

Abstract
Visualizing the physical basis for molecular behavior inside living cells is a grand challenge in biology. RNAs are central to biological regulation, and RNA’s ability to adopt specific structures intimately controls every step of the gene expression program1. However, our understanding of physiological RNA structures is limited; current in vivo RNA structure profiles view only two of four nucleotides that make up RNA2,3. Here we present a novel biochemical approach, In Vivo Click SHAPE (icSHAPE), that enables the first global view of RNA secondary structures of all four bases in living cells. icSHAPE of mouse embryonic stem cell transcriptome versus purified RNA folded in vitro shows that the structural dynamics of RNA in the cellular environment distinguishes different classes of RNAs and regulatory elements. Structural signatures at translational start sites and ribosome pause sites are conserved from in vitro, suggesting that these RNA elements are programmed by sequence. In contrast, focal structural rearrangements in vivo reveal precise interfaces of RNA with RNA binding proteins or RNA modification sites that are consistent with atomic-resolution structural data. Such dynamic structural footprints enable accurate prediction of RNA-protein interactions and N6-methyladenosine (m6A) modification genome-wide. These results open the door for structural genomics of RNA in living cells and reveal key physiological structures controlling gene expression.
Selective 2’-Hydroxyl Acylation followed by Primer Extension (SHAPE) accurately identifies flexible (single stranded) bases in RNA for all four bases. However, current methods are potentially limited by high background (>70% of RNA molecules have no modification due to single hit kinetics) and high false positive rates due to spurious reverse transcription (RT) stops4. We overcome these problems by developing a new SHAPE probe that permits in vivo SHAPE modification and subsequent selective purification of the modified RNAs.
We designed, synthesized, and tested a novel bifunctional chemical probe for in vivo RNA structure profiling genome-wide (NAI-N3, Fig. 1a, 1b, and Extended Data Fig 1). NAI-N3 adds an azide group to NAI, a cell permeable SHAPE reagent5. By using copper-free click chemistry, a biotin moiety is selectively and efficiently added to NAI-N3-modified RNA, providing a stringent purification handle with streptavidin beads (Fig. 1c, Extended Data Fig. 2). NAI-N3 generated identical profiles of RT stops to those obtained using our previously designed SHAPE reagent5. The fidelity of structural measurements was not affected by “clicking” biotin onto the NAI-N3 nor by molecular crowding of proteins, and NAI-N3 showed uniform modification of all bases in denatured RNAs (Extended Data Fig. 3). We term this new chemoaffinity structure probing methodology In Vivo Click SHAPE (icSHAPE); this method can also be applied to any ex vivo preparation of RNA with slight modifications.
Figure 1. icSHAPE is a Novel and Robust Method for Measuring RNA Structure.

a, Chemical scheme for the preparation of acylated RNA, which can be purified by biotin-streptavidin purification. b, Schematic of icSHAPE modification and purification steps to generate a sequencing library. c, Dot blot of biotin-modified RNA from icSHAPE through streptavidin affinity isolation. d, Denaturing gel electrophoresis of icSHAPE on rRNA from mESCs. The corresponding icSHAPE profile, generated from deep sequencing is to the right. e, Pymol representation of rRNA, corresponding to regions of icSHAPE that are more reactive in vivo (PDB3J3D). f, Pymol representation of rRNA, corresponding to regions of icSHAPE that are more reactive in vitro.
icSHAPE of ribosomal RNAs in mouse embryonic stem cells (mESC) indicated that it is quantitative and accurate, reporting the known structures of 18S and 28S rRNAs (Fig. 1d-f and Extended Data Fig. 4). Manual structure-probing gels and deep sequencing results from icSHAPE revealed high correlations (Pearson correlation r=0.93, in vivo; Fig. 1d and Extended Data Fig. 4). Ribosomal RNA is known to require the cellular environment for proper folding, and differences between in vivo and in vitro icSHAPE measurements highlighted important structural elements in the intact ribosome. We mapped nucleotides of icSHAPE difference to the cryo-EM structure of the human 80S ribosome6 and searched for differences between the in vivo and in vitro conditions. Conserved (mouse to human) nucleotides of high icSHAPE signal in vivo were unpaired in the cryo-EM structure (Fig. 1e); conversely residues lacking icSHAPE reactivity in vivo were base-paired or engaged in extensive interactions that may stabilize the RNA backbone in the mature ribosome (Fig. 1f). Overall, these data demonstrate that icSHAPE accurately measures RNA structures, both in and outside of living cells.
We next used icSHAPE to measure RNA structural profiles of polyadenylated transcripts in mESCs and generated ~2.1 billion measurements for over 13,200 RNAs in vivo and in vitro, with high reproducibility (Extended Data Fig. 5 and 6). The nucleotide composition in the transcriptome, mock-treated RNA, and icSHAPE RNA are highly concordant, with a slight enrichment in NAI-N3 samples for A’s and U’s (Fig. 2a). This enrichment is expected given their bias for being located in single stranded or loop regions7. icSHAPE thus affords the first complete RNA structurome of all four nucleotides in vivo.
Figure 2. icSHAPE Reveals Unique Structural Profiles for Nucleobase Reactivity and Posttranscriptional Interactions.

a, RT stop distribution for the transcriptome, DMSO control, or icSHAPE libraries. b, icSHAPE data track and VTD calculation of the MALAT1 RNA (chr19:5796010-5796081). icSHAPE data are scaled from 0 (no reactivity) to 1 (max reactivity). c, The VTD distribution for icSHAPE libraries. d, GINI index of icSHAPE data in vivo vs. in vitro. e, The distribution of VTD profiles for all hexamer motifs across the transcriptome. Bin locations for several motifs for post-transcriptional regulation are highlighted.
icSHAPE data revealed the scale and distribution of RNA structural dynamics between in vitro, where folding is programmed entirely by sequence, versus in vivo where folding occurs in the context of the intracellular environment.8 Recent transcriptome-wide dimethylsulfate probing (DMS-seq), which interrogates two bases with strong bias toward adenosines (68% A’s and 24% C’s)2,4, suggests that RNA structures are largely unfolded in vivo2; however sampling only two of four nucleotides could result in an incomplete picture. We quantified RNA structural dynamics using two metrics: First, we calculated the difference in reactivity between our in vivo and in vitro icSHAPE measurements, termed the Vivo – Vitro Difference (VTD, Fig. 2b and Methods). Adenosine residues have the largest VTD, whereas guanosine and cytidine residues are less variable between environments (Fig. 2c). These observations suggest that utilizing probes that have a broader reactivity profile such as NAI-N3 will give a more complete representation of RNA structure.
Second, we used the GINI index2 to quantify the distribution of icSHAPE reactivity profiles. Structured RNAs have some bases that are reactive and some not, leading to unequal distribution and high GINI, while unfolded RNAs have most bases in a uniformly reactive conformation (low GINI). We found that RNAs are less folded in vivo, consistent with a prior report2, but the extent of unfolding varies in degrees that distinguish different classes of RNAs (Fig. 2d). Protein-coding mRNAs exhibited noticeable but partial unfolding (average GINI of 0.7 in vitro to 0.5 in vivo), with the largest variation noted at 3’ untranslated regions (UTR) compared to coding sequences (CDS) or 5’UTRs. In contrast, noncoding RNAs, such as pseudogenes, long noncoding RNAs, and primary microRNA precursors, retain substantially more of their RNA structure in vivo (p< 2.2×10−16, noncoding vs. coding, Student’s t-test). One exception to this rule are snoRNAs, which exhibit the greatest level of increased reactivity in vivo among all classes of transcripts and may result from extensive rearrangements due to snoRNP binding. Most RNAs in vivo possess a substantial level of RNA structure beyond previous expectation based on DMS-seq2. The current data suggest that RNA structural signatures in vivo can distinguish coding vs. structural or regulatory RNAs, consistent with prior in vitro studies9-12.
The dramatically different environments that RNA experiences when inside a cell compared to in vitro predicts that our VTD parameter could provide insight into functionally important RNA regulatory elements. To assess this possibility, we measured the VTD for all hexamer sequences (Fig. 2e, Supplementary Table 1). We observed unique VTD profiles for sequence motifs driving diverse post-transcriptional processes, including translation initiation, interaction with RNA binding proteins (RBPs, e.g. Rbfox2), RNA modification (m6A), and microRNA seed matches13,14,15 (Supplementary Table 1 and 2). These results show that the VTD may classify RNA regulatory elements as pre-programmed or sensitive to in vivo remodeling. Further, distinctive VTD profiles precisely at sites of post-transcriptional regulatory motifs suggest that RNA structural dynamics may be used to monitor these regulatory events in cells.
We hypothesized that translational regulatory elements may have their icSHAPE profiles conserved between in vivo and in vitro because the Kozak sequence, important for translation initiation16, is among the most stable (low VTD) regions within mRNAs (Fig. 2d). RNA accessibility from −1 to −5 nucleotides upstream of the start codon plays a major role in regulating translational output10,17. We used translation initiation18 and pause sites18, defined by ribosome profiling, to center our structural reactivity analysis across the transcriptome (Fig. 3). Canonical initiation AUG sites are indeed preceded by ~5 nts of increased accessibility, and this pattern is nearly identical to in vitro folded RNA (Fig. 3a and 3b). A similar pattern of conserved upstream accessibility also precedes noncanonical start sites at upstream open reading frames (uORF) and N-terminal truncations (Fig. 3c). Non-start site AUG codons are also associated with increased preceding reactivity, while noncanonical CUG start codons have a different profile, suggesting that RNA accessibility alone is not sufficient to dictate translational start sites (Extended Data Fig. 7). Ribosome profiling also defined ribosome pause sites as having a strong preference for glutamate or aspartate in the A-site, where transfer RNA (tRNA) identity and the nascent peptide sequence are believed to strongly influence translation kinetics18. icSHAPE data at ribosome pause sites revealed a distinctive signature: loss of reactivity at the E and P sites while the A site is more reactive, preceded by strong 3-nt periodic reactivity pattern 5’ to the pause site for ~12 nts (Fig. 3d and 3e). Furthermore, a very similar pattern was observed in vitro under conditions that do not maintain mRNA interactions with the ribosome or tRNAs, suggesting that these structural profiles are programmed by mRNA sequence. Analysis of negative control sites – defined as sites on the same transcripts that match the codon composition, are in frame, and are at least 20 nts away from true pause sites – showed a very similar icSHAPE signature at the presumed ribosome E, P, and A sites, but negative controls lacked the 5’ periodic signal (gray box in Fig. 3e and 3f). This observation suggests that the icSHAPE signature at ribosome pause sites is likely due to the codon bias at such sites, but sequences 5’ to the pause may influence pausing. These results identify several physiological structural signatures of translational control elements, and suggest they may be largely pre-programmed by mRNA sequence.
Figure 3. icSHAPE Reveals Structural Profiles Associated with Translation.

a, Cartoon representation of ribosomes translating an mRNA. The uORF initiation site is represented by ribosome initiation upstream of the canonical start codon. The canonical start position is demarcated by “AUG”. The N-terminal truncation is represented as a ribosome initiating to the 3’-end of the canonical start “AUG”. b, icSHAPE profile at canonical start codon position. c, icSHAPE profiles at uORF and N-terminal truncation sites. d, Cartoon representation of a paused ribosome and its corresponding A-P-E sites. A: acceptor; P: peptidyl-transferase; E: exit. e, The icSHAPE profile at ribosome pause sites. f, icSHAPE profile at negative control sites for pause sequences. Gray box highlights a region of structural difference upstream of true pause sites vs. controls.
In contrast, focal RNA structural rearrangements in vivo can identify sites of RBP interactions, which regulate RNA splicing, localization, and stability19 (Fig. 2d). The forkhead box (Fox) family of RBPs important for tissue-specific control of alternative splicing, with Rbfox2 playing key roles in ES cells.14,20 High VTD at the known Rbfox2 binding motif (UGCAUG)14,20 indicates a strong structural rearrangement in vivo. Alignment with Rbfox:RNA NMR structure21 and Rbfox2 binding sites identified by individual-nucleotide crosslinking immunoprecipitation (iCLIP) in mESC20 showed that differential icSHAPE signal precisely matches the key RNA residues involved in Rbfox interaction (Fig. 4a and 4b). U1, G2, and A4 in the motif showed strong icSHAPE VTD signal. The 2’ hydroxyl groups of these three residues are flipped outward while G2 and A4 base pair upon Rbfox interaction21, consistent with adoption of new structural environments in vivo that we detect at these residues. In principle, dynamic structural footprints of RBPs may enable comprehensive readout of RNA-RBP interactions in vivo. We tested this idea by implementing a Support Vector Machine (SVM) algorithm to learn dynamic icSHAPE signals that are best able to predict sites of RNA regulation, using held out data for cross-validation of prediction accuracy (Extended Data Fig. 8 and Methods). Indeed, the combination of both in vivo and in vitro icSHAPE data increased the ability to predict true Rbfox2 binding sites compared to motif sequence or conservation alone, particularly at lower false positive rates where accuracy is most important (Area Under the Curve [AUC] = 0.74, Extended Data Fig. 8).
Figure 4. icSHAPE Dynamics Reveals and Predicts Post-transcriptional Interactions.

a, Structure of Rbfox1-RNA interaction, highlighting the RNA-protein interface. The RNA is shown with a blue backbone and orange bases; each 2’hydroxyl is green (PDB:2ERR). b, The differential icSHAPE profile at Rbfox2 target mRNAs measured in vivo vs. in vitro maps precisely to the Rbfox binding sites. c, Model of interplay between m6A and RNA structure. d, Differential icSHAPE signal for m6A methylated vs. non-methylated sites with the same underlying sequence motif, both in vivo. icSHAPE signal from unmodified sites are subtracted from m6A-modified sites; asterisks (*) indicate positions with significant differences (p<0.05, FDR<0.05). Data from wild type and Mettl3 KO mESCs are plotted for comparison. e, ROC curve for prediction of m6A sites, incorporating icSHAPE profiles. f, Effect of m6A on RNA structure on Nanog mRNA. Top: location of Mettl3-dependent m6A sites (highlight in yellow); m6A-RIP data from Batista et al25. Bottom: icSHAPE profile of WT and Mettl3 KO cells.
As an independent validation, we used icSHAPE data to predict the binding sites of HuR, a RBP that regulates transcript stability15, and also performed the first HuR iCLIP in mESCs. Comparing in vivo vs. in vitro icSHAPE data precisely identified peaks of structural arrangement at authentic HuR binding sites (defined by iCLIP sites), and enabled reasonably accurate prediction of HuR binding from icSHAPE data alone (AUC = 0.841, Extended Data Fig. 8, HuR iCLIP data in Extended Data Fig. 9). Thus, icSHAPE data can distinguish true binding sites from other sequence motif instances, collectively boosting prediction accuracy.
We also identified a critical connection between RNA structure and RNA modification, a newly appreciated and pervasive mode of post-transcriptional control13. The most prevalent modification in mRNAs, m6A, occurs at GGm6ACU motifs near stop codons, and acts in part to control RNA splicing and stability22,23. It has been hypothesized that m6A methylation occurs at sites that contain un-paired motifs24, but no direct structural evidence has been presented to support this model. Comparison of icSHAPE signals at m6A-modified vs. unmodified instances of the GGACU motif in mESCs25 revealed a specific structural signature, with stronger icSHAPE reactivity (consistent with unpaired RNA) at positions both surrounding and including the modified A (Fig. 4d, Extended Data Fig. 8). m6A sites in different subdomains of mRNAs or in lncRNAs have nearly identical icSHAPE profiles (Extended Data Fig. 10). Evaluation of all predictive features using our SVM algorithm showed that motif conservation or motif position offers some predictive value (AUC=0.617 or 0.824, respectively) as previously reported24, but use of icSHAPE data (AUC = 0.846) or all features together (AUC = 0.914) improved prediction rate (Fig. 4e). These results show that icSHAPE structure profiles can be used to accurately predict post-transcriptional modifications on a transcriptome-wide scale.
The strong RNA structural signature at m6A sites may arise from the ability m6A to destabilize RNA helices26 (represented in Fig. 4c) or the structural selectivity of m6A modification machinery for unpaired bases. In the former scenario, removal of m6A should cause increased basepairing (loss of icSHAPE signal) whereas the latter scenario predicts little change to RNA structural profile. To distinguish between these hypotheses, we determined the icSHAPE profile of mESCs genetically ablated for Mettl325, a key m6A methyltransferase that is required for ESC differentiation. We observed that in Mettl3 knockout cells, canonical motif sites that lost m6A modification also substantially lost icSHAPE signal transcriptome-wide (Fig. 4d), as exemplified by key m6A target sites in Nanog mRNA (Fig. 4f). These results suggest that m6A impacts RNA structure, favoring the transition from paired to unpaired RNA. The ability to couple genetic perturbation with comprehensive, base-resolution structural maps in vivo is a potentially powerful approach to dissect regulators of RNA structure.
Understanding how RNA structures contribute to biological regulation opens the door to understanding a physical dimension of the transcriptome. icSHAPE bridges a gap in RNA sequencing technologies that currently lack the ability to infer a mechanistic basis of biological function. The ability to view the structural dynamics of all four RNA bases in living cells is essential to uncover specific sequence motifs underlying different modes of post-transcriptional regulation27, and has enabled accurate identification and de novo prediction of trans-acting factor binding and chemical modification at single nucleotide resolution. In the future, viewing the RNA structurome when cells are exposed to different stimuli or genetic perturbations should revolutionize our understanding of gene regulation in biology and medicine.
Methods
Methyl 2-(azidomethyl)nicotinate
1.00 g of methyl 2-methylnicotinate was dissolved in 5 mL anhydrous dichloromethane. 2.30 g trichloroisocyanuric acid was added and the resulting suspension stirred overnight at room temperature. The reaction was diluted with dichloromethane and quenched by the addition of saturated sodium bicarbonate solution. The phases were separated and the organic phase was washed once with brine, dried over magnesium sulfate, filtered, and concentrated to afford a yellow oil. NMR data was consistent with literature reports.
The crude product of the above reaction (1.09 g) was dissolved in 12 mL anhydrous N,N-dimethylformamide and 0.77 g sodium azide was added. The reaction was stirred overnight at room temperature then quenched with saturated sodium bicarbonate solution. The aqueous layer was extracted with ethyl acetate, and the organic layer washed 3 times with water and 3 times with brine. The organic layer was dried over magnesium sulfate, filtered, and concentrated to afford 0.91 g (71%, 2 steps) of a yellow oil that solidified upon standing.
1H NMR (400 MHz, CDCl3): 3.94 (3H, s), 4.88 (2H, S), 7.37 (1H, m), 8.29 (1H, dd, J = 8 Hz, 1.6 Hz), 8.76 (1H, dd, J = 4.6 Hz, 1.6 Hz)
13C NMR (100 MHz, CDCl3): 52.8, 54.4, 122.9, 125.0, 139.1, 152.4, 156.7, 166.0
ESI-MS (Calc M-H = 191.06): 191.98
2-(azidomethyl)nicotinic acid
0.50 gram of methyl 2-(azidomethyl)nicotinate was stirred vigorously in 10 mL of 1:1 MeOH:10% aqueous NaOH. After 10 minutes TLC indicated complete consumption of starting material. 25 mL of water were added, the crude reaction mixture was washed once with ether (10 mL) then acidified to pH 4 with 10% aqueous HCl and extracted 5 times with 50 mL ethyl acetate. The organic layer was dried over magnesium sulfate, filtered, and concentrated to afford 0.46 g (99%) of a white solid that was pure by NMR.
1H NMR (400 MHz, DMSO-d6): 4.81 (2H, s), 7.50 (1H, m), 8.28 (1H, dd, J = 7.8 Hz, 1.6 Hz), 8.74 (1H, dd, J = 5 Hz, 1.6 Hz), 13.64 (1H, br. s).
13C NMR (100 MHz, DMSO-d6): 53.4, 123.4, 125.9, 139.0, 151.9, 156.0, 167.1
ESI-MS (Calc M-H = 177.04): 177.05
2-(azidomethyl)nicotinic acid acyl imidazole
0.15 g 2-(azidomethyl)nicotinic acid was dissolved in 0.21 mL anhydrous dimethylsulfoxide. A solution of 0.14 g carbonyldiimidazole in 0.21 mL anhydrous dimethylsulfoxide was added dropwise, creating rapid gas evolution. The reaction was allowed to proceed for one hour and the resulting solution used as a 2M stock solution for RNA SHAPE experiments. For NMR data collection an analytical sample was prepared in dichloromethane as described above. The reaction was stirred over night and the solvent removed in vacuum. The product was then isolated by flash column chromatography on silica (ethyl acetate).
1H NMR (400 MHz, DMSO-d6): 4.62 (2H, s), 7.16 (1H, dd, J = 1.7 Hz, 0.8 Hz), 7.60 (1H, dd, J = 7.9 Hz, 4.9 Hz), 7.66 (1H, m), 8.15 (2H, m), 8.84 (1H, dd, J = 4.9 Hz, 1.7 Hz).
13C NMR (100 MHz, DMSO-d6): 52.8, 117.9, 123.0, 127.3, 130.9, 137.7, 138.5, 151.9, 154.6, 164.7
In Vitro transcription and acylation of RNA
RNA was transcribed from amplified inserts using T7 Megascript kit from Ambion, following manufacturer’s protocol. In a typical in vitro modification protocol, RNA was heated in metal-free water for two minutes at 95°C. The RNA was then flash-cooled on ice. The RNA 3x SHAPE buffer (333 mM HEPES, pH 8.0, 20mM MgCl2, 333mM NaCl) was added and the RNA was allowed to equilibrate at 37°C for ten minutes. To this mixture, 1μL of 10x electrophile stock in DMSO (+) or DMSO (−) was added. The reaction was permitted to continue until the desired time. Reactions were cleaned up using RNeasy columns (Qiagen) following manufactures protocol and eluted RNase-free water.
In Vitro manual SHAPE analysis
32P-end-labeled DNA primer (reverse primer above) was annealed to 3μg of total RNA by incubating at 95°C for two minutes followed by a step-down cooling (2°/sec) to 4°C. To the reaction first-strand buffer, DTT and dNTPs were added. The reaction was pre-incubated at 52 °C for one minute, then Superscript III (2U/μLfinal concentration) was added. Extensions were performed for ten minutes. To the reaction, 1μL of 4M NaOH was added and allowed to react for 5 minutes at 95°C. 10μL of Gel Loading Buffer II (GLBII, Ambion, Inc.) was then added, and cDNA extensions were resolved on 8% denaturing (7M Urea) polyacrylamide gels (29:1 acrylamide:bisacrylamide, 1xTBE). All (−) lanes are those from DMSO control treated cells. In addition, all sequencing lanes are from DMSO control treated cells. cDNA extensions were visualized by phosphorimaging (STORM, Molecular Dynamics). cDNA bands were integrated with SAFA28 SHAPE reactivities were normalized to a scale spanning 0 to 1.5, where 1.0 is defined as the mean intensity of highly reactive nucleotides.29 RNA secondary structures were predicted using mFOLD software.30
Characterization of manual SHAPE-enriched RT stops
Copper-free click chemistry of acylated RNA
In a typical reaction, acylated RNA (1pmol) was reacted with 100eq. of DIBO-biotin (Life Technologies) for two hours, at 37°C, in 1x Phosphate Buffer Saline (PBS). Reactions were extracted once with acid phenol:chloroform (pH 4.5±0.2) and twice with chloroform. RNA was precipitated with 40μL of 3M sodium acetate buffer (pH 5.2) and 1μL of glycogen (20μg/μL). Pellets were washed twice with 70% ethanol and resuspended in 10μL RNase-free water.
Enrichment of NAI-N3 modified RNA
The following protocol was used for manual enrichment protocols used to optimize capture conditions. To 1pmol of precipitated and biotinylated RNA (in 900μL of binding buffer: 50mM Tris-HCl pH 7.0 and 1mM EDTA) was added 50μL (slurry) of DYNAL MyOneC1 beads (Life Technologies) was added. The reaction mixture was then incubated for one hour at room temperature. The beads were then collected on a magnetic plate and the solution decanted. The beads were then resuspended and washed 4 times with Biotin Wash Buffer (10mM Tris-HCl, pH 7.0, 1mM EDTA, 4M NaCl, 0.2% Tween). The beads were then washed 3x with RNase-free water. To elute the purified RNA off the resin beads were incubated in 1X proteinase K buffer with 20U of proteinase K (Life Technologies), 1mM D-Biotin (Sigma Aldrich), and 20U of SUPERaseIn (Life Technologies). The reaction was permitted to run for 30 minutes at 37°C.Beads were then collected by magnet and the supernatant removed and set on ice. This was repeated twice more and elutions were pooled. Reactions were extracted once with acid phenol:chloroform (pH 4.5±0.2) and twice with chloroform. RNA was precipitated with 40μL of 3M sodium acetate buffer (pH 5.2) and 1μL of glycogen (20 μg/μL). Pellets were washed twice with 70% ethanol and resuspended in 10μL RNase-free water.
Dot Blot analysis of enriched NAI-N3 modified RNA
Hybond N+ membranes (GE) were pre-incubated in 1xPBS. Precipitated RNA was dissolved in 100μL of 1xPBS. RNA was added to the Hybond membrane and crosslinked using 254nm UV light. The Hybond membrane was washed three times with 1xPBS. To the membrane was added NorthernLights Streptavidin NL493 (in PBS-Tween20) for visualization. After incubation, the membrane was washed 3x in 1xPBS-Tween20. The membrane was dried and imaged by phosphorimaging (STORM, Molecular Dynamics).
Tissue culture and In Vivo SHAPE modification
Mouse embryonic stem cells (v6.5 line) were grown on gelatinized dishes in serum and LIF. Unmodified total RNA was extracted by removing media, washing once in room temperature 1xPBX, and adding 2mL (10cm dish) or 7mL (15cm dish) of Trizol directly to the cells. Subsequent RNA extract was performed using the miRNeasy mini- or midi-column and protocol (Qiagen) as recommended by the manufacture. In Vivo modification of cellular RNAs was performed as described previously.5 Briefly, cells were rinsed once on the plate in room temperature 1xPBS, decanted, scraped in 1xPBS, and collected into a 15mL tube. Cells were pelleted at room temperature and resuspended in 450μL of 1xPBS. 50μL of 10x electrophile stock in DMSO (+) or DMSO (−) was added drop-wise, immediately mixed by inversion, and incubated at 37°C on end-over-end rotation for 20 minutes. Reactions were pelleted for 1min at 4°C at 10,000 rpm and resuspended in 500μL of 1xPBS. Samples were then transferred to 15mL tubes with 2-7mL of pre-aliquoted Trizol and RNA extracted as described above.
Methods to ensure titrated hit-kinetics of RNA modification
We titrated NAI-N3 for single-hit kinetics that are comparable to those routinely used in chemical probing of RNA structure. For example, we obtained nearly identical secondary structure for 5S rRNA as previously reported with single hit regime.5 After NAI-N3 modification and biotin pulldown, we retrieved approximately 10-20% of the input RNA as modified RNA, consistent with the expected Poisson distribution of single-hit modification.
icSHAPE deep sequencing library preparation
RNA preparation
DMSO (mock) or NAI-N3 (experimental) modified total RNA was used as input for the deep sequencing library preparation. Before library preparation input RNA should be modified (or mock treated) under In Vitro or In Vivo conditions as described above. For “total RNA” libraries, no additional processing was needed. For “poly-A selected” samples, 200μg of total RNA was used per poly-A purist column (Ambion, Inc.), which should yield ~2μg of enriched RNA. Poly-A selection was performed a total of two times using the same poly-dT beads (“double poly-A selection). The NAI-N3 sample may have lower yield post purification so additional starting material could be required.
NAI-N3 biotinylation and RNA fragmentation
All RNA samples (NAI-N3 and DMSO treated) are processed through a copper-free “click” reaction. RNA is brought to 97μL in 1xPBS and 1μL of SUPERaseIn and 2μL of 185mM DIBO-Biotin are added. Samples were mixed by brief vortexing and then incubated at 37°C for 2 hours in a Thermomixer (Eppendorf). Reactions were stopped by adding 350μL of Buffer RLT (Qiagen) and then 900μL of 100% ethanol (EtOH). Each RNA sample was processed by passing over a RNeasy Mini column (Qiagen), two 500μL washes with Buffer RPE (Qiagen), one no-buffer spin to dry the column, and finally two 50μL elutions in RNase-free water (final 100μL). Samples were then frozen for 5 minutes on dry ice and concentrated to 9μL using a lyophilizer (Labconco). Concentrated RNA samples (9μL) were then moved to 0.5mL PCR tubes for fragmentation. Samples were heated to 95°C for 90 seconds and then 1μL of 10x RNA Fragmentation Reagent (Ambion, Inc.) was added and samples were placed back at 95°C for 70-90 seconds. Reactions were quenched by adding 1μL of RNA Fragmentation Stop Solution (Ambion, Inc.) and moved to ice. RNA was cleaned up by adding 35μL of Buffer RLT and 100μL of 100% EtOH and purified using RNeasy Mini columns as above. Samples are then concentrated with a lyophilizer to 5μL.
RNA end repair, RNA ligation, and RNA size selection
To resolve the 3’-end phosphate generated by the fragmentation process T4 PNK is used. To each 5μL sample 2μL of 5x PNK buffer (350mM Tris-HCl pH 6.5, 50mM MgCl2, 25mM DTT), 1μL SUPERaseIn, and 2μL of T4 PNK (NEB) is added, mixed by flicking, and incubated at 37°C for 1 hour. After end-repair samples are moved directly to 3’-end ligation by adding 1μL of 50μM 3’ Adaptor, 1μL of 10x T4 RNL2tr buffer (NEB), 1.5μL of T4 RNL2tr K227Q (NEB), 1μL of 100mM DTT, and 8μL of 50% PEG8000. Mix samples by flicking and incubate at 16°C overnight.
Ligation Note
NAI-N3 samples must use “3’-Adaptor-3’ddc” (/5rApp/AGATCGGA AGAGCGGTTCAG/3ddC/) while DMSO samples must use “3’-Adaptor-3’Biotin” (/5rApp/AGATCGGAAGAGCGGTTCAG/3Bio/). The “click” chemistry will label only the NAI-N3 modified RNAs in the NAI-N3 pool of transcripts with a biotin moiety, thus allowing the selective purification of structurally informative molecules. The DMSO samples are not capable of “click” chemistry and every molecule in this pool is desired for sequencing so addition of a biotin moiety must happen in an unbiased fashion. Thus, DMSO samples have a 3’Biotin modification added specifically to their 3’ Adaptor to allow for downstream processing in parallel of the DMSO and NAI-N3 samples.
After the overnight ligation 30μL of water, 185μL of Buffer RLT, and 400μL of 100% EtOH is added to each sample and purified using RNeasy Mini columns as above. Samples are concentrated to 5μL using a lyophilizer and 5μL of GLBII is added and stored on ice. To size select the RNA samples a mini 6% TBE PAGE gel with 7M Urea is cast and pre-run to 50W for 8 minutes. Samples are loaded without prior heating and the PAGE gel is imaged using a 1:10,000 dilution of SybrGold (Life Technologies). RNA is visualized on a BlueBox (Clare Chemical) and fragmented RNA ranging between 20-120nts (40-140nts with the 3’ Adaptor ligated) are excised with a scalpel. Gel slices are crushed through a 0.75mL tube nested in a 2mL tube by centrifugation and 300μL of Crush Soak Buffer (500mM NaCl, 1mM EDTA) is added with 3μL of SUPERaseIn. RNA is eluted overnight at 4°C on rotation.
RT, streptavidin capture, cDNA elution, and cDNA size selection
RNA samples are purified away from residual PAGE using 0.45μm Spin-X columns (Corning) and the 300μL elutions are transferred to silicionized 1.5mL tubes (Fisher Scientific, used in all subsequent steps). RNA is precipitated by adding 30μL of 3M sodium acetate buffer (pH 5.2), 0.8μL of GlycoBlue (Ambion, Inc.) and 1mL of 100% EtOH. Samples are frozen for 1 hour on dry ice, spun at max speed (15,000 rpm) for 1 hour at 4°C, washed with 800μL of ice-cold 80% EtOH, decanted, air-dried and then resuspended in a 0.5mL PCR tube with 11.5μL of water. To the RNA samples add 1μL of 10μM RT primer (/5phos/DDDNNAACCNNNN AGATCGGAAGAGCGTCGTGAT/iSp18/GGATCC/iSp18 /TACTGAACCGC, /5phos/ = 5’ phosphate, D=A/T/G, /iSp18/ = 18carbon PEG spacer) and 1μL of 10mM dNTPs. Heat the samples to 70°C for 5 minutes and then cool slowly to 25°C (2deg/second) and hold at 25°C for one minute. After primer annealing add 0.5μL of SUPERaseIn, 1μL 100mM DTT, 4μL of 5x First Strand Buffer and 1μL of SuperScript III (Life Technologies). cDNA extension occurs for 3 minutes at 25°C, 7 minutes at 42°C, and finally at 52°C for 15 minutes. After cDNA extension do not raise samples above 37°C to avoid denaturing conditions.
MyOneC1 streptavidin beads for cDNA capture and NAI-N3 modified RNA enrichment are prepared (40μL slurry per sample) by washing 3x in 1mL of Biotin Bind Buffer (100mM Tris-HCl pH 7.0, 10mM EDTA, 1M NaCl) and resuspending the beads in 40μL Biotin Bind Buffer and 1μL SUPERaseIn per reaction. After the reverse transcription reaction completes 40μL of pre-washed beads are added to each sample, mixed by flicking, and incubated at room temperature for 45 minutes. After streptavidin capture samples are washed at room temperature serially with 4×100μL of Biotin Wash Buffer, 2×100μL 1xPBS and finally moved to 1.5mL tubes. cDNA is eluted by adding 1μL RNaseA/T1 cocktail (Ambion, Inc) 1μL RNaseH (Enzymatics), 12.5μL 50mM D-Biotin, 5μL 10x Elution Buffer (500mM HEPES, 750mM NaCl, 30mM MgCl2, 1.25% Sarkosyl, 0.25% Na-deoxycholate, 50mM DTT), 30.5μL water and incubating at 37°C for 30min in a Thermomixer at 800 rpm. Samples are mixed with 1μL 100% DMSO, heated to 95°C for 3 minutes, placed on a magnet, and the 50μL cDNA elution moved to a new tube. The elution is repeated once (total of two times and final of 100μL). cDNA is processed by adding 1mL of Buffer PNI and purifying over a MiniElute columns (Qiagen) following manufactures protocol and eluting twice in 15μL of Buffer EB (final 30μL). cDNAs are concentrated using a lyophilizer to 5μL and equal volume of GLBII is added. Size selection of cDNAs is performed as was done for the RNA size selection. 6% PAGE gel pre-running is critical to achieve denaturing conditions as well as heating the samples to 95°C for 3 minutes prior to PAGE separation. cDNAs are selected for insert sizes of ~20-120nts (~85-205nts with RT primer extension) and depending on the input material amount the libraries may be invisible at this step. Gel slices are crushed as above, 300μL of Crush Soak Buffer is added and cDNAs are eluted at 50°C overnight on rotation.
cDNA circularization, library qPCR, library size selection, sequencing PCR
Purification of eluted cDNA is performed as above for RNA elution. After cDNA precipitation samples are resuspended in 16μL of water, 2μL of 10x CircLigaseII Buffer, 1μL of CircLigaseII (Epicentre) and moved to 0.5mL PCR tubes. cDNA circularization takes place at 60°C for 120 minutes in a PCR machine. Circularized cDNA is purified by adding 200μL of Buffer PNI and processing as above using MiniElute columns, eluting the cDNA twice in 14μL (final ~27μL). Samples are initially amplified in a 60μL qPCR reaction (27μL cDNA, 30μL 2x Phusion HF Master Mix, 0.75μL of 10μM P3_short primer (CTGAACCGCTCTTCCGATCT), 0.75μL of 10μM P5_short primer (ACACGACGCTCTTCCGATCT), 0.72μL of 25x SybrGold). The qPCR machine is programmed as follows: 98°C for 1 minute, 98°C for 15 seconds, 62°C for 30 seconds, 72°C for 45 seconds). After qPCR amplification samples are purified with 600μL of Buffer PNI and MiniElute columns as above. Library DNA is eluted twice in 15μL (total 30μL) and concentrated using a lyophilizer to less than 5μL. A second 6% TBE 7M Urea PAGE gel selection is performed as above to remove any PCR dimer products and all short qPCR primers. Gel slices are crushed as above and eluted overnight at 50°C on rotation. Purification of library DNA is performed as above post PAGE gel elution and after precipitation resuspended in 19μL of water. A final library PCR amplification is performed for three cycles in 40μL reactions (19μL library DNA, 0.5μL of 10μM P3_solexa primer (CAAGCAGAAGACGGCA TACGAGATCGGTCTCGG CATTCCTGCTGAACCGCTCTTCCGATCT), 0.5μL of 10μM P5_solexa primer (AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACA CGACGCTCTTCCGATCT)) and cleaned up using Agencourt AMPure XP beads (Beckman) according to manufactures protocol and eluted the library in 20μL of water. Final library material was quantified on the BioAnalyzer High Sensitivity DNA chip (Agilent) and then sent for deep sequencing on the Illumina HiSeq2500 machine for 1×100bp cycle run.
iCLIP and data analysis
The iCLIP method was performed as described before with the specific modifications below36. v6.5 mESCs were grown as described above and UV-C crosslinked to a total of 0.3J/cm2. Whole cell lysates were generated in CLIP lysis buffer (50mM HEPES, 200mM NaCl, 1mM EDTA, 10% Glycerol, 0.1% NP-40, 0.2% TritonX-100, 0.5% N-lauroylsarcosine) and briefly sonicated using a probe-tip Branson sonicator to solubilize chromatin. Each iCLIP experiment was normalized for total protein amount, typically 2mg, and partially digested with RNaseA (Affymetrix) for 10 minutes at 37°C and quenched on ice. Immunoprecipitations of HuR were carried out with Protien G Dynabeads (Life Technologies) and anti-HuR antibody (3A2, Santa Cruz) for 3 hours at 4°C on rotation. Samples were wash sequentially in 1mL for 5min each at 4°C: 2x high stringency buffer (15mM Tris-HCl pH7.5, 5mM EDTA, 2.5mM EGTA, 1% TritonX-100, 1% Na-deoxycholate, 120mM NaCl, 25mM KCl), 1x high salt buffer (15mM Tris-HCl pH7.5, 5mM EDTA, 2.5mM EGTA, 1% TritonX-100, 1% Na-deoxycholate, 1M NaCl), 1x NT2 buffer (50mM Tris-HCl pH7.5, 150mM NaCl, 1mM MgCl2, 0.05% NP-40). 3’-end RNA dephosphorylation, 3’-end ssRNA ligation, 5’ labeling, SDS-PAGE separation and transfer, autoradiograph, RNP isolation, ProteinaseK treatment, and overnight RNA precipitation took place as previously described36. The 3’-ssRNA ligation adaptor was modified to contain a 3’biotin moiety as a blocking agent. The iCLIP library preparation was performed as described previously36. Final library material was quantified on the BioAnalyzer High Sensitivity DNA chip (Agilent) and then sent for deep sequencing on the Illumina HiSeq2500 machine for 1×75bp cycle run. iCLIP data analysis was performed as previously described.36
RNA Structure Analysis
Sequencing, reads mapping, and data quality control
We generated four replicates for each library (DMSO PolyA, NAI PolyAin vivo and in vitro). We performed single-end sequencing on Illumina’s HiSeq sequencer and obtained approximately 200-million to 600-million raw reads for each replicates, totaling 3.9-billion reads. We collapsed these reads to remove PCR duplicates (only reads that have identical sequences including barcode region are regarded as duplicates). Collapsed reads were then subjected to barcode removal and primer and linker trimming by using Trimmomatic31. We mapped trimmed reads to the mouse transcriptome of the Ensembl annotation (build GRCm38.7432) by using Bowtie233.For reads that can be mapped to multiple locations of the transcriptome, we evenly distribute them to up to ten random hits. Finally, we obtained 2.1 billion mapped reads in total. We define the −1 positions of the 5-prime the mapped reads as RT stops, which correspond to modified positions in the NAI libraries, and intrinsic modified (or fragmentation) positions in the DMSO libraries. We defined RT stop coverage as the number of times a base is mapped as a RT stop.
We calculated the expression level of all transcripts in the mouse transcriptome in terms of RPKM. The correlations of transcript expression value (RPKM>0.1) in different replicates are very high (in the range of 0.96 to 1.00). We constructed the background base density profile for each transcript as the sequencing depth of each base in the DMSO libraries. We also calculated the correlation of RT stops for each transcript in different replicates. As shown in Extended Data Figure 5, the correlation is high for most transcripts if we limit to transcripts of average RT stop coverage higher than 2 and regions of background base density higher than 200. So for each library (DMSO poly-A, NAI poly-A in vivo and in vitro) we combine all four replicates into one for the following analysis.
Reactivity score calculation and construction of structural profile
We performed a 5%-5% normalization for each transcript (i.e., The mean of the RT stops of the second top 5% bases, excluding the 32 bases at the beginning and 32 bases in the end of the transcript, will be normalized to 1. All RT stops will be normalized proportionally).
We defined reactivity score as the subtraction of background RT stops (DMSO libraries) from RT stops of the modified NAI libraries, and then adjusted by the background base density:
R = (RT_stopNAI – αRT_stopDMSO) / background_base_densityDMSO
The score is then scaled into the range of [0, 1], after removing the outliers by 90% Winsorization (the top 5th percentile is set to 1 and the bottom 5th percentile is set to 0. We trained the parameter α on the ribosomal RNA structures, and set it to 0.25 to maximize the correlation of reactively score R determined by deep sequencing and reactivity score measured in low throughput gel shift experiments.
For each transcript, we defined its structural profile as the vector of base-resolution reactivity scores from the beginning to the end. The valid structural profile of a transcript is limited to regions of RT stop coverage higher than 2 and background base density higher than 200. Finally, we obtained valid structural profiles for, respectively, 19,347 and 13,281 transcripts from in vivo and in vitro, polyA-selected RNA libraries, among which the majority are messenger RNAs (Extended Data Figure 6).
Metagene analysis of translation sites, pause site, m6A sites and protein binding sites
We calculated metagene structure profile around different functional sits by averaging all valid reactivity score R: (1), 10 nts upstream and downstream of the RNA methylation m6A site, as determined in by our lab previously34; (2), 25 nts upstream and downstream of the translation pause site determined in the same ribosome profiling experiment; (3), 25 nts upstream and downstream of the RNA methylation m6A site, (4) 25 nts upstream and downstream of the binding sites of RNA binding protein Rbfox235 and HuR (Extended Data Figure 8 and 9. See details below).
In the analysis of differential profiles of icSHAPE reactivity scores around m6A and negative control sites, we retrieved a set of target m6A sites that have icSHAPE reactivity scores in both wild type and Mettl3 knock out cells, and defined a set of similar number of non-methylated m6A site with the same sequence motifs (GGACU). For both wild type and knock out cells, we calculated the profiles of average reactivity scores in target sites and negative controls and subtracted the latter from the former scores to define the differential icSHAPE profiles.
We calculated in vivo and in vitro metagene structure profile separately. For each transcript functional sites and their flanking regions, we demand a stringent R score for thousands of transcripts being compared. We generated roughly the same number of negative controls for each set of functional sites. And whenever a sequence motif exists for a functional site, we use that motif in generating the negative control. For example, the same sequence motif GGACU is used to scan the transcriptome and negative controls are randomly selected from the hits excluding regions that are close to a true m6A site.
The HuR iCLIP experiments are performed and clusters of binding sites are determined with the pipeline as described previously.36 Threshold 9 (at least 9 unique RT stops are each genomic coordinate) is used to filter for the true binding sites. The highest peak and its flanking 50 nucleotides in each cluster were retrieved and used to call sequence motifs by using HOMER38, with random sequences of 50 nucleotides from the same set of transcripts as background. The motifs are used as the anchor point in calculating metagene profiles and also used to generate negative controls, using the same protocol as the m6A negative control generation described above.
VTD (in vivo and in vitro structure difference) analysis
We defined and calculated the VTD profile of a transcript by subtracting its valid in vitro structural profile from the in vivo one. We calculate the average VTD profiles for all 4096 possible hexamers in our structurome. The overall VTD score of each hexamer is defined as the average score across the 6 bases of the hexamer.
We retrieved sequence motifs of important functional sites, including Kozak sequences (GCCRCC), m6A sites (GGACU), miR-290 family 6-mer seed matches (GCACUU, complementary to the seeds), and Rbfox2 binding sites (UGCAUG), and highlighted their VTD scores on the VTD histogram of all hexamers. For sites with ambiguity, for example, m6A sites, we took the average of all hexamers that contain GGACU.
We also compiled a resource (Supplementary Table 1 and 2) of VTD scores for all RNA protein binding motifs studied by RNAcompete experiments39, and all mouse microRNA 6-mer seed matches from miRBase40. In addition to the VTD scores, for every motif or seed match, we asked three questions by using permutation test: 1), whether the absolute value of the motif (or seed match) VTD is significantly less than a random hexamer, i.e., represents a stable region; 2), whether the motif (or seed match) VTD is significantly smaller than a random hexamer, i.e., represents a region that is more structured in vivo; and 3), whether the motif (or seed match) VTD is significantly bigger than a random hexamer, i.e., represents a region that is more structured in vitro.
Structure-based prediction of m6A sites and protein binding sites
We constructed a set of SVM models41 for the prediction of m6A sites and protein binding sites using structural profiles, genomic locations, conservations and their combinations.
The structural profile is limited to the range from the −10 to the +10 position of the m6A site or the motifs of the protein binding sites. We used in vivo and in vitro reactivity score separately and jointly in making predictions. We also retrieved a set of genomic features for the prediction of m6A sites and protein binding sites, including whether the site is in the 5’UTR, CDS, or 3’UTR, whether it is at the last exon, whether it is at the largest exon, the distance to start codon, the distance to stop codon, the distance to 5’ of the splicing junction, etc. In addition, we retrieved the UCSC 60-way phastCons conservation score42 for nucleotides in the range from the −10 to the +10 position of the m6A site or the motifs of the protein binding sites.
We used the same set of positive and negative controls and the best predictor is selected by using a parameter-searching tool coming along with the libsvm package (http://www.csie.ntu.edu.tw/~cjlin/libsvm/). We used a five-fold cross validation and calculate the AUC (area under curve) of the ROC (Receiver operating characteristic) curve to evaluate the performance of the predictors (Extended Data Figure 8).
Sequencing Datasets and Source Code
All genomic dataset are deposited on the Gene Expression Omnibus under the accession number GSE64169. Source code used for the icSHAPE analysis is deposited freely available at: https://github.com/qczhang/icSHAPE
Extended Data
Extended Data Figure 1. Chemical synthesis of NAI-N3.

a, Synthetic scheme for NAI-N3. b, 1HMR of Methyl 2-(azidomethyl)nicotinate c, 1HNMR of 2-(azidomethyl)nicotinic acid d, 13CNMR of 2-(azidomethyl)nicotinic acid e, 1HNMR of 2-(azidomethyl)nicotinic acid acyl imidazole f, 13CNMR of 2-(azidomethyl)nicotinic acid acyl imidazole.
Extended Data Figure 2. NAI-N3 is a novel RNA acylation reagent that enables RNA purification.

a, Chemical schematic of RNA acylation and copper-free ‘click’ chemistry utilizing NAI-N3 and dibenzocyclooxtyne-biotin conjugate. b, ATP acylation gel shift showing ATP acylation and copper-free ‘click’ chemistry utilizing NAI-N3 and dibenzocyclooxtyne-biotin conjugate.
Extended Data Figure 3. NAI-N3 is a novel RNA acylation reagent accurately reads out RNA structure.

a, Comparative denaturing gel of NAI and NAI-N3 RNA acylation. b, Denaturing gel analysis of cDNAs that originate from the biotin-purification protocol (Extended Data Figure 1) c, Secondary structure of the SAM-I Riboswitch with enriched residues highlighted in orange and depleted residues highlighted in blue. d, Denaturing gel analysis of denatured RNA probed with NAI-N3 shows even coverage of 2’-hydroxyl reactivity when RNA is unfolded. e, Protein titration with BSA, demonstrating no difference in the SHAPE pattern as a function of protein concentration.
Extended Data Figure 4. icSHAPE is capable of reproducing RNA acylation profiles obtained by manual RNA modification experiments.

icSHAPE (right panels) profiles of ribosomal RNA and compared them to those obtained by manual SHAPE (left panels).
Extended Data Figure 5.
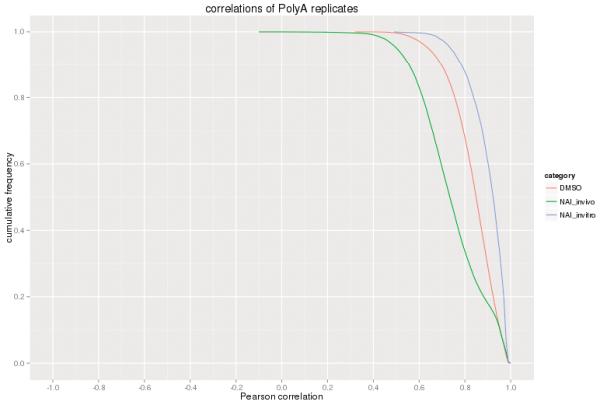
RT stops measured by icSHAPE is very well correlated in different library replicates
Extended Data Figure 6. icSHAPE is capable of measuring the RNA structure profiles of thousands of RNAs, simultaneously.
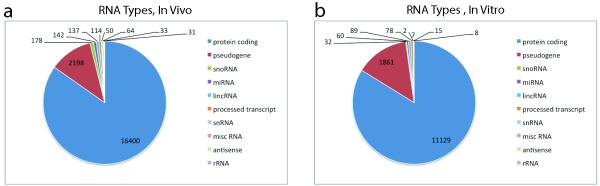
a, The RNAs represented in polyA selected RNA, in vivo. b. The RNAs represented in polyA selected RNA, in vitro.
Extended Data Figure 7. Non-AUG start codons are associated with preceding reactivity, and Non-AUG start codons have a different profile, suggesting that RNA accessibility alone is not a sufficient to drive translation.

a, icSHAPE profile at AUG start codons, in vivo. b, icSHAPE profile at AUG start codons, in vitro. d, icSHAPE profile at CUG start codons, in vivo. d, icSHAPE profile at CUG start codons, in vitro
Extended Data Figure 8. icSHAPE can be used to predict posttranscriptional regulatory elements.

a, icSHAPE profile at Rbfox2 targets, in vivo. b, icSHAPE profile at Rbfox2 targets, in vitro. c, ROC curve of Rbfox2 RNA-protein interactions, predicted using icSHAPE profiles. d, icSHAPE profile at m6A targets, in vivo. The negative control is the set of motif instances that are not m6A modified e, icSHAPE profile at m6A targets, in vitro. f, ROC curve of m6A RNA modification sites, predicted using icSHAPE profiles. g, icSHAPE profile at HuR targets, in vivo. h, icSHAPE profile at HuR targets, in vitro. i, ROC curve of HuR RNA-protein interactions, predicted using icSHAPE profiles.
Extended Data Figure 9. iCLIP analysis of HuR in mESCs.

a, global binding preference of the RBP HuR in mESCs as represented by RT stops across the mouse transcriptome (mm9). HuR mainly binds protein coding, processed, and ribosomal RNAs. b, number of unique RNA transcripts bound by HuR. c, HuR RT stops distributed across protein coding transcript functional domains. HuR prefers intronic and 3’UTR regions. d, Metagene analysis of all HuR bindings sites. Each mRNA region (5’UTR, CDS, or 3’UTR) was scaled to a standard unit width RT stop density across all bound protein coding genes was plotted revealing a clear enrichment for 3’UTR regions in mature protein coding transcripts. e, Individual mRNA binding evens of HuR to genes important for mESC biology including Tet1, β-Actin, Elav1 (HuR itself), and Lin28a. Discrete binding sites are observed focused in 3’UTR and intronic regions.
Extended Data Figure 10.

m6A-associated RNA structure features are preserved, independent of their position along the RNA transcript
Supplementary Material
Figure 5.

Acknowledgement
We thank members of Chang lab, J. Weissman (UCSF), and J. Doudna (UC Berkeley) for comments. Supported by NIH R01HG004361, P50HG007735, CIRM (H.Y.C.), NIH R01068122 (E.T.K.), A.P. Giannini Foundation (R.C.S.), Stanford Dean’s Fellowship (Q.C.Z.), and Stanford Medical Scientist Training Program and NIH F30CA189514 (R.A.F.). H.Y.C. is an Early Career Scientist of the Howard Hughes Medical Institute.
References
- 1.Wan Y, Kertesz M, Spitale RC, Segal E, Chang HY. Understanding the transcriptome through RNA structure. Nat Rev Genet. 2011;12:641–655. doi: 10.1038/nrg3049. doi:10.1038/nrg3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature. 2014;505:701–705. doi: 10.1038/nature12894. doi:10.1038/nature12894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ding Y, et al. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature. 2014;505:696–700. doi: 10.1038/nature12756. doi:10.1038/nature12756. [DOI] [PubMed] [Google Scholar]
- 4.Lucks JB, et al. Multiplexed RNA structure characterization with selective 2'-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq) Proc Natl Acad Sci U S A. 2011;108:11063–11068. doi: 10.1073/pnas.1106501108. doi:10.1073/pnas.1106501108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spitale RC, et al. RNA SHAPE analysis in living cells. Nature chemical biology. 2013;9:18–20. doi: 10.1038/nchembio.1131. doi:10.1038/nchembio.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anger AM, et al. Structures of the human and Drosophila 80S ribosome. Nature. 2013;497:80–85. doi: 10.1038/nature12104. doi:10.1038/nature12104. [DOI] [PubMed] [Google Scholar]
- 7.Weeks KM, Mauger DM. Exploring RNA structural codes with SHAPE chemistry. Accounts of chemical research. 2011;44:1280–1291. doi: 10.1021/ar200051h. doi:10.1021/ar200051h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schroeder R, Grossberger R, Pichler A, Waldsich C. RNA folding in vivo. Curr Opin Struct Biol. 2002;12:296–300. doi: 10.1016/s0959-440x(02)00325-1. [DOI] [PubMed] [Google Scholar]
- 9.Wan Y, et al. Landscape and variation of RNA secondary structure across the human transcriptome. Nature. 2014;505:706–709. doi: 10.1038/nature12946. doi:10.1038/nature12946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wan Y, et al. Genome-wide measurement of RNA folding energies. Mol Cell. 2012;48:169–181. doi: 10.1016/j.molcel.2012.08.008. doi:10.1016/j.molcel.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kertesz M, et al. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010;467:103–107. doi: 10.1038/nature09322. doi:10.1038/nature09322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vandivier L, et al. Arabidopsis mRNA secondary structure correlates with protein function and domains. Plant Signal Behav. 2013;8:e24301. doi: 10.4161/psb.24301. doi:10.4161/psb.24301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jia G, Fu Y, He C. Reversible RNA adenosine methylation in biological regulation. Trends Genet. 2013;29:108–115. doi: 10.1016/j.tig.2012.11.003. doi:10.1016/j.tig.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jangi M, Boutz PL, Paul P, Sharp PA. Rbfox2 controls autoregulation in RNA-binding protein networks. Genes Dev. 2014;28:637–651. doi: 10.1101/gad.235770.113. doi:10.1101/gad.235770.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dai W, Zhang G, Makeyev EV. RNA-binding protein HuR autoregulates its expression by promoting alternative polyadenylation site usage. Nucleic Acids Res. 2012;40:787–800. doi: 10.1093/nar/gkr783. doi:10.1093/nar/gkr783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kozak M. An analysis of 5'-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic acids research. 1987;15:8125–8148. doi: 10.1093/nar/15.20.8125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dvir S, et al. Deciphering the rules by which 5'-UTR sequences affect protein expression in yeast. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E2792–2801. doi: 10.1073/pnas.1222534110. doi:10.1073/pnas.1222534110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. doi:10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lunde BM, Moore C, Varani G. RNA-binding proteins: modular design for efficient function. Nature reviews. Molecular cell biology. 2007;8:479–490. doi: 10.1038/nrm2178. doi:10.1038/nrm2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lovci MT, et al. Rbfox proteins regulate alternative mRNA splicing through evolutionarily conserved RNA bridges. Nat Struct Mol Biol. 2013;20:1434–1442. doi: 10.1038/nsmb.2699. doi:10.1038/nsmb.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Auweter SD, et al. Molecular basis of RNA recognition by the human alternative splicing factor Fox-1. Embo J. 2006;25:163–173. doi: 10.1038/sj.emboj.7600918. doi:10.1038/sj.emboj.7600918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meyer KD, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. doi:10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120. doi: 10.1038/nature12730. doi:10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schwartz S, et al. High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell. 2013;155:1409–1421. doi: 10.1016/j.cell.2013.10.047. doi:10.1016/j.cell.2013.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Batista PJ, et al. m(6)A RNA Modification Controls Cell Fate Transition in Mammalian Embryonic Stem Cells. Cell Stem Cell. 2014;15:707–719. doi: 10.1016/j.stem.2014.09.019. doi:10.1016/j.stem.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kierzek E, Kierzek R. The thermodynamic stability of RNA duplexes and hairpins containing N6-alkyladenosines and 2-methylthio-N6-alkyladenosines. Nucleic Acids Res. 2003;31:4472–4480. doi: 10.1093/nar/gkg633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGinnis JL, Weeks KM. Ribosome RNA assembly intermediates visualized in living cells. Biochemistry. 2014;53:3237–3247. doi: 10.1021/bi500198b. doi:10.1021/bi500198b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das R, Laederach A, Pearlman SM, Herschlag D, Altman RB. SAFA: semi-automated footprinting analysis software for high-throughput quantification of nucleic acid footprinting experiments. Rna. 2005;11:344–354. doi: 10.1261/rna.7214405. doi:10.1261/rna.7214405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gherghe C, et al. Definition of a high-affinity Gag recognition structure mediating packaging of a retroviral RNA genome. Proc Natl Acad Sci U S A. 2010;107:19248–19253. doi: 10.1073/pnas.1006897107. doi:10.1073/pnas.1006897107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flynn RA, et al. Dissecting noncoding and pathogen RNA-protein interactomes. Rna. 2015;21:135–143. doi: 10.1261/rna.047803.114. doi:10.1261/rna.047803.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. doi:10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flicek P, et al. Ensembl 2014. Nucleic Acids Res. 2014;42:D749–755. doi: 10.1093/nar/gkt1196. doi:10.1093/nar/gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. doi:10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. doi:10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jangi M, Boutz PL, Paul P, Sharp PA. Rbfox2 controls autoregulation in RNA-binding protein networks. Genes Dev. 2014;28:637–651. doi: 10.1101/gad.235770.113. doi:10.1101/gad.235770.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. doi:10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ray D, et al. A compendium of RNA-binding motifs for decoding gene regulation. Nature. 2013;499:172–177. doi: 10.1038/nature12311. doi:10.1038/nature12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–73. doi: 10.1093/nar/gkt1181. doi:10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cortes C, Vapnik V. Support-Vector Networks. Machine Learning. 1995;20:273–297. 3. [Google Scholar]
- 41.Siepel A, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. doi:10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.