

Abstract
Overdose of acetaminophen (APAP) can cause acute liver injury that is sometimes fatal, requiring efficient pharmacological intervention. The traditional Chinese herb Bupleurum falcatum has been widely used for the treatment of several liver diseases in eastern Asian countries, and saikosaponin d (SSd) is one of its major pharmacologically-active components. However, the efficacy of Bupleurum falcatum or SSd on APAP toxicity remains unclear. C57BL/6 mice were administered SSd intraperitoneally once daily for five days, followed by APAP challenge. Biochemical and pathological analysis revealed that mice treated with SSd were protected against APAP-induced hepatotoxicity. SSd markedly suppressed phosphorylation of nuclear factor kappa B (NF-kB) and signal transducer and activator of transcription 3 (STAT3) and reversed the APAP-induced increases in the target genes of NF-kB, such as pro-inflammatory cytokine Il6 and Ccl2, and those of STAT3, such as Socs3, Fga, Fgb and Fgg. SSd also enhanced the expression of the anti-inflammatory cytokine Il10 mRNA. Collectively, these results demonstrate that SSd protects mice from APAP-induced hepatotoxicity mainly through down-regulating NF-kB- and STAT3-mediated inflammatory signaling. This study unveils one of the possible mechanisms of hepatoprotection caused by Bupleurum falcatum and/or SSd.
Keywords: saikosaponin d, acetaminophen, hepatotoxicity, NFκB, STAT3
1. Introduction
Acetaminophen (APAP), also known as paracetamol and N-acetyl-p-aminophenol, is an over-the-counter analgesic and antipyretic agent that is widely used in the world. Although safe at therapeutic doses, APAP overdose can cause acute liver failure [1]. Over the past four decades, numerous studies have focused on the molecular mechanism of APAP toxicity and therapeutic strategies to intervene in APAP-induced hepatotoxicity [2-5]. Natural products or their components are promising candidates because of their abundance, diversity, long history of use, and safety.
Bupleurum falcatum is a popular prescribed herb for the treatment of various liver diseases in eastern Asian countries. Saikosaponin d (SSd, Fig. 1A) is considered one of the major active components isolated and identified from this herb [6]. In Sprague-Dawley rats, SSd can decrease transforming growth factor β1 in the liver and attenuate the development of hepatic fibrosis and carcinogenesis induced by dimethylnitrosamine [7]. Supplementation with SSd alone or in combination with curcumin, significantly reduced carbon tetrachloride (CCl4)-induced inflammation and fibrogenesis [8]. In cell culture models, SSd exhibited potent cytotoprotection and anti-proliferation activity against hepatocellular carcinoma cells [9,10]. However, there have been no studies to evaluate the protective effect of SSd against hepatotoxicity induced by APAP.
Fig. 1.
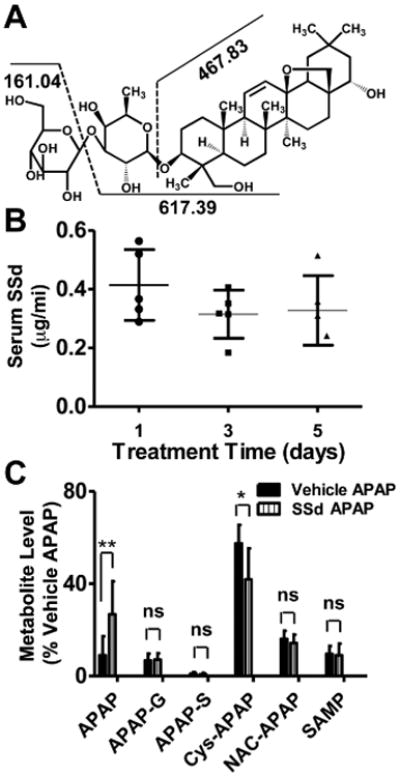
Structure of and fragmentation pattern of SSd, and levels of serum SSd in the mice treated with SSd 2mg/kg twice daily for 5 days. A: SSd structure and its proposed fragmentation pattern. B: SSd concentration 1 h after administration monitored on day 1, 3 and 5. C: Relative abundance of major urinary APAP metabolites involved in APAP-induced liver toxicity. Data were determined by normalizing the single ion counts of each metabolite versus the total ion counts of each urine sample (n=5; **p<0.01, ns: not significant; acetaminophen, APAP; APAP-G, acetaminophen-O-glucuronide; APAP-S, acetaminophen-O-sulfate; Cys-APAP, 3-cysteinylacetaminophen; NAC-APAP, 3-N-acetylcysteinylacetaminophen; SAMP, S-(5-acetylamino-2-hydroxyphenyl)-mercaptopyruvic acid).
SSd was found to modulate inflammatory response. Early studies showed that SSd can activate the phagocytosis of macrophages, modulate T lymphocyte function, and up-regulate interleukin (IL)-2/IL-4 production in thymocytes [11]. It can also elevate corticotropin-releasing factor mRNA levels in the hypothalamus and increase serum corticotropic hormone levels, which are involved in the pro-inflammatory processes. SSd can decrease apoptosis in both p53-postive HepG2 and p53-negative Hep3B cells, as indicated by reduced activation of nuclear factor kappa B (NF-κB) and attenuated expression of Bcl-xl [12]. Protection against CCl4-induced inflammation and fibrogenesis by SSd was correlated with down-regulation of the pro-inflammatory cytokines tumor necrosis factor-α (TNFα), IL-1β, and IL-6, and up-regulation of the anti-inflammatory cytokine IL-10 [8]. Despite the risk of APAP-induced toxicity and the wide application of Bupleurum falcatum for liver diseases in clinic, there are no data on the effect of Bupleurum falcatum or SSd on APAP-induced hepatotoxicity as well as the underlying mechanism. In this study, APAP was injected to SSd-pretreated C57/B6 mice and changes in liver phenotypes and gene expression were examined.
2. Materials and Methods
2.1. Chemicals and reagents
Saikosaponin d (SSd, Fig. 1A), APAP, glutathione (GSH) assay kit, and chlorpropamide were purchased from Sigma–Aldrich (Sigma-Aldrich, St. Louis, MO). Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) assay kits were from Catachem (Bridgeport, CT). Antibodies against NFκB subunit p65 and signal transducer and activator of transcription 3 (STAT3) and their phosphorylated form, p-p65 and p-STAT3, and GAPDH were purchased from Cell Signaling Technologies (Danvers, MA). HPLC grade solvents such as acetonitrile and formic acid were purchased from Fisher Scientific (Hampton, NH). All the other chemicals were of the highest grade from commercial source.
2.2. Animals and drug administration
Male 6- to 7-week-old C57BL6 mice (Jackson Laboratories, Bar Harbor, ME) were maintained in the NCI animal facility under a standard 12 h light/12 h dark cycle with free access to food and water. All procedures were performed in accordance with Institute of Laboratory Animal Resource Guidelines and the animal study protocols approved by the National Cancer Institute Animal Care and Use Committee. Mice were randomly divided into four groups, vehicle/control, SSd/control, vehicle/APAP, and SSd/APAP, and killed 4 h or 24 h after single APAP injection.
For APAP injection, a typical single dose of 200 mg/kg/day was used as described elsewhere [3,13,14]. Considering the published pharmacodynamic and pharmacokinetic information of SSd [6,7], 2 mg/kg once daily was used as the dosing regimen. SSd powder was dissolved in a saline solution supplemented with 0.1% Tween 20 and was administered by intraperitoneal injection at a dose of 2 mg/kg/day once daily for five days. Saline solution containing 0.1% Tween 20 without SSd was administered as a vehicle. APAP was dissolved in warm saline solution (20 mg/mL) and was injected intraperitoneally 30 minutes after the last SSd injection. Saline was injected to mice in the control groups.
Blood was taken from retro-orbital space of the mice in the SSd/control group 1 h after the SSd injection on day 1, 3, and 5 in order to determine circulating SSd concentration. Twenty-four hour urine samples were also collected after APAP administration to measure APAP and its metabolites. Mice were killed at 4 h and 24 h after APAP challenge, following which serum and liver were collected. The liver was fixed in 10% neutral buffered formalin, after briefly washing with phosphate buffered saline. The remaining liver tissue was flash frozen in liquid nitrogen and stored at -80°C for further analysis.
2.3. Biochemical and histological analyses
To measure serum SSd levels, an aliquot of 5 μL serum supernatant was subjected to a Waters ACQUITY ultra-performance liquid chromatography (UPLC) system coupled with a XEVO triple-quadrupole tandem mass spectrometer (Waters, Corp., Milford. MA). The MRM transition 779.5→617.4 in ESI- was monitored after fragmentation analysis (Fig. 1A) and chlorpropamide (277→111) was used as an internal standard. For quantification, a calibration curve constructed using authentic standard had the r2 value above 0.99. The urinary profile was acquired by subjecting an aliquot of 5 μL of urine sample to UPLC coupled to a quadrupole time-of-flight mass spectroscopy (UPLC-ESI-QTOFMS) (Waters, Corp.) and analyzing as previously reported [15].
Serum AST and ALT activities were measured following the manufacturer's instruction with VetSpecTM kits (Catachem, Bridgeport, CT). For hepatic GSH measurement, 50 mg of frozen liver tissues were homogenized in 0.5 mL 5% 5-sulfosalicylic acid using Precellys 24 (Bertin Technologies). After centrifugation at 10,000g for 10 minutes, the supernatant were measured according to the protocol, and results were normalized by the tissue mass and expressed as nmol/mg.
Formalin-fixed liver tissues were subjected to dehydration in serial concentrations of ethanol and xylene and embedded in paraffin. Four-micrometer serial sections were cut and stained with hematoxylin and eosin, followed by histological examination with an Olympus BX41 light microscope.
2.4. Quantitative polymerase chain reaction (qPCR) analysis
Total RNA was extracted from approximately 20 mg frozen liver tissues, using TRIzol reagent (Invitrogen, Carlsbad, CA). cDNA was generated from 1 μg mRNA with a SuperScript II Reverse Transcriptase kit and random oligonucleotides (Invitrogen, Carlsbad, CA). The primer sequences listed in Supplementary Table 1 were designed using qPrimer Depot and their specificity tested by melting curve profiles. qPCR reactions contained 1 μL cDNA, 150 nM of each primer and 5 μL of SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) in a total volume of 10 μL. qPCR was carried out on an ABI-Prism 7900HT Sequence Detection System (Applied Biosystems, Foster City, CA). Measured mRNA abundance was normalized to 18S rRNA and expressed as fold change relative to the vehicle control group.
2.5. Western blot analysis
Liver tissues were homogenized using RIPA buffer (1:10, g/v) in which cocktail inhibitors and mercaptoethanol were freshly added. Protein concentrations were measured using the BCA protein assay kit (Thermo Scientific, Waltham, MA) and normalized to 5 mg/mL. After being mixed with a two-fold dilution of loading buffer (1:1, v/v), 60 μg of protein was loaded on the gel. After electrophoresis, protein was transferred to a PVDF membrane and blocked with 5% bovine serum albumin or skim milk in Tris-buffered saline containing 0.1% Tween 20 for at least 1 h. Membranes were incubated overnight with primary antibodies against p65, p-p65, STAT3, p-STAT3, or GAPDH. Secondary antibodies were incubated for 1 h, and the blotted membranes were prepared with ECL substrate (Thermo Scientific). Band intensities were quantified by densitometry. The ratio p-STAT3/total STAT3 and p-p65/p65 were calculated and normalized by those in vehicle/control group.
2.6. Statistical analysis
All the experimental values were expressed as mean ± SD. Statistical analysis was performed by two-tailed nonparametric Mann-Whitney test for unpaired data using GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA). 95% was set as confidence intervals and difference was considered as significant if the p value was less than 0.05.
3. Results
3.1. SSd concentration, biochemical and toxicological reactions
Injection of 2 mg/kg/day of SSd yielding serum concentrations between 0.3-0.6 μg/mL (Fig. 1B). This level was relatively steady during the administration period. In the SSd/APAP group, more APAP and less 3-cysteinylacetaminophen (Cys-APAP) were excreted in urine compared with vehicle/APAP mice (Fig. 1C).
Serum ALT and AST are reliable indicators of acute hepatic injury. Typically, a time-dependent increase of these markers occurs between 2 to 6 h post-APAP challenge [14]. At 4 h after 200 mg/kg APAP administration, serum ALT was significantly increased with no change in AST (Fig. 2A and B), while at 24 h after APAP treatment, both ALT and AST were increased in the vehicle/APAP group. In contrast, neither enzyme activity increased in mice pretreated with SSd for five days before APAP treatment (Fig. 2C and D).
Fig. 2.
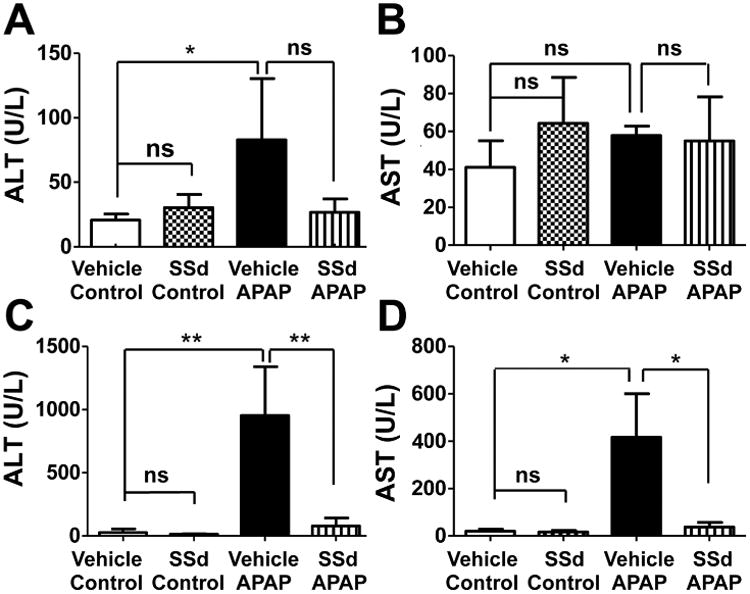
Serum AST and ALT levels in mice treated with APAP and/or SSd. A: 4h ALT level. B: 4h AST level. C: 24h ALT level. D: 24h AST level. SSd was administered by intraperitoneal injection once daily for five days prior to treatment with 200mg/kg/day APAP. AST and ALT in serum collected 4h and 24h after APAP treatment were analyzed. Data were expressed as mean ± SD (n=5; *p<0.05; **p<0.01; ***p<0.001; ns, not significant).
To confirm the changes in serum AST/ALT levels, liver histology was assessed. In the vehicle control and SSd/control groups, the liver tissues were histologically normal, while a dose of 200 mg/kg APAP resulted in large patchy necrosis around the central veins. No overt liver damage was observed in SSd/APAP group (Fig. 3). These results demonstrate a clear protective effect of SSd pre-administration on APAP-induced liver injury.
Fig. 3.
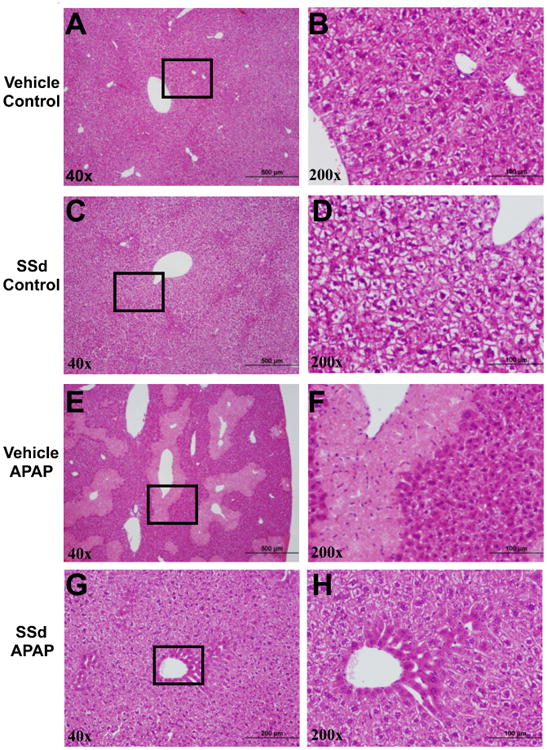
Representative pictures of liver histology in the mice treated with APAP and/or SSd. The patches framed in the pictures in the left column for each group were magnified pictures of the corresponding right column. SSd was administered to non-fasted mice by intraperitoneal injection once daily for five days prior to treatment with 200mg/kg/day APAP.
3.2. Influence of SSd on APAP detoxification, oxidative stress, and peroxisome proliferator-activated receptor α (PPARα) signaling in the liver
In the SSd/APAP group, more APAP and less 3-cysteinylacetaminophen (Cys-APAP) were excreted in urine compared with vehicle/APAP mice (Fig. 1C), suggesting that SSd affects APAP metabolism. APAP is metabolized to the toxic intermediate metabolite N-acetyl-p-benzoquinone imine (NAPQI) in the liver through cytochrome P450 (CYP) 2E1, and to minor extent CYP3A11 (in mice) and CYP1A2. However, expression of Cyp2e1 or Cyp3a11 mRNAs was not changed in any of the groups (Supplementary Fig. S2A and B). NAPQI is catabolized by conjugation with GSH, causing consumption of GSH and enhanced oxidative stress and ensuing mitochondrial dysfunction. Depletion of hepatic GSH usually precedes the increase of AST and ALT activity [14]. Four hours after APAP treatment, a small decrease was seen in both the vehicle/APAP and SSd/APAP groups, but was not significant compared with the vehicle/control. However, 24 h after APAP challenge, GSH levels in the SSd/APAP group were higher than that in the vehicle/APAP group (Supplementary Fig. S1A and B). Expression of superoxide dismutases 1 and 2 (Sod1 and Sod2) mRNAs were analyzed and found to not differ among the four groups, similar to the small variation of GSH as noted above (Supplementary Fig. S2A). Thus, the protective mechanism by traditional antioxidants was apparently not involved in this study.
Activation of peroxisome proliferator-activated receptor α (PPARα) was reported to have an important role in protecting against APAP-induced liver injury through induction of uncoupling protein 2 (UCP2) [3]. Induction of its target genes, such as Acox1, Ehhadh, Cpt1, and Cpt2 were also associated with the protective effect of Schisandra Spehanthera extract against APAP [2]. These PPARα target genes were analyzed and were not significantly altered by SSd (Supplementary Fig. S2C). Thus, PPARα activation was not involved in the protection by SSd, probably due to the lower dose of APAP compared with the previous reports that used doses in excess of 400 mg/kg.
3. 3. Influence of SSd on hepatocyte apoptotic signaling induced by APAP challenge
Apoptosis is an important step in APAP hepatotoxicity [18,19]. Since SSd was reported to inhibit apoptosis, typical genes involved in apoptosis were analyzed. Anti-apoptotic gene Bcl-2 mRNA was higher and pro-apoptotic Bax mRNA was lower in the SSd/APAP group compared with those in the vehicle/APAP group, while Bcl-xl and Bim mRNAs were not changed in any of those groups (Fig. 4A).
Fig. 4.
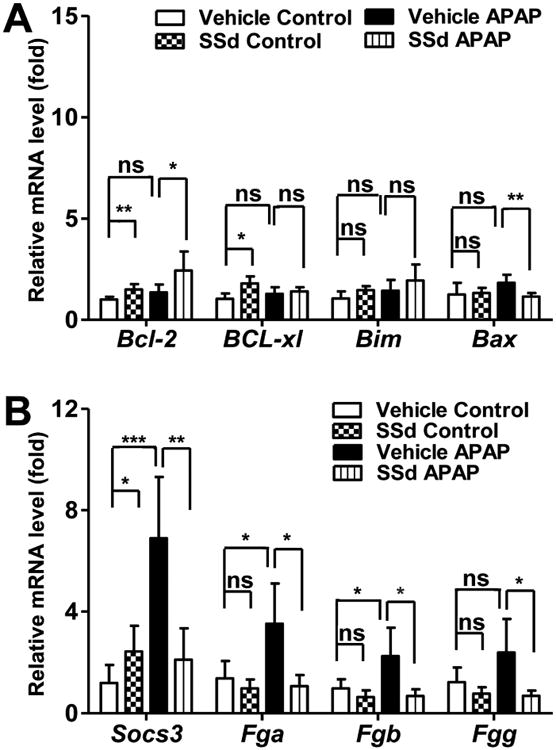
Levels of mRNAs encoding by apoptosis-related factors and STAT3 target genes. A: Bcl-2, BCL-xl, Bim, and Bax mRNAs. B: Socs3, Fga, Fgb, and Fgg mRNAs. Data were from liver samples collected 24h after APAP treatment. The mRNA levels were measured by Q-PCR and normalized by 18S rRNA. Messenger RNA levels in vehicle-treated control mice were arbitrarily set as 1 and results were expressed as mean ± SD (n=5; *p<0.05; **p<0.01; ***p<0.001; ns, not significant).
3.4. SSd suppresses APAP-induced increases in the expression of STAT3 target genes and pro-inflammatory cytokines
STAT3 was reported to be involved in APAP-induced hepatotoxicity, where IL-22, a STAT3-activating cytokine, was shown to attenuate APAP-induced liver injury [20]. Thus, expression of mRNAs encoded by the STAT3 target genes Socs3, Fga, Fgb and Fgg were assessed. These mRNAs were significantly up-regulated in the vehicle/APAP group, and the induction by APAP challenge was reversed by SSd pretreatment (Fig. 4B).
Prevailing evidence indicates that inflammation is induced following production of the toxic metabolite from APAP. Therefore, mRNAs encoded by genes involved in inflammation, including Il-10, Il-6, Tlr4, Ddit3, c-Jun, c-Fos, Ccl2, Icam1, Ripk3 and Tnfα, were analyzed. Among these, Il-6 and Ccl2 mRNAs were markedly increased by APAP injection, and these inductions were reversed by SSd pretreatment. APAP-induced moderate increases in Ddit3, c-Jun, c-Fos, and Icam1 and similar attenuation by SSd treatment were also observed. Additionally, the mRNA encoding the anti-inflammatory cytokine IL-10 was up-regulated in the SSd/APAP group but unchanged in the vehicle/APAP group (Fig. 5).
Fig. 5.
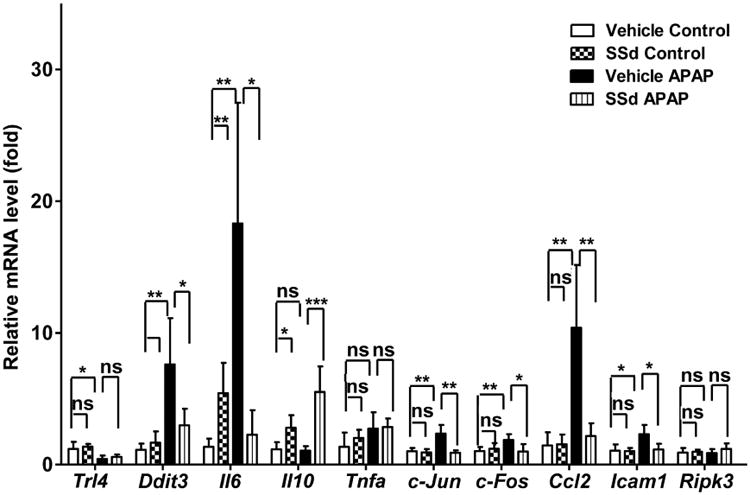
The levels of mRNAs encoded by genes associated with the inflammatory response. Data were from liver samples collected 24h after APAP treatment. The mRNA expression levels were measured by qPCR and normalized by 18S rRNA. Messenger RNA expression level in the vehicle control mice was arbitrarily set as 1 and results expressed as mean ± SD (n=5; *p<0.05; **p<0.01; ***p<0.001; ns, not significant).
Collectively, these results show that attenuation of APAP-induced up-regulation of STAT3 target genes and pro-inflammatory cytokines, such as Il-6 and Ccl2, is associated with amelioration of hepatotoxicity caused by SSd administration.
3.5. SSd inhibits APAP-induced activation of STAT3 and NF-kB
Western blotting was used to assess the activation of STAT3 and NFκB, a master regulator of genes involved in inflammation. The expression levels of phosphorylated STAT3 were increased sharply (5.5±0.93 fold) in the vehicle/APAP group and was markedly attenuated in the SSd/APAP group (2.2±1.19 fold) (Fig. 6). Similarly, the active form p-p65 were also increased moderately (2.08±0.10 fold) in the vehicle/APAP group and decreased in the SSd/APAP group (0.53±0.04 fold) (Fig. 6). Therefore, these results indicate that the protective effect of SSd against APAP hepatotoxicity is mainly derived from inhibition of NF-κB and STAT3 activation.
Fig. 6.
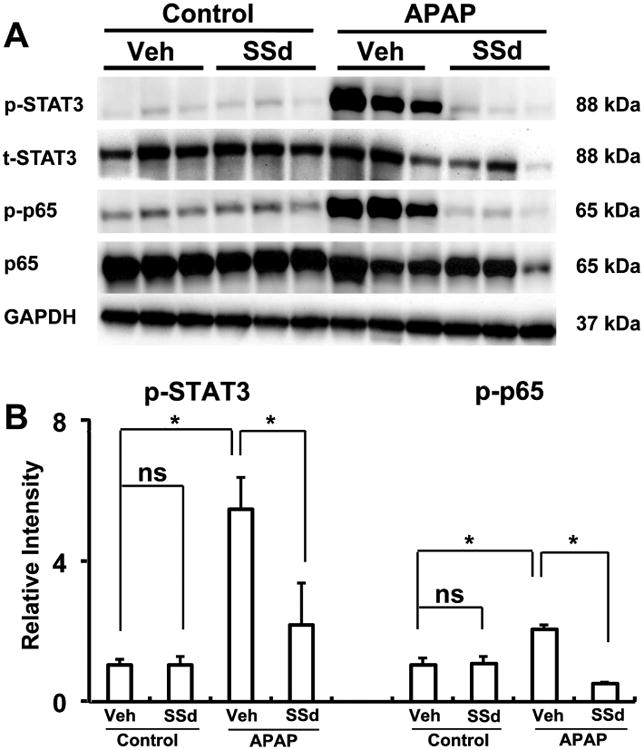
Western blot and densitometry analysis of STAT3 and NFκB in liver extracts. Liver samples were collected 24h after vehicle (Veh) + APAP treatment and SSd + APAP treatment. GAPDH was used as a loading control. The molecular weight was indicated at the right side of the respective band. A: Western blot of STAT3 and NFκB in liver extracts. B: Densitometry analysis of STAT3 and NFκB activation in liver extracts. Results of control group were set as 1 and data were expressed as relative intensity of p-p65 and p-STAT versus p65 and STAT respectively (n=3; *p<0.01; ns, not significant).
4. Discussion
After oral ingestion of APAP, about 90% is directly conjugated with glucuronic acid or sulfate. The remaining 5-10% is metabolized largely by CYP2E1 to NAPQI [15,21]. This reactive metabolite can bind covalently with cellular proteins, leading to irreversible toxicity. After activation of APAP to NAPQI, subsequent propagation and amplification of reactive oxygen species (ROS) and stress-induced signal transduction pathways lead to cell death [21]. Protection against APAP-induced hepatotoxicity by GSH, N-acetylcysteine, taurine, hypotaurine and other antioxidants was reported [4,5,22-24]. The unchanged expression of Cyp2e1, Cyp3a11, Sod1, and Sod2 mRNAs, as well as the slight variation of GSH after SSd administration, indicates the above traditional mechanisms were likely not directly involved in the protective effect of SSd.
The relationship between PPARα, fatty acid β-oxidation and APAP-induced hepatotoxicity was established [14,15], and Ucp2, a PPARα target gene, was recently found to mediate protective effects of PPARα activators [3]. In Cyp2e1-null mice, PPARα activation was much more significant and more persistent than in wild-type mice following a toxic APAP dose, leading to prolonged up-regulation of PPARα target genes (Cpt1, Cpt2, Acot1, and Cyp4a10) involved in fatty acid β-oxidation [14,15]. Inhibition of fatty acid β-oxidation leads to an increase in serum palmitoylcarnitine and other acylcarnitines following high-dose APAP treatment [14]. The Chinese traditional herb Schisandrae Sphenanthera extract was recently shown to protect against APAP-induced hepatotoxicity as revealed by the recovery of fatty acid β-oxidation and lowering serum acylcarnitines [2]. In the present study, none of the typical PPARα target genes or the serum acylcarnitines were altered by SSd. This difference could be due to the lower dose of 200 mg/kg applied in the present study. Thus, protection against APAP hepatotoxicity by SSd does not involve PPARα activation.
Attenuation of IL-6 was observed with increased liver injury induced by APAP in Kupffer cell-depleted mice [25]. In IL-6 knockout mice dosed with APAP, fewer regenerating hepatocytes were present, and thus IL-6 appears to play a role in liver regeneration [26]. Additionally, IL-6 can protect against liver injury by up-regulating the hepatic cytoprotective heat shock proteins [27]. However, considering the differential activation of NFκB between the APAP group and SSd/APAP group, IL-6, the downstream product of NFκB, likely promotes toxicity rather than regeneration in this context.
IL-10 was reported to be a protective factor in limiting formation of TNF-α and IL-1 [25]. In Kupffer cells, binding of IL-10 to its receptor leads to prolonged activation of STAT3, thereby inhibiting the inflammatory response. In IL-10 knockout mice, more severe liver injury was associated with increased formation of pro-inflammatory cytokines and inducible nitric oxide synthase expression after APAP administration [28]. In the present study, IL-10 was unchanged in the vehicle/APAP group, but increased in the SSd/APAP group. Thus, modulation of IL-10 by SSd may offer a protective role against APAP toxicity.
Recently, prophylactic injection of IL-22, a STAT3-activating cytokine, significantly reduced hepatocyte damage due to APAP, suggesting a protective role of STAT3 [20]. However, forced expression of STAT3 target gene Socs3 in T cells was reported to exacerbate APAP-induced hepatotoxicity [29]. In the present study, expression of Socs3, and the other identified STAT3 target genes Fga, Fgb and Fgg was increased in the vehicle/APAP group and these increases were reversed in SSd/APAP group. In accordance with this result, STAT3 was activated in the APAP group but attenuated in SSd/APAP group. Since IL-6 is the result of NFκB activation and an activator of STAT3, the above modifications could be downstream responses to NFκB activation.
In the progression of drug-induced hepatotoxicity and liver disease, it is generally accepted that inflammation plays a critical role [21,30,31]. Among the inflammatory pathways, STAT3 and NFκB were most often reported [31,32], both of which are involved in APAP-induced hepatotoxicity [29]. In the present study, both were activated as revealed by western blot analysis. Additionally, their typical downstream response factor IL-6 and target gene Socs3 were up-regulated. This indicates the NFκB and STAT3 signal transduction pathways are directly involved in hepatotoxicity induced by APAP. Additionally, APAP-induced enhancement of expression of c-Jun and c-Fos was corrected/reversed/attenuated by SSd. This is similar to the protective mechanism of the PPARα activators Wy-14643 and fenofibrate where anti-inflammation mediated by c-Jun and c-Fos is involved [3]. In contrast, the pro-apopototic genes Bim and Bax were only moderately modified in the present study. These data suggest that inflammation is related to the toxic events induced by APAP and that the protective mechanism of SSd is associated with modification of NFκB-IL6-STAT3 signaling.
In conclusion, SSd, the active component of traditional Chinese herb Bupleurum falcatum, can protect against APAP-induced hepatotoxicity via inhibiting of NFκB and STAT3 signaling. These data suggest new insights to understand the role of inflammation underlying APAP hepatotoxicity and clinical application of Bupleurum falcatumas, a hepatoprotectant in eastern Asian countries.
Supplementary Material
Highlights.
Mice treated with saikosaponin d were protected against APAP-induced hepatotoxicity.
Saikosaponin d reversed APAP-induced expression of pro-inflammatory cytokines.
Saikosaponin d enhanced expression of the anti-inflammatory Il10 mRNA.
Saikosaponin d suppressed phosphorylation of NF-kB and STAT3.
Acknowledgments
This work was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health. Financial support was also provided in part by National Natural Science Foundation of China [Grant 81273582 and 81302848], and the K.C.Wong Magna Fund in Ningbo University.
Abbreviations
- SSd
saikosaponin d
- APAP
acetaminophen
- APAP-G
acetaminophen-O-glucuronide
- APAP-S
acetaminophen-O-sulfate
- Cys-APAP
3-cysteinylacetaminophen
- NAC-APAP
3-N-acetylcysteinylacetaminophen
- SAMP
S-(5-acetylamino-2-hydroxyphenyl)-mercaptopyruvic acid
- NFκB
nuclear factor-kappa B,
- STAT3
signal transducer and activator of transcription 3
- CCl4
carbon tetrachloride
- IL
interleukin
- GSH
glutathione
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- qPCR
quantitative real-time PCR
- SOD
superoxide dismutase
- CYP
cytochrome P450
- UCP2
uncoupling protein 2
- PPARα
peroxisome proliferator-activated receptor alpha
- TNFα
tumor necrosis factor alpha
- UPLC
ultra-performance liquid chromatography
- NAPQI
N-acetyl-p-benzoquinone imine
Footnotes
Conflict of interest: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lee WM. Acetaminophen and the U.S. acute liver failure study group: lowering the risks of hepatic failure. Hepatology. 2004;40(1):6–9. doi: 10.1002/hep.20293. [DOI] [PubMed] [Google Scholar]
- 2.Bi H, Li F, Krausz KW, Qu A, Johnson CH, Gonzalez FJ. Targeted metabolomics of Serum acylcarnitines evaluates hepatoprotective effect of Wuzhi Tablet (Schisandra sphenanthera Extract) against Acute acetaminophen toxicity. Evid Based Complement Alternat Med. 2013 doi: 10.1155/2013/985257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patterson AD, Shah YM, Matsubara T, Krausz KW, Gonzalez FJ. Peroxisome proliferator-activated receptor alpha induction of uncoupling protein 2 protects against acetaminophen-induced liver toxicity. Hepatology. 2012;56(1):281–290. doi: 10.1002/hep.25645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oz HS, McClain CJ, Nagasawa HT, Ray MB, de Villiers WJ, Chen TS. Diverse antioxidants protect against acetaminophen hepatotoxicity. J Biochem Mol Toxicol. 2004;18(6):361–368. doi: 10.1002/jbt.20042. [DOI] [PubMed] [Google Scholar]
- 5.Acharya M, Lau-Cam CA. Comparison of the protective actions of N-acetylcysteine, hypotaurine and taurine against acetaminophen-induced hepatotoxicity in the rat. J Biomed Sci. 2010;17(Suppl 1):S35. doi: 10.1186/1423-0127-17-S1-S35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu L, Song R, Tian JX, Tian Y, Liu GQ, Zhang ZJ. Analysis of saikosaponins in rat plasma by anionic adducts-based liquid chromatography tandem mass spectrometry method. Biomed Chromatograph. 2011;26(7):808–815. doi: 10.1002/bmc.1734. [DOI] [PubMed] [Google Scholar]
- 7.Fan J, Li X, Li P, Li N, Wang T, Shen H, et al. Saikosaponin-d attenuates the development of liver fibrosis by preventing hepatocyte injury. Biochem Cell Biol. 2007;85(2):189–195. doi: 10.1139/O07-010. [DOI] [PubMed] [Google Scholar]
- 8.Wu SJ, Tam KW, Tsai YH, Chang CC, Chao JC. Curcumin and saikosaponin a inhibit chemical-induced liver inflammation and fibrosis in rats. Am J Chin Med. 2010;38(1):99–111. doi: 10.1142/S0192415X10007695. [DOI] [PubMed] [Google Scholar]
- 9.Chiang LC, Ng LT, Liu LT, Shieh DE, Lin CC. Cytotoxicity and anti-hepatitis B virus activities of saikosaponins from Bupleurum species. Planta Med. 2003;69(8):705–709. doi: 10.1055/s-2003-42797. [DOI] [PubMed] [Google Scholar]
- 10.Hsu YL, Kuo PL, Lin CC. The proliferative inhibition and apoptotic mechanism of Saikosaponin D in human non-small cell lung cancer A549 cells. Life Sci. 2004;75(10):1231–1242. doi: 10.1016/j.lfs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 11.Kato M, Pu MY, Isobe K, Iwamoto T, Nagase F, Lwin T, et al. Characterization of the immunoregulatory action of saikosaponin-d. Cell Immunol. 1994;159(1):15–25. doi: 10.1006/cimm.1994.1291. [DOI] [PubMed] [Google Scholar]
- 12.Hsu YL, Kuo PL, Chiang LC, Lin CC. Involvement of p53, nuclear factor kappaB and Fas/Fas ligand in induction of apoptosis and cell cycle arrest by saikosaponin d in human hepatoma cell lines. Cancer Lett. 2004;213(2):213–221. doi: 10.1016/j.canlet.2004.03.044. [DOI] [PubMed] [Google Scholar]
- 13.Cheng J, Ma X, Krausz KW, Idle JR, Gonzalez FJ. Rifampicin-activated human pregnane X receptor and CYP3A4 induction enhance acetaminophen-induced toxicity. Drug Metab Dispos. 2009;37(8):1611–1621. doi: 10.1124/dmd.109.027565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen C, Krausz KW, Shah YM, Idle JR, Gonzalez FJ. Serum metabolomics reveals irreversible inhibition of fatty acid beta-oxidation through the suppression of PPARalpha activation as a contributing mechanism of acetaminophen-induced hepatotoxicity. Chem Res Toxicol. 2009;22(4):699–707. doi: 10.1021/tx800464q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen C, Krausz KW, Idle JR, Gonzalez FJ. Identification of novel toxicity-associated metabolites by metabolomics and mass isotopomer analysis of acetaminophen metabolism in wild-type and Cyp2e1-null mice. J Biol Chem. 2008;283(8):4543–4559. doi: 10.1074/jbc.M706299200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raucy JL, Lasker JM, Lieber CS, Black M. Acetaminophen activation by human liver cytochromes P450IIE1 and P450IA2. Arch Biochem Biophys. 1989;271(2):270–283. doi: 10.1016/0003-9861(89)90278-6. [DOI] [PubMed] [Google Scholar]
- 17.Zaher H, Buters JT, Ward JM, Bruno MK, Lucas AM, Stern ST, et al. Protection against acetaminophen toxicity in CYP1A2 and CYP2E1 double-null mice. Toxicol Appl Pharmacol. 1998;152(1):193–199. doi: 10.1006/taap.1998.8501. [DOI] [PubMed] [Google Scholar]
- 18.El-Hassan H, Anwar K, Macanas-Pirard P, Crabtree M, Chow SC, Johnson VL, et al. Involvement of mitochondria in acetaminophen-induced apoptosis and hepatic injury: roles of cytochrome c, Bax, Bid, and caspases. Toxicol Appl Pharmacol. 2003;191(2):118–129. doi: 10.1016/s0041-008x(03)00240-0. [DOI] [PubMed] [Google Scholar]
- 19.Adams ML, Pierce RH, Vail ME, White CC, Tonge RP, Kavanagh TJ, et al. Enhanced acetaminophen hepatotoxicity in transgenic mice overexpressing BCL-2. Mol Pharmacol. 2001;60(5):907–915. doi: 10.1124/mol.60.5.907. [DOI] [PubMed] [Google Scholar]
- 20.Scheiermann P, Bachmann M, Goren I, Zwissler B, Pfeilschifter J, Muhl H. Application of interleukin-22 mediates protection in experimental acetaminophen-induced acute liver injury. Am J Pathol. 2013;182(4):1107–1113. doi: 10.1016/j.ajpath.2012.12.010. [DOI] [PubMed] [Google Scholar]
- 21.Jaeschke H. Role of inflammation in the mechanism of acetaminophen-induced hepatotoxicity. Expert Opin Drug Metab Toxicol. 2005;1(3):389–397. doi: 10.1517/17425255.1.3.389. [DOI] [PubMed] [Google Scholar]
- 22.Bajt ML, Knight TR, Lemasters JJ, Jaeschke H. Acetaminophen-induced oxidant stress and cell injury in cultured mouse hepatocytes: protection by N-acetyl cysteine. Toxicol Sci. 2004;80(2):343–349. doi: 10.1093/toxsci/kfh151. [DOI] [PubMed] [Google Scholar]
- 23.James LP, McCullough SS, Lamps LW, Hinson JA. Effect of N-acetylcysteine on acetaminophen toxicity in mice: relationship to reactive nitrogen and cytokine formation. Toxicol Sci. 2003;75(2):458–467. doi: 10.1093/toxsci/kfg181. [DOI] [PubMed] [Google Scholar]
- 24.Bajt ML, Knight TR, Farhood A, Jaeschke H. Scavenging peroxynitrite with glutathione promotes regeneration and enhances survival during acetaminophen-induced liver injury in mice. J Pharmacol Exp Ther. 2003;307(1):67–73. doi: 10.1124/jpet.103.052506. [DOI] [PubMed] [Google Scholar]
- 25.Ju C, Reilly TP, Bourdi M, Radonovich MF, Brady JN, George JW, et al. Protective role of Kupffer cells in acetaminophen-induced hepatic injury in mice. Chem Res Toxicol. 2002;15(12):1504–1513. doi: 10.1021/tx0255976. [DOI] [PubMed] [Google Scholar]
- 26.James LP, Lamps LW, McCullough S, Hinson JA. Interleukin 6 and hepatocyte regeneration in acetaminophen toxicity in the mouse. Biochem Biophys Res Commun. 2003;309(4):857–863. doi: 10.1016/j.bbrc.2003.08.085. [DOI] [PubMed] [Google Scholar]
- 27.Masubuchi Y, Bourdi M, Reilly TP, Graf ML, George JW, Pohl LR. Role of interleukin-6 in hepatic heat shock protein expression and protection against acetaminophen-induced liver disease. Biochem Biophys Res Commun. 2003;304(1):207–212. doi: 10.1016/s0006-291x(03)00572-2. [DOI] [PubMed] [Google Scholar]
- 28.Bourdi M, Masubuchi Y, Reilly TP, Amouzadeh HR, Martin JL, George JW, et al. Protection against acetaminophen-induced liver injury and lethality by interleukin 10: role of inducible nitric oxide synthase. Hepatology. 2002;35(2):289–298. doi: 10.1053/jhep.2002.30956. [DOI] [PubMed] [Google Scholar]
- 29.Numata K, Kubo M, Watanabe H, Takagi K, Mizuta H, Okada S, et al. Overexpression of suppressor of cytokine signaling-3 in T cells exacerbates acetaminophen-induced hepatotoxicity. J Immunol. 2007;178(6):3777–3785. doi: 10.4049/jimmunol.178.6.3777. [DOI] [PubMed] [Google Scholar]
- 30.Maeda S, Kamata H, Luo JL, Leffert H, Karin M. IKKbeta couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell. 2005;121(7):977–990. doi: 10.1016/j.cell.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 31.Sun B, Karin M. Inflammation and liver tumorigenesis. Front Med. 2013;7(2):242–254. doi: 10.1007/s11684-013-0256-4. [DOI] [PubMed] [Google Scholar]
- 32.Wang H, Lafdil F, Kong X, Gao B. Signal transducer and activator of transcription 3 in liver diseases: a novel therapeutic target. Int J Biol Sci. 2011;7(5):536–550. doi: 10.7150/ijbs.7.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.