

Abstract
Objective
We tested the hypothesis that endothelial proliferator-activated receptor-gamma (PPARγ) protects against vascular thrombosis using a transgenic mouse model expressing a PPARγ mutant (E-V290M) selectively in endothelium.
Approach and Results
The time to occlusive thrombosis of the carotid artery was significantly shortened in E-V290M mice compared with non-transgenic (non-Tg) littermates after either chemical injury with ferric chloride (5.1±0.2 vs. 10.1±3.3 minutes; P = 0.01) or photochemical injury with rose bengal (48±9 vs. 74±9 minutes; P = 0.04). Gene Set Enrichment Analysis demonstrated upregulation of NF-κB target genes, including P-selectin, in aortic endothelial cells from E-V290M mice (P < 0.001). Plasma P-selectin and carotid artery P-selectin mRNA were elevated in E-V290M mice (P < 0.05). P-selectin-dependent leukocyte rolling on mesenteric venules was increased in E-V290M mice compared with non-Tg mice (53±8 vs. 25±7 per minute; P = 0.02). The shortened time to arterial occlusion in E-V290M mice was reversed by administration of P-selectin blocking antibodies or neutrophil-depleting antibodies (P = 0.04 and P = 0.02, respectively) prior to photochemical injury.
Conclusions
Endothelial PPARγ protects against thrombosis through a mechanism that involves downregulation of P-selectin expression and diminished P-selectin-mediated leukocyte-endothelial interactions.
Keywords: PPARγ, Thrombosis, Endothelium, P-Selectin, NF-κB
Introduction
Peroxisome proliferator-activated receptor-gamma (PPARγ) is a ligand-activated transcription factor that regulates lipid metabolism, adipocyte differentiation, blood pressure, and insulin sensitivity.1 PPARγ is widely expressed in adipose tissue, liver, muscle, heart, macrophages, and bone, as well as in vascular endothelial and smooth muscle cells.2 In patients with type 2 diabetes, treatment with therapeutic PPARγ agonists such as thiazolidinediones (TZDs) improves glucose control and lowers blood pressure,3 and also may protect from progression of atherosclerosis.4-6 The observation that TZDs exert protective metabolic and vascular effects is consistent with genetic evidence indicating that subjects who carry dominant-negative PPARγ mutations develop severe insulin resistance and hypertension.7
Vascular thrombosis is a major complication of cardiovascular disease and is strongly associated with risk factors such as diabetes, metabolic syndrome, atherosclerosis, and hypertension that are known to be modulated by PPARγ.8 However, the influence of PPARγ on thrombotic susceptibility is not well understood. Some studies performed with cultured endothelial cells have suggested that PPARγ activation may protect against thrombosis by repressing the activation of the transcription factor NF-κB, downregulating the expression of proinflammatory cell adhesion molecules, and enhancing endothelial nitric oxide production.9-13 In contrast, PPARγ agonists have been found to stimulate increased generation of procoagulant microparticles from monocytes and macrophages,14 an observation that might help explain the paradoxical increase in risk of myocardial infarction seen in diabetic patients treated with some TZDs.15 Thus, PPARγ agonists may exert opposing effects on thrombotic susceptibility via actions on different target cells. Clearly, a better understanding of the tissue-specific effects of PPARγ in regulating antithrombotic capacity is needed.16
We previously developed transgenic mouse models in which dominant-negative mutations in the ligand-binding domain of human PPARγ (V290M or P467L) are selectively expressed in endothelium under the control of the VE-cadherin promoter.17, 18 These mutations interfere with basal and agonist-induced PPARγ transcriptional activity and repress PPARγ target genes.19, 20 We demonstrated that endothelium-specific V290M PPARγ (E-V290M) transgenic mice exhibit increased susceptibility to endothelial vasomotor dysfunction when fed a high fat diet17 or crossed to apolipoprotein E-deficient mice.21 In the current study, we used the E-V290M murine model to test the hypothesis that endothelial PPARγ protects against arterial thrombosis in vivo and examine the mechanistic role of the endothelial cell adhesion molecule P-selectin.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
Carotid artery thrombosis is accelerated in E-V290M transgenic mice
To investigate the potential antithrombotic functions of PPARγ specifically in endothelium, we studied transgenic mice expressing a dominant-negative human PPARγ mutant (V290M) targeted to vascular endothelium. Experimental thrombosis of the carotid artery was induced in male E-V290M and non-Tg mice by either transmural chemical injury with ferric chloride (Figure 1A) or luminal injury with the photo-activatable dye, rose bengal (Figure 1B). Compared with non-Tg mice, E-V290M mice exhibited a prothrombotic phenotype with both methods of carotid artery injury. After ferric chloride injury, the time to stable occlusion of the carotid artery was significantly shorter in E-V290M mice than non-Tg mice (P = 0.01; Figure 1A). The time to stable occlusion also was shorter in E-V290M mice compared with non-Tg mice after photochemical injury (P = 0.04; Figure 1B). Immunohistochemical staining demonstrated the presence of cells expressing the neutrophil antigen Ly-6 and tissue factor within the thrombosed lumen of the carotid artery after photochemical injury (Figure 2). The Ly-6 and tissue factor-positive cells were localized near the intimal layer of the vessel wall, which suggested that activated neutrophils were interacting with the damaged endothelium or subendothelium at the site of injury.
Figure 1.

Carotid artery thrombosis is accelerated in E-V290M transgenic mice. Carotid artery thrombosis was induced by either chemical injury with (A) 7% FeCl3 (N = 5 to 7) or (B) photochemical injury with rose bengal (N = 7 to 8) in male non-Tg or E-V290M mice at 14-16 weeks of age. The time to stable occlusion was measured using a Doppler flow probe. Values are mean ± SE. The P-values were determined using the rank sum test.
Figure 2.
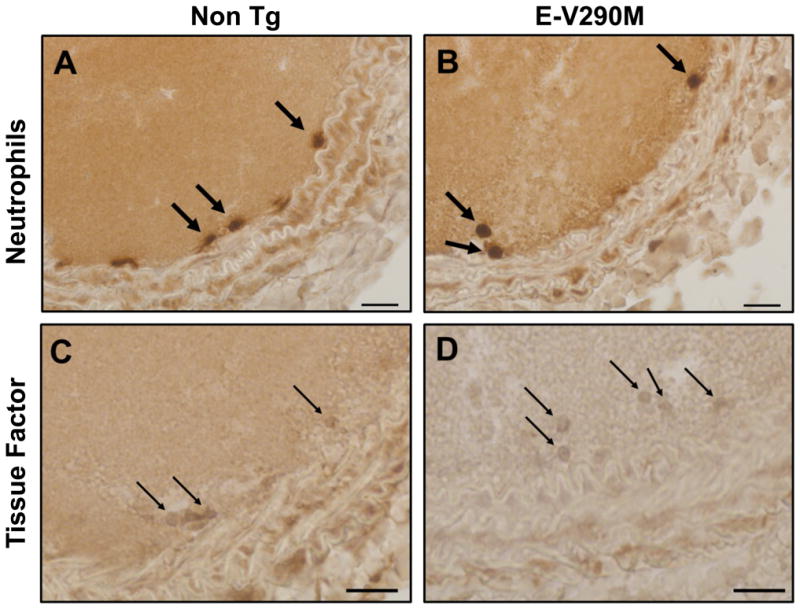
Immunohistochemical detection of neutrophils and tissue factor in carotid artery thrombi. Carotid artery thrombosis was induced by photochemical injury with rose bengal in male non-Tg and E-V290M mice, and the carotid arteries were harvested and subjected to immunohistochemical staining for neutrophils (Ly-6) or tissue factor (PAA524Mu01). Cells staining positively for neutrophils (thick arrows) and tissue factor (thin arrows) were detected within the thrombus adjacent to the intima. Bar indicates 20 μm.
Venous thrombosis is not enhanced in E-V290M mice
Venous thrombosis was induced by ligation of the inferior vena cava (IVC). There were no significant differences in the weight or length of venous thrombi isolated from E-V290M mice compared with non-Tg mice 48 hours after IVC ligation (Supplemental Figure I).
Dominant-negative PPARγ upregulates endothelial NF-κB target genes, including P-selectin
To determine if genes known to be important in the regulation of vascular thrombosis are altered by endothelial PPARγ interference, we analyzed an existing mRNA microarray dataset (available from NCBI-GEO at accession GSE11870) generated from gene expression profiling of endothelial cells derived from E-V290M mice and their non-Tg littermates.17 We first queried the dataset for genes with established roles in vascular thrombosis (Table 1). Several of these genes exhibited a significant change in expression in endothelial cells of E-V290M mice, with the largest increase observed in the Selp gene encoding P-selectin (6.9-fold upregulation; P < 0.01). The highly significant upregulation of Selp, a known NF-κB target gene,22 suggested the possibility that the NF-κB pathway was activated in the endothelium of E-V290M mice. To further address this possibility, we queried the dataset using a list of experimentally validated NF-κB target genes (http://bioinfo.lifl.fr/NF-KB/). In addition to statistically significant and robust increases in several individual NF-κB target genes (see Supplemental Table I), the complete set of NF-κB targets, as a group, displayed a significant increase in expression by GSEA (P < 0.001, normalized enrichment score 2.14).
Table 1. Microarray dataset analysis. Changes in expression of thrombosis-related genes in endothelial cells of E-V290M mice compared with non-Tg mice.
| Gene* | Probe | Fold Change | P-Value | Description |
|---|---|---|---|---|
| Selp | 1420558_at | 6.90 | 0.0061 | P-selectin |
| F2rl1 | 1448931_at | 5.29 | 0.0856 | protease-activated receptor-2 |
| Serpine1 | 1419149_at | 4.15 | 0.0665 | plasminogen activator inhibitor-1 |
| Entpd1 | 1450939_at | 1.72 | 0.0050 | CD39 |
| Thbd | 1448529_at | 1.29 | 0.1104 | thrombomodulin |
| Sele | 1421712_at | 1.25 | 0.1743 | E-selectin |
| Icam1 | 1424067_at | 1.22 | 0.1967 | Intercellular adhesion molecule-1 |
| F2r | 1437308_s_at | 1.19 | 0.3228 | protease activate receptor-1 |
| Plat | 1415806_at | 1.15 | 0.2174 | tissue plasminogen activator |
| Serpine2 | 1416666_at | 1.15 | 0.5358 | protease nexin-1 |
| Anxa5 | 1425567_a_at | 1.08 | 0.3687 | annexin A5 |
| F3 | 1417408_at | 0.95 | 0.6423 | tissue factor |
| Vwf | 1435386_at | 0.92 | 0.4708 | von Willebrand factor |
| Vcam1 | 1451314_a_at | 0.88 | 0.3016 | vascular cell adhesion molecule-1 |
| Tfpi | 1438530_at | 0.84 | 0.2418 | tissue factor pathway inhibitor |
| Adamts1 | 1450716_at | 0.81 | 0.1452 | ADAMTS1 |
| Procr | 1420664_s_at | 0.77 | 0.2049 | endothelial protein C receptor |
Gene: Official gene symbol from NCBI; Probe: probe set from Affymetrix MOE430 array; Fold change: fold change relative to non-transgenic littermates. For genes with multiple probe sets, the probe set with the lowest p-value was selected. Data are from an existing microarray dataset (available from NCBI-GEO at accession GSE11870).
E-V90M mice have elevated levels of P-selectin mRNA and protein
To confirm that the altered expression of the Selp gene observed in the microarray dataset analysis was associated with increased expression of P-selectin in E-V90M mice, we measured levels of P-selectin mRNA in the carotid artery by qPCR. We found that P-selectin mRNA was elevated 2.3-fold in E-V290M mice compared with non-Tg mice (P = 0.03; Figure 3A). Similarly, E-V290M mice had significantly elevated levels of circulating soluble P-selectin antigen in plasma compared with non-Tg mice (P = 0.004; Figure 3B). Because plasma soluble P-selectin can originate from platelets as well as endothelial cells,23 we also measured platelet P-selectin surface expression by flow cytometry. No differences in platelet surface P-selectin were observed between E-V290M and non-Tg mice at baseline or after activation of platelet alpha granule release with thrombin (Figure 4A). Additionally, there were no differences in the level of fibrinogen binding at baseline or after activation with thrombin between E-V290M and non-Tg mice (Figure 4B). These findings suggest that the elevation of plasma soluble P-selectin in E-V290M mice was due to increased expression of P-selectin in endothelial cells rather than release of P-selectin from platelets. These observations also demonstrate that the endothelium-targeted dominant-negative V290M transgene does not have any appreciable effect on platelet activation in E-V290M mice.
Figure 3.

Elevated levels of P-selectin mRNA and protein in E-V290M mice. (A) Levels of P-selectin mRNA in carotid arteries from E-V290M or non-Tg mice were measured by qPCR (N = 5 to 6). (B) Plasma levels of soluble P-selectin were measured in E-V290M or non-Tg mice (N = 6 to 8). Values are mean ± SE. The P-values were determined using the Student's t-test.
Figure 4.

Platelet activation responses and P-selectin expression do not differ between E-V290M and non-Tg mice. Platelet surface P-selectin (A) and fibrinogen binding (B) were measured by flow cytometry at baseline and after stimulation with 0.05 U/ml thrombin (N = 5 mice in each group). Values are mean ± SE. **P < 0.01; *P < 0.05 vs. baseline by two-way ANOVA. MFI: mean fluorescence intensity.
E-V290M mice have increased P-selectin-mediated leukocyte-endothelial interactions
To assess the functional activity of endothelial P-selectin, we visualized leukocyte rolling on unstimulated mesenteric veins in real time using phase contrast video microscopy. We observed a 2-fold increase in rolling leukocytes per minute in E-V290M mice (53 ± 8) compared with non-Tg mice (25±7; P = 0.02) (Figure 5 and Supplemental Movies 1 and 2). Leukocyte rolling was almost completely inhibited by pre-treatment with a P-selectin blocking antibody in both E-V290M and non-Tg mice (Figure 5 and Supplemental Movie 3), demonstrating that leukocyte rolling was dependent on P-selectin.
Figure 5.
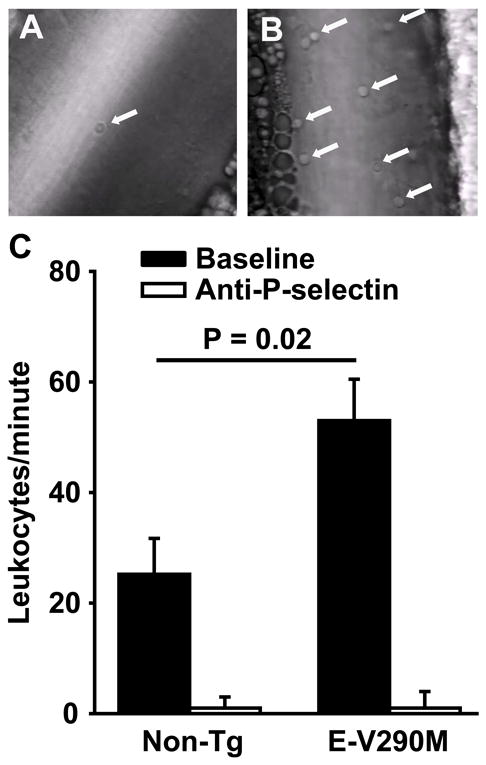
Increased P-selectin-mediated leukocyte rolling in E-V290M mice. Leukocyte rolling on unstimulated mesenteric veins of male non-Tg (A) or E-V290M (B) mice (4 to 6 weeks of age; N = 6 to 10 per group) was measured in real time using phase contrast video microscopy. Arrows indicate rolling leukocytes. (C) The extent of leukocyte rolling was quantitated by counting the number of cells passing a fixed point per minute (leukocytes/minute). Leukocyte rolling was abolished after pre-treatment with a P-selectin antibody in both E-V290M or non-Tg mice (N = 4 per group). Values are mean ± SE. The P-values were determined using one-way ANOVA. See Supplemental Online Material for video files.
Accelerated carotid artery thrombosis in E-V290M mice is dependent on P-selectin and neutrophils
To determine the mechanistic role of P-selectin-mediated endothelial-leukocyte interactions in the prothrombotic phenotype of E-V290M mice, mice were pre-treated with either the P-selectin blocking antibody or a neutrophil depleting antibody (anti-Ly-6G) prior to inducing carotid artery thrombosis by photochemical injury. Treatment with anti-Ly-6G resulted in a 95% decrease in peripheral blood neutrophil count, from 1.39±0.65 to 0.07±0.04 K/μl (P = 0.04). Compared with E-V290M mice pre-treated with a control IgG, E-V290M mice pre-treated with either the P-selectin blocking antibody or the neutrophil-depleting antibody exhibited prolonged times to thrombotic occlusion (P = 0.04 and P = 0.02, respectively; Figure 6). Antibody pre-treatment did not have any significant effects on the time to occlusion in non-Tg mice (Figure 6). These findings suggest that the accelerated carotid artery thrombosis in E-V290M mice is dependent on both P-selectin and neutrophils.
Figure 6.

Accelerated carotid artery thrombosis in E-V290M mice is dependent on both P-selectin and neutrophils. Carotid artery thrombosis was induced by photochemical injury with rose bengal in male non-Tg or E-V290M mice after pre-treatment with either a control IgG, an anti-P-selectin blocking antibody, or a neutrophil-depleting antibody (anti-Ly-6G) (N = 4 to 10 in each group). The time to stable occlusion was measured using a Doppler flow probe. Values are mean ± SE. The P-values were determined using the rank sum test.
Human PPARγ transgene expression in blood cells
We have shown previously that the VE-cadherin promoter confers endothelial-selective expression of dominant-negative human PPARγ in E-V290M mice.18 Because VE-cadherin is also expressed in hematopoietic stem cells,24 we measured the expression of human PPARγ V290M mRNA by qPCR in platelets and leukocytes, as well as in lung, which is rich in endothelium (Supplemental Figure II). As expected, strong expression of human PPARγ V290M mRNA was found in lung. Trace expression of human PPARγ V290M mRNA also was detected in peripheral blood leukocytes, at a level that was >15-fold lower than that in lung. No expression of human PPARγ V290M mRNA was detected in platelets.
Discussion
PPARγ is well known to have anti-inflammatory properties mediated by altered expression of pro- and anti-inflammatory genes.1, 16, 21 However, despite an established association between inflammatory conditions and thrombotic risk,25 the potential role of PPARγ in protecting from thrombosis remains poorly defined. Administration of high-affinity PPARγ agonists has been reported to protect against thrombosis in a mouse model of insulin resistance26 and to have anti-platelet effects in vitro.27, 28 In contrast, some PPARγ agonists have been reported to stimulate increased generation of procoagulant microparticles from monocytes and macrophages.14 Moreover, the cell-specific influences of PPARγ in vascular endothelium on thrombotic susceptibility remain poorly defined. With this uncertainty in mind, and in consideration of the growing public heath burden of thrombotic complications associated with obesity and insulin resistance,29 we utilized the E-V290M transgenic mouse model to delineate the effects on thrombosis of endogenous PPARy expressed in endothelium. The major findings from our study are that 1) cell-specific interference with endogenous PPARγ in vascular endothelium increases susceptibility to arterial thrombosis in mice, and 2) the mechanism of accelerated thrombosis in E-V290M mice is mediated by upregulation of P-selectin and increased P-selectin-dependent endothelial-leukocyte interactions.
We chose to use a dominant negative approach, rather than a PPARγ gene knockout approach for a number of reasons. First, dominant negative mutations such as V290M, while rare, are bonafide mutations that cause disease in human patients.7 Second, the PPARγ null mouse has an embryonic lethal phenotype30 and there are no null mutations known to exist in humans. Finally, the alternative approach of using an endothelial tissue-specific knockout model31 has a major limitation. In the unliganded state, PPARγ binds to DNA and actively represses genes by recruiting a co-repressor complex to the gene being silenced32. When a ligand is present, the co-repressor complex is replaced by a co-activator complex, leading to gene activation. Deleting PPARγ has a similar effect as ligand activation because there is a small induction of gene expression due to the loss of active repression. This does not occur with the use of dominant negative PPARγ mutants. As expected, we observed strong expression of the PPARγ V290M transgene in endothelium. We also detected trace expression of the transgene by qPCR in peripheral blood mononuclear cells (Supplemental Figure II). It is possible that the low-level expression of the transgene in peripheral blood cells may be due to residual activity of the VE-cadherin promoter in cells of myeloid lineage.24 Alternatively, it may represent the presence of trace amounts of circulating endothelial cells in the mononuclear cell fraction.
PPARγ has been reported to inhibit the activation of NF-κB in endothelial cells,16 and our gene expression analysis confirmed that known NF-κB target genes are significantly upregulated in aortic endothelial cells of E-V290M mice. In particular, we identified P-selectin as an NF-κB target gene that is highly upregulated in the endothelium of E-V290M mice compared with non-Tg mice. P-selectin (CD62P) is an inducible cell-surface leukocyte adhesion molecule that mediates initial interactions between circulating neutrophils and the activated endothelium.23 In accordance with the upregulation of P-selectin seen in the microarray dataset analysis, we observed increased levels of P-selectin mRNA in the carotid artery and increased circulating soluble P-selectin in the plasma of E-V290M mice (Figure 3). Importantly, the increased expression of endothelial P-selectin was accompanied by increased leukocyte rolling, indicating that the P-selectin expressed in E-V290M mice was functional in mediating increased leukocyte-endothelial interactions. In addition to mediating leukocyte recruitment to activated endothelium, P-selectin can promote thrombosis by facilitating platelet adhesion and inducing the generation of procoagulant microparticles and neutrophil extracellular traps.23, 33 We therefore reasoned that P-selectin may be a key mediator of the prothrombotic phenotype of E-V290M mice.
The prothrombotic phenotype of E-V290M mice was apparent when carotid artery thrombosis was induced by either chemical injury with ferric chloride or photochemical injury with rose bengal (Figure 1). For subsequent mechanistic experiments, we chose to focus on the photochemical injury model because it is more endothelium-dependent34 whereas the ferric chloride injury model is partially mediated by red blood cells.35 To determine the role of P-selectin-mediated leukocyte-endothelial interactions in the enhanced thrombotic susceptibility of E-V290M mice, we pre-treated mice with a P-selectin blocking antibody that was shown to almost completely eliminate leukocyte rolling (Figure 5). We found that the P-selectin blocking antibody largely reversed the prothrombotic effect of endothelium-specific PPARγ interference (Figure 6). We also observed a similar protective anti-thrombotic effect after depletion of >95% of the circulating neutrophils in E-V290M mice (Figure 6). Together, these findings suggest that endogenous endothelial PPARγ protects from thrombosis through a mechanism that involves downregulation of P-selectin expression, perhaps by antagonizing the transcriptional effects of NF-κB, and diminished P-selectin mediated leukocyte-endothelial interactions.
Both ferric chloride and rose bengal induce vascular injury via oxidative mechanisms, leading to endothelial damage and denudation.34 The thrombotic response to oxidative injury is thought to be initiated by the adhesion of platelets and leukocytes to the exposed subendothelium. However, recent findings suggest that, at least for the ferric chloride model, the injured endothelium may be retained after ferric chloride exposure and may contribute to thrombosis.35, 36 Our findings suggest that expression of P-selectin on the retained, injured endothelium contributes to leukocyte adhesion and thrombosis after oxidative injury. We acknowledge that it is possible that additional endothelial PPARγ and/or NF-κB target genes, such as E-selectin, VCAM-1, ICAM-1, and tissue factor also may contribute to the prothrombotic phenotype of E-V290M mice. However, none of these genes emerged from the gene expression dataset analysis as significantly upregulated in endothelial cells from E-V290M mice. We note that genes encoding protease-activated receptor 2 (F2rl1) and plasminogen activator inhibitor 1 (Serpine1) were upregulated 5-fold and 4-fold, respectively, with a borderline level of statistical significance (P < 0.1) (Table 1). These may be attractive target genes for future study.
The lack of a venous thrombosis phenotype in E-V290M mice using the IVC stasis model is surprising, because P-selectin is thought to play a mechanistic role in venous thrombosis.37 We consider these findings to be very interesting, however, because they suggest that non-endothelial sources of P-selectin (e.g. platelet P-selectin) may play a greater role than endothelial P-selectin in driving venous thrombosis, In contrast, endothelial P-selectin may contribute more directly to arterial thrombosis. In support of this idea, it has been suggested that multiple pools of P-selectin promote venous thrombogenesis.38
Our findings may have implications for the clinical observation that, despite their generally protective metabolic and cardiovascular effects in diabetic patients, some TZDs have been found to paradoxically increase the risk of thrombotic vascular complications such as myocardial infarction.15 Our results suggest that activation of PPARγ specifically in vascular endothelium is antithrombotic and protective, raising the possibility that the apparent prothrombotic effects of systemically administered PPARγ agonist may be mediated through effects in other cell types. The protective antithrombotic effect of the P-selectin blocking antibody observed in E-V290M mice suggests that antagonism of P-selectin may be potential therapeutic approach to prevent thrombosis in patients with impaired PPARγ function due to obesity or insulin resistance. Oral P-selectin blocking agents are being developed for several different clinical indications.39-42
In summary, this study demonstrates that selective inactivation of PPARγ in vascular endothelium results in a prothrombotic phenotype characterized by upregulation of P-selectin and other NF-κB target genes. Our data further demonstrate that interference with endothelial PPARγ leads to accelerated thrombosis through a mechanism in which increased expression of P-selectin causes enhanced leukocyte recruitment to the vessel wall. These findings suggest that one mechanism for protection against thrombosis by endogenous endothelial PPARγ is by suppressing P-selectin-mediated leukocyte-endothelial interactions.
Supplementary Material
Significance.
Vascular thrombosis is a major complication of cardiovascular disease. Peroxisome proliferator-activated receptor-gamma (PPARγ) is a ligand-activated transcription factor that regulates lipid metabolism, adipocyte differentiation, blood pressure, and insulin sensitivity. The influence of PPARγ on thrombotic susceptibility is not well understood, and PPARγ agonists may exert opposing effects on thrombotic susceptibility via actions on different target cells. Using a transgenic mouse model expressing a dominant-negative PPARγ mutant selectively in endothelium, we tested the hypothesis that PPARγ protects against vascular thrombosis by altering the gene expression profile in endothelial cells. The novel findings of this study are: 1) selective interference with the transcription factor PPARγ in endothelium causes a prothrombotic phenotype in transgenic mice, and 2) endothelial PPARγ protects against thrombosis by downregulating P-selectin-mediated leukocyte-endothelial interactions.
Acknowledgments
We thank Severine Groh, Jeff Stevens, and Prem Prakash for technical assistance. The Sysmex automatic hematology analyzer used in this study was provided on an on-loan basis from Sysmex Corporation, Kobe, Japan.
Sources of Funding: The authors gratefully acknowledge the generous research support of the Roy J. Carver Trust (to C.D.S.) and the American Society of Hematology (to S.R.L.). This study was supported in part by research funding from National Institutes of Health (NIH) grants HL063943 and HL062984 to S.R.L., NIH grants HL048058, HL061446, HL062984, HL084207 to C.D.S., NIH grants HL118246 and HL118742 to A.K.C., NIH grant T32 HL007344 to M.A.G., an American Heart Association (AHA) postdoctoral award to H.J., and an AHA predoctoral award to I.O.B.
Nonstandard Abbreviations and Acronyms
- E-V290M
transgenic mouse model expressing the V290M PPARγ mutant selectively in endothelium
- FITC
fluorescein isothiocyanate
- GSEA
gene set enrichment analysis
- IVC
inferior vena cava
- PPARγ
proliferator-activated receptor-gamma
- non-Tg
non-transgenic
- PE
phycoerythrin
- TZD
thiazolidinedione
Footnotes
Disclosures: S.R.L. is a consultant to Novo Nordisk. The authors declare no competing financial interests.
References
- 1.Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- 2.Sigmund CD. Endothelial and vascular muscle PPARgamma in arterial pressure regulation: lessons from genetic interference and deficiency. Hypertension. 2010;55:437–44. doi: 10.1161/HYPERTENSIONAHA.109.144170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sharma AM, Staels B. Peroxisome proliferator-activated receptor gamma and adipose tissue--understanding obesity-related changes in regulation of lipid and glucose metabolism. J Clin Endocrinol Metab. 2007;92:386–95. doi: 10.1210/jc.2006-1268. [DOI] [PubMed] [Google Scholar]
- 4.Mazzone T, Meyer PM, Feinstein SB, Davidson MH, Kondos GT, D'Agostino RB, Sr, Perez A, Provost JC, Haffner SM. Effect of pioglitazone compared with glimepiride on carotid intima-media thickness in type 2 diabetes: a randomized trial. Jama. 2006;296:2572–81. doi: 10.1001/jama.296.21.joc60158. [DOI] [PubMed] [Google Scholar]
- 5.Nissen SE, Nicholls SJ, Wolski K, Nesto R, Kupfer S, Perez A, Jure H, De Larochelliere R, Staniloae CS, Mavromatis K, Saw J, Hu B, Lincoff AM, Tuzcu EM, Investigators P. Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: the PERISCOPE randomized controlled trial. Jama. 2008;299:1561–73. doi: 10.1001/jama.299.13.1561. [DOI] [PubMed] [Google Scholar]
- 6.Hodis HN, Mack WJ, Zheng L, Li Y, Torres M, Sevilla D, Stewart Y, Hollen B, Garcia K, Alaupovic P, Buchanan TA. Effect of peroxisome proliferator-activated receptor gamma agonist treatment on subclinical atherosclerosis in patients with insulin-requiring type 2 diabetes. Diabetes Care. 2006;29:1545–53. doi: 10.2337/dc05-2462. [DOI] [PubMed] [Google Scholar]
- 7.Barroso I, Gurnell M, Crowley VE, Agostini M, Schwabe JW, Soos MA, Maslen GL, Williams TD, Lewis H, Schafer AJ, Chatterjee VK, O'Rahilly S. Dominant negative mutations in human PPARgamma associated with severe insulin resistance, diabetes mellitus and hypertension. Nature. 1999;402:880–883. doi: 10.1038/47254. [DOI] [PubMed] [Google Scholar]
- 8.Roach RE, Lijfering WM, Flinterman LE, Rosendaal FR, Cannegieter SC. The increased risk of arterial cardiovascular disease after venous thrombosis is determined by common etiologic factors. Blood. 2013 doi: 10.1182/blood-2013-01-479238. [DOI] [PubMed] [Google Scholar]
- 9.Jackson SM, Parhami F, Xi XP, Berliner JA, Hsueh WA, Law RE, Demer LL. Peroxisome proliferator-activated receptor activators target human endothelial cells to inhibit leukocyte-endothelial cell interaction. Arterioscler Thromb Vasc Biol. 1999;19:2094–104. doi: 10.1161/01.atv.19.9.2094. [DOI] [PubMed] [Google Scholar]
- 10.Wang N, Verna L, Chen NG, Chen J, Li H, Forman BM, Stemerman MB. Constitutive activation of peroxisome proliferator-activated receptor-gamma suppresses pro-inflammatory adhesion molecules in human vascular endothelial cells. J Biol Chem. 2002;277:34176–81. doi: 10.1074/jbc.M203436200. [DOI] [PubMed] [Google Scholar]
- 11.Sasaki M, Jordan P, Welbourne T, Minagar A, Joh T, Itoh M, Elrod JW, Alexander JS. Troglitazone, a PPAR-gamma activator prevents endothelial cell adhesion molecule expression and lymphocyte adhesion mediated by TNF-alpha. BMC physiology. 2005;5:3. doi: 10.1186/1472-6793-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calnek DS, Mazzella L, Roser S, Roman J, Hart CM. Peroxisome proliferator-activated receptor gamma ligands increase release of nitric oxide from endothelial cells. Arterioscler Thromb Vasc Biol. 2003;23:52–7. doi: 10.1161/01.atv.0000044461.01844.c9. [DOI] [PubMed] [Google Scholar]
- 13.Ricote M, Glass CK. PPARs and molecular mechanisms of transrepression. Biochim Biophys Acta. 2007;1771:926–35. doi: 10.1016/j.bbalip.2007.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Neri T, Cordazzo C, Carmazzi Y, Petrini S, Balia C, Stefanelli F, Amoruso A, Brunelleschi S, Breschi MC, Pedrinelli R, Paggiaro P, Celi A. Effects of peroxisome proliferator-activated receptor-gamma agonists on the generation of microparticles by monocytes/macrophages. Cardiovasc Res. 2012;94:537–44. doi: 10.1093/cvr/cvs125. [DOI] [PubMed] [Google Scholar]
- 15.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–71. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 16.Duan SZ, Usher MG, Mortensen RM. Peroxisome proliferator-activated receptor-gamma-mediated effects in the vasculature. Circ Res. 2008;102:283–294. doi: 10.1161/CIRCRESAHA.107.164384. [DOI] [PubMed] [Google Scholar]
- 17.Beyer AM, de Lange WJ, Halabi CM, Modrick ML, Keen HL, Faraci FM, Sigmund CD. Endothelium-specific interference with peroxisome proliferator activated receptor gamma causes cerebral vascular dysfunction in response to a high-fat diet. Circ Res. 2008;103:654–61. doi: 10.1161/CIRCRESAHA.108.176339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beyer AM, Baumbach GL, Halabi CM, Modrick ML, Lynch CM, Gerhold TD, Ghoneim SM, de Lange WJ, Keen HL, Tsai YS, Maeda N, Sigmund CD, Faraci FM. Interference with PPARgamma signaling causes cerebral vascular dysfunction, hypertrophy, and remodeling. Hypertension. 2008;51:867–871. doi: 10.1161/HYPERTENSIONAHA.107.103648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Keen HL, Halabi CM, Beyer AM, de Lange WJ, Liu X, Maeda N, Faraci FM, Casavant TL, Sigmund CD. Bioinformatic analysis of gene sets regulated by ligand-activated and dominant-negative peroxisome proliferator-activated receptor gamma in mouse aorta. Arterioscler Thromb Vasc Biol. 2010;30:518–25. doi: 10.1161/ATVBAHA.109.200733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li G, Leff T. Altered promoter recycling rates contribute to dominant-negative activity of human peroxisome proliferator-activated receptor-gamma mutations associated with diabetes. Mol Endocrinol. 2007;21:857–64. doi: 10.1210/me.2006-0401. [DOI] [PubMed] [Google Scholar]
- 21.Pelham CJ, Keen HL, Lentz SR, Sigmund CD. Dominant Negative PPARgamma Promotes Atherosclerosis, Vascular Dysfunction and Hypertension Through Distinct Effects in Endothelium and Vascular Muscle. Am J Physiol Regul Integr Comp Physiol. 2013 doi: 10.1152/ajpregu.00607.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–66. doi: 10.1038/sj.onc.1203239. [DOI] [PubMed] [Google Scholar]
- 23.Cambien B, Wagner DD. A new role in hemostasis for the adhesion receptor P-selectin. Trends in molecular medicine. 2004;10:179–86. doi: 10.1016/j.molmed.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 24.Dzierzak E. The emergence of definitive hematopoietic stem cells in the mammal. Curr Opin Hematol. 2005;12:197–202. doi: 10.1097/01.moh.0000160736.44726.0e. [DOI] [PubMed] [Google Scholar]
- 25.Levi M, van der Poll T, Schultz M. Infection and inflammation as risk factors for thrombosis and atherosclerosis. Seminars in Thrombosis and Hemostasis. 2012;38:506–14. doi: 10.1055/s-0032-1305782. [DOI] [PubMed] [Google Scholar]
- 26.Bodary PF, Vargas FB, King SA, Jongeward KL, Wickenheiser KJ, Eitzman DT. Pioglitazone protects against thrombosis in a mouse model of obesity and insulin resistance. J Thromb Haemost. 2005;3:2149–2153. doi: 10.1111/j.1538-7836.2005.01551.x. [DOI] [PubMed] [Google Scholar]
- 27.Akbiyik F, Ray DM, Gettings KF, Blumberg N, Francis CW, Phipps RP. Human bone marrow megakaryocytes and platelets express PPARgamma, and PPARgamma agonists blunt platelet release of CD40 ligand and thromboxanes. Blood. 2004;104:1361–8. doi: 10.1182/blood-2004-03-0926. [DOI] [PubMed] [Google Scholar]
- 28.Moraes LA, Spyridon M, Kaiser WJ, Jones CI, Sage T, Atherton RE, Gibbins JM. Non-genomic effects of PPARgamma ligands: inhibition of GPVI-stimulated platelet activation. J Thromb Haemost. 2010;8:577–87. doi: 10.1111/j.1538-7836.2009.03732.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blokhin IO, L SR. Mechanisms of thrombosis in obesity. Current Opinion in Hematology. 2013 doi: 10.1097/MOH.0b013e3283634443. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, Koder A, Evans RM. PPAR gamma is required for placental, cardiac, and adipose tissue development. Molecular cell. 1999;4:585–95. doi: 10.1016/s1097-2765(00)80209-9. [DOI] [PubMed] [Google Scholar]
- 31.Qu A, Shah YM, Manna SK, Gonzalez FJ. Disruption of endothelial peroxisome proliferator-activated receptor gamma accelerates diet-induced atherogenesis in LDL receptor-null mice. Arterioscler Thromb Vasc Biol. 2012;32:65–73. doi: 10.1161/ATVBAHA.111.239137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sigmund CD. Endothelial and vascular muscle PPARgamma in arterial pressure regulation: lessons from genetic interference and deficiency. Hypertension. 2010;55:437–444. doi: 10.1161/HYPERTENSIONAHA.109.144170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD, Jr, Wrobleski SK, Wakefield TW, Hartwig JH, Wagner DD. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci U S A. 2010;107:15880–5. doi: 10.1073/pnas.1005743107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Westrick RJ, Winn ME, Eitzman DT. Murine models of vascular thrombosis. Arterioscler Thromb Vasc Biol. 2007;27:2079–2093. doi: 10.1161/ATVBAHA.107.142810. [DOI] [PubMed] [Google Scholar]
- 35.Barr JD, Chauhan AK, Schaeffer GV, Hansen JK, Motto DG. Red blood cells mediate the onset of thrombosis in the ferric chloride murine model. Blood. 2013;121:3733–41. doi: 10.1182/blood-2012-11-468983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eckly A, Hechler B, Freund M, Zerr M, Cazenave JP, Lanza F, Mangin PH, Gachet C. Mechanisms underlying FeCl3-induced arterial thrombosis. J Thromb Haemost. 2011;9:779–89. doi: 10.1111/j.1538-7836.2011.04218.x. [DOI] [PubMed] [Google Scholar]
- 37.Myers DD, Hawley AE, Farris DM, Wrobleski SK, Thanaporn P, Schaub RG, Wagner DD, Kumar A, Wakefield TW. P-selectin and leukocyte microparticles are associated with venous thrombogenesis. J Vasc Surg. 2003;38:1075–89. doi: 10.1016/s0741-5214(03)01033-4. [DOI] [PubMed] [Google Scholar]
- 38.Culmer DL, Diaz JA, Hawley AE, Jackson TO, Shuster KA, Sigler RE, Wakefield TW, Myers DD., Jr Circulating and vein wall P-selectin promote venous thrombogenesis during aging in a rodent model. Thrombosis research. 2013;131:42–8. doi: 10.1016/j.thromres.2012.10.013. [DOI] [PubMed] [Google Scholar]
- 39.Myers DD, Jr, Rectenwald JE, Bedard PW, Kaila N, Shaw GD, Schaub RG, Farris DM, Hawley AE, Wrobleski SK, Henke PK, Wakefield TW. Decreased venous thrombosis with an oral inhibitor of P selectin. J Vasc Surg. 2005;42:329–36. doi: 10.1016/j.jvs.2005.04.045. [DOI] [PubMed] [Google Scholar]
- 40.Kutlar A, Ataga KI, McMahon L, Howard J, Galacteros F, Hagar W, Vichinsky E, Cheung AT, Matsui N, Embury SH. A potent oral P-selectin blocking agent improves microcirculatory blood flow and a marker of endothelial cell injury in patients with sickle cell disease. Am J Hematol. 2012;87:536–9. doi: 10.1002/ajh.23147. [DOI] [PubMed] [Google Scholar]
- 41.Wang P, Yang Y, Hong H, Zhang Y, Cai W, Fang D. Aptamers as therapeutics in cardiovascular diseases. Current medicinal chemistry. 2011;18:4169–74. doi: 10.2174/092986711797189673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Woollard KJ, Chin-Dusting J. P-selectin antagonism in inflammatory disease. Curr Pharm Des. 2010;16:4113–8. doi: 10.2174/138161210794519192. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.