

Abstract
The poor solubility of many newly discovered drugs has resulted in numerous challenges for the time-controlled release of therapeutics. In this study, an advanced drug delivery platform to encapsulate and deliver hydrophobic drugs, consisting of poly (lactic-co-glycolic acid) (PLGA) nanoparticles incorporated within poly (ethylene glycol) (PEG) microgels, was developed. PLGA nanoparticles were used as the hydrophobic drug carrier, while the PEG matrix functioned to slow down the drug release. Encapsulation of the hydrophobic agents was characterized by fluorescence detection of the hydrophobic dye Nile Red within the microgels. In addition, the microcomposites prepared via the droplet-based microfluidic technology showed size tunability and a monodisperse size distribution, along with improved release kinetics of the loaded cargo compared with bare PLGA nanoparticles. This composite system has potential as a universal delivery platform for a variety of hydrophobic molecules.
I. INTRODUCTION
Technological advances in the pharmaceutical industry have led to numerous discoveries and the development of novel therapeutic compounds over the years. Unfortunately, many of the new bioactive entities are highly hydrophobic with an intrinsically poor water solubility, hence exhibiting low oral bioavailability.1,2 Faced with the constant challenge to either discard or develop a new compound, a reliable hydrophobic drug delivery system would provide an alluring alternative for pharmaceutical companies. A popular delivery strategy, especially for drugs with a wide therapeutic window and long-term low dosage requirements,3 is to encapsulate the active substances in micro- and nano-vehicles because of the capability to protect the cargo from degradation, reduce toxicity, and improve bioavailability and absorption by the tissue.3,4
Various synthetic or naturally occurring materials, such as phospholipids, liposomes,5 amphiphilic micelles, and polymersomes,6 or biodegradable polymeric spheres,7 have been explored as carriers for the encapsulated entities. Among these systems, poly (lactic-co-glycolic acid) (PLGA) nanoparticles emerge due to their excellent biocompatibility and approval by the US Food and Drug Administration (FDA) and European Medicine Agency (EMA).8,9 PLGA nanoparticles also exhibit a high loading capacity of various insoluble therapeutics10 due to their hydrophobic nature. The effectiveness of PLGA nanoparticles as nanocarriers has been established in various studies for the encapsulation of poor water-soluble drugs, such as paclitaxel,11 haloperidol,12 and estradiol,13 among others. However, a significant biological barrier for drug-loaded PLGA nanoparticles exists because the body identifies the hydrophobic particles as foreign and mass eliminates them to the liver or the spleen.8,14 Therefore, surface modifications, such as coating with a layer of hydrophilic materials, are typically required.8 Additionally, an undesirable high burst release is often observed from the PLGA nanoparticles, potentially elevating the local drug concentration to near or above toxic levels.15,16 Because the burst release is typically linked to the surface concentration of the encased drugs,17 surface-related strategies can also control the initial burst release. Unfortunately, PLGA nanoparticles lack surface-active functional groups, complicating the surface modifying process.17
In this study, using a relatively simple fabrication process, a multistage drug delivery platform consisting of highly biocompatible18,19 poly (ethylene glycol) (PEG) microgels impregnated with drug-loaded PLGA nanoparticles was explored. The hydrophilic nature of the PEG hydrogel surface has been proven non-inflammatory and prolongs the drug availability in the body.19,20 Furthermore, the synergistic effect of the multistage delivery system endowed the carriers with reduced burst release and subsequently demonstrated the sustained release profile of the hydrophobic model drug ibuprofen. Although the success of similar multistage systems utilizing porous silicon particles in various polymer matrices has been encouraging,21,22 the absolute safety of the silicon particles in those studies remains a concern.23 In contrast, PEG-PLGA multistage vehicles offer additional benefits, such as excellent biocompatibility and biodegradability.
Another important parameter of these delivery systems is the particle size, which plays important roles in determining the cargo release rates.24–27 Droplet microfluidic technology provides a method to generate highly monodisperse particles with a precisely controllable size and morphology.21,28–30 In addition to fabricating uniformly sized spheres that might be crucial for physiological interactions of the encased therapeutics,25 microfluidics provide added benefits, such as the flexibility in material selection and chemical compositions, the ability to simultaneously encapsulate different cargos, and a nearly 100% theoretical encapsulation efficiency.25,29,31–34 Equipped with the numerous advantages afforded by droplet-based microfluidics, the multistage design described in this study can be widely applied in long-term and multi-functional delivery systems.
II. EXPERIMENTAL METHODS
Poly (DL-lactide-co-glycolide) 50:50 (PLGA) (IV = 1.03 dl/g) was obtained from Purac Biochem (Gorinchem, Netherlands). Dichloromethane (DCM), poly (vinyl alcohol) (PVA) (Molecular weight of 9000–10 000, 80% hydrolyzed), PEG diacrylate (PEG-DA) (average Mn of 575), photoinitiator 2-hydroxy-4′-(2-hydroxyethoxy)-2-methylpropiophenone, demulsifier 1H,1H,2H,2H-perfluoro-1-octanol, ibuprofen and Nile red were purchased from Sigma-Aldrich (Singapore). The fluorocarbon oil used was HFE-7500 (3M Novec TM, Singapore).
The PLGA nanoparticles were fabricated using the emulsion-solvent evaporation method. Briefly, 100 mg PLGA was dissolved in the DCM organic solvent at room temperature for 15 min. Subsequently, this organic phase was emulsified in 10 ml of the aqueous phase containing PVA as a stabilizer (2% w/v) using a Misonix sonicator (XL-2000) in remote operation for approximately 20 times. The resulting o/w emulsion was stirred for approximately 5 h to facilitate the evaporation of the volatile DCM solvent. The PLGA nanoparticles were then collected after centrifugation at 11 000 rpm for 6 min. Loading of ibuprofen or Nile red was achieved by dissolving the respective cargo with PLGA in DCM (10%, w/w of polymer), and the remaining procedure was performed as previously described. The ibuprofen loading efficiency to the PLGA nanoparticles was examined by releasing the cargo to 40 ml of phosphate-buffered saline (PBS; pH 7.2) at 37 °C. The release study was performed until no further ibuprofen was detected in the buffer after incubation for a day. Furthermore, in addition to scanning electron microscopy (SEM) observations, the size of the nanoparticles was determined by a Zetasizer (Nano ZS, Malvern Instruments, Malvern, UK), which is based on the dynamic light scattering principle. The polydispersity index (PDI) was also obtained as an indication of the extent of the nanoparticle size distribution. A PDI value of 0 suggests that the sample is perfectly homogeneous, while a PDI value of 1 indicates that the sample has a broad size range, possibly containing aggregates.
Ibuprofen-loaded PLGA nanoparticles were initially mixed with the PEG-DA solution (PEG-DA to PLGA ratio of 3:1 v/v) containing the photoinitiator (20 mg in 1 ml of pre-gel solution). The total PEG-DA concentration was 40% v/v. Subsequently, monodisperse aqueous emulsions in HFE-7500 oil were formed using a microfluidic cross junction droplet generation device. The resulting droplets were collected and UV-crosslinked with a UV light-emitting diode (LED) curing system (Uvata, Shanghai) at 50 W for 1 min. The remaining oil phase was washed off using the demulsifier to obtain the structural microgels. Subsequently, the homogeneous microcomposites were washed again at least three times with distilled water. To verify the encapsulation of the PLGA nanoparticles in the PEG microgels, Nile red-loaded nanospheres were used. Observations through the fluorescence microscope should confirm the presence of the nanoparticles within the gel matrix.
The release behaviors of ibuprofen from bare PLGA nanoparticles and the structural microgels were determined in 1 ml of PBS at 37 °C. At pre-defined intervals, the samples were centrifuged, and the supernatant was collected and replaced with fresh PBS solution. The collected solution was analyzed with a UV-vis spectrophotometer (UV-2450, Shimadzu, Kyoto, Japan) at a wavelength of 222 nm. The initial loading quantity of ibuprofen in the bare PLGA nanoparticles and PEG-PLGA composites was maintained constant.
III. RESULTS AND DISCUSSION
To deliver hydrophobic therapeutics, PLGA nanoparticles were incorporated within the PEG microgel matrix. The process scheme of the demonstrated structural microparticles is presented in Fig. 1. Briefly, ibuprofen was loaded into PLGA nanoparticles prior to mixing with the PEG-DA pre-gel solution. Subsequently, at the cross-junction of the droplet generation device, the aqueous pre-gel mixture was sheared by the oil phase to generate monodisperse droplets. The collected pre-gel droplets were then polymerized by exposure to UV light. Finally, multistage delivery microcomposites were obtained after washing with the demulsifier and water. Diffusion-driven ibuprofen release studies were subsequently performed in a PBS buffer solution.
FIG. 1.

(a) Schematic illustration of the preparation procedure of the PEG-PLGA microcomposites. The hydrophobic drug was pre-uploaded into PLGA nanoparticles. These nanoparticles were encapsulated into PEG hydrogel micro particles for sustained drug delivery. (b) The SEM image of the monodispersed PLGA nanoparticles was recorded. To quantify the particle size distribution, the size distribution spectrum of the prepared PLGA nanoparticles was analyzed using the dynamic light scattering technique. (c) The PEG hydrogel particles were manipulated through microfluidics to encapsulate the PLGA nanoparticles.
First, PLGA nanoparticles were fabricated using the emulsion-solvent evaporation method. Loading of the hydrophobic ibuprofen was achieved by co-dissolving the therapeutics with the polymer in the organic solvent. Nanosized emulsions of the organic phase in water, which would harden to form the nanospheres, were induced by sonication. The energy output of the sonication step primarily determined the sizes of the resulting PLGA nanoparticles.17 A narrow size distribution was preferred to obtain consistent release behaviors, and the SEM image of the nanoparticles in Fig. 1(b) showed relatively uniformly sized spheres. The average diameter of the prepared nanoparticles was 187 nm, as determined by the dynamic light scattering technique using the Zetasizer. The spectrum showed that the nanoparticles were relatively monodispersed, which was further supported by a low PDI value of 0.14. UV-vis analysis showed that high ibuprofen loading efficiency (∼98%) was achieved in the nanospheres. The cargo-loaded nanoparticles were then encapsulated within the PEG microgel matrix (Fig. 1(c)) by blending with the PEG-DA solution prior to the formation of droplets at the cross junction of the microfluidic device.
The flow rate of the dispersed aqueous pre-gel phase was maintained at 1 μl/min in all of the experiments, while the flow rates of the continuous oil phase were varied at 2 μl/min, 4 μl/min, and 8 μl/min. As shown in Fig. 2, as the flow ratio (dispersed phase to continuous phase) was reduced, the size of the droplets formed was also reduced. Increasing the oil flow rate with respect to the aqueous flow rate strengthens the shear force generated to pull and break the main aqueous stream into droplets, resulting in smaller uniform droplets.30,31 This tunability of the particle size offers a significant advantage to the developed delivery platform because the delivery vehicle size plays many important roles in cargo release and in vivo uptake rates.24,25 Gelation of the collected pre-gel droplets was achieved via crosslinking under a UV light before observing the particles under a microscope.
FIG. 2.

Changing the flow ratios of the oil to aqueous phase resulted in different diameter microgels. When the oil-aqueous flow ratios were increased, small gel particles were generated. In the experiments, the aqueous flow rate was maintained at 1 μl/min for all of the experiments.
The presence of PLGA nanoparticles caused the prepared microgels to appear rougher and darker under bright field observation, as illustrated in Figs. 3(a) and 3(b). To further confirm the presence of PLGA nanoparticles, Nile red was loaded into the nanoparticles, and the resulting microcomposites were observed under a fluorescence microscope. Spherical red fluorescence microspheres were observed (Fig. 3(c)), thus demonstrating that the hydrophobic dye-loaded PLGA nanoparticles were incorporated within the PEG matrix to ensure delivery of the hydrophobic compounds.
FIG. 3.
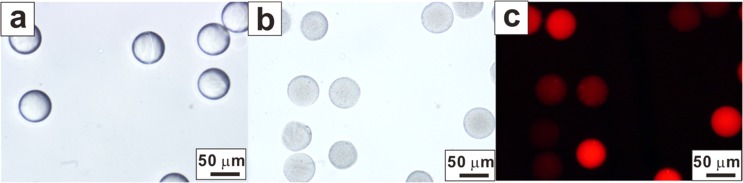
Microcomposites formed with a flow ratio of 4:1. (a) PEG microgels with no cargo. (b) Bright field image of the PEG-PLGA microcomposites. (c) Fluorescence image of the PEG-PLGA microcomposites.
To investigate the drug release capability of the multistage system, ibuprofen was loaded into the PLGA nanoparticles. Ibuprofen is a non-steroidal anti-inflammatory drug with antipyretic and analgesic properties.4 However, the poor solubility and bioavailability of ibuprofen may limit the full realization of its maximum therapeutic potential. Therefore, sustained delivery of the drug would provide a significant benefit. Release kinetics from the bare PLGA nanoparticles and the PEG-PLGA composites were investigated and compared. The release profiles were of a typical shape commonly obtained by other polymeric-based nano- or micro-particle delivery systems in which an initial burst release was followed by a slower release.35 Often, a high burst release is accompanied by detrimental effects and can be considered a disadvantage for the delivery platforms. Toxicity and waste problems of the encapsulated compounds would emerge, or the drug might even fail to reach the target tissues or cells.8
Bare PLGA nanoparticles without the PEG polymer matrix demonstrated a high burst release in which 67.5% of the loaded ibuprofen was released within 5 h of incubation. In contrast, the PEG-PLGA microcomposites had a lower burst release of 49.3% within the same interval, indicating that the PEG matrix possessed the barrier properties that reduced the diffusion of ibuprofen into the surroundings (Fig. 4(a)). The reduced initial burst also indicated a prolonged delivery period. Ibuprofen continued to be released into the PBS buffer solution after two months (more than 42 days) (Fig. 4(b)). In addition to curbing the burst release, the subsequent linear release phase should maintain a slower and controlled release rate. The linear ibuprofen release rates for the microcomposites were approximately 1.2 times lower compared with the bare nanoparticles. The release rates can be further fine-tuned by the precise control of the composite vehicle sizes with droplet microfluidic technology. It was observed that smaller sized particles exhibit faster release rates due to larger surface area to volume ratio (Fig. S1 in the supplementary material).36 This in vitro release behavior reflects the sustained release capabilities of the multistage design proposed in this study compared with single stage bare nanoparticle hydrophobic drug delivery methods.
FIG. 4.

Cumulative ibuprofen release profiles from the bare PLGA nanoparticles (Bare NPs) and the multi-stage PEG-PLGA microcomposite system. (a) Ibuprofen release within the first 1.5 days. (b) Ibuprofen release in 40 days. The data points represent the mean ± standard deviation of the data from three independent samples.
IV. CONCLUSIONS
This study introduced a multistage hydrophobic drug delivery system that utilizes the advantages offered by PLGA nanoparticle carriers and the hydrophilic surface properties of PEG microgels while maintaining a relatively simple fabrication process. This microcomposite design suppressed the initial burst release of ibuprofen. Moreover, slower sustained release rates were obtained following the burst for up to two months. This multistage design coupled with the additional advantages offered by droplet microfluidics enable more controllable and consistent release behaviors. Therefore, the concept of manipulating a multistage drug releasing platform through a microfluidic assembly is a promising alternative to other hydrophobic drug release systems to enrich drug delivery applications.
ACKNOWLEDGMENTS
We gratefully acknowledge the funding provided by the NUS start-up Grant No. R-397-000-137-133, the NUS Medicine-Engineering Seed Grant No. R-397-000-152-112, MOE AcRF Tier-1 R-397-000-153-112, the Singapore MIT Alliance for Research and Technology (SMART) research Grant No. R-397-000-146-592, and the facilities provided by the Singapore Institute for Neurotechnology (SINAPSE). We thank Muhammad Zahin Ramlan from Temasek Polytechnic, Singapore for his assistance to characterize the correlation between particle sizes and drug release profiles.
References
- 1.Williams H. D., Trevaskis N. L., Charman S. A., Shanker R. M., Charman W. N., Pouton C. W., and Porter C. J., Pharmacol. Rev. 65(1), 315 (2013). 10.1124/pr.112.005660 [DOI] [PubMed] [Google Scholar]
- 2.Edwards G. and Krishna S., Eur. J. Clin. Microbiol. Infect. Dis. 23(4), 233 (2004). 10.1007/s10096-004-1113-9 [DOI] [PubMed] [Google Scholar]
- 3.Hung L.-H., Teh S.-Y., Jester J., and Lee A. P., Lab Chip 10(14), 1820 (2010). 10.1039/c002866e [DOI] [PubMed] [Google Scholar]
- 4.Wischke C. and Schwendeman S. P., Int. J. Pharm. 364(2), 298 (2008). 10.1016/j.ijpharm.2008.04.042 [DOI] [PubMed] [Google Scholar]
- 5.Hurler J., Berg O. A., Skar M., Conradi A. H., Johnsen P. J., and Škalko-Basnet N., J. Pharm. Sci. 101(10), 3906 (2012). 10.1002/jps.23260 [DOI] [PubMed] [Google Scholar]
- 6.Jain J. P., Ayen W. Y., and Kumar N., Curr. Pharm. Des. 17(1), 65 (2011). 10.2174/138161211795049822 [DOI] [PubMed] [Google Scholar]
- 7.Fernández-Carballido A., Herrero-Vanrell R., Molina-Martınez I., and Pastoriza P., Int. J. Pharm. 279(1), 33 (2004). 10.1016/j.ijpharm.2004.04.003 [DOI] [PubMed] [Google Scholar]
- 8.Danhier F., Ansorena E., Silva J. M., Coco R., Le Breton A., and Préat V., J. Controlled Release 161(2), 505 (2012). 10.1016/j.jconrel.2012.01.043 [DOI] [PubMed] [Google Scholar]
- 9.Sahana D., Mittal G., Bhardwaj V., and Kumar M., J. Pharm. Sci. 97(4), 1530 (2008). 10.1002/jps.21158 [DOI] [PubMed] [Google Scholar]
- 10.Fredenberg S., Wahlgren M., Reslow M., and Axelsson A., Int. J. Pharm. 415(1), 34 (2011). 10.1016/j.ijpharm.2011.05.049 [DOI] [PubMed] [Google Scholar]
- 11.Fonseca C., Simões S., and Gaspar R., J. Controlled Release 83(2), 273 (2002). 10.1016/S0168-3659(02)00212-2 [DOI] [PubMed] [Google Scholar]
- 12.Budhian A., Siegel S. J., and Winey K. I., J. Microencapsulation 22(7), 773 (2005). 10.1080/02652040500273753 [DOI] [PubMed] [Google Scholar]
- 13.Mittal G., Sahana D. K., Bhardwaj V., and Kumar M. N. V. R., J. Controlled Release 119(1), 77 (2007). 10.1016/j.jconrel.2007.01.016 [DOI] [PubMed] [Google Scholar]
- 14.Kumari A., Yadav S. K., and Yadav S. C., Colloids Surf., B 75(1), 1 (2010). 10.1016/j.colsurfb.2009.09.001 [DOI] [PubMed] [Google Scholar]
- 15.Huang X. and Brazel C. S., J. Controlled Release 73(2–3), 121 (2001). 10.1016/S0168-3659(01)00248-6 [DOI] [PubMed] [Google Scholar]
- 16.Shaillender M., Luo R., Venkatraman S. S., and Neu B., Int. J. Pharm. 415(1), 211 (2011). 10.1016/j.ijpharm.2011.06.011 [DOI] [PubMed] [Google Scholar]
- 17.Luo R., Neu B., and Venkatraman S. S., Small 8(16), 2585 (2012). 10.1002/smll.201200398 [DOI] [PubMed] [Google Scholar]
- 18.Zhang L., Jeong Y.-I., Zheng S., Jang S. I., Suh H., Kang D. H., and Kim I., Polym. Chem. 4(4), 1084 (2013). 10.1039/C2PY20755A [DOI] [Google Scholar]
- 19.Nolan C. M., Reyes C. D., Debord J. D., García A. J., and Lyon L. A., Biomacromolecules 6(4), 2032 (2005). 10.1021/bm0500087 [DOI] [PubMed] [Google Scholar]
- 20.Yang T., Long H., Malkoch M., Gamstedt E. K., Berglund L., and Hult A., J. Polym. Sci., Part A: Polym. Chem. 49(18), 4044 (2011). 10.1002/pola.24847 [DOI] [Google Scholar]
- 21.Zhang H., Liu D., Shahbazi M. A., Mäkilä E., Herranz-Blanco B., Salonen J., Hirvonen J., and Santos H. A., Adv. Mater. 26(26), 4497 (2014). 10.1002/adma.201400953 [DOI] [PubMed] [Google Scholar]
- 22.Liu D., Zhang H., Herranz-Blanco B., Mäkilä E., Lehto V. P., Salonen J., Hirvonen J., and Santos H. A., Small 10(10), 2029 (2014). 10.1002/smll.201303740 [DOI] [PubMed] [Google Scholar]
- 23.Shahbazi M.-A., Herranz B., and Santos H. A., Biomatter 2(4), 296 (2012). 10.4161/biom.22347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shakweh M., Besnard M., Nicolas V., and Fattal E., Eur. J. Pharm. Biopharm. 61(1–2), 1 (2005). 10.1016/j.ejpb.2005.04.006 [DOI] [PubMed] [Google Scholar]
- 25.Duncanson W. J., Lin T., Abate A. R., Seiffert S., Shah R. K., and Weitz D. A., Lab Chip 12(12), 2135 (2012). 10.1039/c2lc21164e [DOI] [PubMed] [Google Scholar]
- 26.Luo R., Lim Z. H., Li W., Shi P., and Chen C. H., Chem. Commun. 50, 7052 (2014). 10.1039/c4cc02216e [DOI] [PubMed] [Google Scholar]
- 27.Luo R., Cao Y., Shi P., and Chen C. H., Small 10(23), 4886 (2014). 10.1002/smll.201401312 [DOI] [PubMed] [Google Scholar]
- 28.Mou C.-L., Ju X.-J., Zhang L., Xie R., Wang W., Deng N.-N., Wei J., Chen Q., and Chu L.-Y., Langmuir 30(5), 1455 (2014). 10.1021/la4046379 [DOI] [PubMed] [Google Scholar]
- 29.Herranz-Blanco B., Arriaga L. R., Mäkilä E., Correia A., Shrestha N., Mirza S., Weitz D. A., Salonen J., Hirvonen J., and Santos H. A., Lab Chip 14(6), 1083 (2014). 10.1039/c3lc51260f [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luo R., Ranjan S., Zhang Y., and Chen C.-H., Chem. Commun. 49(72), 7887 (2013). 10.1039/c3cc44111c [DOI] [PubMed] [Google Scholar]
- 31.Abbaspourrad A., Carroll N. J., Kim S.-H., and Weitz D. A., J. Am. Chem. Soc. 135(20), 7744 (2013). 10.1021/ja401960f [DOI] [PubMed] [Google Scholar]
- 32.Lagus T. P. and Edd J. F., J. Phys. D: Appl. Phys. 46(11), 114005 (2013). 10.1088/0022-3727/46/11/114005 [DOI] [Google Scholar]
- 33.Garstecki P., Gitlin I., DiLuzio W., Whitesides G. M., Kumacheva E., and Stone H. A., Appl. Phys. Lett. 85(13), 2649 (2004). 10.1063/1.1796526 [DOI] [Google Scholar]
- 34.Luo R., Venkatraman S. S., and Neu B. R., Biomacromolecules 14(7), 2262 (2013). 10.1021/bm4003915 [DOI] [PubMed] [Google Scholar]
- 35.Reis C. P., Neufeld R. J., Ribeiro A. J., and Veiga F., Nanomed.: Nanotechnol., Biol. Med. 2(1), 8 (2006). 10.1016/j.nano.2005.12.003 [DOI] [Google Scholar]
- 36.See supplementary material Fig. S1 at http://dx.doi.org/10.1063/1.4916230E-BIOMGB-9-001597 for characterization of the correlation between particle sizes and drug release profiles.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- See supplementary material Fig. S1 at http://dx.doi.org/10.1063/1.4916230E-BIOMGB-9-001597 for characterization of the correlation between particle sizes and drug release profiles.