

Abstract
Bile acids have been shown to be important hormones during the feed/fast cycle, allowing the liver to coordinately regulate nutrient metabolism. How they accomplish this has not been fully elucidated. Conjugated bile acids have been shown to activate both the ERK1/2 and AKT signaling pathways via S1PR2 in rodent hepatocytes and in vivo. Here, we report that feeding mice a high fat diet, infusion of taurocholate into the chronic bile fistula rat, or overexpression of the gene encoding S1PR2 in mouse hepatocytes significantly up-regulated hepatic SphK2, but not SphK1. Key genes encoding nuclear receptors/enzymes involved in nutrient metabolism were significantly down-regulated in livers of S1PR2−/− and SphK2−/− mice. In contrast, overexpression of the gene encoding S1PR2 in primary mouse hepatocytes differentially increased SphK2, but not SphK1, and mRNA levels of key genes involved in nutrient metabolism. Nuclear levels of S1P, an endogenous inhibitor of HDAC 1/2, as well as the acetylation of H3K9, H4K5 and H2BK12, were significantly decreased in hepatocytes prepared from S1PR2−/− and SphK2−/− mice. Both S1PR2−/− and SphK2−/− mice rapidly developed fatty livers on a high fat diet suggesting the importance of conjugated bile acids, S1PR2 and SphK2 in regulating hepatic lipid metabolism.
Keywords: Bile acid, Sphingosine Kinase, S1PR2, Hepatic Lipid Metabolism
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is currently one of the most common liver diseases and affects a third of adults and an increasing number of children in the United States (1). It has emerged as a major public health concern due to its progression to non-alcoholic steatohepatitis, liver cirrhosis and liver cancer (2). NAFLD accounted for 47% of chronic liver disease cases from 1988 to 1994; but from 2005 to 2008 its prevalence increased to 75% (3). An increasing body of evidence suggests that the development and disease progression of NAFLD are closely associated with obesity, inflammation and insulin resistance (4–6). Dysregulation of hepatic sterol and lipid metabolism represents an important pathological factor of NAFLD (7).
Bile acids are pivotal to regulating metabolic pathways in the liver. During the last decade, researchers have discovered that bile acids not only act as detergents for solubilization of lipids and fat-soluble vitamins in the intestines, but also serve as important nutrient signaling molecules during the feed/fast cycle. They have been shown to activate specific nuclear receptors [farnesoid X receptor (FXR), pregnane x receptor (PXR), vitamin D receptor], TGR5, a Gαs protein coupled receptor (GPCR), and multiple signaling pathways in the cells of the liver and gastrointestinal tract. Activation of nuclear receptors and cell signaling pathways by bile acids regulates the expression of numerous genes encoding enzymes/proteins involved in the metabolism/synthesis of bile acids, glucose, fatty acids, lipoproteins as well as energy metabolism (8–14). However, we do not have a complete understanding of how bile acid signals are integrated to regulate genes involved in hepatic metabolism.
We have previously reported that sphingosine-1-phosphate (S1P) is an endogenous inhibitor of histone deacetylases (HDAC) 1 and 2 (15). The acetylation/deacetylation of the lysine residues of histones is a critical component of the epigenetic system of gene regulation in mammalian cells. Increased acetylation of histones is correlated with chromatin remodeling and increased transcriptional activity (16). S1P is a lipid mediator involved in the regulation of fundamental cellular responses and is synthesized inside the cell from sphingosine by either sphingosine kinase 1 (SphK1) or sphingosine kinase 2 (SphK2). S1P, a regulatory metabolite, is then transported out of the cell in a regulated manner (17). Extracellular S1P has been shown to activate 5 different G-protein coupled receptors (GPCRs) located on the surface of mammalian cells. The GPCRs activated by S1P have been linked to the activation of various cell signaling pathways, including ERK1/2 and AKT (18, 19). SphK2 is primarily located in the cell nucleus. In previous studies, we reported that S1P and SphK2 enhance histone acetylation (15). Expression of SphK2 in these cells increased the acetylation of histones H3 (H3K9), H4 (H4K5) and H2B (H2BK12) without altering the acetylation of histone H2A. However, the physiological importance of the regulation of specific histone acetylation by S1P in the liver is currently unknown.
Our previous study reported that conjugated bile acids (CBAs) activate the AKT (insulin signaling pathway) and ERK1/2 signaling pathways via Gαi-coupled GPCRs (20). Recently, we reported that the sphingosine-1-phosphate receptor 2 (S1PR2) is activated by CBAs in primary rodent hepatocytes and in vivo (21). Indeed, S1PR2 has been previously reported to have physiological roles in the liver both in vitro and in vivo (22–24). In the current study, we report that activation of S1PR2 by CBAs is essential for the regulation of SphK2 and nuclear S1P levels, which regulate histone acetylation and global gene expression. Finally, S1PR2−/− and SphK2−/− mice rapidly develop fatty livers on a high fat diet (HFD) suggesting the importance of this novel link between S1PR2 and SphK2 in regulating hepatic lipid metabolism.
EXPERIMENTAL PROCEDURES
Animals
Male Sprague Dawley rats, 100 to 150 gms, were purchased from Harlan Laboratories (Frederick, MD). SphK2−/− and S1PR2−/− mice were a gift from Dr. R. Proia (NIDDK), which have been well-characterized and used in a number of studies. All mice were bred in pathogen-free conditions with normal lighting and wild type and knockout mice were from the same litters. All procedures were approved by the VCU IACUC committee that is accredited by AAALAC. The mice were fed with normal diet, HFD (TD.88137, Harlan Teklad) or HFD+0.5% Cholic Acid (HFD+CA) (TD.02028, Harlan Teklad) for two weeks.
Bile fistula (BF) rat model
Biliary fistulas and intraduodenal cannulas were placed in male Sprague-Dawley rats under brief anesthesia as previously described (25). After surgery, they were placed in individual metabolic cages with water and normal chow ad libitum. All animals received continuous infusion of glucose-electrolyte replacement solution. After 48 h of chronic biliary diversion, taurocholate was infused at a rate of 1.05 ml per 100 g rat per h and at a concentration of 36 μmoles per 100 g rat per h for the time indicated. At the end of the experiment, 0.1 g of liver was harvested to make total cell lysates for Western blot analysis. The rest was snap-frozen.
Primary rat and mouse hepatocyte cultures
Primary mouse hepatocyte monolayer cultures were prepared from mice by the collagenase-perfusion technique of Bissell and Guzelian as described previously (26). Single cell suspension was used for nuclear extraction for western blot and quantification of sphingolipids by mass spectrometry. Otherwise, cells were plated at 2 × 106 cells per collagen-coated 60-mm dish in serum-free Williams E medium containing penicillin, dexamethasone (0.1 μM), and thyroxine (1 μM).
Transfection of mouse hepatocytes with the gene encoding S1PR2
The primary mouse hepatocytes were plated on 6-well plated coated with collagen. After the cells were attached to the plate, a mammalian expressing vector containing mouse S1PR2 cDNA (pcDNA3-mS1PR2) was transfected into the cells using a PolyJet in vitro DNA transfection reagent according to the instruction provided by manufacturer (SignaGen Laboratories, Rockville, MD).
Nuclear extracts
Cells were washed with cold PBS and re-suspended in buffer containing 10 mM Hepes (pH 7.8), 10 mM KCl, 0.1 mM EDTA, 1 mM Na3VO4, 1 mM DTT, 1:500 protease inhibitors and 0.2 mM PMSF, and incubated on ice for 15 min. NP-40 was added (0.75%) and cells were vortexed for 10 sec. Nuclei and supernatant (“cytoplasm”) were separated by centrifugation at 3000 rpm for 3 min at 4 °C. Nuclei were resuspended in buffer containing 20 mM Hepes (pH 7.8), 0.4 M NaCl, 1 mM EDTA, 1 mM Na3VO4, 1 mM DTT and 1:500 protease inhibitors and incubated on ice for 15 min.
Quantification of sphingolipids by mass spectrometry
Lipids were extracted from primary hepatocytes after nuclear extracts, and the sphingolipids were quantified by liquid chromatography, electrospray ionization-tandem mass spectrometry (LC-ESI-MS/MS, 4000 QTRAP, AB Sciex), as previously described (15, 17, 18).
Oil red O and Nile Red staining
Frozen liver was sectioned serially at 5 μm thick with a cryostat and placed on slides. The sections then were fixed in neutral buffered 10% formalin for 10 min. To detect neutral lipid accumulation, the sections were incubated with a freshly prepared color substrate solution for 15 min at 37°C, followed by rinsing in distilled water. The sections were counter-stained with hematoxylin. Primary hepatocytes were also stained with Nile Red and Oil Red O in order to detect cellular lipids (27, 28).
Quantitation of total cholesterol and triglycerides in livers of wild type and SphK2−/− mice on different diets
Wild type and SphK2−/− mice were fed a normal diet (ND), high fat diet (HFD) or high fat diet plus 1% cholic acid (HFD+CA) for 2 weeks. Animals were sacrificed, pieces of liver extracted by Folch extraction (29) and total cholesterol and triglycerides quantitated by Wako kit assays, as described by the manufacturer (30).
In vitro assays
Methods for western blot and quantitative RT- PCR (RT-PCR) were performed as previously described (15, 20, 21, 28). The rabbit polyclonal antibodies were: histone H3 and H4K5ac from Millipore, H3K9ac and H2BK12ac from Cell Signaling, and HDAC1 and HDAC2 from Santa Cruz Biotechnology. Murine C-terminal anti-SphK2 antibody was kindly provided by Dr. Richard Proia, National Institutes of Health (NIH), Bethesda, MD, USA.
Sphingosine kinase activity assays
SphK1 and SphK2 activity assays were performed exactly as previously described (31).
Chromatin immunoprecipitation (ChIP) assay
Chromatin crosslinking was performed in mouse primary hepatocytes isolated from wild type and SphK2−/− mice. Nuclei were isolated and ChIP assay was performed with Pierce™ Magnetic ChIP kit (Thermo Scientific) and EpiTect Mouse H3K9ac ChIP Antibody Kit (QIAGEN) according to the instructions provided by the manufacturers. Real-time PCR was used to quantify ChIP assay results. The following SYBR primers were used to detect mouse srebp1c and cyp7a1 promoter regions. srebp1c: forward: 5′-ACCAACTGCCACTATCTA-3′, reverse: 5′-GGACCATTTTAGCCTCAG-3′; cyp7a1: forward: 5′-ACCTTCGGCTTATCGACTATTGC-3′, reverse:5′-TATCTGGCCTTGAACTAAGTCCATCT-3′. Control IgG was used as a negative control for substracting background. ChIP DNA amount for gene promoters of interest was normalized to that of a housekeeping gene GAPDH.
Statistical analysis of data
All the experiments were repeated at least three times and the results were expressed as mean ± SE. One-way analysis of variance (ANOVA) was employed to analyze the differences between sets of data using GraphPad Prism (Graph-Pad, San Diego, CA). A value of P < 0.05 was considered statistically significant.
Results
Histone Acetylation is Decreased and Genes Encoding Enzymes and Nuclear Receptors Involved in Hepatic Sterol and Lipid Metabolism are Down-regulated in SphK2−/− Mice
We have previously reported that S1P is a specific inhibitor of histone deacetylases 1 and 2 (HDAC 1/2) in human breast cancer cell lines (15). In the current study, we employed the SphK2−/− mouse model to determine if S1P regulates histone acetylation and gene expression in the liver. We measured the level of acetylation of histone H3K9, H4K5 and H2BK12 in primary hepatocytes isolated from SphK2−/− mice and compared them with the wild type control animals. The data presented in Fig. 1A shows a dramatic decrease in the acetylation of H3K9, H4K5 and H2BK12 in SphK2−/− mice. Messenger RNA was isolated from livers of wild type and SphK2−/− mice, and the levels of mRNA encoding key enzymes and nuclear receptors involved in sterol and lipid metabolism were determined by quantitative PCR. The data presented in Fig. 1B shows a significant decrease in the mRNA levels of a number of key genes involved in sterol and lipid metabolism such as SREBP-1c, FAS and LDLR, but has no significant effect on CYP7A1. The chromatin immuneprecipitation (ChIP) assay further confirmed that SphK2-mediated signaling is involved in regulating of key genes of hepatic lipid metabolism. As shown in Fig. 1C, decrease of acetylated H3K9 level in SphK2−/− mice primary hepatocytes is correlated to the low abundance of SREBP-1c promoter DNA associated with H3K9ac, but not CYP7A1. Furthermore, a chemical inhibitor of HDACs (SAHA) significantly up-regulated the mRNA levels of key enzymes and nuclear receptors in primary hepatocytes isolated from wild type mice suggesting that histone acetylation maybe a key regulator of gene expression in the liver (Fig. 1D).
Fig. 1. Effect of SphK2 on hepatic histone acetylation levels and mRNA levels of key genes involved in hepatic lipid metabolism.

(A) Nuclear extracts of primary hepatocytes isolated from Wild type (WT) and SphK2−/− mice were prepared as described in “Methods”. The protein levels of SphK2, H3K9ac, H4K5ac, H2BK12ac and total H3 were determined by Western blot analysis. Representative images are shown. (B) Total RNA was isolated from the livers of WT (solid bar) or SphK2−/− mice (checkered bar) (male, 20-week old). The mRNA levels of key genes involved in lipid metabolism were determined using real time RT-PCR and normalized using GAPDH. Abbs. SREBP-1c: sterol regulatory element-binding protein-1c; FAS: fatty acid synthase; LDLR: low-density lipoprotein receptor; CYP27A1: sterol 27-hydroxylase; CYP7A1: cholesterol 7 α-hydroxylase; BSEP: bile salt export pump; FXR: farnesoid X receptor; BECN1: autophagy-related gene (Atg) 6; PPARγ: peroxisome proliferator-activated receptor γ. *P<0.05, **P<0.01, ***P<0.01, statistical significance relative to WT mice, n=5. (C) Effect of SphK2 on association of acetylated H3K9 in SREBP1c and CYP7A1 chromatins. Mouse primary hepatocytes were isolated from wild type (WT) and SphK2−/− mice. ChIP assay and real-time PCR were used to quantify the DNA amount of SREBP1c and CYP7A1 associated with H3K9ac as described in “Methods”. Results were expressed as relative abundance of chromatin DNA associated with H3K9ac. Values are mean ± S.E. **P<0.01, statistical significance relative to WT, n=3. (D) The wild type mouse primary hepatocytes (solid bar) were treated with a chemical inhibitor of HDAC, SAHA (1 μM) for 8h (checkered bar). The total RNA was isolated. The mRNA levels of key genes involved in lipid metabolism were determined using real time PCR and normalized using β-actin. CPT-1α: Carnitine palmitoyltransferase I α; PCG1α: Peroxisome proliferator-activated receptor gamma coactivator 1-alpha; ACC1: Acetyl-CoA carboxylase 1.
Feeding a High Fat Diet (HFD) or HFD+CA Up-regulated Hepatic SphK2
We next wanted to determine if feeding a wild type mouse a HFD, which increases the cycling of the bile acid pool, or a HFD plus cholic acid (HFD+CA) would up-regulate hepatic SphK2. The data presented in Fig. 2 shows that SphK2 mRNA, protein and enzyme activity were all significantly increased by HFD and HFD+CA when compared to a normal diet in wild type mice. SphK2−/− mice were used as a control in order to show lack of SphK2 protein and activity (Fig. 2). The small amount of baseline SphK2 activity is due to the small overlap of SphK1 activity using standard assay conditions for these two enzymes.
Fig. 2. Effect of high fat diet and cholic acid on SphK2 expression and enzymatic activity in liver.
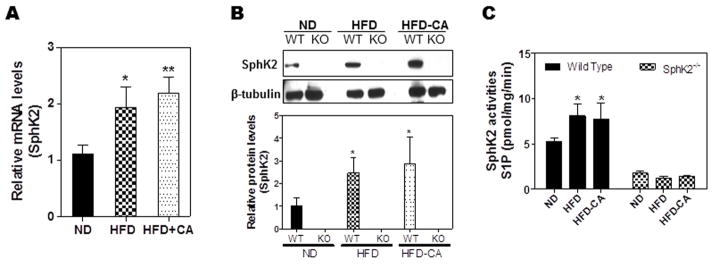
Wild type (WT) and SphK2−/− (KO) mice (male, 20-week old) were fed normal chow diet (ND; solid bar), high fat diet (HFD; checkered bar), or HFD containing 1% cholic acid (HFD+CA; shaded bar) for two weeks. (A) The mRNA levels of SphK2 in the liver of WT mice were determined by real time RT-PCR and normalized using GAPDH as internal control. (B) The protein expression levels of SphK2 in the liver of WT and SphK2−/− mice were determined by Western Blot analysis and normalized using β-tubulin as loading control. (C) The enzyme activities of SphK2 were measured using [γ32P]-ATP. *P<0.05, **P<0.01, statistical significance relative to control group.
Taurocholate (TCA) Increases Hepatic Levels of SphK2, but not SphK1, via S1PR2
Our previous studies showed that CBAs activate the AKT and ERK1/2 signaling pathways via S1PR2 in primary rodent hepatocytes and in vivo in the chronic bile fistula rat (21). Because SphK2 has been reported to be activated by ERK1/2 (15), we hypothesized that bile acids may regulate SphK2. To test this hypothesis, the chronic bile fistula rat model was used. TCA was infused for 4 hours into the intestines of the chronic bile fistula rat at a concentration previously shown to be non-toxic (25). Animals were sacrificed, livers harvested, total protein extracts prepared, and SphK1 and SphK2 protein levels and specific activity measured. TCA infusion significantly increases the protein (4-fold) and enzyme specific activity (~70%) of SphK2 (Fig. 3A and 3C). The difference between protein and activity may be due to lack of activation via phosphorylation (32). In contrast, there was no significant effect of TCA infusion on SphK1 protein or enzyme specific activity in these same extracts (Fig. 3A and 3B). Finally, we measured the effect of JTE-013, a chemical antagonist of S1PR2, on the activation of SphK2 activity by TCA in primary rat hepatocytes from wild type animals. The data show a significant inhibition of TCA-induced activation of SphK2 by JTE-013, but no effect on SphK1 activity (Online Fig. S1).
Fig. 3. Induction of SphK2 by TCA in the chronic bile fistula rat.

Bile fistulas were placed in rats and the bile was drained for 48 hrs. TCA was infused intraduodenally at a rate of 1.05 ml/h/100 g rat and a concentration of 36 μmol/h/100 g rat for 4h (checkered bar). Animals were harvested and the liver pieces from each animal were isolated and snap-frozen. The protein levels of SphK1 and SphK2 were detected by Western blot analysis using antibodies against SphK1 and SphK2 as described in “Methods”. (A) Representative images of immunoblots for SphK1, SphK2 and actin are shown. Relative densities of SphK1 and SphK2 were determined by scanning laser density spectrometry and actin was used as a loading control. (B and C) The specific enzyme activities of SphK1 and SphK2 were determined using [γ32P]-ATP and sphingosine as described in “Methods”. *p<0.05, statistical significance relative to control group, n=5.
S1PR2−/− Mice Have Lower Levels of Hepatic SphK2, but not SphK1
Next, we wanted to determine if there was a physiological link between S1PR2 and SphK2 using S1PR2−/− mice. SphK2 mRNA, protein and enzyme activity in livers and primary hepatocytes prepared from S1PR2−/− mice were measured. As shown in Fig. 4A–D, the expression levels of SphK2 were significantly down-regulated in extracts from whole liver and primary hepatocytes prepared from S1PR2−/− mice. There was no significant effect on SphK1 expression in either liver or primary hepatocytes (MPH) of S1PR2−/− mice. Moreover, when TCA was added to culture medium of primary hepatocytes prepared from wild type mice, there was a rapid activation of SphK2, but not SphK1, activity (Fig. 4E and F). In contrast, hepatocytes prepared from S1PR2−/− mice showed a significantly lower SphK2 activity compared to wild type mice and no increase of the SphK2 activity by TCA (Fig. 4F). In total, all the current data suggest that hepatic SphK2 expression and enzyme activity is regulated by CBAs via S1PR2.
Fig. 4. Effect of S1PR2 on SphK2 expression and enzymatic activity.

(A) Total RNA was isolated from the liver of Wild type (WT) and S1PR2−/− mice. The mRNA levels of SphK2 were determined using real-time RT-PCR. (B) The total liver protein lysates of WT and S1PR2−/− mice were prepared and protein levels of SphK1 and SphK2 were determined by Western blot analysis. Representative images are shown. (C) The relative densities of SphK1 (solid bar) and SphK2 (checkered bar) were determined using β-tubulin as a loading control. (D) Primary mouse hepatocytes were isolated from Wild type (WT) and S1PR2−/− mice. The protein levels of SphK1 and SphK2 were determined by Western Blot analysis and the representative images are shown. β-tubulin was used as a loading control. (E and F) Primary mouse hepatocytes isolated from WT (closed circle) and S1PR2−/− (closed diamond) mice were treated with TCA (100 μM) for 0–30 min). The enzyme activities of SphK1 and SphK2 were measured using [γ32P]-ATP and sphingosine.
Histone Acetylation, Gene Expression and Nuclear S1P are Decreased in the Livers of S1PR2−/− Mice
Because SphK2 was down-regulated in the S1PR2−/− mice, we wanted to determine if these mice had a similar histone acetylation and gene expression pattern as SphK2−/− mice. In order to investigate this question, we measured histone acetylation and steady-state mRNA levels of key genes encoding enzymes and nuclear receptors involved in sterol and lipid metabolism. In addition, we used mass spectrometry to measure the levels of S1P and DHS1P in the nucleus and cytosol of primary hepatocytes prepared from wild type and S1PR2−/− mice. The data in Fig. 5A shows that the specific histone acetylation is markedly down-regulated in primary hepatocytes from S1PR2−/− mice. Moreover, similar to SphK2−/− mice, there was a significant down-regulation of key genes encoding enzymes and nuclear receptors involved in sterol and lipid metabolism (Figs. 1B and 5B). Finally, the levels of S1P and DHS1P were significantly decreased in the nucleus, but not in the cytosol, of S1PR2−/− mice (Fig. 5C and D).
Fig. 5. Effect of S1PR2 on hepatic histone acetylation levels and mRNA levels of key genes involved in hepatic lipid metabolism.

(A) Nuclear extracts of primary hepatocytes isolated from Wild type (WT) and S1PR2−/− mice were prepared as described in “Methods”. The protein levels of SphK2, H3K9ac, H4K5ac, H2BK12ac and total H3 were determined by Western blot analysis. Representative images are shown. (B) Total RNA was isolated from the livers of WT (solid bar) or S1PR2−/− (checkered bar) mice (male, 20-week old). The mRNA levels of key genes involved in lipid metabolism were determined using real time RT-PCR and normalized using β-actin. Abbs: SREBP-1c: sterol regulatory element-binding protein-1c; FAS: fatty acid synthase; LDLR: low-density lipoprotein receptor; CYP27A1: sterol 27-hydroxylase; CYP7A1: cholesterol 7 α-hydroxylase; BSEP: bile salt export pump; FXR: farnesoid X receptor. BECN1: autophagy-related gene (Atg) 6; PPARγ: peroxisome proliferator-activated receptor γ. *P<0.05, **P<0.01, ***P<0.01, statistical significance relative to WT mice, n=5–8. (C and D) The S1P and DHS1P levels in the cytosol and nucleus of mouse primary hepatocytes isolated from WT and S1PR2−/− mice were measured using mass spectrometry as described in “Methods”. *P<0.05, statistical significance relative to WT.
Overexpression of the Gene Encoding S1PR2 Differentially Up-regulates mRNA Levels of Genes Involved in Nutrient Metabolism in Mouse Hepatocytes
Does an increase in S1PR2 up-regulate SphK2 and other hepatic genes? To answer this question, primary mouse hepatocytes prepared from wild type and S1PR2−/− mice were transfected with an expression plasmid, encoding mouse S1PR2 cDNA, using PolyJet™ DNA In Vitro Transfection Reagent. After 48 h, the total cellular RNA was isolated and the mRNA levels of specific genes were quantitated by real-time RT-PCR. As shown in Fig. 6A, there was approximately a 300 to 400-fold increase in the mRNA levels of S1PR2 in transfected cells. As hypothesized, there was a 5 to 10-fold increase in SphK2 mRNA, but no significant effect on SphK1 mRNA, in cells over-expressing S1PR2 (Fig. 6B). Surprisingly, in cells over-expressing S1PR2, there was a differential up-regulation of specific genes involved in nutrient metabolism. For example, mRNA levels of ApoB-100, SHP, CYP7A1, FXRα, and SREBP1c were up-regulated ~15-fold, ~15-fold, ~8-fold, ~5-fold and ~5-fold, respectively, in cells over-expressing S1PR2. In contrast, there was a less noticeable effect on the mRNA levels of genes encoding LDLR (~3-fold increase) and BSEP (~2-fold increase), and no effect on CYP27A mRNA levels, suggesting specificity in the effects S1PR2 on gene expression in hepatocytes (Fig. 6C). Finally, when the gene encoding S1PR2 was over-expressed in hepatocytes prepared from SphK2−/− mice, there was no significant up-regulation of genes involved in nutrient metabolism, suggesting the importance of SphK2 in their regulation (Fig. 6D).
Fig. 6. Effect of overexpression of the gene encoding S1PR2 on gene regulation in mouse primary hepatocytes.

The mouse primary hepatocytes isolated from wild type (WT) or S1PR2−/− mice were transfected with a control vector or pcDNA3-mS1PR2 as described in “Methods”. The total cellular RNA was isolated after 48h. The mRNA levels of specific target genes were determined using real-time RT-PCR and normalized using GAPDH as an internal control. (A) The mRNA levels of S1PR2; (B) The mRNA levels of SphK1 and SphK2; (C) The mRNA levels of key genes involved in lipid metabolism. *P<0.05, **P<0.01, ***P<0.001, statistical significance relative to control vector transfected cells. (D) Effect of overexpression of the gene encoding S1PR2 on gene regulation in SphK2−/− mouse primary hepatocytes. The mouse primary hepatocytes isolated from SphK2−/− mice were transfected with a control vector or pcDNA3-mS1PR2 as described above. The total cellular RNA was isolated after 48h. The mRNA levels of specific target genes were determined using real-time RT-PCR and normalized using GAPDH as an internal control. The mRNA levels of key genes involved in lipid metabolism. *P<0.05, **P<0.01, ***P<0.001, statistical significance relative to control vector transfected cells.
SphK2−/− and S1PR2−/− Mice Rapidly Develop Fatty Livers
Because TCA significantly increases hepatic SphK2 via S1PR2, and SphK2 regulates histone acetylation and genes encoding nuclear receptors/enzymes involved in hepatic sterol and lipid metabolism, we wondered what physiological effects this would have on SphK2−/− and S1PR2−/− mice. We examined the livers and primary hepatocytes of SphK2−/− and S1PR2−/− mice by H&E staining of liver sections for histology and for lipids using Oil Red O or Nile Red from animals on a normal diet and HFD. Interestingly, SphK2−/− and S1PR2−/− mice fed a normal diet showed increased levels of hepatic lipids as indicated by Oil Red O staining (Online Fig. S2A and B). Similarly, hepatocytes prepared from either SphK2−/− or S1PR2−/− mice and stained with either Oil Red O or Nile Red showed increased levels of intracellular lipids (Online Fig. S2C and D). There was a marked increase in serum triglycerides in SphK2−/− mice possibly due to a down regulation of hepatic lipase (HL) and hormone sensitive lipase (HSL) (See Online Table 1 and Online Fig. S3). Moreover, SphK2−/− and S1PR2−/− mice fed with HFD showed enlarged livers and a dramatic increase in hepatic lipid accumulation (Fig. 7). Quantitation of cholesterol and triglycerides from livers of SphK2−/− mice showed significantly increased levels on a HFD and HFD-CA (Online Fig. S4). Similar data were obtained for S1PR2−/− (not shown).
Fig. 7. Effect of HFD on hepatic lipid accumulation in S1PR2−/− SphK2−/− mice.

Wild type (WT), S1PR2−/− and SphK2−/− mice (male, 20-week old) were fed HFD for two weeks. The liver sections were stained using Oil Red O or HE. The images were taken with an Olympus microscope equipped with image recorder using a 400x lens. Representative images of livers, HE staining and Oil Red O staining are shown. (A) S1PR2−/− mice; (B) SphK2−/− mice.
Discussion
We have previously reported that S1P inhibits HDAC1/2 in cancer cells (15). However, the physiological relevance of the inhibition of HDAC1/2 by nuclear S1P in the liver has never been investigated. Our recent studies using the chronic bile fistula rat model reported that CBAs activate S1PR2 in primary hepatocytes and in vivo (21). Activation of the S1PR2 by CBAs or S1P activates the ERK1/2 and AKT pathways in hepatocytes (20, 21). We proposed that the S1PR2 may regulate nuclear SphK2 in a physiologically relevant manner and control the expression of key genes encoding enzymes and nuclear receptors involved in regulating nutrient metabolism (Fig. 8). In the current study, we show that intraduodenal infusion of TCA into the chronic bile fistula rat, feeding mice a HFD, or overexpression of the gene encoding S1PR2 in mouse hepatocytes significantly up-regulated SphK2, but not SphK1 mRNA levels (Figs. 2, 3 and 6). Moreover, our data suggest that hepatic SphK2 expression and enzyme activity are regulated by CBAs via S1PR2 (Fig. 4).
Fig. 8. Model of CBA regulation of hepatic genes encoding enzymes involved in nutrient metabolism.

CBA returning from the intestines following a meal activates S1PR2. Activation of this GPCR then activates nuclear SphK2 via cell signaling pathways increasing the levels of S1P in the nucleus. Nuclear S1P inhibits specific histone deacetylases (HDACs) causing an increase in acetylation of histones and up-regulation of genes encoding nuclear receptors and enzymes involved in nutrient metabolism. This CBA-activated nutrient signaling pathway is hypothesized to allow the liver to be more efficient in metabolizing a bolus of incoming nutrients allowing the liver to maintain “nutrient homeostasis”.
What is the physiological relevance of up-regulation of SphK2 by CBAs or HFD in the liver? Our data show that nuclear SphK2 is significantly down-regulated in S1PR2−/− mice (Fig. 4). The down-regulation of SphK2 results in a significant decrease in the levels of S1P in nuclei in S1PR2−/− hepatocytes (Fig. 5C and D). In this regard, there was a similar decrease in the levels of acetylation of specific histones in both SphK2−/− and S1PR2−/− mice (Figs. 1 and 5). The steady-state mRNA levels of key genes encoding enzymes and nuclear receptors involved in the regulation of metabolism were significantly down-regulated in both SphK2−/− and S1PR2−/− mice in a similar manner (Figs. 1 and 5). Interestingly, the expression of the gene encoding S1PR2 in primary mouse hepatocytes differentially up-regulated genes encoding enzyme/nuclear receptors involved in sterol and lipid metabolism (Fig. 6). S1PR2−/− and SphK2−/− mice rapidly develop fatty livers on a HFD with the accumulation of cholesterol and triglycerides (Fig. 7 and Online Fig. S4).Moreover, S1PR2−/− and SphK2−/− mice also accumulated lipid in the liver on a normal diet, but to a much lesser extent than on a HFD (Fig. 7, Online Fig. S2). We speculate that genes required for the transport and metabolism of lipids (i.e. ApoB-100 and CPT-1α) failed to be up-regulated in livers of animals deficient in SphK2 or S1PR2 leading to fatty livers (See Online Fig. S5). In total, these results suggest that S1PR2 is a crucial GPCR that allows CBAs to regulate hepatic nutrient metabolism by regulating SphK2 which controls nuclear S1P levels.
Nuclear S1P levels regulate histone acetylation and the transcriptional activity of key genes encoding enzymes and nuclear receptors involved in sterol and lipid metabolism in the liver (Fig. 8). It is of interest that overexpression of the gene encoding S1PR2 differentially up-regulated the mRNA levels of specific genes involved in sterol and lipid metabolism in mouse primary hepatocytes (Fig. 6). In this regard, the same pattern of gene regulation was reproducibly observed in both wild-type and S1PR2−/− mouse hepatocytes following overexpression of the gene encoding S1PR2, suggesting specificity in gene induction by this epigenetic system. Surprisingly, both SHP and CYP7A1 were both up-regulated by overexpression of the gene encoding S1PR2 as SHP has been reported to repress the gene encoding CYP7A1 (Fig. 6C). SHP is a nuclear receptor without a DNA binding domain that acts as a co-repressor by interacting with liver related homologue-1 (LRH-1) and hepatocyte nuclear factor 4α (HNF-4α) that bind and activate the CYP7A1 promoter (8, 33, 34). However, SHP probably also plays a minor role in regulating CYP7A1 gene expression as bile acids and FXR agonist still repress CYP7A1 in SHP−/− mice (35). FGF-15/19 is induced in an FXR-dependent manner in the ileum and appears to be the major regulator of CYP7A1 in the liver (36). FXR15/19 binds FGF receptor 4 (FGFR4) in the liver activating cell signaling pathways that down-regulate the gene encoding CYP7A1. SHP has also been reported to coordinately repress CYP7A1 in the liver by regulating epigenetic mechanisms (16). In this regard, SHP has been reported to interact with HDAC1/2 in the nucleus to decrease histone acetylation and increase histone methylation by interacting with G9A histone lysine methyltransferase allowing chromatin remodeling by interacting with Swi/Snf Brm, which represses CYP7A1. However, one might hypothesize that up-regulation and activation of nuclear SphK2, which increases nuclear S1P levels, could counteract the repression effects of the SHP: HDAC1/2 complex by inhibiting HDAC1/2 activity. Therefore, the relative nuclear levels of S1P and SHP complexes may determine histone acetylation/methylation levels and transcriptional activity of key genes encoding enzymes and nuclear receptors involved in metabolism.
In summary, the current study reports on the elucidation of CBA-activated epigenetic mechanism regulating hepatic gene expression encoding enzymes and nuclear receptors involved in sterol and lipid metabolism (Fig. 8). The data highlight the important role that S1PR2, nuclear SphK2, S1P and histone acetylation play in this mechanism. Dysregulation of this cell signaling system may have significance in the development of fatty liver and related diseases especially in individuals on a high fat diet.
Supplementary Material
Acknowledgments
We thank Dr. David Mangelsdorf for reviewing our manuscript and helpful suggestions. This work was supported by National Institutes of Health Grant R01 DK-057543-11(to PBH, HZ and KT), R37GM043880 (to SS), R01CA160688 (to KT); VA Merit Award (to HZ and WP); Masayuki Nagahashi was a Japan Society for the Promotion of Science Postdoctoral Fellow for Research Abroad.
Abbreviations
- BF
bile fistula
- CBA
conjugated bile acids
- HFD
high fat diet
- HFD+CA
high fat diet plus cholic acid
- NAFLD
nonalcoholic fatty liver disease
- ND
normal diet
- SphK1
sphingosine kinase 1
- SphK2
sphingosine kinase 2
- SphK2−/−
sphingosine kinase 2 knock out
- S1P
sphingosine-1-phosphate
- S1PR2
sphingosine 1-phosphate receptor 2
- S1PR2−/−
sphingosine 1-phosphate receptor 2 knock out
- TCA
taurocholate
- ChIP
chromatin immunoprecipitation
Footnotes
Contributions: PBH, KT and HZ conceived the original ideas, designed the study, analyzed the data and wrote the manuscript; WMP and PC carried out the analysis of gallbladder bile acid, serum analysis and data analysis; MN, animal breeding and genotyping, pathology, Western blotting, writing of manuscript; YW, RL, and XW carried out lipid staining, RNA isolation and real-time RT-PCR; NCH, JL, Western blotting; JCA measured sphingolipids; AY and TA, animal breeding, genotyping and pathology. SS provided animals, helped design the study, and analyzed data.
Competing Financial interest: The authors declare no competing financial interest.
References
- 1.Lewis JR, Mohanty SR. Nonalcoholic fatty liver disease: a review and update. Dig Dis Sci. 2010;55:560–578. doi: 10.1007/s10620-009-1081-0. [DOI] [PubMed] [Google Scholar]
- 2.Malaguarnera M, Di Rosa M, Nicoletti F, Malaguarnera L. Molecular mechanisms involved in NAFLD progression. J Mol Med (Berl) 2009;87:679–695. doi: 10.1007/s00109-009-0464-1. [DOI] [PubMed] [Google Scholar]
- 3.Younossi ZM, Stepanova M, Afendy M, Fang Y, Younossi Y, Mir H, Srishord M. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin Gastroenterol Hepatol. 2011;9:524–530. e521. doi: 10.1016/j.cgh.2011.03.020. quiz e560. [DOI] [PubMed] [Google Scholar]
- 4.Almeda-Valdes P, Cuevas-Ramos D, Aguilar-Salinas CA. Metabolic syndrome and non-alcoholic fatty liver disease. Ann Hepatol. 2009;8 (Suppl 1):S18–24. [PubMed] [Google Scholar]
- 5.Polyzos SA, Kountouras J, Zavos C. Nonalcoholic fatty liver disease: the pathogenetic roles of insulin resistance and adipocytokines. Curr Mol Med. 2009;9:299–314. doi: 10.2174/156652409787847191. [DOI] [PubMed] [Google Scholar]
- 6.Tilg H, Moschen AR. Insulin resistance, inflammation, and non-alcoholic fatty liver disease. Trends Endocrinol Metab. 2008;19:371–379. doi: 10.1016/j.tem.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 7.Alkhouri N, Dixon LJ, Feldstein AE. Lipotoxicity in nonalcoholic fatty liver disease: not all lipids are created equal. Expert Rev Gastroenterol Hepatol. 2009;3:445–451. doi: 10.1586/egh.09.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiang JY. Bile acids: regulation of synthesis. J Lipid Res. 2009;50:1955–1966. doi: 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, Dent P. Bile acids as regulatory molecules. J Lipid Res. 2009;50:1509–1520. doi: 10.1194/jlr.R900007-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lefebvre P, Cariou B, Lien F, Kuipers F, Staels B. Role of bile acids and bile acid receptors in metabolic regulation. Physiol Rev. 2009;89:147–191. doi: 10.1152/physrev.00010.2008. [DOI] [PubMed] [Google Scholar]
- 11.Scotti E, Gilardi F, Godio C, Gers E, Krneta J, Mitro N, De Fabiani E, et al. Bile acids and their signaling pathways: eclectic regulators of diverse cellular functions. Cell Mol Life Sci. 2007;64:2477–2491. doi: 10.1007/s00018-007-7280-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008;7:678–693. doi: 10.1038/nrd2619. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y, Edwards PA. FXR signaling in metabolic disease. FEBS Lett. 2008;582:10–18. doi: 10.1016/j.febslet.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 14.de Aguiar Vallim TQ, Tarling EJ, Edwards PA. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013;17:657–669. doi: 10.1016/j.cmet.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, Luo C, et al. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science. 2009;325:1254–1257. doi: 10.1126/science.1176709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith Z, Ryerson D, Kemper JK. Epigenomic regulation of bile acid metabolism: emerging role of transcriptional cofactors. Mol Cell Endocrinol. 2013;368:59–70. doi: 10.1016/j.mce.2012.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takabe K, Kim RH, Allegood JC, Mitra P, Ramachandran S, Nagahashi M, Harikumar KB, et al. Estradiol induces export of sphingosine 1-phosphate from breast cancer cells via ABCC1 and ABCG2. J Biol Chem. 2010;285:10477–10486. doi: 10.1074/jbc.M109.064162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strub GM, Maceyka M, Hait NC, Milstien S, Spiegel S. Extracellular and intracellular actions of sphingosine-1-phosphate. Adv Exp Med Biol. 2010;688:141–155. doi: 10.1007/978-1-4419-6741-1_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takabe K, Paugh SW, Milstien S, Spiegel S. “Inside-out” signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev. 2008;60:181–195. doi: 10.1124/pr.107.07113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao R, Cronk ZX, Zha W, Sun L, Wang X, Fang Y, Studer E, et al. Bile acids regulate hepatic gluconeogenic genes and farnesoid X receptor via G(alpha)i-protein-coupled receptors and the AKT pathway. J Lipid Res. 2010;51:2234–2244. doi: 10.1194/jlr.M004929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Studer E, Zhou X, Zhao R, Wang Y, Takabe K, Nagahashi M, Pandak WM, et al. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology. 2012;55:267–276. doi: 10.1002/hep.24681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ikeda H, Watanabe N, Ishii I, Shimosawa T, Kume Y, Tomiya T, Inoue Y, et al. Sphingosine 1-phosphate regulates regeneration and fibrosis after liver injury via sphingosine 1-phosphate receptor 2. J Lipid Res. 2009;50:556–564. doi: 10.1194/jlr.M800496-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Serriere-Lanneau V, Teixeira-Clerc F, Li L, Schippers M, de Wries W, Julien B, Tran-Van-Nhieu J, et al. The sphingosine 1-phosphate receptor S1P2 triggers hepatic wound healing. FASEB J. 2007;21:2005–2013. doi: 10.1096/fj.06-6889com. [DOI] [PubMed] [Google Scholar]
- 24.Kageyama Y, Ikeda H, Watanabe N, Nagamine M, Kusumoto Y, Yashiro M, Satoh Y, et al. Antagonism of sphingosine 1-phosphate receptor 2 causes a selective reduction of portal vein pressure in bile duct-ligated rodents. Hepatology. 2012;56:1427–1438. doi: 10.1002/hep.25780. [DOI] [PubMed] [Google Scholar]
- 25.Heuman DM, Hernandez CR, Hylemon PB, Kubaska WM, Hartman C, Vlahcevic ZR. Regulation of bile acid synthesis. I. Effects of conjugated ursodeoxycholate and cholate on bile acid synthesis in chronic bile fistula rat. Hepatology. 1988;8:358–365. doi: 10.1002/hep.1840080228. [DOI] [PubMed] [Google Scholar]
- 26.Heuman DM, Pandak WM, Hylemon PB, Vlahcevic ZR. Conjugates of ursodeoxycholate protect against cytotoxicity of more hydrophobic bile salts: in vitro studies in rat hepatocytes and human erythrocytes. Hepatology. 1991;14:920–926. doi: 10.1002/hep.1840140527. [DOI] [PubMed] [Google Scholar]
- 27.Wu X, Zhang L, Gurley E, Studer E, Shang J, Wang T, Wang C, et al. Prevention of free fatty acid-induced hepatic lipotoxicity by 18beta-glycyrrhetinic acid through lysosomal and mitochondrial pathways. Hepatology. 2008;47:1905–1915. doi: 10.1002/hep.22239. [DOI] [PubMed] [Google Scholar]
- 28.Zhou H, Pandak WM, Jr, Lyall V, Natarajan R, Hylemon PB. HIV protease inhibitors activate the unfolded protein response in macrophages: implication for atherosclerosis and cardiovascular disease. Mol Pharmacol. 2005;68:690–700. doi: 10.1124/mol.105.012898. [DOI] [PubMed] [Google Scholar]
- 29.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- 30.Wang Y, Zhang L, Wu X, Gurley EC, Kennedy E, Hylemon PB, Pandak WM, et al. The role of CCAAT enhancer-binding protein homologous protein in human immunodeficiency virus protease-inhibitor-induced hepatic lipotoxicity in mice. Hepatology. 2013;57:1005–1016. doi: 10.1002/hep.26107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maceyka M, Milstien S, Spiegel S. Sphingosine kinases, sphingosine-1-phosphate and sphingolipidomics. Prostaglandins Other Lipid Mediat. 2005;77:15–22. doi: 10.1016/j.prostaglandins.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 32.Hait NC, Bellamy A, Milstien S, Kordula T, Spiegel S. Sphingosine kinase type 2 activation by ERK-mediated phosphorylation. J Biol Chem. 2007;282:12058–12065. doi: 10.1074/jbc.M609559200. [DOI] [PubMed] [Google Scholar]
- 33.Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, Moore LB, Galardi C, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–526. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 34.Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, Auwerx J, Mangelsdorf DJ. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell. 2000;6:507–515. doi: 10.1016/s1097-2765(00)00050-2. [DOI] [PubMed] [Google Scholar]
- 35.Wang L, Lee YK, Bundman D, Han Y, Thevananther S, Kim CS, Chua SS, et al. Redundant pathways for negative feedback regulation of bile acid production. Dev Cell. 2002;2:721–731. doi: 10.1016/s1534-5807(02)00187-9. [DOI] [PubMed] [Google Scholar]
- 36.Potthoff MJ, Kliewer SA, Mangelsdorf DJ. Endocrine fibroblast growth factors 15/19 and 21: from feast to famine. Genes Dev. 2012;26:312–324. doi: 10.1101/gad.184788.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.