

Abstract
Background:
Preclinical and clinical data suggest that cannabidiol (CBD), a major non-psychotomimetic compound from Cannabis sativa, induces antipsychotic-like effects. However, the antipsychotic properties of repeated CBD treatment have been poorly investigated. Behavioral changes induced by repeated treatment with glutamate N-methyl-D-aspartate receptor (NMDAR) antagonists have been proposed as an animal model of schizophrenia-like signs. In the present study, we evaluated if repeated treatment with CBD would attenuate the behavioral and molecular modifications induced by chronic administration of one of these antagonists, MK-801.
Methods:
Male C57BL/6J mice received daily i.p. injections of MK-801 (0.1, 0.5, or 1mg/kg) for 14, 21, or 28 days. Twenty-four hours after the last injection, animals were submitted to the prepulse inhibition (PPI) test. After that, we investigated if repeated treatment with CBD (15, 30, and 60mg/kg) would attenuate the PPI impairment induced by chronic treatment with MK-801 (1mg/kg; 28 days). CBD treatment began on the 6th day after the start of MK-801 administration and continued until the end of the treatment. Immediately after the PPI, the mice brains were removed and processed to evaluate the molecular changes. We measured changes in FosB/ΔFosB and parvalbumin (PV) expression, a marker of neuronal activity and a calcium-binding protein expressed in a subclass of GABAergic interneurons, respectively. Changes in mRNA expression of the NMDAR GluN1 subunit gene (GRN1) were also evaluated. CBD effects were compared to those induced by the atypical antipsychotic clozapine.
Results:
MK-801 administration at the dose of 1mg/kg for 28 days impaired PPI responses. Chronic treatment with CBD (30 and 60mg/kg) attenuated PPI impairment. MK-801 treatment increased FosB/ΔFosB expression and decreased PV expression in the medial prefrontal cortex. A decreased mRNA level of GRN1 in the hippocampus was also observed. All the molecular changes were attenuated by CBD. CBD by itself did not induce any effect. Moreover, CBD effects were similar to those induced by repeated clozapine treatment.
Conclusions:
These results indicate that repeated treatment with CBD, similar to clozapine, reverses the psychotomimetic-like effects and attenuates molecular changes observed after chronic administration of an NMDAR antagonist. These data support the view that CBD may have antipsychotic properties.
Keywords: antipsychotic, cannabidiol, cannabinoid, clozapine, NMDA receptor hypofunction, schizophrenia
Introduction
Cannabidiol (CBD), a major compound from Cannabis sativa, is devoid of the typical psychoactive effects induced by ∆9-tetrahydrocannabinol (THC). In fact, CBD attenuates the psychotomimetic effects induced by high doses of THC in humans (Zuardi et al., 1982; Bhattacharyya et al., 2010). These findings led to the hypothesis that CBD could have antipsychotic properties. Accordingly, several pre-clinical studies have indicated that CBD induces antipsychotic-like effects (for review see Campos et al., 2012). These effects have also been described in open-label clinical studies (Zuardi et al., 1995, 2006) and recently confirmed in a controlled, randomized, double-blind clinical trial (Leweke et al., 2012). Nevertheless, the mechanism of action of CBD is poorly understood.
Animal models based on acute and chronic administration of N-methyl-D-aspartate-receptor (NMDAR) antagonists, such as phencyclidine (PCP), ketamine, and MK-801, are widely used to investigate the neurobiology of schizophrenia and the effects induced by compounds with antipsychotic properties (Javitt and Zukin, 1991; Rujescu et al., 2006; Bubenikova-Valesova et al., 2008). Although the pathophysiology of schizophrenia is quite complex, involving several neurotransmitters, evidence indicates the participation of the glutamatergic system. For example, administration of NMDAR antagonists in rodents and humans can evoke positive and negative symptoms, as well as cognitive deficits that resemble those seen in schizophrenia patients, supporting the proposal that this treatment could be a valid animal model of this disorder (Krystal et al., 1994, 2005; Lisman et al., 2008). Moreover, in laboratory animals, the effects induced by chronic administration of these antagonists, unlike those seen after acute injection, are proposed to more closely resemble the behavioral, neurochemical, and neuroanatomical changes observed in schizophrenia patients (Jentsch and Roth, 1999). Among these changes, a decrease in parvalbumin (PV) expression, a calcium-binding protein expressed in a subclass of fast-spiking GABAergic interneurons centrally involved in information processing in the brain, has been observed after chronic treatment with NMDAR antagonists in rodents (Abdul-Monim et al., 2007). This finding has been consistently observed in the post-mortem brains of schizophrenia patients (Lewis et al., 2005) and has been suggested to be a consequence of an NMDAR signaling hypofunction (Gonzalez-Burgos and Lewis, 2012).
The prepulse inhibition (PPI) test is a behavioral paradigm used to assess sensorimotor gating mechanisms. In a PPI test, a weaker non-startling stimulus (prepulse) presented at a short interval before a startling acoustic stimulus (pulse) reduces the processing of, and attenuates the response to, the latter stimulus (Braff and Geyer, 1990; Geyer et al., 2001). It was originally proposed by Graham (1975) that the prepulse triggers sensory-neuronal processing routines that need to be protected against disruption, leading to inhibition of the subsequent response to the startling stimulus. A disruption in this inhibitory processes is frequently observed in schizophrenia patients (Braff, Geyer, Light, et al., 2001), and in other psychiatric disorders as well (Braff, Geyer, and Swerdlow, 2001). Thus, although PPI deficit, even if experimentally induced, does not constitute an animal model of schizophrenia per se, it is a valid model to investigate sensorimotor gating disruption similar to that seen in schizophrenia patients, with face, predictive, and construct validity (Swerdlow et al., 2000; Geyer et al., 2001).
Although there are acute studies indicating positive effects of CBD in NMDAR-based animal models of schizophrenia (Long et al., 2006; Gururajan et al., 2011, 2012), at the moment no study has investigated the effects of this drug in models based on repeated administration of NMDAR antagonists. Therefore, in the present study we investigated whether repeated treatment with CBD would attenuate behavioral changes in the PPI test induced by chronic administration of the NMDAR antagonist MK-801. We also measured changes in PV expression in brain structures related to the neurobiology of schizophrenia, such as the medial prefrontal cortex (mPFC), dorsal striatum (dSTR), nucleus accumbens (NAc) core and shell, and dorsal hippocampus (dentate gyrus; DG, CA1, and CA3). Since a decrease in PV expression has been related to hypofunction of NMDAR-mediated neurotransmission, we evaluated the mRNA expression of the NMDAR GluN1 subunit gene (GRN1). The GluN1 subunit is an obligatory subunit of NMDAR and, although changes in its expression have been associated with schizophrenia (Weickert et al., 2013), few studies have analyzed the effects of chronic blockade of these receptors on mRNA expression of this gene. Finally, as the brain sites of CBD antipsychotic-like effects are unknown, we decided to evaluate changes in FosB/ΔFosB protein expression, a marker of neuronal activity. FosB/ΔFosB accumulates and persists in a region-specific manner in the brain for relatively long periods due to its stability in response to different kinds of chronic stimuli, including repeated administration of antipsychotics (Atkins et al., 1999).
Material and Methods
Animals
The experiments were performed using male C57BL/6J mice that were 6 weeks of age at the beginning of the treatment. Animals were housed in groups of four per cage (41 x 33 x 17cm) in a temperature-controlled room (243±1°C) under standard laboratory conditions with free access to food and water and a 12h light/dark cycle (lights on at 6:00 AM). Procedures were conducted in conformity with the Brazilian Society of Neuroscience and Behavior guidelines for the care and use of laboratory animals, which are in compliance with international laws and politics. The institution’s Animal Ethics Committee approved the housing conditions and experimental procedures (process number: 165/2010).
Drugs
The following drugs were used: CBD (THC Pharm), clozapine (Tocris), and MK-801 (Sigma-Aldrich). CBD was diluted in 2% Tween 80 in saline, while clozapine was diluted in saline supplemented with 30 μl of 0.1M hydrochloric acid; the pH was adjusted to a value close to neutrality when necessary. MK-801 was diluted in saline. The drugs were injected intraperitoneally (i.p.) in a 10mL/kg volume.
Experimental Design
Animals received daily i.p. injections of saline or MK-801 (0.1, 0.5, or 1mg/kg) for 14, 21, or 28 days (n = 6–8/group). Twenty-four hours after the last injection, the animals were submitted to the PPI test. Based on the results of this experiment, we investigated whether repeated treatment with CBD (15, 30, and 60mg/kg) or clozapine (1mg/kg) would attenuate the PPI impairment induced by chronic treatment with MK-801 (1mg/kg) for 28 days (Figure 1). CBD or clozapine treatment began on the 6th day after the start of MK-801 administration and continued until the end of the treatment (n = 14/group). One day later, animals were submitted to the PPI. CBD and clozapine were administered 30min before MK-801 or saline. Immediately after the PPI test, animals were euthanized and their brains processed to assess changes in FosB/ΔFosB and PV protein expression (n = 7/group) or NMDA-receptor GluN1 subunit gene mRNA expression (n = 5–7/group). The doses and the experimental design were based on previous studies (Long et al., 2006; Vigano et al., 2009; Casarotto et al., 2010; Elhardt et al., 2010; Guidali et al., 2011). We also tested the effects of a single CBD or clozapine administration in the last (28th) day of treatment with MK-801 in the PPI to test whether repeated CBD or clozapine administration would be required for the observed drug effects (n = 7/group; Supplementary Figure 1).
Figure 1.

Experimental design. The animals received daily i.p. injections of saline (SAL) or MK-801 (1mg/kg) for 28 days. CBD (15, 30, and 60mg/kg) or vehicle (VEH) treatment began on the 6th day after the start of MK-801 administration and continued until the end of the treatment. One day later, animals were submitted to the prepulse inhibition (PPI) test. Immediately after the PPI, their brains were removed and processed to assess immunohistochemical changes in FosB/ΔFosB and parvalbumin (PV) expression and changes in mRNA expression of the NMDAR GluN1 subunit gene by RT-PCR. CBD effects were compared to those induced by the atypical antipsychotic clozapine (CLZ; 1mg/kg).
Mice showed a normal increase in body weight (measured every day) that was independent of treatment (Supplementary Figure 2).
Procedure
Pre-pulse Inhibition Test
The PPI test was performed according to a protocol previously described by our group (Issy et al., 2009). Briefly, it was conducted simultaneously in two identical startle response systems (Med Associates). A continuous acoustic signal provided a background white noise level of 65±1 dB. The pulse (pulse alone) was a burst of white noise of 105 dB with a rise/decay of 5ms and duration of 20ms, and the prepulse intensities were set to 80, 85, and 90 dB of pure tone, 7 KHz frequency, and duration of 10ms. The cages were calibrated before each test to ensure equal sensitivity of both response platforms throughout the procedure. The platform calibration was done by adjusting the gain on the load cell amplifier to 150 arbitrary units at a standard weight appropriated for mice (40g). The limits of the load cell were −2047 to +2047 arbitrary units. After a 5min acclimatization period in which mice received no stimuli except the background noise, they were presented with a series of 10 stimuli (pulse alone). The first 10 pulse-alone trials allowed the within-session habituation to the startle stimulus and were not considered for statistical analysis of the percentage of PPI. The PPI test consisted of 64 trials pseudorandomly divided into eight different categories presented with an inter-stimulus interval of 30 s: pulse alone (105 dB); prepulse alone (80, 85, or 90 dB); prepulse + pulse with 100ms interval between prepulse and pulse; and null, where no stimulus was presented. Prepulse stimulus did not elicit an acoustic startle response. Mean acoustic startle response to pulse-alone (P) trials and each prepulse + pulse (PP + P) trial was calculated for each subject. These data were used in the statistical analysis to assess drug-induced changes in startle amplitude and in PPI. The level of PPI was determined by expressing the PP + P startle amplitude as a percentage decrease from P startle amplitude, according to the following formula: %PPI = 100–[100 × (PP + P/P)]. This transformation reduces statistical variability attributable to differences between animals and is a direct measure of the PPI level.
Immunohistochemical Detection of FosB/ΔFosB and PV
Some animals, immediately after the exposure to the PPI, were deeply anesthetized with a lethal dose of urethane (25%, 5mL/kg; i.p.) and transcardially perfused with phosphate buffered saline (PBS) followed by 4% paraformaldehyde in 0.1M phosphate buffer. Then, the brains were removed and post-fixed in 4% paraformaldehyde for 2h. After that they were immersed in 30% sucrose in 0.1M phosphate buffer for cryoprotection over 48h. The brains were frozen at −40ºC on dry ice and isopentane and 25 µm-thick serial coronal sections were obtained using a cryostat microtome at -20ºC (CM-1900, Leica). The free-floating sections were rinsed (3 times, 5min each) in PBS 0.1M + 0.15% Triton-X (pH 7.4; washing buffer) and then pre-incubated for 30min with 1% hydrogen peroxide in PBS to remove endogenous peroxidase activity. To avoid unspecific activity, free-floating sections were also incubated in a solution containing 5% bovine serum albumin, washing buffer, and 5% normal goat (FosB/ΔFosB) or horse (PV) serum for 1h. Sections were incubated overnight at 21ºC with polyclonal rabbit anti-FosB/ΔFosB antibodies (1:1000, H-75, sc-7203, Santa Cruz Biotechnology) or monoclonal mouse anti-PV antibodies (1:1000, P227, Sigma-RBI). Subsequent to the primary antibody incubation, sections were successively washed (washing buffer) and incubated for 1h in a secondary antibody solution (PBS) containing biotinylated goat anti-rabbit or horse anti-mouse antibodies (1:400; Vector Laboratories). The sections were then incubated with the avidin-biotin immunoperoxidase complex for 2h (1:300, Vectastain ABC kit, Vector Lab). Afterwards, FosB/∆FosB and PV-like immunoreactivity were revealed by the addition of chromogen diaminobenzidine (DAB; Sigma-Aldrich) into acetate buffer, H2O2 0.02%, and nickel ammonium sulfate 1% or DAB into Tris-buffered saline and H2O2 0.02%, respectively. All the reactions were performed at the temperature of 21ºC.
Slides immunostaining for PV or FosB/∆FosB were observed under phase contrast microscopy (Olympus BX50, Olympus). The microscope has an attached camera (Olympus DP72, Olympus) that captured the images for analysis with the help of the Image Pro-Plus 6.0 software (Mediacybernetics). The follow brain structures were evaluated: mPFC, dSTR, NAc core, and shell and dorsal hippocampus (DG, CA1, and CA3).
Quantification of PV or FosB/∆FosB-positive cells was made with the 10X or 20X objective, respectively. An observer blinded to group assignment performed the analysis. For each brain structure, 3–4 tissue sections from each animal were analyzed. All stained cells in the whole area of each brain region of interest were recorded (FosB/∆FosB-mPFC: approximately 0.068mm2; dSTR and NAc core and shell: 0.076mm2; DG: 0.062mm2; CA1: 0.020mm2; CA3: 0.024mm2; PV-mPFC: approximately 0.27mm2; dSTR and NAc core and shell: 0.30mm2; DG: 0.062mm2; CA1: 0.020mm2; CA3: 0.024mm2). Immunoreactive cells were counted using ImageJ software (Research Services Branch, National Institutes of Health). Those cells with purple or brown impregnations with areas between 10 and 80 µm2 were considered FosB/ΔFosB- and PV-positive cells, respectively. As the brain structures evaluated are bilateral; the mean value was calculated. The results were expressed as the number of positive cells/0.1mm2. Neuroanatomical sites were identified with the help of the Franklin and Paxinos mouse brain atlas (Franklin and Paxinos, 2008). The anterior–posterior localization from bregma of the analyzed regions was: mPFC, 1.98–1.70mm; dSTR and NAc core and shell, 1.54–0.98mm; and dorsal hippocampus, -1.82 to -2.30mm.
Real-Time Quantitative Polymerase Chain Reaction (RT-PCR)
Animals were deeply anesthetized with a lethal dose of urethane (25%, 5mL/kg; i.p.) and decapitated immediately after the exposure to the PPI. After decapitation, the frontal cortex, striatum, and hippocampus were collected by microdissection in RNAse-free conditions and stored at -80ºC until RNA isolation. Total RNA was extracted using Trizol (Invitrogen), and cDNA reaction was performed using 1 µg of total RNA in a high-capacity cDNA kit (Applied Biosystem) according to the manufacturer’s instructions. The relative level of mRNA expression of the NMDAR GluN1 subunit gene (GRN1) was evaluated in the StepOne real-time PCR system, using Applied Biosystems real-time master mix with Taqman® gene expression probes (Assay ID Mm01212171_s1). Total RNA was normalized based on Ct values for the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) housekeeping gene (Assay ID Mm99999915_g1). All reactions were duplicated, and fold change was calculated using the 2−ΔΔCt method. Data are shown as a relative percentage of mRNA expression to the control group.
Statistical Analysis
The PPI results were analyzed by mixed-design analysis of variance (ANOVA) with treatment as the main independent factor and prepulse intensity (80, 85, and 90 dB) as the repeated factor. The immunohistochemical data were analyzed by two-way ANOVA using the first (vehicle, CBD, or clozapine) and the second (MK-801 or saline) treatment as main factors. In cases of significant interactions between factors, specific one-way ANOVAs were performed. The Student-Newman-Keuls (S-K-N) test was used for post hoc analysis. mRNA expression results failed to reach homogeneity of variances and were analyzed by Kuskal-Wallis followed by the Dunn test. All data are represented as the mean ± the standard error of mean (SEM). Results of statistical tests with p < 0.05 were considered significant. Since there was no difference in PPI and immunohistochemical results between animals that received Tween80 2% in saline (used to dissolve CBD) + saline or saline supplemented with 30 μl of 0.1M hydrochloric acid (used to dissolve clozapine) + saline, they were joined together in a control group (vehicle + saline).
Results
Effects of NMDAR Antagonist MK-801 for 14, 21 or 28 days on PPI
There was a significant treatment effect only after 28 days of repeated drug administration, where animals treated with MK-801 1mg/kg had a greater PPI impairment compared to all other groups (F3,25 = 5.36, p = 0.005; S-N-K., p < 0.05; Figure 2). There was also a significant effect of intensity at all measurements, with louder prepulse stimuli causing greater PPI (F2,50 = 40.39–40.9, p < 0.001; Figure 2), but there was no significant interaction between treatment and prepulse intensity (F6,50 = 1.55, p > 0.05). MK-801 treatment for 14, 21, or 28 days did not modify the acoustic startle response to the pulse-only trials, which would be indicative of a motor-impairing effect (Supplementary Table 1).
Figure 2.

Mice received daily i.p. injections of saline or MK-801 (0.1, 0.5, or 1mg/kg) for 14, 21, or 28 days. Twenty-four hours after the last injection, the animals were submitted to the PPI test. MK-801 (1mg/kg) disrupted PPI only after 28 days of treatment (n = 6–8/group). The data are presented as the mean ± SEM. *A general treatment effect: p < 0.05 vs. all other groups using a mixed-design ANOVA followed by S-N-K.
CBD and Clozapine Effects on PPI Impairment Induced by MK-801
Both CBD (30 and 60mg/kg) and clozapine attenuated the PPI disruption induced by treatment with MK-801 for 28 days (Figure 3). Mixed-design ANOVA indicated significant effects of prepulse intensity (F2,208= 103.4, p < 0.001) and treatment (F7,104 = 4.6, p < 0.001). There was also an interaction between prepulse intensity and treatment (F14,208 = 2.35, p = 0.005). One-way ANOVA analyses conducted at each prepulse intensity showed significant effects at 85 dB (F7,104 = 5.75, p < 0.001) and 80 dB (F7,104 = 4.09, p = 0.001). At 85 dB animals treated with vehicle + MK-801 showed a significant impairment of PPI compared to control (vehicle + saline), an effect not prevented by clozapine or CBD (S-N-K, p < 0.05). At 80 dB, however, PPI impairment induced by MK-801 was attenuated by clozapine and CBD (30mg/kg). Moreover, animals treated with CBD (60mg/kg) + MK-801 presented a significantly lower PPI impairment compared to those receiving vehicle + MK-801 (S-N-K, p < 0.05).
Figure 3.
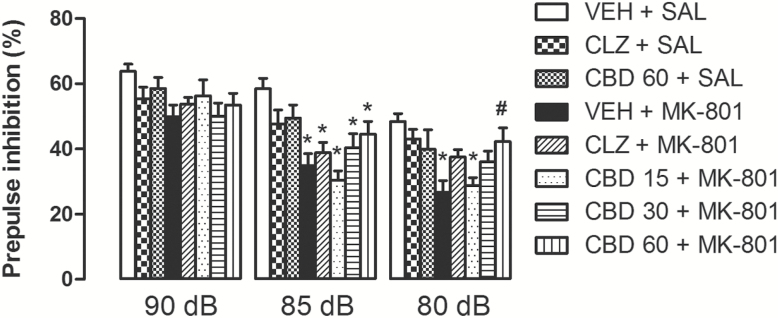
CBD (30 and 60mg/kg) attenuated the PPI impairment induced by repeated treatment with MK-801 (1mg/kg) for 28 days. Similar to CBD, clozapine (CLZ; 1mg/kg) attenuated the MK-801-induced PPI disruption (n = 14/group). The data are presented as the mean ± SEM. *p < 0.05 vs. VEH + SAL group, #p < 0.05 vs. VEH + MK-801 group; mixed-design ANOVA followed by S-N-K.
The treatments did not modify the acoustic startle response to the pulse-only trials (Supplementary Table 2).
We also observed that CBD or clozapine administration given once on the last day of MK-801 treatment did not attenuate the chronic MK-801-induced PPI impairment (Supplementary Figure 3), indicating that CBD and clozapine effects seem to depend on the repeated treatment and are not due to the last injection of these drugs.
Changes in FosB/ΔFosB Expression in Specific Brain Regions
Quantification of FosB/ΔFosB-positive cells in the mPFC revealed significant effects of the first (vehicle, clozapine, or CBD; F2,36 = 4.00, p = 0.02) and second treatments (saline or MK-801; F1,36 = 4.84, p = 0.034) and an interaction between them (F2,36 = 4.39, p = 0.02; Figure 4A and B). Post hoc analysis showed that animals treated with vehicle + MK-801 had a significantly higher number of FosB/ΔFosB-positive cells compared to all other groups (S-N-K, p < 0.05). Neither CBD (60mg/kg) nor clozapine affected FosB/ΔFosB expression in the mPFC per se (p > 0.05).
Figure 4.

Effects of chronic MK-801 (1mg/kg), clozapine (CLZ; 1mg/kg), and CBD (60mg/kg) treatment on FosB/ΔFosB protein expression in the mice mPFC (A and B) and NAc core (C and D). MK-801 induced a significant increase in the number of FosB/ΔFosB-positive cells in the mPFC (A) and NAc core (C). CBD and clozapine blocked FosB/ΔFosB increase in the mPFC, but did not modify FosB/ΔFosB increase in the NAc core. Clozapine also induced an increase in the number of FosB/ΔFosB-positive cells in the NAc core (C). The data are presented as the mean ± SEM (n = 7/group). *p < 0.05 vs. VEH + SAL group; two-way ANOVA followed by S-K-N test. Photomicrographs of FosB/ΔFosB-like immunoreactivity (20X; Bar = 100 μm) in the mPFC (B) and NAc core (D).
In the NAc core, there were also significant effects of the first (vehicle, clozapine, or CBD; F2,36 = 5.11, p = 0.01) and second treatments (saline or MK-801; F1,36 = 14.23, p = 0.001) and an interaction between them (F2,36 = 3.93, p = 0.03; Figure 4C and D). It was observed an increase in the number of FosB/ΔFosB-positive cells in the NAc core of vehicle + MK-801–treated mice compared to control (vehicle + saline group, S-N-K, p < 0.05). Different from the mPFC, clozapine and CBD were unable to attenuate the MK-801-induced changes in the NAc core (S-N-K, p < 0.05). Clozapine per se also induced an increase in the number of FosB/ΔFosB-positive cells (S-N-K, p < 0.05), but CBD did not induce any change by itself (S-N-K, p > 0.05).
No change in FosB/ΔFosB expression was observed in the dSTR, NAc shell, DG, CA1, or CA3 (Supplementary Figure 4).
Changes in PV Expression in Specific Brain Regions
MK-801 induced a decrease in PV expression in the mPFC. This change was attenuated by concomitant treatment with CBD or clozapine. There was a tendency for a significant effect of the first treatment (F2,36 = 3.06, p = 0.060), a significant effect of the second treatment (F1,36 = 10.41, p = 0.003), and an interaction between the treatments (F2,36 = 4.76, p = 0.015; Figure 5). Animals treated with vehicle + MK-801 presented a decrease in the number of PV-positive cells compared to control (vehicle + saline; S-N-K, p < 0.05), a change attenuated by CBD and clozapine. Neither CBD nor clozapine affected PV expression in the mPFC (S-N-K, p > 0.05).
Figure 5.

Effects of chronic MK-801 (1mg/kg), clozapine (CLZ; 1mg/kg), and CBD (60mg/kg) treatment on PV protein expression in mouse mPFCs (n = 7/group). (A) MK-801 induced a decrease in the number of PV-positive cells in the mPFC. CBD and clozapine attenuated PV decrease in the mPFC. The data are presented as the mean ± SEM. *p < 0.05 vs. VEH + SAL group, #p < 0.05 vs. VEH + MK-801 group; two-way ANOVA followed by S-K-N test. (B) Photomicrographs of PV-like immunoreactivity in the mPFC (10X; Bar = 200 μm). CC: corpus callosum; mPFC: medial prefrontal cortex.
No change in the PV expression was observed in the dSTR, DG, CA1, or CA3 (Supplementary Figure 5). Moreover, PV immunostaining was poorly detected in the NAc, indicating that PV may be expressed at levels below the limit of detection within the NAc core and shell. Indeed, low levels of PV-positive cells in the NAc were observed by Todtenkopf et al. (2004) in rats.
Changes in mRNA Expression of GRN1 in Specific Brain Regions
Animals treated with vehicle + MK-801 presented a significantly lower mRNA expression in the hippocampus compared to controls (vehicle + saline; χ2 = 16.4, d.f. = 5, p = 0.006; Dunn test, p < 0.05; Figure 6). These effects were attenuated by CBD and clozapine. The treatments did not modify GluR1 mRNA expression in the frontal cortex and striatum (p > 0.05; Figure 6).
Figure 6.

Effects of chronic MK-801 (1mg/kg), clozapine (CLZ; 1mg/kg), and CBD (60mg/kg) treatment on mRNA expression of the NMDAR GluN1 subunit gene (GRN1) in the (A) frontal cortex, (B) striatum, and (C) hippocampus. The data are presented as the mean ± SEM (n = 5–7/group). *p < 0.05 vs. VEH + SAL group; Kuskal-Wallis followed by the Dunn test.
Discussion
Corroborating the proposal that repeated administration of NMDAR antagonists could be a useful animal model of schizophrenia, the results of the present study show that repeated administration of the NMDAR antagonist MK-801 for 28 days impaired PPI performed 24h after the end of the treatment. No effect was observed after 14 or 21 days of MK-801 treatment. These results are consistent with those indicating a lack of PPI impairment after repeated treatment with the NMDAR antagonist PCP for 14 days (Martinez et al., 1999) and those showing cognitive deficits after 28 days of chronic intermittent PCP treatment (Vigano et al., 2009; Guidali et al., 2011). Besides the PPI disruption, chronic MK-801 treatment also decreased the number of PV-positive cells in the mPFC and the mRNA levels of the NMDAR GluN1 subunit gene (GRN1) in the hippocampus. These molecular changes are proposed to be similar to those observed in schizophrenia patients (Lewis et al., 2005; Weickert et al., 2013). In addition, MK-801 treatment also increased the number of FosB/ΔFosB-positive cells in the mPFC and NAc core. Both the behavioral and molecular changes induced by MK-801 treatment were attenuated by repeated treatment with CBD or the atypical antipsychotic clozapine.
The effects of clozapine and CBD on improving the PPI deficits were seen only at the 80 dB prepulse intensity. This is not an uncommon finding, since drug effects may vary across prepulse intensities in the PPI session. Significant effects of the prepulse intensity are usually seen (Blumenthal, 1996; Swerdlow et al., 2003, 2007; Yee et al., 2005; Larrauri and Schmajuk, 2006; Issy et al., 2009). The biological significance of this sound-parameter dependence is not completely understood, but the novelty or the context associated with the preceding signal (prepulse) appears to be a critical factor (Blumenthal and Levey, 1989; Lane et al., 1991).
The present results are consistent with previous studies showing a beneficial effect of CBD on PPI deficits in schizophrenia-like models that employ acute drug administration (Long et al., 2006; Levin et al., 2014). They also indicate, however, that repeated treatment with CBD is necessary to counteract the PPI deficit induced by chronic MK-801. Moreover, contrary to the study by Long et al. (2012) showing tolerance after chronic CBD treatment for the PPI deficits presented by transgenic mice (Nrg1 TM HET), our results suggest that CBD repeated treatment does not induce tolerance.
Typical antipsychotics, such as haloperidol, seem to be unable to reverse behavioral changes induced by NMDAR antagonists in rodents. Atypical antipsychotics such as clozapine, olanzapine, and quetiapine, on the other hand, are effective in attenuating those changes (Geyer et al., 2001). Accordingly, CBD attenuation of PPI impairment induced by repeated MK-801 administration was similar to that produced by clozapine. Indeed, previous studies from our group suggest that CBD exhibits a profile similar to atypical antipsychotic drugs. For example, it does not induce catalepsy (Zuardi et al., 1991; Moreira and Guimaraes, 2005), a characteristic extrapyramidal motor effect induced by typical antipsychotics. Moreover, similar to clozapine, a single CBD administration in rats induced c-Fos protein expression, a marker of neuronal activation, in the NAc and mPFC, but not in the striatum (Guimaraes et al., 2004). Despite these results, no study had investigated the pattern of neuronal activation induced by repeated treatment with CBD. Moreover, even if NMDAR antagonists alter cell activity in several structures involved in the neurobiology of schizophrenia and modulation of sensorimotor gating, such as the PFC, hippocampus, and NAc (Duncan et al., 1998), the brain regions involved in the effects induced by repeated administration of these drugs are not completely known. Therefore, we decided to investigate FosB/ΔFosB protein expression in the aforementioned brain structures. Whereas c-Fos and most of the other Fos-related proteins are highly unstable and therefore rapidly disappear after the stimulus, FosB and its truncated splice variant ΔFosB accumulate in specific brain regions after repeated exposure to several stimuli (Nestler et al., 1999). Once induced, these proteins are stable and persist in the brain for weeks after the original stimulus (Nestler et al., 1999). Thus, levels of FosB/ΔFosB may vary regionally (depending on neuronal activity in normal animals) and/or be markedly influenced by drug treatment or neurological disorders.
We observed an increase in FosB/ΔFosB expression in the mPFC and NAc core of animals repeatedly treated with MK-801. Concomitant treatment with CBD or clozapine attenuated the changes induced by MK-801 in the mPFC, but not in the NAc core. However, even if CBD exhibits a pre-clinical and clinical profile similar to clozapine from a phenotypic point of view, the mechanisms involved could be rather distinct. This could help to explain some differences observed in our study. For example, while CBD did not change FosB/ΔFosB expression per se, clozapine increased this expression in the NAc core. This latter effect partially corroborated previous findings in the literature (Rodriguez et al., 2001; Grande et al., 2004). Contrary to other studies (Kontkanen et al., 2002), however, clozapine failed to increase FosB/ΔFosB expression in the mPFC and NAc shell. The reasons for these differences are unknown but could depend on different species, treatment schedules, and doses.
The mechanisms involved in the increase of FosB/ΔFosB expression in the mPFC induced by MK-801 are not completely understood. However, increased glutamate levels after long-term treatment with NMDAR antagonists have been described in this brain region (Moghaddam et al., 1997; Wang et al., 2000). It may depend on the blockade of a tonic inhibitory influence of GABAergic interneurons over excitatory projections to the mPFC (Moghaddam et al., 1997; Krystal et al., 2003; Jodo et al., 2005), leading to a disinhibition of glutamate release from neurons targeted by those interneurons (Olney and Farber, 1995; Adams and Moghaddam, 1998). NMDAR blockade, therefore, may cause overall cortical excitation by disinhibiting glutamatergic pyramidal neurons. Indeed, neuroimaging data suggest that the schizophrenia-like symptoms caused by NMDAR antagonists are associated with frontal cortical activation (Lahti et al., 1995; Breier et al., 1997).
Increased FosB/ΔFosB expression has also been associated with neuroplastic/adaptive changes induced by chronic stimuli that promote synaptic remodeling, including repeated administration of antipsychotics, stress stimuli, and reward stimuli (Atkins et al., 1999; Nestler et al., 1999; Lobo et al., 2013). Moreover, viral-mediated overexpression of ΔFosB in mouse PFC induced cognitive deficits (Dietz et al., 2014). These results suggest that changes in FosB/ΔFosB expression, in addition to indicating cellular activation after repeated stimuli, could also have functional consequences. Additional studies are clearly needed to address this question.
Another potentially important finding of the present study was the significant reduction in PV immunoreactivity induced by repeated MK-801 treatment in the mPFC, an effect attenuated by CBD. A more robust attenuation was induced by clozapine. Decreased GABAergic signaling is among the most consistent postmortem pathological changes observed in schizophrenia (Reynolds et al., 2002; Lewis et al., 2005). These deficits are largely restricted to GABAergic PV-positive interneurons (Lewis et al., 2005). It has been suggested that the PV interneuron dysfunction in schizophrenia could result from a hypofunction of NMDAR-mediated signaling (Belforte et al., 2010; Moreau and Kullmann, 2013). These neurons synapse on the cell body or the axon initial segment of glutamatergic neurons, where they regulate pyramidal cell output. Furthermore, it is likely that a reduction in PV interneuron functionality results not only in a decreased inhibitory control over pyramidal cell activity, but also a reduction in the coordinated activity of large brain networks leading, for example, to a loss of the dopaminergic neurotransmission regulation (Schwartz et al., 2012). Changes in PV expression in the mPFC induced by MK-801, therefore, could contribute to increased neuronal activity in the mPFC and NAc core, reflected by the increase in the number of FosB/ΔFosB-positive cells. Importantly, a decrease in PV immunoreactivity appears to be associated with reduced expression of PV protein, but not with a reduced number of PV interneurons (Hashimoto et al., 2003).
A decrease in the number of PV-positive cells is also consistently observed in a diverse variety of animal models of schizophrenia (Penschuck et al., 2006; Harte et al., 2007), including repeated treatment with other NMDAR antagonists (Abdul-Monim et al., 2007; Amitai et al., 2012). Repeated ketamine treatment decreases PV levels in GABAergic interneurons in vitro and in the mouse PFC (Behrens et al., 2007). mRNA levels of PV gene were also reduced in the PFCs of mice (Cochran et al., 2003) and monkeys (Morrow et al., 2007) after repeated PCP administration. Similar to our findings, Amitai et al. (2012) observed that repeated treatment with clozapine prevented the cognitive deficits and reduced PV expression in the PFC following repeated PCP treatment. Based on these pieces of evidence, it is possible to speculate that the attenuation of PV changes and MK-801-induced PPI impairment by CBD and clozapine could be related. Moreover, the decrease in PV-positive cells was restricted to the mPFC. Since this region is thought to modulate, rather than mediate, PPI (Swerdlow et al., 2001), this result suggests that CBD effects depend on changes in modulatory PPI processes.
Even if changes in mRNA expression are difficult to interpret in the absence of information on the levels of the corresponding protein, our results suggest that chronic blockade of NMDAR decreases mRNA expression of the NMDAR obligatory GluN1 subunit. Changes in mRNA expression of the GluN1 subunit gene have already been reported in the post mortem brains of schizophrenia patients (Gao et al., 2000; Law and Deakin, 2001; Weickert et al., 2013). Few studies, however, have investigated the effects of repeated treatment with NMDAR antagonists on these changes. In our study, repeated MK-801 treatment decreased GluN1 mRNA expression in the hippocampus, but not in the frontal cortex and striatum, an effect that was also attenuated by CBD and clozapine. Even if contradictory results exist in the literature (Rujescu et al., 2006), reduction in mRNA expression of this subunit in the hippocampus of schizophrenia patients has been reported (Gao et al., 2000; Law and Deakin, 2001). In addition, a study using a single photon emission tomography ligand for NMDAR revealed that medication-free schizophrenia patients had lower NMDAR binding in the left hippocampus, a change not observed in clozapine-treated patients (Pilowsky et al., 2006).
In conclusion, the present results indicate specific behavioral and molecular changes induced by chronic administration of the NMDAR antagonist MK-801. Repeated CBD treatment, similar to the atypical antipsychotic clozapine, reversed or attenuated MK-801-induced behavioral psychotomimetic-like effects and the associated molecular changes. These results are consistent with the proposal that CBD may have antipsychotic properties.
Supplementary Material
For supplementary material accompanying this paper, visit http://www.ijnp.oxfordjournals.org/
Statement of Interest
The authors declare no conflicts of interest.
Acknowledgments
The authors thank José Carlos de Aguiar and Celia A. da Silva for technical assistance. FVG has a FAPESP fellowship (2010/17343-0). Research was supported by grants from FAPESP and CNPq.
References
- Abdul-Monim Z, Neill JC, Reynolds GP. (2007). Sub-chronic psychotomimetic phencyclidine induces deficits in reversal learning and alterations in parvalbumin-immunoreactive expression in the rat. J Psychopharmacol 21(2):198–205. [DOI] [PubMed] [Google Scholar]
- Adams B, Moghaddam B. (1998). Corticolimbic dopamine neurotransmission is temporally dissociated from the cognitive and locomotor effects of phencyclidine. J Neurosci 18(14):5545–5554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amitai N, Kuczenski R, Behrens MM, Markou A. (2012). Repeated phencyclidine administration alters glutamate release and decreases GABA markers in the prefrontal cortex of rats. Neuropharmacology 62(3):1422–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins JB, Chlan-Fourney J, Nye HE, Hiroi N, Carlezon WA, Jr., Nestler EJ. (1999). Region-specific induction of deltaFosB by repeated administration of typical versus atypical antipsychotic drugs. Synapse 33(2):118–128. [DOI] [PubMed] [Google Scholar]
- Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, Dugan LL. (2007). Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science 318(5856):1645–1647. [DOI] [PubMed] [Google Scholar]
- Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, Quinlan EM, Nakazawa K. (2010). Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci 13(1):76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Morrison PD, Fusar-Poli P, Martin-Santos R, Borgwardt S, Winton-Brown T, Nosarti C, O’Carroll CM, Seal M, Allen P, Mehta MA, Stone JM, Tunstall N, Giampietro V, Kapur S, Murray RM, Zuardi AW, Crippa JA, Atakan Z, McGuire PK. (2010). Opposite effects of delta-9-tetrahydrocannabinol and cannabidiol on human brain function and psychopathology. Neuropsychopharmacology 35(3):764–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal TD. (1996). Inhibition of the human startle response is affected by both prepulse intensity and eliciting stimulus intensity. Biol Psychology 44(2):85–104. [DOI] [PubMed] [Google Scholar]
- Blumenthal TD, Levey BJ. (1989). Prepulse rise time and startle reflex modification: Different effects for discrete and continuous prepulses. Psychophysiology 26(2):158–165. [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA. (1990). Sensorimotor gating and schizophrenia. Human and animal model studies. Arch Gen Psychiatry 47(2):181–188. [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Light GA, Sprock J, Perry W, Cadenhead KS, Swerdlow NR. (2001). Impact of prepulse characteristics on the detection of sensorimotor gating deficits in schizophrenia. Schizophr Res 49(1–2):171–178. [DOI] [PubMed] [Google Scholar]
- Braff DL, Geyer MA, Swerdlow NR. (2001). Human studies of prepulse inhibition of startle: normal subjects, patient groups, and pharmacological studies. Psychopharmacology (Berl) 156(2–3):234–258. [DOI] [PubMed] [Google Scholar]
- Breier A, Malhotra AK, Pinals DA, Weisenfeld NI, Pickar D. (1997). Association of ketamine-induced psychosis with focal activation of the prefrontal cortex in healthy volunteers. Am J Psych 154(6):805–811. [DOI] [PubMed] [Google Scholar]
- Bubenikova-Valesova V, Horacek J, Vrajova M, Hoschl C. (2008). Models of schizophrenia in humans and animals based on inhibition of NMDA receptors. Neurosci Biobehav Rev 32(5):1014–1023. [DOI] [PubMed] [Google Scholar]
- Campos AC, Moreira FA, Gomes FV, Del Bel EA, Guimaraes FS. (2012). Multiple mechanisms involved in the large-spectrum therapeutic potential of cannabidiol in psychiatric disorders. Phil Trans R Soc B 367(1607):3364–3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casarotto PC, Gomes FV, Resstel LB, Guimaraes FS. (2010). Cannabidiol inhibitory effect on marble-burying behaviour: involvement of CB1 receptors. Behav Pharmacol 21(4):353–358. [DOI] [PubMed] [Google Scholar]
- Cochran SM, Kennedy M, McKerchar CE, Steward LJ, Pratt JA, Morris BJ. (2003). Induction of metabolic hypofunction and neurochemical deficits after chronic intermittent exposure to phencyclidine: differential modulation by antipsychotic drugs. Neuropsychopharmacology 28(2):265–275. [DOI] [PubMed] [Google Scholar]
- Dietz DM, Kennedy PJ, Sun H, Maze I, Gancarz AM, Vialou V, Koo JW, Mouzon E, Ghose S, Tamminga CA, Nestler EJ. (2014). DeltaFosB induction in prefrontal cortex by antipsychotic drugs is associated with negative behavioral outcomes. Neuropsychopharmacology 39(3):538–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan GE, Leipzig JN, Mailman RB, Lieberman JA. (1998). Differential effects of clozapine and haloperidol on ketamine-induced brain metabolic activation. Brain Res 812(1–2):65–75. [DOI] [PubMed] [Google Scholar]
- Elhardt M, Martinez L, Tejada-Simon MV. (2010). Neurochemical, behavioral and architectural changes after chronic inactivation of NMDA receptors in mice. Neurosci Lett 468(2):166–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. (2008). The mouse brain in stereotaxic coordinates, 3th ed. New York: Academic Press. [Google Scholar]
- Gao XM, Sakai K, Roberts RC, Conley RR, Dean B, Tamminga CA. (2000). Ionotropic glutamate receptors and expression of N-methyl-D-aspartate receptor subunits in subregions of human hippocampus: effects of schizophrenia. Am J Psych 157(7):1141–1149. [DOI] [PubMed] [Google Scholar]
- Geyer MA, Krebs-Thomson K, Braff DL, Swerdlow NR. (2001). Pharmacological studies of prepulse inhibition models of sensorimotor gating deficits in schizophrenia: a decade in review. Psychopharmacology (Berl) 156(2–3):117–154. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Burgos G, Lewis DA. (2012). NMDA receptor hypofunction, parvalbumin-positive neurons, and cortical gamma oscillations in schizophrenia. Schizophr Bull 38(5):950–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham FK. (1975). The more or less startling effects of weak prestimulation. Psychophysiology 12(3):238–248. [DOI] [PubMed] [Google Scholar]
- Grande C, Zhu H, Martin AB, Lee M, Ortiz O, Hiroi N, Moratalla R. (2004). Chronic treatment with atypical neuroleptics induces striosomal FosB/DeltaFosB expression in rats. Biol Psychiatry 55(5):457–463. [DOI] [PubMed] [Google Scholar]
- Guidali C, Vigano D, Petrosino S, Zamberletti E, Realini N, Binelli G, Rubino T, Di Marzo V, Parolaro D. (2011). Cannabinoid CB1 receptor antagonism prevents neurochemical and behavioural deficits induced by chronic phencyclidine. Int J Neuropsychopharmacology 14(1):17–28. [DOI] [PubMed] [Google Scholar]
- Guimaraes VM, Zuardi AW, Del Bel EA, Guimaraes FS. (2004). Cannabidiol increases Fos expression in the nucleus accumbens but not in the dorsal striatum. Life Sci 75(5):633–638. [DOI] [PubMed] [Google Scholar]
- Gururajan A, Taylor DA, Malone DT. (2011). Effect of cannabidiol in a MK-801-rodent model of aspects of schizophrenia. Behav Brain Res 222(2):299–308. [DOI] [PubMed] [Google Scholar]
- Gururajan A, Taylor DA, Malone DT. (2012). Cannabidiol and clozapine reverse MK-801-induced deficits in social interaction and hyperactivity in Sprague-Dawley rats. J Psychopharmacol 26(10):1317–1332. [DOI] [PubMed] [Google Scholar]
- Harte MK, Powell SB, Swerdlow NR, Geyer MA, Reynolds GP. (2007). Deficits in parvalbumin and calbindin immunoreactive cells in the hippocampus of isolation reared rats. J Neural Transm 114(7):893–898. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Volk DW, Eggan SM, Mirnics K, Pierri JN, Sun Z, Sampson AR, Lewis DA. (2003). Gene expression deficits in a subclass of GABA neurons in the prefrontal cortex of subjects with schizophrenia. J Neurosci 23(15):6315–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issy AC, Salum C, Del Bel EA. (2009). Nitric oxide modulation of methylphenidate-induced disruption of prepulse inhibition in Swiss mice. Behav Brain Res 205(2):475–481. [DOI] [PubMed] [Google Scholar]
- Javitt DC, Zukin SR. (1991). Recent advances in the phencyclidine model of schizophrenia. Am J Psych 148(10):1301–1308. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Roth RH. (1999). The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 20(3):201–225. [DOI] [PubMed] [Google Scholar]
- Jodo E, Suzuki Y, Katayama T, Hoshino KY, Takeuchi S, Niwa S, Kayama Y. (2005). Activation of medial prefrontal cortex by phencyclidine is mediated via a hippocampo-prefrontal pathway. Cereb Cortex 15(5):663–669. [DOI] [PubMed] [Google Scholar]
- Kontkanen O, Lakso M, Wong G, Castren E. (2002). Chronic antipsychotic drug treatment induces long-lasting expression of fos and jun family genes and activator protein 1 complex in the rat prefrontal cortex. Neuropsychopharmacology 27(2):152–162. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD, Heninger GR, Bowers MB, Jr., Charney DS. (1994). Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51(3):199–214. [DOI] [PubMed] [Google Scholar]
- Krystal JH, D’Souza DC, Mathalon D, Perry E, Belger A, Hoffman R. (2003). NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology (Berl) 169(3–4):215–233. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Perry EB, Jr., Gueorguieva R, Belger A, Madonick SH, Abi-Dargham A, Cooper TB, Macdougall L, Abi-Saab W, D’Souza DC. (2005). Comparative and interactive human psychopharmacologic effects of ketamine and amphetamine: implications for glutamatergic and dopaminergic model psychoses and cognitive function. Arch Gen Psychiatry 62(9):985–994. [DOI] [PubMed] [Google Scholar]
- Lahti AC, Holcomb HH, Medoff DR, Tamminga CA. (1995). Ketamine activates psychosis and alters limbic blood flow in schizophrenia. Neuroreport 6(6):869–872. [DOI] [PubMed] [Google Scholar]
- Lane SJ, Ornitz EM, Guthrie D. (1991). Modulatory influence of continuous tone, tone offset, and tone onset on the human acoustic startle response. Psychophysiology 28(5):579–587. [DOI] [PubMed] [Google Scholar]
- Larrauri J, Schmajuk N. (2006). Prepulse inhibition mechanisms and cognitive processes: a review and model. EXS 98:245–278. [DOI] [PubMed] [Google Scholar]
- Law AJ, Deakin JF. (2001). Asymmetrical reductions of hippocampal NMDAR1 glutamate receptor mRNA in the psychoses. Neuroreport 12(13):2971–2974. [DOI] [PubMed] [Google Scholar]
- Levin R, Peres FF, Almeida V, Calzavara MB, Zuardi AW, Hallak JE, Crippa JA, Abílio VC. (2014). Effects of cannabinoid drugs on the deficit of prepulse inhibition of startle in an animal model of schizophrenia: the SHR strain. Front Pharmacol 5:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leweke FM, Piomelli D, Pahlisch F, Muhl D, Gerth CW, Hoyer C, Klosterkotter J, Hellmich M, Koethe D. (2012). Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl Psychiatry 2:e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. (2005). Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci 6(4):312–324. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, Grace AA. (2008). Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci 31(5):234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobo MK, et al. (2013). DeltaFosB Induction in Striatal Medium Spiny Neuron Subtypes in Response to Chronic Pharmacological, Emotional, and Optogenetic Stimuli. J Neurosci 33(47):18381–18395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long LE, Malone DT, Taylor DA. (2006). Cannabidiol reverses MK-801-induced disruption of prepulse inhibition in mice. Neuropsychopharmacology 31(4):795–803. [DOI] [PubMed] [Google Scholar]
- Long LE, Chesworth R, Huang XF, Wong A, Spiro A, McGregor IS, Arnold JC, Karl T. (2012). Distinct neurobehavioural effects of cannabidiol in transmembrane domain neuregulin 1 mutant mice. PLOS ONE 7(4):e34129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez ZA, Ellison GD, Geyer MA, Swerdlow NR. (1999). Effects of sustained phencyclidine exposure on sensorimotor gating of startle in rats. Neuropsychopharmacology 21(1):28–39. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Adams B, Verma A, Daly D. (1997). Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci 17(8):2921–2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau AW, Kullmann DM. (2013). NMDA receptor-dependent function and plasticity in inhibitory circuits. Neuropharmacology 74:23–31. [DOI] [PubMed] [Google Scholar]
- Moreira FA, Guimaraes FS. (2005). Cannabidiol inhibits the hyperlocomotion induced by psychotomimetic drugs in mice. Eur J Pharmacol 512(2–3):199–205. [DOI] [PubMed] [Google Scholar]
- Morrow BA, Elsworth JD, Roth RH. (2007). Repeated phencyclidine in monkeys results in loss of parvalbumin-containing axo-axonic projections in the prefrontal cortex. Psychopharmacology (Berl) 192(2):283–290. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Kelz MB, Chen J. (1999). DeltaFosB: a molecular mediator of long-term neural and behavioral plasticity. Brain Res 835(1):10–17. [DOI] [PubMed] [Google Scholar]
- Olney JW, Farber NB. (1995). Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry 52(12):998–1007. [DOI] [PubMed] [Google Scholar]
- Penschuck S, Flagstad P, Didriksen M, Leist M, Michael-Titus AT. (2006). Decrease in parvalbumin-expressing neurons in the hippocampus and increased phencyclidine-induced locomotor activity in the rat methylazoxymethanol (MAM) model of schizophrenia. Eur J Neurosci 23(1):279–284. [DOI] [PubMed] [Google Scholar]
- Pilowsky LS, Bressan RA, Stone JM, Erlandsson K, Mulligan RS, Krystal JH, Ell PJ. (2006). First in vivo evidence of an NMDA receptor deficit in medication-free schizophrenic patients. Mol Psychiatry 11(2):118–119. [DOI] [PubMed] [Google Scholar]
- Reynolds GP, Beasley CL, Zhang ZJ. (2002). Understanding the neurotransmitter pathology of schizophrenia: selective deficits of subtypes of cortical GABAergic neurons. J Neural Transm 109(5–6):881–889. [DOI] [PubMed] [Google Scholar]
- Rodriguez JJ, Garcia DR, Nakabeppu Y, Pickel VM. (2001). Enhancement of laminar FosB expression in frontal cortex of rats receiving long chronic clozapine administration. Exp Neurol 168(2):392–401. [DOI] [PubMed] [Google Scholar]
- Rujescu D, Bender A, Keck M, Hartmann AM, Ohl F, Raeder H, Giegling I, Genius J, McCarley RW, Moller HJ, Grunze H. (2006). A pharmacological model for psychosis based on N-methyl-D-aspartate receptor hypofunction: molecular, cellular, functional and behavioral abnormalities. Biol Psychiatry 59(8):721–729. [DOI] [PubMed] [Google Scholar]
- Schwartz TL, Sachdeva S, Stahl SM. (2012). Glutamate neurocircuitry: theoretical underpinnings in schizophrenia. Front Pharmacol 3:195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Braff DL, Geyer MA. (2000). Animal models of deficient sensorimotor gating: what we know, what we think we know, and what we hope to know soon. Behav Pharmacol 11(3–4):185–204. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA, Braff DL. (2001). Neural circuit regulation of prepulse inhibition of startle in the rat: current knowledge and future challenges. Psychopharmacology (Berl) 156:194–215. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Stephany N, Wasserman LC, Talledo J, Shoemaker J, Auerbach PP. (2003). Amphetamine effects on prepulse inhibition across-species: replication and parametric extension. Neuropsychopharmacology 28(4):640–650. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Blumenthal TD, Sutherland AN, Weber E, Talledo JA. (2007). Effects of prepulse intensity, duration, and bandwidth on perceived intensity of startling acoustic stimuli. Biol Psychology 74:389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todtenkopf MS, Stellar JR, Williams EA, Zahm DS. (2004). Differential distribution of parvalbumin immunoreactive neurons in the striatum of cocaine sensitized rats. Neuroscience 127(1):35–42. [DOI] [PubMed] [Google Scholar]
- Vigano D, Guidali C, Petrosino S, Realini N, Rubino T, Di Marzo V, Parolaro D. (2009). Involvement of the endocannabinoid system in phencyclidine-induced cognitive deficits modelling schizophrenia. Int J Neuropsychop 12(5):599–614. [DOI] [PubMed] [Google Scholar]
- Wang HD, Takigawa M, Hamada K, Shiratani T, Takenouchi K, Wang G. (2000). Reciprocal information flow between prefrontal cortex and ventral tegmental area in an animal model of schizophrenia. Neuroreport 11(9):2007–2011. [DOI] [PubMed] [Google Scholar]
- Weickert CS, Fung SJ, Catts VS, Schofield PR, Allen KM, Moore LT, Newell KA, Pellen D, Huang XF, Catts SV, Weickert TW. (2013). Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol Psychiatry 18(11):1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee BK, Chang T, Pietropaolo S, Feldon J. (2005). The expression of prepulse inhibition of the acoustic startle reflex as a function of three pulse stimulus intensities, three prepulse stimulus intensities, and three levels of startle responsiveness in C57BL6/J mice. Behav Brain Res 163(2):265–276. [DOI] [PubMed] [Google Scholar]
- Zuardi AW, Shirakawa I, Finkelfarb E, Karniol IG. (1982). Action of cannabidiol on the anxiety and other effects produced by delta 9-THC in normal subjects. Psychopharmacology (Berl) 76(3):245–250. [DOI] [PubMed] [Google Scholar]
- Zuardi AW, Rodrigues JA, Cunha JM. (1991). Effects of cannabidiol in animal models predictive of antipsychotic activity. Psychopharmacology (Berl) 104(2):260–264. [DOI] [PubMed] [Google Scholar]
- Zuardi AW, Morais SL, Guimarães FS, Mechoulam R. (1995). Antipsychotic Effect of Cannabidiol. J Clin Psychiatry 56(10):485–486. [PubMed] [Google Scholar]
- Zuardi AW, Hallak JE, Dursun SM, Morais SL, Sanches RF, Musty RE, Crippa JA. (2006). Cannabidiol monotherapy for treatment-resistant schizophrenia. J Psychopharmacol 20(5):683–686. [DOI] [PubMed] [Google Scholar]