

Abstract
Osteonectin/SPARC is one of the most abundant non-collagenous extracellular matrix proteins in bone, regulating collagen fiber assembly and promoting osteoblast differentiation. Osteonectin-null and –haploinsufficient mice have low turnover osteopenia, indicating that osteonectin contributes to normal bone formation. In male idiopathic osteoporosis patients, osteonectin 3’ UTR single nucleotide polymorphism (SNP) haplotypes that differed only at SNP1599 (rs1054204) were previously associated with bone mass. Haplotype A (containing SNP1599G) was more frequent in severely affected patients, whereas haplotype B (containing SNP1599C) was more frequent in less affected patients and healthy controls. We hypothesized that SNP1599 contributes to variability in bone mass by modulating osteonectin levels. Osteonectin 3’UTR reporter constructs demonstrated that haplotype A has a repressive effect on gene expression compared to B. We found that SNP1599G contributed to a miR-433 binding site and miR-433 inhibitor relieved repression of the haplotype A, but not B, 3’ UTR reporter construct.
We tested our hypothesis in vivo, using a knock-in approach to replace the mouse osteonectin 3’ UTR with human haplotype A or B 3’ UTR. Compared to haplotype A mice, bone osteonectin levels were higher in haplotype B mice. B mice displayed higher bone formation rate and gained more trabecular bone with age. When parathyroid hormone was administered intermittently, haplotype B mice gained more cortical bone area than A mice. Cultured marrow stromal cells from B mice deposited more mineralized matrix and had higher osteocalcin mRNA compared with A mice, demonstrating a cell-autonomous effect on differentiation. Altogether, SNP1599 differentially regulates osteonectin expression and contributes to variability in bone mass, by a mechanism that may involve differential targeting by miR-433. This work validates the findings of the previous candidate gene study, and it assigns a physiological function to a common osteonectin allele, providing support for its role in the complex trait of skeletal phenotype.
Keywords: Osteoporosis, Molecular pathways – remodeling, Genetic animal models, Non-collagenous proteins, Human association studies
Introduction
Osteoporosis is a prevalent disorder characterized by low bone mineral density (BMD), deterioration of bone microarchitecture and increased incidence of fracture;(1) and genetic factors account for 60-80% of total variability in BMD.(2) Understanding the genetic determinants underlying bone mass may improve prognosis and provide novel targets for therapeutic intervention. In this regard, genome wide association studies (GWAS) and candidate gene studies have associated allelic variants of genes such as estrogen receptor (ER)-α, transforming growth factor (TGF)-β, osteoprotegerin, and type I collagen A1 with bone mass.(3-6) However, only a few studies have demonstrated a mechanism whereby a polymorphism could contribute to bone mass phenotype.
Indeed, a previous GWAS study in premenopausal women linked variations in BMD to genomic regions including 5q33-35.(7) Although candidate genes in the 5q33-35 interval were not identified, this region contains the gene for osteonectin/Sparc (secreted protein acidic and rich in cysteine), one of the most abundant non-collagenous extracellular matrix proteins in bone. In osteoblasts, osteonectin promotes commitment, differentiation, and survival. Osteonectin also suppresses adipogenic differentiation of mesenchymal precursor cells. In vivo, osteonectin-null and –haploinsufficient mice develop low turnover osteopenia, characterized by reduced osteoblast and osteoclast number and surface, and low bone formation rate.(8-12) Moreover, osteonectin-null mice accumulate less bone in response to intermittent administration of parathyroid hormone (PTH), the best bone-anabolic treatment clinically available at this time.(12)
Based on these findings, we had previously performed a candidate gene study, to determine whether 3 single nucleotide polymorphisms (SNPs) in the 3’ untranslated region (UTR) of osteonectin (Figure 1A) were associated with bone mass in a cohort of men with low turnover idiopathic osteoporosis, a disorder primarily attributed to genetic determinants.(13) Briefly, this cohort consisted of middle aged Caucasian men with a BMD T score of less than −2.0 at the lumbar spine, who lacked known secondary causes for osteoporosis. The control subjects were age and body mass index matched to the patients, and had BMD T scores of more than 1.0 at the lumbar spine. As a group, the idiopathic osteoporosis patients had mean serum PTH and IGF1 levels in the low normal range. Their indices of bone formation were significantly reduced, although eroded surface was not different between patients and their matched controls. (14, 15) In the osteoporotic cohort, prevalence of fragility fracture was 23%. (13)
Figure 1. Osteonectin 3’ UTR SNP 1599 regulates 3’ UTR function.

(A) Schematic representation of the human osteonectin cDNA, and the characterized SNPs at bases 1046, 1599 and 1970 (grey triangles). (B) Human ON 3’ UTR (cDNA bases 1018-2123) representing haplotypes A and B were cloned into pMIR-REPORT Luciferase vector. Luciferase activity was quantified in hFOB1.19 cells and normalized to β-galactosidase activity (*p<0.05 different from vector; # p<0.05 different from haplotype A, N=6).
In this cohort, one of the two most common osteonectin 3’ UTR haplotypes that we identified, haplotype A, was found at a higher frequency in the most severely affected osteoporotic patients, whereas the second most common haplotype, B, was found at a higher frequency in the healthy controls. In addition, haplotype B was associated with higher BMD in the patient population.(13) Osteonectin 3’ UTR haplotype A contained the SNPs at cDNA bases 1046C_1599G_1970T, whereas haplotype B consisted of SNPs 1046C_1599C_1970T (Figure 1A). Since these BMD-associated haplotypes differed only at cDNA base 1599, we hypothesized that SNP 1599G/C (rs1054204) may impact osteonectin expression, and affect bone mass.
The 3’ UTR represents a powerful regulatory region, with the potential to modulate mRNA stability, translation and localization.(16) Polymorphisms in the 3’ UTR have the potential to alter the secondary structure of the mRNA, as well as its interaction with trans-acting factors, such as microRNAs (miRNAs, miRs). miRNAs are small, endogenous non-coding RNAs that, for the most part, decrease the stability and/or translation of protein-encoding mRNAs.
Recent studies have associated mutations or SNPs in miRNA binding with skeletal disorders. For example, a SNP in the 3’ UTR of Fgf2 (fibroblast growth factor 2), that abrogates miR-146a and -146b binding sites, was linked to low BMD in osteoporotic patients.(17) Another study attributed a mutation in the binding site for miR-433 in the 3’ UTR of Hdac6 (histone deacetylase 6) to the pathogenesis of X-linked chondrodysplasia.(18)
In this study we show that human osteonectin SNP1599 differentially regulates gene expression and contributes to a miR-433 binding site. Specifically, 1599G, found in haplotype A, has a repressive effect on gene expression and osteoblastic differentiation compared to 1599C, which is found in haplotype B. Moreover, using novel knock-in mouse models, we demonstrate that compared to mice carrying human osteonectin haplotype A 3’ UTR (SNP 1599G), mice with the haplotype B 3’ UTR knock-in (SNP 1599C) have higher levels of osteonectin in bone, higher bone formation rate, increased trabecular bone volume with age, and a greater increase in cortical bone volume in response to the bone-anabolic PTH treatment. These data substantiate the relationship between osteonectin 3’ UTR SNP 1599 and skeletal phenotype, validate our initial observations in the cohort of idiopathic osteoporosis patients, and suggest that SNP 1599 affects bone mass by modulating osteonectin expression.
Materials and Methods
Generation of osteonectin 3’ UTR haplotype constructs
Human osteonectin 3’ UTR (cDNA bases 1018-2123) representing haplotypes A and B found in idiopathic osteoporosis patients were PCR amplified from genomic DNA and cloned downstream of the Luciferase gene in pMIR-REPORT vector (Life Technologies). Details of cloning are described in Supporting Information (SI) text.
Cell culture, transfection and luciferase activity
Human fetal osteoblastic 1.19 cell line (hFOB1.19) was purchased from American Type Culture Collection (ATCC number CRL-11372) and were grown at 33.5° C and differentiated into osteoblast cultures at 39.5° C in the presence of differentiation cocktail: 100 μg/mL ascorbic acid, 10−8 M menadione (Vitamin K), 5 mM β-glycerolphosphate (β-GP), and 10−7 M 1-25(OH)2-Vitamin D3 (all from Sigma, St Louis, MO).(19, 20) Bone marrow stromal cells (BMSCs) harvested from long bones of mice were differentiated in presence of 5 mM β-Glycerolphosphate and 50 μg/mL ascorbic acid. (8, 11)
Haplotype A and B 3’ UTR luciferase constructs were transfected into hFOB1.19 cells using Fugene6 (Roche, Indianapolis, IN). For co-transfection of miRNA inhibitors or mimics and luciferase constructs into hFOB1.19 cells, X-tremeGENE reagent (X-tremeGENE; Roche) was used. Details of cell culture and transfection are described in SI text.
Generation of osteonectin knock-in mice
A knock-in strategy was used to replace the mouse osteonectin 3’ UTR with human haplotype A or haplotype B 3’ UTR. These mouse models were generated at the Gene Targeting and Transgenic Facility at UCHC. Osteonectin haplotype A (ON+/A) and haplotype B (ON+/B) mice were generated using 129 embryonic stem cells and were back crossed into C57BL/6J. Mice heterozygous for A and B alleles (ONA/B) were bred to create homozygous A and B mice (ONA/A and ONB/B).
Quantitative RT-PCR analysis
RNA was extracted using the miRNeasy mini kit (Qiagen, Valencia, CA). miR-433 levels were determined using the TaqMan MicroRNA assay (Life Technologies, Grand Island, NY) and normalized to RNU48 for human tissue or sno202 RNA for mouse tissue. Osteonectin, alkaline phosphatase, osteocalcin, and bone sialoprotein (Ibsp) mRNA levels were determined using MMLV-Reserve Transcriptase (Invitrogen) and iQ SYBR Green Supermix (BioRad) and normalized to 18sRNA. Primer sequences and details of quantitative RT-PCR are described in SI text.
Western blot analysis
Equal amounts of long bone extract were subjected to Western blot analysis for osteonectin and β-actin. Rabbit anti-bovine osteonectin primary antibody (BON-1; gift of L. Fisher, NIDCR, NIH) (1:4000), rabbit anti-β-actin primary antibody (Abcam, 1:1000), and goat anti-rabbit-horseradish peroxidase conjugated secondary antibody (1:20,000) were used. Experimental details of this are described in SI text.
Osteoblast differentiation, mineralization and proliferation analysis
BMSCs harvested from long bones were cultured in osteoblast differentiation medium. Cultures were stained with either 1% Alizarin red S pH 6.45 (Sigma) or 0.05% Crystal violet stain (Fisher Scientific). Alizarin red stain was quantified at absorbance 405 nm, while Crystal Violet stain was measured at 570 nm, as described in SI text. Cell proliferation was assessed using MTS CellTiter 96® AQueous One Solution cell proliferation assay kit (Promega, Madison, USA), according to the manufacturer's instructions.
Morphological analysis
To study bone-anabolic response, 10-week old male mice were injected subcutaneously with 40 μg/kg/day rhPTH (1–34) (PTH) (Bachem, Torrance, CA) or vehicle alone (2% heat inactivated mouse serum in acidified saline), 5 days per week for 4 weeks.(12) Details of μCT imaging and histomorphometry of femurs are described in SI text. All procedures were approved by the Institutional Animal Care and Use Committee at UCHC.
Statistics
All the quantitative data are expressed as a mean ± SEM. Statistical significance was determined by one-way ANOVA with Bonferroni post-hoc test or Student's t-test. Allele frequencies were evaluated using the Freeman-Halton extension of Fisher's exact test.
Results
SNP 1599 modulates osteonectin 3’ UTR function and represses haplotype A 3’ UTR
To determine the role of SNP 1599 in regulating the osteonectin 3’ UTR, we cloned the 1 kb osteonectin 3’ UTR, representing human haplotype A or B, into a Luciferase reporter construct. In these reporter constructs, the cloned sequence functioned as 3’ UTR for the Luciferase gene, the transcription of which was constitutively driven by a strong promoter. The constructs were transiently transfected into hFOB1.19 cells, a conditionally immortalized human osteoblastic cell line. We found that the haplotype A 3’ UTR construct had lower luciferase activity compared to haplotype B, suggesting that haplotype A, which was found at a higher frequency in the most severely affected osteoporosis patients, had a more repressive effect on gene expression (Figure 1B). These data indicate that SNP 1599 contributed to differences in osteonectin 3’ UTR function.
SNP 1599 introduces a novel miR-433 binding site in haplotype A 3’ UTR
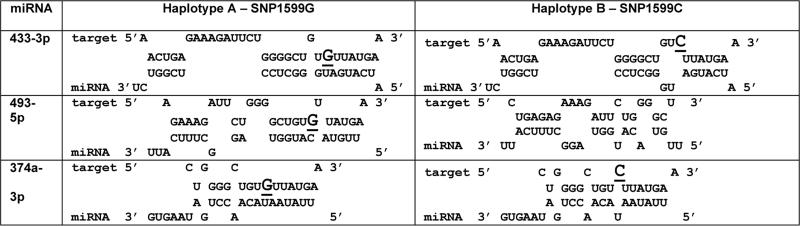
To determine whether miRNAs may mediate the differential regulation of osteonectin by SNP 1599G/C, we used RNAhybrid (http://bibiserv.techfak.uni-bielefeld.de/bibi/Tools.html) and miRbase v15.0 to assemble a panel of candidate miRNAs with the potential to interact with the region containing SNP 1599. (21) The list of candidate miRNAs was further refined, based on whether or not SNP 1599 would facilitate or disrupt the interaction of the miRNA with the osteonectin 3’ UTR. We observed the potential for differential interaction of miR-433-3p, miR-493-5p and miR-374a-3p in presence of 1599G (haplotype A), compared with 1599C (haplotype B) (Table 1). Moreover, these miRNAs are expressed in hFOB1.19 cells.
Table 1.
RNAhybrid analysis of putative miR-433, -493 and -374 binding sites in osteonectin 3’ UTR. SNP 1599 G/C is underlined and indicated by larger font in the seed binding region of haplotype A or B 3’. UTR.
|
To test the hypothesis that the candidate miRNAs may differentially target haplotypes A and B in vitro, hFOB1.19 cells were transiently co-transfected with either haplotype A or B 3’ UTR reporter constructs and inhibitor for miR-433-3p, miR-493-5p or miR-374a-3p. In the presence of miR-433 inhibitor, luciferase activity of the haplotype A construct was significantly increased compared to the non-targeting control, whereas activity of the haplotype B construct was not affected. In contrast, inhibitor for miR-374a-3p or miR-493-5p did not significantly increase luciferase expression from either haplotype A or B constructs (Figure 2A). miR-433 targeting of haplotype A 3’ UTR was also evaluated in hFOB1.19 cells using miR-433 mimic and a scramble mimic control. In the presence of miR-433 mimic, haplotype A 3’ UTR luciferase activity was inhibited compared to the control, whereas haplotype B 3’ UTR activity was not significantly affected (Figure 2B).
Figure 2. miR-433 represses haplotype A 3’ UTR and decreases during osteoblastic differentiation in vitro.

Luciferase activity of haplotype A and B pMIR-report constructs in hFOB1.19 cells co-transfected with specific miRNA inhibitors or scramble control. Inhibitors for miR-433, -493 and -374a (A) and miR-433 mimic or scramble control mimic (B) were tested; luciferase activity was normalized to β-galactosidase activity (* p<0.05 different from scramble control, N=6). (C) miR-433, alkaline phosphatase (ALP) and osteocalcin (OC) RNA in hFOB 1.19 cells at confluence (0), and during osteoblastic differentiation. miR-433 normalized to RNU48; ALP and OC mRNA normalized to 18s RNA (*p<0.05 different from confluence, N=3). (D) ALP and OC RNA in hFOB1.19 cells transfected with miR-433 or scramble inhibitor and cultured in osteoblast differentiation medium for 3 days (*p<0.05 different from scramble inhibitor, N=3).
These results suggest that SNP 1599 introduces a novel miR-433 binding site in haplotype A 3’ UTR, which may contribute to differential regulation of osteonectin. However, our data do not preclude the possibility that other miRNAs or trans-acting factors could also demonstrate differential regulation of the haplotype A and B 3’ UTRs.
miR-433 expression decreases during osteoblastic differentiation
Since miR-433 could differentially regulate the human osteonectin 3’ UTR, we determined whether the expression of this miRNA was altered during osteoblastic differentiation, using hFOB1.19 cells as a model.(19, 20) Osteoblastic differentiation was induced using a vitamin D-containing cocktail, and after 3 or 6 days, mRNAs for the early osteoblastic marker alkaline phosphatase (ALP), and the mature osteoblast marker, osteocalcin (OC), were dramatically increased. In contrast, expression of miR-433 decreased during differentiation, such that miR-433 levels were lowest when osteoblastic differentiation markers were highest (Figure 2C). A similar phenomenon was reported by others when miR-433 levels were evaluated in a BMP2-treated murine pre-osteoblast cell line.(22)
To determine the role of miR-433 in human osteoblastic differentiation, hFOB1.19 cells were transfected with either a miR-433 inhibitor or a scramble control inhibitor and subjected to osteoblastic differentiation for 3 days. Here, miR-433 inhibitor increased mRNA for the osteoblast markers ALP and OC, compared to the scramble control, confirming that miR-433 is a negative regulator of osteoblast maturation in vitro (Figure 2D).
SNP 1599 affects osteonectin expression in vivo
Many mature miRNAs display sequence conservation across species, and the sequence of mature miR-433 is identical between mouse and human (miRBase). Whereas selected regions of the mouse and human osteonectin 3’ UTR are highly conserved, such as the miR-29 binding sites in the proximal portion of the UTR,(23) mouse and human osteonectin are not well conserved in the region containing human SNP1599 (UCSD Genome Browser). Therefore, to examine the impact of SNP 1599 on osteonectin expression in bone in vivo, we used a knock-in strategy to replace the mouse osteonectin 3’ UTR with the 1 kb human osteonectin 3’ UTR, representing either haplotype A (ONA/A) or haplotype B (ONB/B) (Figure S1). In relation to other mouse strains, C57Bl/6 mice have a low bone mass phenotype.(24,25) We chose to examine the function of the human osteonectin 3’ UTR haplotypes in the C57Bl/6 genetic background because our candidate gene study revealed an association between osteonectin haplotype and bone mass only in the idiopathic osteoporosis patient group. We reasoned that the effect of the osteonectin 3’ UTR haplotype might be most apparent in a low bone mass background, where other gene variants may not be sufficient to rescue a low bone mass phenotype. Moreover, we chose to limit our present analysis to males, to further mimic our previous study in male idiopathic osteoporosis patients.
Osteonectin transcript and protein levels in femur of homozygous haplotype A and B (ONA/A and ONB/B) male mice were examined. Western blot analysis of protein extracts demonstrated 2-3 fold lower osteonectin levels in ONA/A femur in comparison to ONB/B, indicating that SNP 1599 contributed to differential osteonectin accumulation in bone in vivo (Figure 3B). At the same time, qRT-PCR revealed a ~2 fold increase in osteonectin mRNA in femur of ONA/A mice compared with ONB/B (Figure 3A), while miR-433 levels were similar (Figure S2). This apparent discrepancy may be related to the fact that the protein data represent the accumulation of osteonectin in bone tissue, whereas the RNA data represent a window of gene expression at 6-8 weeks of age.
Figure 3. Osteonectin levels and gain in trabecular bone is higher in haplotype B knock-in mice.

(A) Osteonectin mRNA was quantified by qRT-PCR using RNA isolated from the femurs of 6-8 week old ONA/A and ONB/B mice. Osteonectin mRNA levels were normalized to 18s RNA (N=4 mice/ haplotype, * p<0.05 different from haplotype A). (B) Western blotting for osteonectin and β-actin in lysates from long bones of ONA/A and ONB/B mice (*p<0.05 different from haplotype A, N=3 mice/group). (C, D) μCT analysis of changes in femoral bone from 10 to 20 weeks of age in ONA/A and ONB/B mice. Trabecular bone volume (BVF), trabecular bone number (Tb.N) trabecular bone spacing (Tb.Sp.). Total (periosteal) area (Tt.Ar), marrow area (Ma.Ar), cortical area (Ct.Ar). *p<0.05 different from haplotype A, N=4-9 mice/group.
SNP 1599 affects changes in bone mass with age
Using μCT, we analyzed the skeletal phenotype of male ONA/A and ONB/B mice at 10 and 20 weeks of age. The femurs of 10-week old mice ONA/A and ONB/B displayed similar trabecular and cortical bone parameters (Table 2). At 20 weeks of age, trabecular bone volume and trabecular thickness were significantly higher in ONB/B mice compared with ONA/A (Table 2). When the percentage change in trabecular bone parameters between 10 and 20 weeks was examined, the differences between the two genotypes became more apparent (Figure 3C). For example, between 10 and 20 weeks of age, ONB/B mice realized a 20-25% gain in trabecular bone volume fraction (BVF), while ONA/A mice did not. Trabecular number (Tb.N) decreased with age in both the genotypes, and the decrease was significantly less in ONB/B mice. Trabecular spacing (Tb.Sp.) increased between 10 and 20 weeks of age in ONA/A mice, but not in ONB/B animals (Figure 3C). In contrast, cortical bone area (Ct.Ar.) increased from 10 to 20 weeks of age in both genotypes, to a similar extent (Figure 3D). This was not unexpected, as the osteonectin-null and haploinsufficient mice display primarily defects in trabecular bone volume.(10) Overall, these data suggest that human osteonectin 3’ UTR haplotypes A and B differentially affect the trabecular bone compartment, and that ONB/B gained more trabecular bone with age than ONA/A mice.
Table 2.
MicroCT analysis of trabecular and cortical bone parameters in femur of 10-week and 20-week old male ONA/A and ONB/B mice.
| Trabecular parameters | AA | BB | ||
|---|---|---|---|---|
| 10 weeks | 20 weeks | 10 weeks | 20 weeks | |
| BVF (%) | 0012.60±0.010 | 0012.20±0.00 | 0011.70±0.02 | 0014.80±0.01* |
| Tb.Th.(μm) | 045.48±3.72 | 0046.97±1.23 | 0053.06±2.76 | 0055.24±1.89* |
| Tb.N.(/mm) | 005.20±0.33 | 0004.20±0.18 | 004.47±0.30 | 0004.23±0.14 |
| Tb.Sp.(μm) | 192.25±11.97 | 237.56±9.59 | 225.06±17.56 | 232.34±9.46 |
| Cortical parameters | AA | BB | ||
|---|---|---|---|---|
| 10 weeks | 20 weeks | 10 weeks | 20 weeks | |
| Tt.Ar.(mm2) | 1.96±0.10 | 2.24±0.04 | 1.89±0.03 | 2.06±0.11 |
| Ma.Ar.(mm2) | 1.08±0.04 | 1.24±0.02 | 1.00±0.03 | 00001.08±0.08 |
| Ct.Ar.(mm2) | 0.88±0.06 | 0.96±0.03 | 0.88±0.02 | 0.98±0.04 |
Trabecular bone volume fraction (BVF), trabecular bone number (Tb.N) trabecular bone spacing (Tb.Sp.) and trabecular thickness (Tb.Th.). Total (periosteal) area (Tt.Ar.), marrow area (Ma.Ar.), cortical area (Ct.Ar.). (10 weeks; N = 4-5 mice per group) (20 weeks; N = 8-9 mice per group). Mean ± SEM.
p<0.05 different from haplotype A
SNP 1599 modifies the bone anabolic effect of intermittent PTH
Since intermittent administration of PTH is the best bone anabolic therapy currently available, we studied the response of ONA/A and ONB/B mice to this treatment. 10-week old male mice were injected daily with 40 μg/kg PTH (1-34) or vehicle, 5 days per week, for 4 weeks. μCT analysis showed that after 4 weeks of treatment, PTH significantly increased cortical bone area in both ONA/A and ONB/B mice, however the gain in cortical bone in ONB/B mice was nearly twice than that observed in ONA/A mice (Figure 4A, Table 3). The PTH mediated increase in the total cross-sectional area (Tt.Ar.) was greater in ONB/B mice compared with ONA/A mice, providing a mechanism for increased cortical bone area. PTH-mediated changes in marrow area (Ma.Ar.) did not reach significance (p=0.07) in either genotype (Figure 4A and Table 3). These data indicate that the bone anabolic effect of intermittent PTH was greater in ONB/B mice compared to ONA/A.
Figure 4. PTH induced gain of cortical bone area is greater in haplotype B knock-in mice, and BMSCs from haplotype B mice have enhanced osteoblastic differentiation in vitro.

μCT and histomorphometric analysis of femoral trabecular bone from 14-week old ONA/A and ONB/B mice that had been injected for 4 weeks with 40 μg/kg/day PTH or vehicle. (A) Percent change in cortical bone parameters vs. vehicle in femur. Total (periosteal) area (Tt.Ar), marrow area (Ma.Ar), cortical area (Ct.Ar). (B) Percent change in trabecular bone parameters vs. vehicle in femur. Trabecular bone volume (BVF), trabecular thickness (Tb.Th), trabecular bone number (Tb.N). Histomorphometric analysis of osteoblast number and bone formation rate (C and D) in femoral trabecular bones of vehicle and PTH treated ONA/A and ONB/B mice (*p<0.05 different from ONA/A mice, N=4-6/group). Osteocalcin (E) and Bone sailoprotein (F) RNA in BMSCs from ONA/A and ONB/B mice undergoing osteoblastic differentiation for 2 weeks (*p<0.05 different from ONA/A, N=3). Quantified alizarin red (G) and crystal violet (H) staining, and alizarin red stain normalized to crystal violet (I) for BMSCs from ONA/A and ONB/B mice undergoing osteoblastic differentiation for up to 4 weeks (*p<0.05 different from ONA/A, N = 4).
Table 3.
MicroCT analyses of trabecular and cortical bone parameters in femur of vehicle and PTH injected ONA/A and ONB/B mice.
| Trabecular parameters | AA | BB | ||
|---|---|---|---|---|
| Vehicle | PTH | Vehicle | PTH | |
| BVF (%) | 12.00±0.01 | 14.00±0.01 | 12.00±0.01 | 16.00±0.01 |
| Tb.Th.(μm) | 47.03±1.16 | 59.54±1.94# | 46.40±2.54 | 61.65±3.81# |
| Tb.N.(/mm) | 4.43±0.04 | 4.10±0.16 # | 4.72±0.16 | 4.25±0.21 |
| Cortical parameters | AA | BB | ||
|---|---|---|---|---|
| Vehicle | PTH | Vehicle | PTH | |
| Tt.Ar. (mm2) | 2.15±0.07 | 2.17±0.08 | 2.00±0.05 | 2.36±0.06 # |
| Ma.Ar. (mm2) | 1.22±0.06 | 1.15±0.05 | 1.10±0.03 | 1.21±0.07 |
| Ct.Ar. (mm2) | 0.93±0.02 | 1.02±0.02 # | 0.90±0.02 | 1.09±0.02 # |
14-week old male haplotype A and B knock-in mice had been injected for 4 weeks with 40 [μg/kg/day PTH or vehicle. Trabecular bone volume fraction (BVF), trabecular bone number (Tb.N) trabecular bone spacing (Tb.Sp.) and trabecular number (Tb.N.). Total (periosteal) area (Tt.Ar.), marrow area (Ma.Ar.), cortical area (Ct. Ar.). (N=4-6 mice per group) Mean ± SEM.
p<0.05 different from corresponding vehicle injected mice of the same genotype.
We also assessed changes in the trabecular bone at the femoral metaphysis of PTH or vehicle injected mice using microCT. Although the PTH-mediated increase in trabecular bone volume was not statistically significant, PTH did increase trabecular thickness and number to a similar extent in mice of both genotypes (Figure 4B, Table 3). Histomorphometry was used to evaluate bone remodeling parameters and bone formation rate in femoral trabecular region of vehicle and PTH injected ONA/A and ONB/B mice.(12) Interestingly, osteoblast number was lower in vehicle treated ONB/B mice compared to ONA/A, whereas bone formation rate (BFR) was higher in the vehicle treated ONB/B mice (Figure 4C,and D; Table 4, Figure S4). These data suggest greater bone forming activity, per cell, in ONB/B mice. With PTH treatment, osteoblast number and bone formation rate in both ONA/A and ONB/B mice were significantly increased, such that they were equivalent. Differences in osteoclast number and eroded surface were not seen between ONA/A and ONB/B mice, in the presence or absence of PTH treatment (Table 4). Altogether, these data indicate that ONA/A mice have decreased bone formation compared with ONB/B mice, providing an explanation for the failure of ONA/A mice to increase trabecular bone volume from 10 to 20 weeks of age (Figure 4D).
Table 4.
Histomorphometric analysis of femoral trabecular bone of vehicle and PTH injected ONA/A and ONB/B mice.
| Histomorphometry | AA | BB | ||
|---|---|---|---|---|
| Formation | Vehicle | PTH | Vehicle | PTH |
| Osteoblast number (N.Ob/B.Pm, /mm2) | 1.64±0.26 | 6.28±1.16# | 0.91±0.23* | 6.39±1.52# |
| Mineralizing surface (MS/BS, %) | 4.77±0.55 | 10.58±1.63# | 7.34±0.90* | 8.86±1.16 |
| Bone formation rate (μm3/μm2/ day) | 93.29±1.17 | 317.27±35.02# | 168.36±21.90* | 196.81±21.34 |
| AA | BB | |||
|---|---|---|---|---|
| Resorption | Vehicle | PTH | Vehicle | PTH |
| Osteoclast number (N.Oc/B.Pm; /mm2) | 0.91±0.22 | 0.81±0.25 | 0.72±0.07 | 0.80±0.21 |
| Osteoclast surface (Oc.S/BS, %) | 2.54±0.64 | 2.27±0.66 | 1.97±0.21 | 2.26±0.10 |
| Eroded surface (ES/BS, %) | 5.67±1.14 | 5.63±1.05 | 4.76±0.53 | 5.29±0.38 |
14-week old haplotype A and B knock-in mice had been injected for 4 weeks with 40 μg/kg/day PTH or vehicle (N = 4-6 mice per group)
p±0.05 different from the corresponding vehicle injected mice of the same genotype. Mean ± SEM.
p<0.05 different from haplotype A mice in the same treatment group.
To determine whether PTH might have a direct effect on miR-433 expression, wild type mouse BMSCs were cultured to confluence, serum-deprived, and treated with PTH or vehicle for 3, 6, 12 or 24 hours of treatment (Figure S6). However, PTH treatment did not regulate miR-433 levels at any time point tested, suggesting that the actions of PTH on bone in vivo may not be related to direct effects on miR-433.
Cell autonomous effect of SNP 1599 on osteoblastic differentiation and mineralized matrix deposition
To determine whether the effect of SNP 1599 on bone formation was cell autonomous, we monitored osteoblastic differentiation markers in BMSCs from ONA/A and ONB/B mice cultured for up to 2 weeks. After 1 week of culture in osteoblast differentiation medium, ONB/B cells displayed significantly more osteocalcin and bone sialoprotein mRNA compared to ONA/A cultures (Figure 4E and F). After 2 weeks of culture, bone sialoprotein mRNA levels were no longer significantly different between genotypes, whereas osteocalcin mRNA remained elevated in ONB/B cultures.
We also assessed mineralized matrix deposition in BMSCs undergoing osteoblastic differentiation in vitro. Mineralized matrix deposition was quantified using alizarin red staining, whereas differences in cell density were monitored by crystal violet staining (Figure S5). The stains were then solubilized and quantified; and alizarin red staining was normalized to crystal violet (Figure 4G-I). Although both cultures were plated at the same density, crystal violet staining was significantly greater in ONA/A compared to ONB/B cultures (Figure 4H). Crystal violet staining peaked at week 2 in ONA/A cultures, and at week 3 in ONB/B cultures. However, despite the lower cell number, ONB/B cultures showed a greater alizarin red staining at weeks 3 and 4 of differentiation (Figure 4G). After normalizing alizarin red staining by crystal violet, the difference in mineralized matrix deposition between ONA/A and ONB/B cultures became more apparent (Figure 4I). This suggests that ONB/B osteoblasts deposited significantly more mineralized matrix per cell compared to ONA/A cultures, and support the in vivo observations (Figure 4C and D).
To determine whether there were inherent differences in the growth rate of ONA/A and ONB/B BMSCs, MTS assay was used to monitor the growth of sub-confluent cultures. We found that the growth rate for ONA/A and ONB/B stromal cells did not differ (Figure S5A). Similarly, previous in vitro studies showed that osteonectin-null osteoblasts displayed decreased osteoblast maturation and mineralized matrix deposition, but no defects in cell growth.(11) The present in vitro studies support the concept that, in BMSCs, osteonectin levels do not impact cell growth, but have effects on osteoblastic differentiation.
Discussion
Skeletal phenotype is a complex genomic trait, and only a minute fraction of the genetic variants contributing to this phenotype have been identified. Further, the in vivo function of most osteoporosis-associated polymorphisms identified through GWAS and candidate gene studies is not known.(1) In this study, we developed a novel knock-in mouse model to determine the in vivo impact of a human regulatory region polymorphism on skeletal phenotype. This model demonstrated that a SNP in the osteonectin 3’ UTR can regulate osteonectin levels and bone volume, essentially validating the association between osteonectin 3’ UTR SNP haplotypes and bone mass first identified in a cohort of Caucasian men with idiopathic osteoporosis.(13) Moreover, we identified a potential molecular mechanism by which this SNP regulates osteonectin expression, through differential targeting of a miRNA.
Bone matrix is enriched in osteonectin. This integrin-binding matricellular protein is important for regulating collagen matrix assembly and organization.(26-28) In fact, many connective tissue pathologies detected in osteonectin-null mice have been attributed to defective extracellular matrix composition.(27-29) In vitro and in vivo studies demonstrate that collagen fibril organization is impaired in bone matrices of osteonectin-null mice, likely contributing to decreased bone mineralization.(27) In vitro, osteonectin promotes osteoblast survival, differentiation and matrix mineralization.(11) It is possible that extracellular matrix organization differs between ONA/A and ONB/B mice, which may contribute to differences in osteoblastic differentiation and mineralization; studies to address these questions are ongoing.
Although we did not analyze osteonectin mRNA or protein levels in ONA/A and ONB/B mice after PTH administration, studies by Turner et al. (2007) showed that intermittent PTH administration increased osteonectin mRNA in bone of rats subjected to hind limb unloading.(30) Previously, we reported that although osteonectin-null and haploinsufficient mice gain bone in response to intermittent PTH therapy, their bone-anabolic response was less than that seen in wild type mice. For the most part, the response of these mice to PTH could be related to osteonectin gene dosage.(12) Although the dose of PTH used in our previous study was higher than the one used in this report (80 vs 40 μg/kg/day), our results suggest that higher levels of osteonectin in bone are associated with greater PTH-mediated bone gain, particularly with regard to the cortical compartment (Figure 4A). It is possible that the higher levels of osteonectin in ONB/B mice could facilitate the anabolic response of bone to intermittent PTH treatment.
The expression of osteonectin is tightly regulated through mechanisms that alter transcription, mRNA stability, and translation.(20, 23, 31-36) In osteoblasts, the osteonectin transcript is quite stable, with a half-life of >24 hours under conditions of transcription arrest.(31) Therefore, regulation of translation would likely provide the most rapid means of decreasing osteonectin synthesis. Recently, 2 evolutionarily conserved binding sites for the miR-29 family of miRNAs were found in the proximal region of the osteonectin 3’ UTR.(23) Induction of miR-29 expression by canonical Wnt signaling provided potent repression of osteonectin protein synthesis, within one hour of treatment.(20) This example illustrates the efficiency by which miRNAs could regulate osteonectin levels.
In this study, we identified miR-433 as a miRNA that could play a role in regulating osteonectin expression. The mature miR-433 sequence is identical between mice and humans, and the organization of the miR-433 genomic locus conserved. Previously, miR-433 was shown to decrease during BMP2-induced osteoblastic differentiation of C3H10T1/2 cells, and to target the Runx2 3’ UTR.(22) Our study confirms that miR-433 expression decreases during osteoblastic differentiation in human cells, and that miR-433 has an inhibitory effect on differentiation (Figure 2). We also demonstrated that SNP 1599 modulates the ability of miR-433 to regulate the human osteonectin 3’ UTR (Figure 2). Although the effect of the miR-433 inhibitor on the haplotype A construct was modest, it is consistent with effects reported in other studies, and may reflect the relatively low level of miR-433 expression in the human osteoblast cell line.(18) Moreover, osteonectin is expressed in multiple tissues, many of which likely have a complement of miRNAs that are distinct from that found in the bone cells. Other miRNAs might bind to the osteonectin 3’ UTR, and differential binding of these miRNAs to the region containing SNP 1599 must be strongly considered.
Others have examined the potential association of osteonectin SNPs with disease phenotype in systemic sclerosis, hepatocellular carcinoma, glaucoma, and keratoconus.(29, 37-40) Although some studies have associated particular osteonectin 3’ UTR SNPs and disease, these reports have not involved SNP 1599, nor have they described potential mechanisms. Osteonectin SNP 1599G is a common variant (Table 5).(41) In a sample population of North Americans that are of African descent, frequency of the SNP1599G allele, present in haplotype A, is less than that for populations of European or Chinese descent (dbSNP summary for ss24686914). Studies have shown that, as a group, African Americans have higher bone mineral density compared to Caucasians.(42, 43) Although several candidate genes have been associated with BMD within these groups, it is not clear whether decreased frequency of SNP 1599G could play a part in this effect.(44)
Table 5.
Osteonectin SNP 1599 (rs1054204) allele and genotype frequencies (from dbSNP summary for ss24686914). (41)
| Population ID | Ethnicity | Allele Frequency | Genotype Frequency |
|---|---|---|---|
| AFD EUR Panel-North America | European | C=0.521 G=0.479 |
C/G=0.542 C/C=0.208 G/G=0.250 |
| AFD CHN Panel-North America | Asian | C=0.604 G=0.396 |
C/G=0.542 C/C=0.333 G/G=0.125 |
| AFD AFR Panel-North America | African | C=0.783 G=0.217 |
C/G=0.348 C/C=0.609 G/G=0.043 |
In the literature, there has been argument about whether the majority of phenotypic variance is driven by rare variants with large effects or by common variants with small effects.(45) Most likely, rare and common variants work together, in conjunction with gene-environment interactions, to specify phenotype. The present study was performed in an inbred mouse strain, and the animals were housed in well-controlled environmental conditions. These experimental parameters allowed us to decrease the impact of genetic variance and gene-environment interactions on trabecular and cortical bone phenotype. This permitted us to assign a physiological function to a common osteonectin allele, providing support for its contribution to the complex trait of skeletal phenotype.
Presently, estimation of a patient's risk of fracture is performed using models based on clinical, demographic and anthropomorphic information, such as BMD, previous fracture, a parent with hip fracture, smoking, glucocorticoid and alcohol use. Although valuable, the prognostic performance of these models could be improved especially in case of idiopathic osteoporosis. Including genetic profiling data for an individual could help improve accuracy of risk assessment and better inform treatment decisions.(46) Osteonectin SNP 1599 could be of importance to consider in investigations of idiopathic osteoporosis. However, the impact of a single variant on fracture risk is small, and identification of many more gene variants with an impact on the skeleton is necessary before real gains in fracture prediction can be realized.(46, 47) Data such as those reported here will contribute to the pool of SNP variants needed for individualized risk assessment and fracture prevention.
Supplementary Material
Figure 5. Working model for osteonectin 3’ UTR SNP mediated effect on bone.

In haplotype A knock-in mice, osteonectin levels in bone are lower compared to haplotype B knock-in mice. The osteonectin 3’ UTR haplotype A is targeted by miRNA (miR-433), which may contribute to lower osteonectin and lower bone volume.
Acknowledgements
We thank Dr. Larry Fisher (NIDCR, NIH) for the gift of the BON-1 antibody, Dr. Andrea Alford (Univ. Michigan, Ann Arbor) for advice on osteonectin protein extraction, Dr. Peter Maye for BSP primers and Dr. Remegius Jackson for contributing to Luciferase reporter construction. Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health under Award Number AR044877 (to AMD) and by the Center for Molecular Medicine at UConn Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Author contributions: NSD, KK, SPY and AMD designed the research. NSD, KK, CBK, DJA, RCR, and AMD performed the research and analyzed the data. NSD and AMD wrote the manuscript and take responsibility for the data analysis. All authors approved of manuscript submission.
Disclosure page.
All authors state that they have no conflicts of interest.
References
- 1.Stewart TL, Ralston SH. Role of genetic factors in the pathogenesis of osteoporosis. J Endocrinol. 2000;166(2):235–45. doi: 10.1677/joe.0.1660235. [DOI] [PubMed] [Google Scholar]
- 2.Peacock M, Turner CH, Econs MJ, Foroud T. Genetics of osteoporosis. Endocr Rev. 2002;23(3):303–26. doi: 10.1210/edrv.23.3.0464. [DOI] [PubMed] [Google Scholar]
- 3.Kurt O, Yilmaz-Aydogan H, Uyar M, Isbir T, Seyhan MF, Can A. Evaluation of ERalpha and VDR gene polymorphisms in relation to bone mineral density in Turkish postmenopausal women. Mol Biol Rep. 2012;39:6723–30. doi: 10.1007/s11033-012-1496-0. [DOI] [PubMed] [Google Scholar]
- 4.Yamada Y. Association of polymorphisms of the transforming growth factor-beta1 gene with genetic susceptibility to osteoporosis. Pharmacogenetics. 2001;11:765–71. doi: 10.1097/00008571-200112000-00004. [DOI] [PubMed] [Google Scholar]
- 5.Shen L, Qiu Y, Xing S, et al. Association between osteoprotegerin genetic variants and bone mineral density in Chinese women. Int Immunopharmacol. 2013;16(2):275–8. doi: 10.1016/j.intimp.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 6.Falcon-Ramirez E, Casas-Avila L, Miranda A, Diez P, Castro C, Rubio J, et al. Sp1 polymorphism in collagen I alpha1 gene is associated with osteoporosis in lumbar spine of Mexican women. Mol Biol Rep. 2011;38:2987–92. doi: 10.1007/s11033-010-9963-y. [DOI] [PubMed] [Google Scholar]
- 7.Koller DL, Econs MJ, Morin PA, et al. Genome screen for QTLs contributing to normal variation in bone mineral density and osteoporosis. J Clin Endocrinol Metab. 2000;85(9):3116–20. doi: 10.1210/jcem.85.9.6778. [DOI] [PubMed] [Google Scholar]
- 8.Kapinas K, Kessler CB, Delany AM. miR-29 suppression of osteonectin in osteoblasts: regulation during differentiation and by canonical Wnt signaling. J Cell Biochem. 2009;108(1):216–24. doi: 10.1002/jcb.22243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boskey AL, Moore DJ, Amling M, Canalis E, Delany AM. Infrared analysis of the mineral and matrix in bones of osteonectin-null mice and their wildtype controls. J Bone Miner Res. 2003;18(6):1005–11. doi: 10.1359/jbmr.2003.18.6.1005. [DOI] [PubMed] [Google Scholar]
- 10.Delany AM, Amling M, Priemel M, Howe C, Baron R, Canalis E. Osteopenia and decreased bone formation in osteonectin-deficient mice. J Clin Invest. 2000;105(9):1325. doi: 10.1172/JCI7039C1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delany AM, Kalajzic I, Bradshaw AD, Sage EH, Canalis E. Osteonectin-null mutation compromises osteoblast formation, maturation, and survival. Endocrinology. 2003;144(6):2588–96. doi: 10.1210/en.2002-221044. [DOI] [PubMed] [Google Scholar]
- 12.Machado do Reis L, Kessler CB, Adams DJ, Lorenzo J, Jorgetti V, Delany AM. Accentuated osteoclastic response to parathyroid hormone undermines bone mass acquisition in osteonectin-null mice. Bone. 2008;43(2):264–73. doi: 10.1016/j.bone.2008.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delany AM, McMahon DJ, Powell JS, Greenberg DA, Kurland ES. Osteonectin/SPARC polymorphisms in Caucasian men with idiopathic osteoporosis. Osteoporos Int. 2008;19(7):969–78. doi: 10.1007/s00198-007-0523-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurland ES, Rosen CJ, Cosman F, McMahon D, Chan F, et al. Insulin-like growth factor-I in men with idiopathic osteoporosis. J Clin Endocrinol Metab. 1997;82:2799–2805. doi: 10.1210/jcem.82.9.4253. [DOI] [PubMed] [Google Scholar]
- 15.Kurland ES, Chan FK, Rosen CJ, Bilezikian JP. Normal growth hormone secretory reserve in men with idiopathic osteoporosis and reduced circulating levels of insulin-like growth factor-I. J Clin Endocrinol Metab. 1998;83:2576–2579. doi: 10.1210/jcem.83.7.4971. [DOI] [PubMed] [Google Scholar]
- 16.Mendell JT, Dietz HC. When the message goes awry: disease-producing mutations that influence mRNA content and performance. Cell. 2001;107(4):411–4. doi: 10.1016/s0092-8674(01)00583-9. Review. [DOI] [PubMed] [Google Scholar]
- 17.Lei SF, Papasian CJ, Deng HW. Polymorphisms in predicted miRNA binding sites and osteoporosis. J Bone Miner Res. 2011;26(1):72–8. doi: 10.1002/jbmr.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simon D, Laloo B, Barillot M, et al. A mutation in the 3'-UTR of the HDAC6 gene abolishing the post-transcriptional regulation mediated by hsa-miR-433 is linked to a new form of dominant X-linked chondrodysplasia. Hum Mol Genet. 2010;19(10):2015–27. doi: 10.1093/hmg/ddq083. [DOI] [PubMed] [Google Scholar]
- 19.Harris SA, Enger RJ, Riggs BL, Spelsberg TC. Development and characterization of a conditionally immortalized human fetal osteoblastic cell line. J Bone Miner Res. 1995;10(2):178–86. doi: 10.1002/jbmr.5650100203. [DOI] [PubMed] [Google Scholar]
- 20.Kapinas K, Kessler C, Ricks T, Gronowicz G, Delany AM. miR-29 modulates Wnt signaling in human osteoblasts through a positive feedback loop. J Biol Chem. 2010;285(33):25221–31. doi: 10.1074/jbc.M110.116137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rehmsmeier M, Steffen P, Hochsmann M, Giegerich R. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10(10):1507–17. doi: 10.1261/rna.5248604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim EJ, Kang IH, Lee JW, Jang WG, Koh JT. MiR-433 mediates ERRγ-suppressed osteoblast differentiation via direct targeting to Runx2 mRNA in C3H10T1/2 cells. Life Sci. 2013;92(10):562–8. doi: 10.1016/j.lfs.2013.01.015. [DOI] [PubMed] [Google Scholar]
- 23.Kapinas K, Kessler CB, Delany AM. miR-29 suppression of osteonectin in osteoblasts: regulation during differentiation and by canonical Wnt signaling. J Cell Biochem. 2009;108(1):216–24. doi: 10.1002/jcb.22243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sheng MH, Lau KH, Beamer WG, Baylink DJ, Wergedal JE. In vivo and in vitro evidence that the high osteoblastic activity in C3H/HeJ mice compared to C57BL/6J mice is intrinsic to bone cells. Bone. 2004;35:711–719. doi: 10.1016/j.bone.2004.05.013. [DOI] [PubMed] [Google Scholar]
- 25.Shultz KL, Donahue LR, Bouxsein ML, Baylink DJ, Rosen CJ, Beamer WG. Congenic strains of mice for verification and genetic decomposition of quantitative trait loci for femoral bone mineral density. J Bone Miner Res. 2003;18:175–185. doi: 10.1359/jbmr.2003.18.2.175. [DOI] [PubMed] [Google Scholar]
- 26.Bradshaw AD, Sage EH. SPARC, a matricellular protein that functions in cellular differentiation and tissue response to injury. J Clin Invest. 2001;107(9):1049–54. doi: 10.1172/JCI12939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kapinas K, Lowther KM, Kessler CB, et al. Bone matrix osteonectin limits prostate cancer cell growth and survival. Matrix Biol. 2012;31(5):299–307. doi: 10.1016/j.matbio.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rentz TJ, Poobalarahi F, Bornstein P, Sage EH, Bradshaw AD. SPARC regulates processing of procollagen I and collagen fibrillogenesis in dermal fibroblasts. J Biol Chem. 2007;282:22062–22071. doi: 10.1074/jbc.M700167200. [DOI] [PubMed] [Google Scholar]
- 29.Chen LJ, Tam PO, Tham CC, et al. Evaluation of SPARC as a candidate gene of juvenile-onset primary open-angle glaucoma by mutation and copy number analyses. Mol Vis. 2010;16:2016–2025. [PMC free article] [PubMed] [Google Scholar]
- 30.Turner RT, Evan GL, Lotinun S, Lapke PD, Iwaniec UT, et al. Dose-Response effects of intermittent PTH on cancellous bone in hindlimb unloaded rats. J Bone Miner Res. 2007;22:64–71. doi: 10.1359/jbmr.061006. [DOI] [PubMed] [Google Scholar]
- 31.Delany AM, Canalis E. Basic fibroblast growth factor destabilizes osteonectin mRNA in osteoblasts. Am J Physiol. 1998;274(3 Pt. 1):734–40. doi: 10.1152/ajpcell.1998.274.3.C734. [DOI] [PubMed] [Google Scholar]
- 32.Chamboredon S, Briggs J, Vial E, et al. v-Jun downregulates the SPARC target gene by binding to the proximal promoter indirectly through Sp1/3. Oncogene. 2003;22:4047–4061. doi: 10.1038/sj.onc.1206713. [DOI] [PubMed] [Google Scholar]
- 33.Dominguez P, et al. Expression of the osteonectin gene potentially controlled by multiple cis- and trans-acting factors in cultured bone cells. J Bone Miner Res. 1991;6:1127–1136. doi: 10.1002/jbmr.5650061015. [DOI] [PubMed] [Google Scholar]
- 34.Ibaraki K, Robey PG, Young MF. Partial characterization of a novel ‘GGA’ factor which binds to the osteonectin promoter in bovine bone cells. Gene. 1993;130:225–232. doi: 10.1016/0378-1119(93)90423-z. [DOI] [PubMed] [Google Scholar]
- 35.Ng KW, Manji SS, Young MF, Findlay DM. Opposing influences of glucocorticoid and retinoic acid on transcriptional control in preosteoblasts. Mol Endocrinol. 1989;3:2079–2085. doi: 10.1210/mend-3-12-2079. [DOI] [PubMed] [Google Scholar]
- 36.Sauk JJ, Norris K, Kerr JM, Somerman MJ, Young MF. Diverse forms of stress result in changes in cellular levels of osteonectin/SPARC without altering mRNA levels in osteoligament cells. Calcif Tissue Int. 1991;49:58–62. doi: 10.1007/BF02555904. [DOI] [PubMed] [Google Scholar]
- 37.De Bonis P, Laborante A, Pizzicoli C, et al. Mutational screening of VSX1, SPARC, SOD1, LOX, and TIMP3 in keratoconus. Mol Vis. 2011;17:2482–2494. [PMC free article] [PubMed] [Google Scholar]
- 38.Lagan AL, Pantelidis P, Renzoni EA, et al. Single-nucleotide polymorphisms in the SPARC gene are not associated with susceptibility to scleroderma. Rheumatology. 2005;44:197–201. doi: 10.1093/rheumatology/keh460. [DOI] [PubMed] [Google Scholar]
- 39.Segat L, Milanese M, Pirulli D, et al. Secreted protein acidic and rich in cysteine (SPARC) gene polymorphism association with hepatocellular carcinoma in Italian patients. J Gastroenterol Hepatol. 2009;24:1840–1846. doi: 10.1111/j.1440-1746.2009.06009.x. [DOI] [PubMed] [Google Scholar]
- 40.Zhou X, Tan FK, Reveille JD, et al. Association of novel polymorphisms with the expression of SPARC in normal fibroblasts and with susceptibility to scleroderma. Arthritis Rheum. 2002;46:2990–2999. doi: 10.1002/art.10601. [DOI] [PubMed] [Google Scholar]
- 41.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hochberg MC. Racial differences in bone strength. Trans Am Clin Climatol Assoc. 2007;118:305–15. [PMC free article] [PubMed] [Google Scholar]
- 43.George A, Tracy JK, Meyer WA, Flores RH, Wilson PD, Hochberg MC. Racial differences in bone mineral density in older men. J Bone Miner Res. 2003;18:2238–44. doi: 10.1359/jbmr.2003.18.12.2238. [DOI] [PubMed] [Google Scholar]
- 44.Gong G, Haynatzki G, Haynatzka V, Howell R, Kosoko-Lasaki S, Fu YX, Yu F, Gallagher JC, Wilson MR. Bone mineral density-affecting genes in Africans. J Natl Med Assoc. 2006;98(7):1102–8. [PMC free article] [PubMed] [Google Scholar]
- 45.Gibson G. Rare and common variants: twenty arguments. Nat Rev Genet. 2012;13:135–145. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nguyen TV, Eisman JA. Genetics and the individualized prediction of fracture. Curr Osteoporos Rep. 2012;10:236–244. doi: 10.1007/s11914-012-0113-4. [DOI] [PubMed] [Google Scholar]
- 47.Mitchell BD, Streeten EA. Clinical impact of recent genetic discoveries in osteoporosis. Appl Clin Genet. 2013;6:75–85. doi: 10.2147/TACG.S52047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.