

Abstract
Multiple Sclerosis (MS) is an autoimmune disease characterized by inflammation and neurodegeneration. Current MS treatments were designed to reduce inflammation in MS, rather than to directly prevent neurodegeneration. Estrogen has well-documented neuroprotective effects in a variety of disorders of the CNS, including experimental autoimmune encephalomyelitis (EAE), the most widely used mouse model of MS. Treatment with an estrogen receptor-beta (ERβ) ligand is known to effectively ameliorate clinical disease and provide neuroprotection in EAE. However, the protective effects of this ERβ ligand have only been demonstrated when administered prior to disease (prophylactically). Here, we tested whether ERβ ligand treatment could provide clinical protection when treatment was initiated after onset of disease (therapeutically). We found that therapeutic treatment effectively ameliorated clinical disease in EAE. Specifically, ERβ ligand-treated animals exhibited preserved axons and myelin as compared to vehicle treated animals. We observed no difference in the number of T lymphocytes, macrophages, or microglia in the CNS of vehicle versus ERβ ligand-treated animals. Our findings show that therapeutically administered ERβ ligand successfully treats clinical EAE, bearing translational relevance to MS as a candidate neuroprotective agent.
Introduction
Multiple sclerosis is an autoimmune disease that is characterized by inflammation, demyelination, and axonal loss in the CNS. Environmental, genetic, and immunological factors have been associated with susceptibility to MS, but its cause is still unknown (Milo & Kahana 2010). Current MS treatments such as copaxone and interferon beta are immunomodulatory treatments (Johnson 2012; Wolinsky et al. 2001; Hartung et al. 2002; Hartung et al. 2002). Most MS therapeutics target the inflammatory component of the disease and do not directly protect against neurodegeneration. They reduce relapses by approximately half but do not halt permanent disability accumulation. Therefore, there is a need for a neuroprotective MS treatment(Ulzheimer et al. 2010).
The steroid hormone estrogen has well-documented neuroprotective effects in many models of neurodegenerative disorders of the CNS, such as Parkinson’s and Alzheimer’s diseases, as well as traumatic brain injury and stroke (Tiwari-Woodruff et al. 2007; Brann et al. 2007; Cho et al. 2003; Garcia-Segura et al. 2001; Suzuki et al. 2009). Estrogens have also been shown to be beneficial in experimentally induced autoimmune encephalomyelitis (EAE), the most commonly used mouse model of MS (Spence & Voskuhl 2012; Gold & Voskuhl 2009; Croxford et al. 2011). Namely, estrogen treatment is both anti-inflammatory and neuroprotective in EAE (Kim et al. 1999; Spence & Voskuhl 2012; Bebo et al. 2001; Morales et al. 2006; Offner 2004; Subramanian et al. 2003). Thus, selective estrogen receptor ligands are potential candidates for neuroprotective MS treatments.
Estrogen receptor-beta (ERβ) ligands have promising therapeutic potential. Initial studies have demonstrated that treatment with an ERβ-specific ligand was effective at ameliorating disease progression, and pathology demonstrated neuroprotection in mice with EAE (Du, Sandoval, Trinh, Umeda, et al. 2010b; Du, Sandoval, Trinh & Voskuhl 2010a; Tiwari-Woodruff & Voskuhl 2009; Tiwari-Woodruff et al. 2007). However, in all of these studies ERβ ligand treatment commenced before EAE induction. This is inconsistent with the potential clinical applications of ERβ ligand treatment in human MS, since patients are unlikely to receive treatment prior to disease onset. Therefore, we sought to determine whether ERβ ligand could mediate neuroprotection in EAE when administered after disease induction. We also sought to understand whether the effects of ERβ ligand treatment persist after cessation of treatment. This experimental design more closely reflects a potential treatment paradigm for human MS patients.
Materials and Methods
Animals
Female WT C57BL/6 mice age 6–8 weeks at the time of disease onset (not ovariectomized), were obtained from Jackson Laboratories (Bar Harbor, ME, USA). Animals were maintained in accordance with guidelines set by the National Institute of Health and as mandated by the University of California Los Angeles Office for the Protection of Research Subjects and the Chancellor’s Animal Research Committee and the PHS Policy on Human Care and Use of Laboratory Animals.
Induction of EAE
For active EAE, mice were immunized by subcutaneous injections into the left flank of 200 μg of MOG peptide, amino acids 35–55, and 200 μg of Mycobacterium tuberculosis in complete Freund’s adjuvant. Immediately after immunization, mice received an intraperitoneal injection of 500 ng pertussis toxin dissolved in 400 μL PBS. Two days later, mice received another intraperitoneal injection of pertussis toxin of the same quantity. Seven days after the initial immunization, the MOG immunization was repeated. MOG peptide, amino acids 35–55, was synthesized to > 98% purity by Mimitopes (Clayton, Victoria, Australia).
Treatment
Animals were treated with either the ERβ agonist diarylpropionitrile (DPN), purchased from Tocris, Cookson Inc. and diluted with 10% molecular-grade ethanol and 90% Miglylol 812N liquid oil (Sasol North America), or with vehicle treatment (ethanol and Miglylol). Animals were treated at the first clear signs of clinical disease (day 10 or 11) and treated until day 35 (inclusive). EAE mice were sacrificed either 35 or 61 days after induction of disease. Animals that were sacrificed day 61 received no treatment from days 37–61 of the experiment. n=4 in each treatment group for figures 1–4, n=5 in each treatment group for figure 5 and 6.
Figure 1.
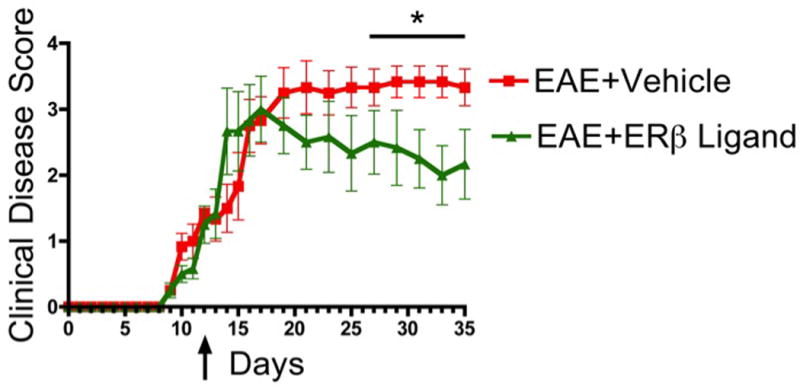
Therapeutically administered ERβ ligand ameliorates clinical disease in EAE. EAE mice treated with ERβ ligand had significantly reduced clinical scores compared with EAE mice treated with vehicle. Arrow reflects the first score after beginning treatment. n=4 per group. * p < 0.05 between EAE+Vehicle and EAE+ ERβ Ligand.
Figure 4.
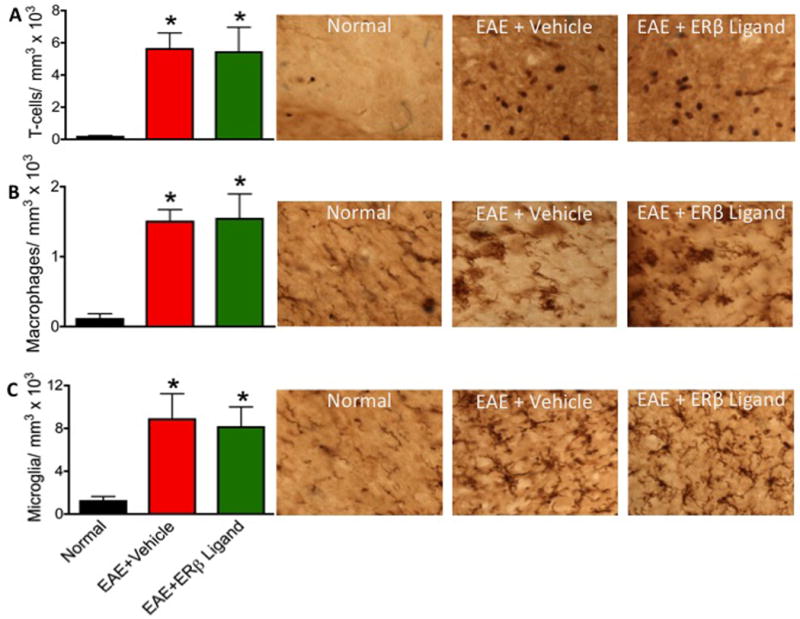
Therapeutic ERβ ligand treatment does not significantly alter numbers of T lymphocytes, macrophages, or microglia in the CNS. (A) T lymphocyte counts were elevated in both vehicle and ERβ ligand treated EAE mice. (B) Iba-1 globoid cells were significantly increased in both vehicle and ERβ ligand treated EAE mice. (C) Iba-1 ramified microglia numbers significantly increased in both vehicle and ERβ ligand treated EAE mice. * p < 0.05 vs. Normal (no EAE). 60x magnification for all images.
Figure 5.
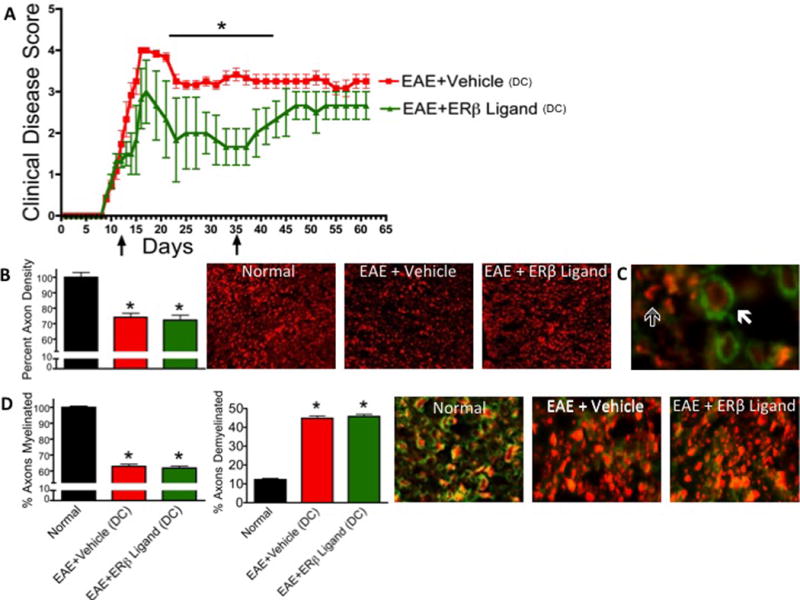
Clinical protection and neuroprotective effects of therapeutic ERβ ligand treatment does not persist after cessation of treatment. (A) Arrows indicate initiation and discontinuation (DC) of treatment. n=5 per group. * p < 0.05 between EAE+Vehicle and EAE+ERβ Ligand. (B) NF200+ axon counts were significantly decreased in EAE mice treated with vehicle or ERβ ligand. 10x magnification. (C) Solid arrow indicates a myelinated axon, outlined arrow indicates demyelinated axon. 40x magnification. (D) The percent of myelinated axons was significantly decreased in both EAE groups compared to normal mice. There was no difference between ERβ ligand and vehicle treated EAE mice with respect to percent of myelinated axons. The percent of demyelinated axons was significantly increased in both EAE treatment groups compared to normal mice. There was no difference between ERβ ligand and vehicle treated mice with respect to percent of demyelinated axons. n=5 per group. * p < 0.05 vs Normal. 40x magnification.
Figure 6.
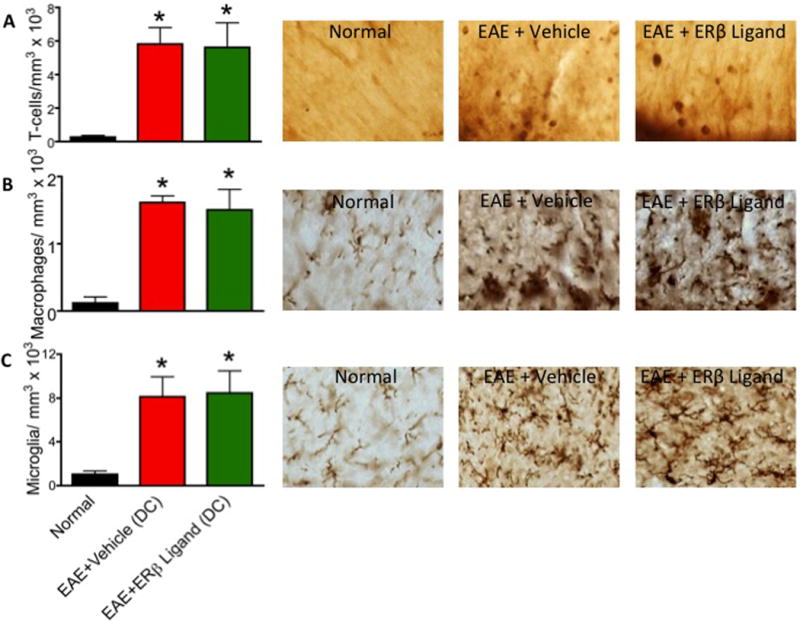
Cessation of therapeutic ERβ ligand treatment does not significantly alter numbers of T lymphocytes, macrophages, or microglia in the CNS. (A) T lymphocyte counts were elevated in both vehicle and ERβ ligand treated EAE mice. (B) Iba-1 globoid cells were significantly increased in both vehicle and ERβ ligand treated EAE mice. (C) Iba-1 ramified microglia numbers significantly increased in both vehicle and ERβ ligand treated EAE mice. * p < 0.05 vs. Normal (no EAE). 60x magnification for all images.
Clinical Scoring
Animals were monitored daily for EE signs based on a standard EAE 0 to 5 scale scoring system: 0, healthy; 1, complete loss of tail tonicity; 2, loss of righting reflex; 3, partial paralysis; 4, complete paralysis of one or both hind limbs; and 5, moribund.
Histological preparation
Female mice were deeply anesthetized in isoflurane and perfused transcardially with ice-cold 1× PBS for 20 to 30 min, followed by 10% formalin for 10 to 15 min. Spinal cords were dissected and submerged in 10% formalin overnight at 4 °C, followed by 30% sucrose for 24 h. Spinal cords were cut in thirds and embedded in optimal cutting temperature compound (Tissue Tek) and frozen at −80 °C. The 40-μm thick free-floating spinal cord cross-sections were obtained with a microtome cryostat (model HM505E) at −20 °C. Tissues were collected serially and stored in 0.1 M PBS with 1% sodium azide at 4 °C until immunohistochemistry.
Immunohistochemistry
Before histological staining, 40-μm thick free-floating sections were thoroughly washed with 0.1 M PBS to remove residual sodium azide. For tissues to be treated with diaminobenzidine (DAB), sections were permeabilized with 0.5% Triton X-100 in 0.1 M TBS and 10% normal goat serum (NGS) for 60 min at room temperature. The following primary antibodies were used: anti-CD3 at 1:2,000 (BD Pharmigen), anti–neurofilament (NF200) at 1:750 dilutions (Sigma), and anti–Iba-1 at 1:10,000 (Wako Chemicals). Tissues were then washed three times for 10 min in 0.1 M TBS. and labeled with secondary anti- bodies conjugated to Cy5 (Vector Labs and Chemicon) for 1 h for NF-200 and MBP. Tissues were labeled with biotin secondary antibodies for CD3, NF200, and Iba-1, followed by ABC/DAB treatment (Vector Labs). Fluorescent sections were mounted on slides, allowed to semidry, and coverslipped in fluoromount G (Fisher Scientific). DAB sections were dried overnight and then dehydrated in 70, 95, and 100% ethanol, followed by 5 min of Citrasolve and coverslipped with Permount (Fisher).
Quantification
To quantify immunohistochemical staining, we examined three spinal cord cross-sections at the T1 to T5 level from each mouse and captured 4 planes per tissue section, for a total of 12 cross-sectional planes from the spinal cord of each mouse. Images were captured under microscope at 10× or 40× magnifications using the DP70 Image software and a DP70 camera (both from Olympus). All images in each experimental set were captured under the same light intensity and exposure limits. Image analysis was performed using ImageJ Software v1.45s. Three sections from each animal were then quantified to calculate the mean per animal. Axonal densities were calculated by counting the number of NF200+ cells in a 40× image over the area of the captured tissue section. Demyelination was quantified by counting the number of NF200+ axons fully encircled by myelin and dividing it by the total number of NF200+ axons. Axons that were not fully enclosed by myelin were considered demyelinated. Inflammatory infiltrates were quantified by counting the number of DAB-positive cells in the dorsal column of the thoracic spinal cord at 40× under a light microscope.
Microscopy
Stained sections were examined and photographed using a confocal microscope (Leica TCS-SP) or a fluorescence microscope (BX51WI; Olympus) equipped with Plan Fluor objectives connected to a camera (DP70, Olympus). Digital images were collected and analyzed using Leica confocal and DP70 camera software. Images were assembled using Adobe Photoshop (Adobe Systems) and Microsoft PowerPoint. DAB sections were examined at the light level at 40× (Nikon Alphaphot-2 YS2).
Statistical Methods
Differences in EAE clinical scores were determined by repeated measures one-way ANOVA and subsequent Post Hoc analysis for multiple comparisons. Immunohistochemical data were analyzed using one-way ANOVA. For these analyses, Bonferroni post hoc analysis was performed on F-stat values (Prism). All statistical tests were two tailed, and a value of P < 0.05 was considered statistically significant.
Results
Therapeutic ERβ ligand treatment ameliorates clinical signs of EAE
In order to determine whether late treatment with an ERβ ligand has a therapeutic effect, active EAE was induced in C57BL/6 mice by immunization with MOG 35–55 peptide. Treatment with ERβ ligand and vehicle was then started after the development of clinical signs. Standard EAE clinical scores revealed that all mice in both the vehicle and ERβ ligand treatment groups developed severe motor deficits about 14 days after immunization (Fig 1). The ERβ ligand and vehicle treatment groups were not significantly different in day of peak disease or the level of peak disease, which occurred 18–20 days after immunization. However, ERβ ligand treatment was able to successfully promote recovery later in the chronic phase of disease, beginning at day 27.
Therapeutic ERβ ligand treatment protects against axonal loss and demyelination in EAE mice
After final measurements of clinical disease scores (35 days post-immunization), all mice were assessed for EAE neuropathology, which is generally characterized by demyelination, axonal loss, and inflammation in white matter of the spinal cord. Axonal loss has previously been shown to correlate most strongly with clinical disease severity (Wujek et al. 2002). In order to evaluate axon counts in the spinal cord, we used immunofluorescence staining for neurofilament 200 (NF200) and semiautomated counting software (Image J). The average number of axons in vehicle treated EAE mice was significantly less than those of normal healthy controls. Axon counts in ERβ ligand treated animals were significantly higher than those in vehicle-treated EAE, demonstrating that ERβ ligand treatment started after disease onset can protect against axonal loss in EAE (Fig 2A).
Figure 2.
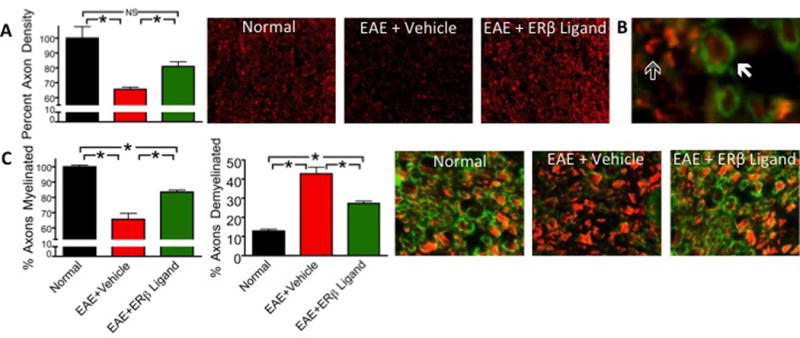
Therapeutic ERβ ligand treatment protects against axonal loss and demyelination. (A) NF200+ axon counts were significantly decreased in EAE mice treated with vehicle. ERβ ligand treatment preserved axons as compared to vehicle treatment. 10x magnification. (B) Solid arrow indicates a myelinated axon, outlined arrow indicates demyelinated axon. 40x magnification. (C) The percent of myelinated axons was significantly decreased in both EAE groups compared to normal mice. ERβ ligand treatment significantly increased the percent of myelinated axons compared to EAE mice treated with vehicle. The percent of demyelinated axons was significantly increased in both EAE treatment groups compared to normal mice. ERβ ligand treatment significantly decreased the percent of demyelinated axons compared to EAE mice treated with vehicle. 40x magnification. n=4 per group. * p < 0.05.
In order to quantify demyelination, we used immunofluorescence to stain spinal cord sections for myelin basic protein (MBP) and NF200. NF200+ axons were considered myelinated if they were encircled completely by a ring of myelin and unmyelinated if they were not encircled or partially encircled by myelin (Fig 2B). We then quantified the percent of myelinated NF200+ axons and compared it across experimental groups (Fig 2C). EAE mice displayed areas lacking MBP staining, indicating the irregular demyelination that is characteristic of EAE. The percent of myelinated axons was significantly reduced in vehicle treated EAE mice compared to normal. In contrast, ERβ ligand treated EAE mice had a significant increase in the percent myelinated axons as compared to vehicle treated mice. The effect on myelin was confirmed by quantification of the percent of demyelinated axons. Vehicle treated EAE mice had a significantly increased percent of demyelinated axons as compared to normal controls, while ERβ ligand treated EAE mice had a smaller percent of demyelinated axons as compared to vehicle treated EAE. Together, these data show that ERβ ligand treatment started after disease onset can both preserve axon numbers as well as reduce demyelination.
Therapeutic ERβ ligand treatment does not alter the level of CNS inflammation in EAE
In EAE, inflammation of the CNS includes infiltration of T lymphocytes and macropahges (Voskuhl et al. 2009). Therefore, sections from the thoracic dorsal column of the spinal cord were collected and analyzed for the infiltration of T lymphocytes, Iba-1 globoid cells (macrophages), and Iba-1 ramified cells (microglia). In EAE, immune cells were detected as discrete infiltrates and were not diffusely scattered throughout the dorsal column (Fig 3). ERβ ligand treatment did not change the level or distribution of these infiltrates.
Figure 3.
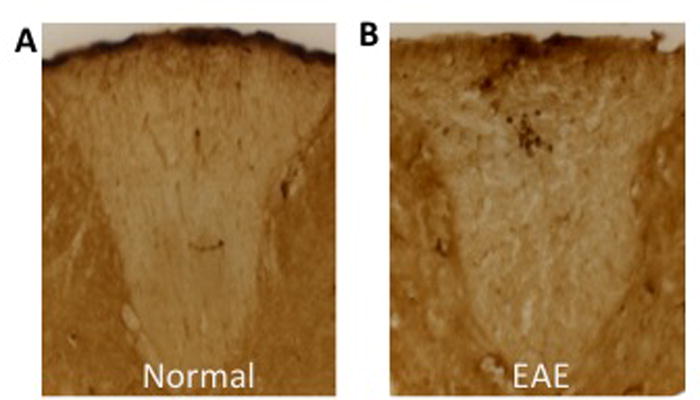
T lymphocyte inflammation in the CNS is concentrated in active lesions, rather than scattered diffusely throughout the white matter of the dorsal column. (A) CD3+ T lymphocytes were not present in the dorsal column of healthy animals. (B) CD3+ T lymphocytes were located in discrete active lesions in EAE mice.
T lymphocytes were identified using immunohistochemistry with an anti-CD3 antibody (Fig 4A). Vehicle and ERβ ligand treated EAE mice showed significantly increased numbers of CD3 positive T cells in spinal cord white matter compared to healthy normal controls. However, T cell counts were indistinguishable between ERβ ligand treated and vehicle treated EAE mice, suggesting that ERβ ligand treatment does not reduce T cell inflammation.
In order to identify Iba-1 cells, immunohistochemistry was used to stain for Iba-1 and cells were separated into two groups as previously described (Voskuhl et al. 2009). Iba-1 globoid cells (Fig 4B) are of the morphology typically associated with monocytes and phagocytic macrophages, while Iba-1 ramified cells (Fig 4C) are of the morphology typically associated with activated microglia, the resident immune cells of the CNS. Both macrophages and microglia were increased in vehicle treated EAE as compared to normals. ERβ ligand treated EAE mice also had high levels of macrophages and microglia in spinal cords, with levels no different than in the vehicle treated EAE mice. Together, this shows that ERβ ligand treatment did not reduce levels of macrophages or activated microglia.
Effects of therapeutic ERβ ligand treatment are not permanent
After observing that therapeutically administered ERβ ligand treatment reduced clinical disease, spared axons, and preserved myelin, we next wanted to address whether this neuroprotective effect could persist after treatment was discontinued. The clinical disease scores for both vehicle and ERβ ligand treated animals reached a peak 17 days after immunization. As in the previous experiment, clinical disease scores of vehicle and ERβ ligand treated animals began to show significant differences after the peak acute phase, during the chronic phase, here at day 21. However, after stopping treatment on day 35 (inclusive), the clinical disease scores of the ERβ ligand treated mice gradually began to increase until, by day 43, they were indistinguishable from vehicle treated EAE mice (Fig 5A).
Because a significant difference had been observed in the neuropathology of therapeutically treated ERβ ligand and vehicle EAE animals, we again assessed neuropathology, this time after cessation of treatment, at day 61 after EAE induction. Using the methods described above, we saw that axonal loss again correlated with clinical disease score. Specifically, the ERβ ligand and vehicle treated EAE animals had reduced axon counts compared to normal mice (Fig 5B). The two treatment groups also had reduced myelin staining as compared to normal mice (Fig 5D). However, in contrast to pathology done during continuous treatment, pathology done after treatment cessation revealed no difference in axonal count or demyelination when comparing ERβ ligand versus vehicle treated EAE mice. Together, the clinical and neuropathology data demonstrate that the neuroprotective effects of therapeutically administered ERβ ligand treatment do not persist after termination of treatment, but rather require continuous treatment.
It was possible that the exacerbation of clinical signs after cessation of ER ligand treatment was due to a relapse, as defined by enhancement of inflammation. In order to assess the inflammatory response, we quantified the infiltration of T lymphocytes, macrophages, and microglia as described above. ERβ ligand and vehicle treated EAE mice were indistinguishable with respect to number of T lymphocytes, macrophages, and microglia in the thoracic dorsal column (Fig 6). Both the ERβ ligand and vehicle treated mice had significantly increased immune cell infiltration compared to normal mice. These data demonstrate that the increase in clinical disease score after cessation of ERβ ligand treatment was not associated with enhancement of CNS inflammation.
Discussion
Although previous studies have demonstrated the efficacy of ERβ ligands in EAE, here we have shown that they are useful when administered after the onset of clinical signs. Specifically, previous studies have demonstrated that ERβ ligands administered 7–10 days before EAE induction provided clinical protection, as well as preserved axons and myelin in the spinal cords (Tiwari-Woodruff et al. 2007). Here, we have shown that therapeutic administration of ERβ ligands initiated after the onset of clinical signs of EAE can still provide such clinical protection. This clinical protection was accompanied by neuroprotection, as ERβ ligand treated mice exhibited decreased axonal loss and demyelination. However, the neuroprotection observed with therapeutic treatment was dependent on continuous administration of the ERβ ligand, as it did not persist after treatment had been terminated. EAE disease progression, like MS, involves a continued evolution of a neurodegenerative process over time. It appears that ERβ ligand treatment keeps this neurodegenerative process in check when present. However, when treatment is halted, the natural progression of the neurodegenerative process resumes, resulting in accumulation of disability, as reflected by clinical disease score. It is nevertheless possible that if ERβ ligand treatment were maintained for long enough periods of time that ultimate disability might be decreased in ERβ ligand treated as compared to vehicle.
In this therapeutic treatment design, we observed no significant difference in CNS inflammation when comparing vehicle and ERβ ligand treated EAE mice. Previous studies involving prophylactic treatment also showed that ERβ ligand treatment in EAE did not reduce overall levels of CNS inflammation or modulate peripheral cytokine production (Du, Sandoval, Trinh, Umeda, et al. 2010b). Despite no quantitative difference in CNS inflammation, previous studies showed that treatment with an ERβ ligand qualitatively altered the nature of inflammation by decreasing the percentage of CNS immune infiltration comprised of dendritic cells and decreasing production of TNF-α by these dendritic cells. Together this suggests that while ERβ ligand treatment is not anti-inflammatory in the peripheral immune system, it can nevertheless modulate both inflammation and neurodegeneration in the CNS during EAE. Furthermore, the mechanism for neuroprotection offered by ERβ ligand treatment in EAE could be either direct or indirect. Direct neuroprotection would entail ERβ ligand binding to CNS cells directly, while indirect neuroprotection would entail ERβ ligand binding to cells in the peripheral immune system to reduce CNS inflammation and thereby provide less of an inflammatory attack on CNS cells. Notably, direct and indirect neuroprotective mechanisms by estrogens in EAE are not mutually exclusive. When a drug suppresses the peripheral immune system, it is impossible to state whether or not it is directly or indirectly neuroprotective on CNS cells unless additional experiments are carried out such as a conditional knockout (CKO) of ERβ in cells of the CNS such as astrocytes, oligodendrocytes, or neurons with subsequent observations that the beneficial effects of ERβ ligand treatment are lost. We have done these CKO experiments with ERα ligand treatment and have demonstrated direct neuroprotection since therapeutic effects of ERα ligand treatment were lost when astrocytes were devoid of ERα (Spence et al. 2011). CNS cell specific knock outs of ERβ are currently being pursued in order to address this issue.
Other studies support ERβ ligands as promising therapeutic agents in MS. Estriol is known to be a relatively “weak” estrogen as compared to estradiol. Nevertheless, estriol has a higher affinity for ERβ than for the classic ERα (Kuiper et al. 1997). Estriol is not significantly detectable during nonpregnant states, but rises to high levels during pregnancy because it is made by the fetal placental unit. When estriol was given at relatively high doses to approximate those which occur during late pregnancy, it was protective in EAE in both female and male mice (Bebo et al. 2001; Kim et al. 1999; Liu et al. 2003; Palaszynski et al. 2004). At this dose, one presumes that estriol has activated both ERα and ERβ, albeit ERβ more. In contrast, ERβ ligand treatment would activate only ERβ receptors, with no significant activation of ERα. Consistent with this proposed action of estriol on both ERα and ERβ is the observation that treatment with estriol in both EAE mice and MS patients induces a modulation of peripheral immune responses (Liu et al. 2003; Soldan et al. 2003; Gold et al. 2009), since modulation of peripheral immune responses is known to occur with ERα, but not ERβ, ligand treatment (Du, Sandoval, Trinh, Umeda, et al. 2010b) (Du, Sandoval, Trinh & Voskuhl 2010a; Tiwari-Woodruff et al. 2007). Also, estradiol, estriol and ERα ligand each significantly decrease the peak severity of EAE at acute onset, while ERβ ligand decreases disease significantly only later in disease after the initial acute onset (Du, Sandoval, Trinh, Umeda, et al. 2010b; Du, Sandoval, Trinh & Voskuhl 2010a; Tiwari-Woodruff et al. 2007). Thus, estriol treatment is both anti-inflammatory and potentially neuroprotective with more ubiquitous effects on EAE, while ERβ ligand treatment would not be principally anti-inflammatory, but rather more strictly neuroprotective with effects on EAE only late. It would theoretically be desirable to combine ERβ ligand treatment with a standard anti-inflammatory treatment in MS, as has been done in EAE (Du, Sandoval, Trinh & Voskuhl 2010a), while such combination with an anti-inflammatory may not be necessary with estriol treatment.
Therapeutic estriol treatment has shown promise in human MS. In a pilot clinical trial of females with relapsing-remitting multiple sclerosis (RRMS), estriol treatment reduced gadolinium enhancing lesions on MRI by approximately 75% as compared to pre-treatment (Sicotte et al. 2002), and there are now two ongoing trials of estriol treatment in MS. One trial (http://www.clinicaltrials.gov/ct2/show/NCT00451204) has a decrease in relapse rates as the primary outcome measure in an attempt to recapitulate the known decrease in relapses that occurs during late pregnancy when estriol levels are high. The other trial (http://clinicaltrials.gov/ct2/show/NCT01466114) has cognitive testing as the primary outcome measure based on the known neuroprotective effect of estrogens on cognition and most recently in EAE (Ziehn et al. 2012).
In summary, estrogen treatments in MS are promising as neuroprotective candidates even when administered after disease onset. Since most of the deleterious side effects of estradiol treatment on breast and uterus are mediated through ERα, novel treatment with ERβ ligands may be viewed as a “next generation” estrogen to optimize efficacy and minimize toxicity.
Acknowledgments
The support for this work was provided by National Institutes of Health [grant number K24 NS062117] to R.R.V. and by National Multiple Society [grant numbers RG4362 and RG4033] to R.R.V., as well as by the Conrad Hilton Foundation, the Jack Skirball Foundation, and the Sherak Family Foundation. A.J.W. is funded in part by a grant to UCLA from the Howard Hughes Medical Institute through the Precollege and Undergraduate Science Education Program. R.D.S. is supported by an NIH Neuroendocrinology, Sex Differences, and Reproduction (LNE) training grant [grant number 5T32HD07228].
Grant information: The support for this work was provided by National Institutes of Health [grant number K24 NS062117] to R.R.V. and by National Multiple Society [grant numbers RG4362 and RG4033] to R.R.V. R.D.S. is supported by an NIH Neuroendocrinology, Sex Differences, and Reproduction (LNE) training grant [grant number 5T32HD07228]. AJW is supported by a grant to UCLA from the Howard Hughes Medical Institute through the Precollege and Undergraduate Science Education Program.
References
- Bebo BF, et al. Low-dose estrogen therapy ameliorates experimental autoimmune encephalomyelitis in two different inbred mouse strains. Journal of immunology (Baltimore, Md. : 1950) 2001;166(3):2080–2089. doi: 10.4049/jimmunol.166.3.2080. [DOI] [PubMed] [Google Scholar]
- Brann DW, et al. Neurotrophic and neuroprotective actions of estrogen: basic mechanisms and clinical implications. Steroids. 2007;72(5):381–405. doi: 10.1016/j.steroids.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JJ, et al. The role of the estrogen in neuroprotection: implications for neurodegenerative diseases. Neuro endocrinology letters. 2003;24:3–4. 141–147. [PubMed] [Google Scholar]
- Croxford AL, Kurschus FC, Waisman A. Mouse models for multiple sclerosis: historical facts and future implications. Biochimica et biophysica acta. 2011;1812(2):177–183. doi: 10.1016/j.bbadis.2010.06.010. [DOI] [PubMed] [Google Scholar]
- Du S, Sandoval F, Trinh P, Voskuhl RR. Additive effects of combination treatment with anti-inflammatory and neuroprotective agents in experimental autoimmune encephalomyelitis. Journal of neuroimmunology. 2010a;219:1–2. 64–74. doi: 10.1016/j.jneuroim.2009.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du S, Sandoval F, Trinh P, Umeda E, et al. Estrogen receptor-β ligand treatment modulates dendritic cells in the target organ during autoimmune demyelinating disease. European Journal of Immunology. 2010b;41(1):140–150. doi: 10.1002/eji.201040796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Segura LM, Azcoitia I, DonCarlos LL. Neuroprotection by estradiol. Progress in neurobiology. 2001;63(1):29–60. doi: 10.1016/s0301-0082(00)00025-3. [DOI] [PubMed] [Google Scholar]
- Gold SM, Voskuhl RR. Estrogen and testosterone therapies in multiple sclerosis. Progress in brain research. 2009;175:239–251. doi: 10.1016/S0079-6123(09)17516-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold SM, et al. Estrogen treatment decreases matrix metalloproteinase (MMP)-9 in autoimmune demyelinating disease through estrogen receptor alpha (ERalpha) Laboratory investigation; a journal of technical methods and pathology. 2009;89(10):1076–1083. doi: 10.1038/labinvest.2009.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung HP, et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. 2002;360(9350):2018–2025. doi: 10.1016/S0140-6736(02)12023-X. [DOI] [PubMed] [Google Scholar]
- Johnson KP. Glatiramer acetate for treatment of relapsing-remitting multiple sclerosis. Expert review of neurotherapeutics. 2012;12(4):371–384. doi: 10.1586/ern.12.25. [DOI] [PubMed] [Google Scholar]
- Kim S, et al. Estriol ameliorates autoimmune demyelinating disease: implications for multiple sclerosis. Neurology. 1999;52(6):1230–1238. doi: 10.1212/wnl.52.6.1230. [DOI] [PubMed] [Google Scholar]
- Kuiper GG, et al. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. 1997;138(3):863–870. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- Liu HB, et al. Estrogen receptor alpha mediates estrogen’s immune protection in autoimmune disease. Journal of immunology (Baltimore, Md. : 1950) 2003;171(12):6936–6940. doi: 10.4049/jimmunol.171.12.6936. [DOI] [PubMed] [Google Scholar]
- Milo R, Kahana E. Multiple sclerosis: geoepidemiology, genetics and the environment. Autoimmunity reviews. 2010;9(5):A387–94. doi: 10.1016/j.autrev.2009.11.010. [DOI] [PubMed] [Google Scholar]
- Morales LBJ, et al. Treatment with an estrogen receptor alpha ligand is neuroprotective in experimental autoimmune encephalomyelitis. Journal of Neuroscience. 2006;26(25):6823–6833. doi: 10.1523/JNEUROSCI.0453-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offner H. Neuroimmunoprotective effects of estrogen and derivatives in experimental autoimmune encephalomyelitis: therapeutic implications for multiple sclerosis. Journal of neuroscience research. 2004;78(5):603–624. doi: 10.1002/jnr.20330. [DOI] [PubMed] [Google Scholar]
- Palaszynski KM, et al. Estriol treatment ameliorates disease in males with experimental autoimmune encephalomyelitis: implications for multiple sclerosis. Journal of neuroimmunology. 2004;149:1–2. 84–89. doi: 10.1016/j.jneuroim.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Sicotte NL, et al. Treatment of multiple sclerosis with the pregnancy hormone estriol. Annals of neurology. 2002;52(4):421–428. doi: 10.1002/ana.10301. [DOI] [PubMed] [Google Scholar]
- Soldan SS, et al. Immune modulation in multiple sclerosis patients treated with the pregnancy hormone estriol. Journal of immunology (Baltimore, Md. : 1950) 2003;171(11):6267–6274. doi: 10.4049/jimmunol.171.11.6267. [DOI] [PubMed] [Google Scholar]
- Spence RD, Voskuhl RR. Neuroprotective effects of estrogens and androgens in CNS inflammation and neurodegeneration. Frontiers in Neuroendocrinology. 2012;33(1):105–115. doi: 10.1016/j.yfrne.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence RD, et al. Neuroprotection mediated through estrogen receptor-alpha in astrocytes. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(21):8867–8872. doi: 10.1073/pnas.1103833108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S, et al. Oral feeding with ethinyl estradiol suppresses and treats experimental autoimmune encephalomyelitis in SJL mice and inhibits the recruitment of inflammatory cells into the central nervous system. Journal of immunology (Baltimore, Md. : 1950) 2003;170(3):1548–1555. doi: 10.4049/jimmunol.170.3.1548. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Brown CM, Wise PM. Neuroprotective effects of estrogens following ischemic stroke. Frontiers in Neuroendocrinology. 2009;30(2):201–211. doi: 10.1016/j.yfrne.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari-Woodruff S, Voskuhl RR. Neuroprotective and anti-inflammatory effects of estrogen receptor ligand treatment in mice. Journal of the neurological sciences. 2009;286:1–2. 81–85. doi: 10.1016/j.jns.2009.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari-Woodruff S, et al. Differential neuroprotective and antiinflammatory effects of estrogen receptor (ER)alpha and ERbeta ligand treatment. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(37):14813–14818. doi: 10.1073/pnas.0703783104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulzheimer JC, et al. Therapeutic approaches to multiple sclerosis: an update on failed, interrupted, or inconclusive trials of immunomodulatory treatment strategies. BioDrugs : clinical immunotherapeutics, biopharmaceuticals and gene therapy. 2010;24(4):249–274. doi: 10.2165/11537160-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Voskuhl RR, et al. Reactive Astrocytes Form Scar-Like Perivascular Barriers to Leukocytes during Adaptive Immune Inflammation of the CNS. Journal of Neuroscience. 2009;29(37):11511–11522. doi: 10.1523/JNEUROSCI.1514-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolinsky JS, et al. United States open-label glatiramer acetate extension trial for relapsing multiple sclerosis: MRI and clinical correlates. Multiple Sclerosis Study Group and the MRI Analysis Center. Multiple sclerosis (Houndmills, Basingstoke, England) 2001;7(1):33–41. doi: 10.1177/135245850100700107. [DOI] [PubMed] [Google Scholar]
- Wujek JR, et al. Axon loss in the spinal cord determines permanent neurological disability in an animal model of multiple sclerosis. Journal of neuropathology and experimental neurology. 2002;61(1):23–32. doi: 10.1093/jnen/61.1.23. [DOI] [PubMed] [Google Scholar]
- Ziehn MO, et al. Estriol preserves synaptic transmission in the hippocampus during autoimmune demyelinating disease. 2012. pp. 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]