crystals. J Struct Biol. 2007;160(3):324–331. doi: 10.1016/j.jsb.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Potter CS, Chu H, Frey B, Green C, Kisseberth N, Madden TJ, Miller KL, Nahrstedt K, Pulokas J, Reilein A, Tcheng D, Weber D, Carragher B. Leginon: a system for fully automated acquisition of 1000 electron micrographs a day. Ultramicroscopy. 1999;77(3-4):153–161. doi: 10.1016/s0304-3991(99)00043-1. [DOI] [PubMed] [Google Scholar]
- 26.Potter CS, Pulokas J, Smith P, Suloway C, Carragher B. Robotic grid loading system for a transmission electron microscope. J Struct Biol. 2004;146(3):431–440. doi: 10.1016/j.jsb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 27.Stagg SM, Lander GC, Pulokas J, Fellmann D, Cheng A, Quispe JD, Mallick SP, Avila RM, Carragher B, Potter CS. Automated cryoEM data acquisition and analysis of 284742 particles of GroEL. J Struct Biol. 2006;155(3):470–481. doi: 10.1016/j.jsb.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 28.Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B. Automated molecular microscopy: the new Leginon system. J Struct Biol. 2005;151(1):41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 29.Suloway C, Shi J, Cheng A, Pulokas J, Carragher B, Potter CS, Zheng SQ, Agard DA, Jensen GJ. Fully automated, sequential tilt-series acquisition with Leginon. J Struct Biol. 2009;167(1):11–18. doi: 10.1016/j.jsb.2009.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lander GC, Stagg SM, Voss NR, Cheng A, Fellmann D, Pulokas J, Yoshioka C, Irving C, Mulder A, Lau PW, Lyumkis D, Potter CS, Carragher B. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J Struct Biol. 2009;166(1):95–102. doi: 10.1016/j.jsb.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Carlo S, Harris JR. Negative staining and cryo-negative staining of macromolecules and viruses for TEM. Micron. 2011;42(2):117–131. doi: 10.1016/j.micron.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milne JL, Borgnia MJ, Bartesaghi A, Tran EE, Earl LA, Schauder DM, Lengyel J, Pierson J, Patwardhan A, Subramaniam S. Cryo-electron microscopy--a primer for the non-microscopist. Febs J. 2013;280(1):28–45. doi: 10.1111/febs.12078. [DOI] [PMC free article] [PubMed] [Google Scholar]