

Abstract
Protein metalloenzymes use various modes for functions where metal–dependent global conformational change is required in some cases, but not in others. In contrast, most ribozymes appear to require a global folding that almost always precedes enzyme reactions. Herein we studied metal–dependent folding and cleavage activity of the 8–17 DNAzyme using single molecule fluorescence resonance energy transfer (FRET). Addition of Zn2+ and Mg2+ resulted in a folding step followed by cleavage reaction, suggesting that the DNAzyme may require metal–dependent global folding for activation. In the presence of Pb2+, however, cleavage reaction occurred without a precedent folding step, suggesting that the DNAzyme may be prearranged to accept Pb2+ for the activity. This feature may contribute to the remarkably fast Pb2+–dependent reaction of the DNAzyme. These results suggest that DNAzymes can use all modes of activation that metalloproteins use.
Metal ion–dependent folding can play a critical role in metalloenzyme function. Understanding the relationship between folding and reaction is important in obtaining deeper insight into the enzyme mechanism. For protein metalloenzymes, an active–site metal–dependent global folding precedes enzymatic reaction in some cases while such a folding is not required in others1. These different modes of activation may fulfill different functions. For example, a reaction preceded by a folding step may be responsible for an allosteric effect in many enzyme functions, or such a folding step may contribute to the overall reaction.
In the early 1980s, RNA molecules that can catalyze enzymatic reactions were discovered and named ribozymes2,3. This discovery was then followed by demonstrations in the 1990s that DNA can also act as enzymes, termed deoxyribozymes or DNAzymes4–7. With only four nucleotides as building blocks versus twenty in proteins, nucleic acid enzymes may need to recruit cofactors to perform some functions. Metal ions are a natural choice and indeed most nucleic acid enzymes require metal ions for function under physiological conditions and therefore, are metalloenzymes. Even though DNAzymes constitute the newest metalloenzyme family, they have already been used in a number of applications such as therapeutic agents8, biosensors9–12, and nanomaterials assembly13, often because DNAzymes are more stable against hydrolysis and more cost–effective to produce than proteins or ribozymes. A primary example is the 8–17 DNAzyme which cleaves a DNA substrate containing one RNA base at the cleavage site (Fig. 1a),8,14–17 and depends on metal ions with the following order of potency: Pb2+ >> Zn2+ >> Cd2+ >> Ca2+ > Mg2+ 15,18,19. Based on this finding, the DNAzyme has been converted into highly sensitive and selective fluorescent9,11, colorimetric10 and electrical sensors20.
Figure 1.

The proposed secondary structure of the 8–17 DNAzyme and its fluorophore attachments for the single molecule FRET studies. (a) The original 8–17 DNAzyme with enzyme (17E) and substrate (17S) strands. For the bulk activity assays described here, the enzyme strand was labeled with a dabcyl (Dy) quencher at the 3′ end, and the substrate strand was labeled with a Alexa fluoro 488 flurophore (Alx) at the 5′ end and a dabcyl quencher at the 3′ end. The arrow indicates a cleavage site. (b) Modified 8–17 DNAzyme and its variants for the single molecule experiments described here. The enzyme strand was extended by 2 nts at the 5′ end and by 2 nts and (dT)5 at the 3′ end, and modified with Cy5 at the 5′ end and a biotin at the 3′ end. The substrate is extended by 2 nts at the 3′ end, and labeled with Cy3 at the 5′ end. For control experiments, two mutant enzymes and two mutant substrates were used. An inactive enzyme has a GC base pair instead of GT wobble pair right next to the cleavage site and a less active 17E(sda5) enzyme has one more T base inserted on the hairpin loop. Two more substrates were used. A non–cleavable substrate has a ribonucleotide A (rA) at the cleavage site instead of a deoxyribonucleotide A, rendering it non–cleavable. Another substrate is extended by 2 more nts at the 5′ end for further thermal stability, and is called long substrate.
Despite the progress made in DNAzyme applications, information about the structure and dynamics of the DNAzyme is surprisingly limited in comparison to those of protein and ribozymes. FRET is one of the most widely used tools for studying the structures and dynamics of biomolecules21,22, including metal ion–dependent folding and catalysis of ribozymes23–26. All studies indicate that global folding is required before enzymatic reaction can occur. Recently, a FRET study of metal–dependent folding of the 8–17 DNAzyme showed no global folding of the DNAzyme in the presence of Pb2+, the most effective cofactor, while the DNAzyme folds into a compact conformation in the presence of Mg2+ and Zn2+ 27. These results left several issues to be resolved. First, to separate folding from subsequent cleavage, the FRET study 27 used an inactive DNAzyme variant where the scissile ribonucleotide A (rA) in the substrate was replaced with deoxyribonucleotide A, which may change metal–binding and thus folding properties. In fact, significant effects of similar modifications on the hairpin ribozyme folding have been reported28. Second, the metal–dependent folding could be heterogeneous, and bulk FRET measurements can detect only average folding behavior and may miss certain conformational changes that are low in population, but important for activity.
To address these issues, it is necessary to observe active cleavable enzymes individually, and in real time along the reaction pathway. FRET at the single molecule level or smFRET is a powerful method to probe dynamic conformational changes and molecular level interactions of biomolecules29,30. For example, smFRET studies of the hairpin ribozyme have revealed heterogeneous folding kinetics and have dissected the cleavage and ligation reactions coupled with folding and unfolding reactions28,31–33. Here we applied smFRET to the study of the conformational changes and cleavage reactions of a DNAzyme. While global folding of the catalytically active 8–17 DNAzyme was observed prior to cleavage with Mg2+ and Zn2+, no global folding was observed with the most active metal ion, Pb2+. No ligation reaction of the cleaved substrates nor dynamic changes between folded and unfolded states were observed, indicating the combined reaction of folding and cleavage of the 8–17 DNAzyme is unidirectional.
DNAzyme constructs
The original 8–17 DNAzyme was labeled with a fluorophore (Alexa fluoro 488) and two quenchers (Dabcyl) for bulk cleavage activity assays (Fig. 1a). To carry out the smFRET studies of the 8–17 DNAzymes on a surface, the substrate and enzyme of the 8–17 DNAzyme were extended to enhance the thermal stability of the complex under conditions of this study and were terminally modified by fluorophores, and biotin (Fig. 1b). The substrate was extended by 2 nucleotides (nts) at the 3′ end. Another substrate was extended by additional 2 nts at the 5′ end, and is called the long substrate. Non–cleavable substrate contains deoxyribonucleotide A instead of rA at the cleavage site. The enzyme strand was extended by 2 nts at both the 5′ end and the 3′ end for base pairing with the extended substrates and had (dT)5–biotin at the 3’ end as a linker. Bulk activity assays of these modified enzyme–substrate complexes indicate that the modifications did not perturb the enzyme activity significantly (Supplementary Fig. 1 online). For the FRET study, the substrates were labeled with Cy3 (donor) at the 5′ end while the enzyme was labeled with Cy5 (acceptor) at the 5′ end. Two mutant enzymes with weakened and abolished activities were also designed. A less active mutant, 17E(sda5), has an extra T base on the hairpin loop16,27. An inactive mutant has GC base pair instead of GT wobble pair right next to the cleavage site, which abolishes the cleavage activity completely19.
Zn2+ and Mg2+–dependent folding and cleavage reactions
Zn2+–dependent activity was observed from individual DNAzymes tethered to a quartz surface using a prism–based total internal reflection (TIR) fluorescence microscope34. The reaction was initiated via flow delivery of an imaging buffer (Materials and Methods) containing 100 µM Zn2+ 21 seconds after starting the fluorescence imaging of more than one hundred molecules. 100 µM Zn2+ is enough to saturate the folding (Supplementary Fig. 2 online). Buffer flow without divalent metal ions showed no FRET changes and no significant photobleaching.
Single molecule time traces of the donor and acceptor intensities, ID and IA respectively, and the corresponding FRET efficiency, EFRET≡ ID/( ID+ IA), shown in Fig. 2a, are typical of the majority molecules (65%, 339 out of 552). Before addition of Zn2+, EFRET was ~ 0.16. Upon adding 100 µM Zn2+, EFRET transited to a higher value of ~ 0.25 immediately, and then to a low value of ~ 0.11, after which the fluorescence signals disappeared. Histograms of smFRET were obtained by measuring EFRET values at each level from 339 individual molecules (Fig. 2b). The unfolding and fluorescence signal disappearing rates were measured from dwell time histograms on the folded and unfolded states, respectively. (Fig. 2d and e) The remaining population includes ~ 10% of molecules showing only a transition to EFRET ~ 0.25 without further change and ~ 25% of molecules without any transition at all in the observation time of 180 seconds. These populations probably represent inactive molecules, consistent with the bulk activity assays (Supplementary Fig. 1 online).
Figure 2.

Zn2+–dependent conformational changes and cleavage reaction. (a) Time traces of donor and acceptor fluorescence signals and FRET changes along the 100 µM Zn2+–dependent reaction. The arrow at 21 s denotes the addition of Zn2+ ions. The activity of the acceptor was checked six times during this data acquisition using excitation at 633 nm. (b) Histograms of the FRET at the three different levels obtained from the individual FRET time traces. (c) Variants’ time traces of donor and acceptor fluorescence signals. Top: a wild type molecule with the long cleavable substrate in the presence of 200 µM Zn2+. Middle: a wild type molecule with the non–cleavable substrate in the presence of 100 µM Zn2+. Bottom: an inactive mutant enzyme with the cleavable substrate in the presence of 100 µM Zn2+. The arrows denote the time of Zn2+ flow. Dwell time histograms of (d) the folded state (EFRET ~ 0.25) and (e) the unfolded state (EFRET ~ 0.11) of the wild type (top) and the 17E(sda5) mutant (bottom) in the presence of 100 µM Zn2+. Data for the wild type and the 17E(sda5) were obtained from three sets and two sets of experiments, respectively. (f) Dwell time histogram of the unfolded state of the wild type molecules with the long substrate in the presence of 200 µM Zn2+. Data was obtained from one set of experiment.
To aid the assignment of different FRET states, we tested a long substrate, the 5’ cleavage product of which should form a more stable complex with the enzyme strand by virtue of two additional base pairs. The long substrate showed similar transition to EFRET ~ 0.25 upon Zn2+ addition and then a subsequent transition to EFRET ~ 0.11 (Fig. 2c). However, in contrast to the short substrate which showed rapid fluorescence disappearance from the EFRET ~ 0.11 state with an average lifetime of 5.5 s (Fig. 2e), very few molecules showed fluorescence disappearance during the measurement time of 70 s (Fig. 2f) Therefore, fluorescence signal disappearance is primarily due to the release of the short 5′ cleavage product carrying the donor rather than donor photobleaching. Next, the same study was carried out using a non–cleavable substrate. We observed the same EFRET ~ 0.25 state induced by the Zn2+ ions but not the subsequent lowering of FRET or fluorescence disappearance (Fig. 2c), supporting our assignment of the EFRET ~ 0.11 state to the cleaved state. In addition, another inactive mutant enzyme, which has GC base pair instead GT wobble pair right next to the cleavage site showed the exactly same behavior (Fig. 2c). Based on the above observations, we assigned the EFRET ~ 0.25 state to a folded state, the subsequent EFRET ~ 0.11 state to a cleaved and unfolded state, and the final fluorescence signal disappearance to the dissociation of the 5’ cleavage product from the enzyme strand (Scheme 1).
Scheme 1.

Proposed reaction pathways of the 8–17 DNAzyme in the presence of Zn2+ or Mg2+ and Pb2+.
Mg2+ ion–dependent FRET changes were also studied. Since Mg2+ is a less effective cofactor in both folding and cleavage19, 10 mM or 50 mM Mg2+, by which complete folding was observed in bulk FRET measurements (Supplementary Fig. 2 online), were used. Upon addition of 10 mM Mg2+, molecules showed immediate folding (EFRET = 0.17 → 0.29), cleavage and unfolding (EFRET = 0.29 → 0.12), and then release of the cleavage product (Fig. 3a and b), similar to those observed with Zn2+ ions. The same behavior was observed with 50 mM Mg2+ except that 50 mM Mg2+ showed a shorter duration of the folded state and a longer duration of the cleaved and unfolded state (Fig. 3c and d). These results are consistent with the bulk activity assays which showed that a higher Mg2+ concentration induces faster cleavage reaction19, yet slower release of the cleaved product due to the stabilized base pairs in a higher ionic strength condition. These observations further support our assignments of the different FRET levels.
Figure 3.
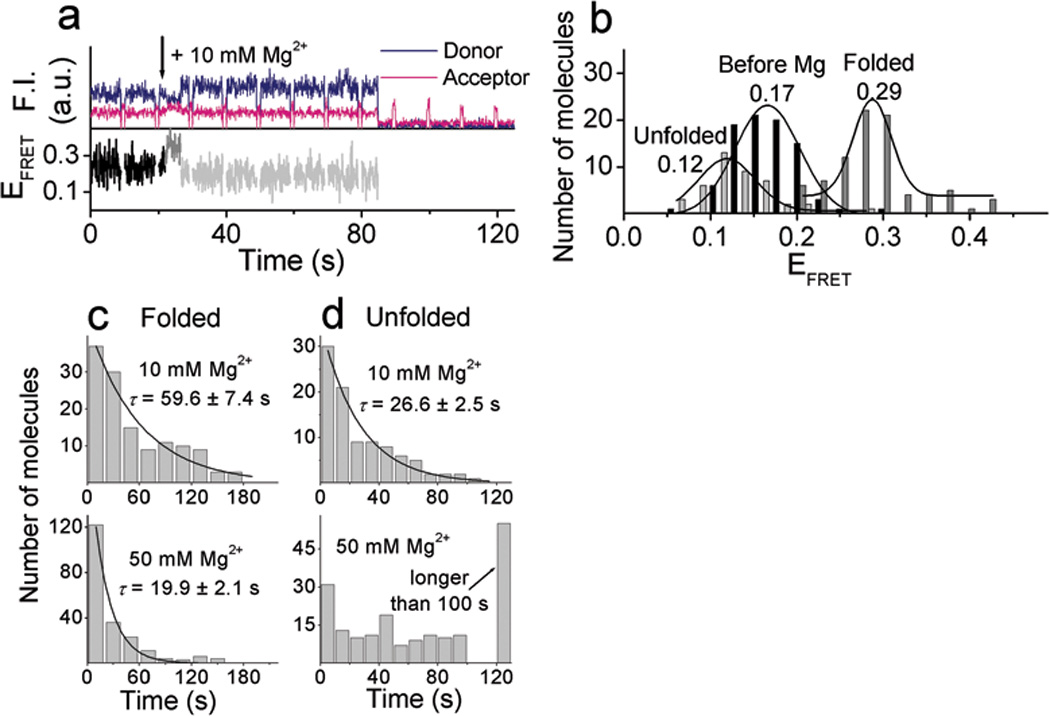
Mg2+–dependent conformational changes and cleavage reaction. (a) Time traces of donor and acceptor fluorescence signals and FRET changes along the 10 mM Mg2+–dependent reaction with the cleavable substrate. (b) Histograms of FRET at the three different levels obtained from the individual FRET time traces in the presence of 10 mM Mg2+. Dwell time histograms of (c) folded state (EFRET ~ 0.29) and (d) unfolded state (EFRET ~ 0.12) in the presence of 10 mM Mg2+ (top) and 50 mM Mg2+ (bottom). Histograms were fitted to a single exponential decay curve. Data for 10 mM and 50 mM Mg2+ were obtained from three and two sets of experiments, respectively.
Pb2+–dependent cleavage reaction
Pb2+–dependent FRET changes along the reaction pathway were measured in the same way in the presence of 20 µM of Pb2+ (Fig. 4a). EFRET started at ~ 0.18. In contrast to what observed with Zn2+ or Mg2+, however, EFRET stayed at the same level upon Pb2+ addition at 21 seconds. Lowering of FRET to EFRET ~ 0.10 was observed later, followed by fluorescence signal disappearance. Histograms of smFRET before and after the drop in EFRET are shown in Fig. 4b. The average time from the Pb2+ addition until the transition to the EFRET ~ 0.10 state is very similar for the short (18.7 s) and long (15.7 s) substrates (Fig. 4c). The dwell time at the EFRET ~ 0.10 state was much longer for the long substrate (Fig. 4d). In addition, reactions with the non–cleavable substrate and with the inactive enzyme did not show the low FRET state nor fluorescence signal disappearance (Supplementary Fig. 3 online). Based on these observations, we assigned the low FRET state to the cleaved state and the disappearance of the donor signal to the release of the 5′ cleavage product (Scheme 1).
Figure 4.

Pb2+–dependent conformational changes and cleavage reaction. (a) Time traces of the fluorescence signals and FRET changes upon injection of 20 µM Pb2+ at 21 s with the cleavable substrate. (b) FRET histograms obtained by measuring the FRET values at the two different states. (c) and (d). Comparison between the cleavable substrate (top) and the long cleavable substrate (bottom) with the wild type enzyme. Dwell time histograms of (c) the Pb2+ injection to FRET change to EFRET ~ 0.10 state and (d) of the unfolded state (EFRET ~ 0.10). The glucose oxidase concentration was 33 U ml−1. Data were obtained from one each set of experiment. (e) and (f) Comparison between the wild type (top) and 17E(sda5) mutant (bottom). Dwell time histograms of (e) the Pb2+ injection to FRET change to EFRET ~ 0.10 state and (f) of the unfolded state (EFRET ~ 0.10). The glucose oxidase concentration was 16 U ml−1. Data for the wild type and the 17E(sda5) were obtained from two and one sets of experiments, respectively. Average reaction times, τ, were measured by fitting the histograms to a single exponential curve.
Studies on a variant with less activity
We next studied a less active 17E(sda5) mutant. A reduced glucose oxidase concentration (16 U ml−1) was used since the glucose oxidase was more detrimental to the 17E(sda5) mutant activity. The 17E(sda5) showed immediate folding upon addition of 100 µM Zn2+, followed by unfolding and disappearing fluorescence signal (Supplementary Fig. 4 online). However, no FRET change was observed in the presence of 20 µM Pb2+, and only later unfolding was observed (Supplementary Fig. 4 online). These observations are highly similar to those with the wild type enzyme except for the difference in the reaction rates. The mutant reaction in the presence of 100 µM Zn2+ was 6.4 fold slower than the wild type reaction (τ = 146 s vs. 22.8 s) (Fig. 2d) and was not affected by reduction in glucose concentration. For the case of 20 µM Pb2+, the two–fold reduction of glucose oxidase concentration reduced the reaction time of the wild type enzyme from τ = 18.7 s to 8.40 s and the mutant had 7 fold slower reaction (τ = 60.3 s) compared to the wild type (Fig. 4c and e). These results are comparable to the bulk activity assay results showing 5.2 fold and 9 fold lower activity for the mutant at 100 µM Zn2+ and 20 µM Pb2+, respectively (Supplementary Fig. 1 online). No difference on the product release rate was observed (Fig. 2e and 4f). These results indicate that the 17E(sda5) shares the same reaction pathway as the wild type.
Pb2+–dependent activity of the folded DNAzyme
The above studies raise an interesting question of whether the Zn2+ or Mg2+–induced folded state of the DNAzyme is an active conformation for the cleavage activity or is rather an off–pathway trapped state with a higher energy barrier toward a cleavage reaction. Therefore, Pb2+–dependent reaction of the wild type DNAzyme was carried out in the presence of Mg2+. We chose 10 mM Mg2+ at which the folding is saturated (Kd = 0.7 mM, see Supplementary Fig. 2 online), but activity is much less in comparison to that of 20 µM Pb2+ (τ 10mM Mg2+ = 59.6 s vs. τ 20 µM Pb2+ = 8.4 s, see Fig. 3c and 4e). Upon flow of a mixture of 20 µM Pb2+ and 10 mM Mg2+, identical FRET changes were observed as those when only Mg2+ was present (Supplementary Fig. 5 online). The cleavage and unfolding reaction was τ = 15.2 s (Supplementary Fig. 5 online) which was two–fold slower than that in the presence of 20 µM Pb2+ only (τ = 8.4 s, Fig. 4e)), but about four–fold faster than that in the presence of 10 mM Mg2+ only (τ = 59.6 s, Fig. 3c)).
Comparisons with the bulk assay results
Finally, we compared the reaction rates between the single molecule fluorescence measurements of immobilized molecules and bulk solution activity measurements of molecules without fluorescent modifications and found good agreements (Supplementary Fig. 6 online).
In this report, the folding and cleavage catalyzed by a DNAzyme has been monitored at the single molecule level for the first time. An initial FRET increase followed by a FRET decrease upon addition of Zn2+ or Mg2+ were assigned to the initial metal–dependent folding followed by the cleavage–induced unfolding through a series of control experiments (Scheme 1). Interestingly, we did not detect any dynamic folding and unfolding transitions once folding occurs or any re–ligation reaction after cleavage in contrast to the hairpin ribozyme which underwent several cycles of folding and unfolding events after cleavage until re-ligation28. However, since 100 µM concentration of Zn2+ used in this study is only two to five–fold higher than the apparent Kd of 18.9 µM for folding of the wild type (Supplementary Fig. 2 online) and 52.6 µM the slow mutant 17E(sda5)27, it remains possible that the DNAzyme is in dynamic equilibrium between the folded and unfolded states on a faster time scale than our time resolution of 100 ms. Fluorescence anisotropy measurements of terminally attached fluorophores suggested that the 3′ product stays bound on the enzyme strand after cleavage (unpublished observation). Thus, we can exclude the possibility that the absence of re–folding and ligation events after cleavage–induced unfolding is due to the fast product release. It is most likely that once the enzyme unfolds after cleavage, it can no longer obtain the active conformation required for catalytic reaction due to the strain relief, rendering the combined reaction of folding and cleavage for the 8–17 DNAzyme essentially unidirectional.
In contrast to single molecule studies of ribozymes and even the same DNAzyme with Mg2+ and Zn2+, we could not detect any evidence of global folding of the 8–17 DNAzyme with Pb2+ ion, the most active metal ion27. It is technically possible that Pb2+–dependent cleavage from the folded state is much faster than our time resolution (100 ms) so that the folded state goes undetected. However, this would also mean that folding itself is very slow, taking 15–18 seconds, which we suggest is less likely because folding was faster than our time resolution with less optimal ions such as Zn2+ and Mg2+. Furthermore, the less active 17E(sda5) mutant did not show any difference from the wild type regarding the pattern of FRET changes. Therefore, we suggest that the 8–17 DNAzyme carries out catalysis along a different reaction pathway in the presence of Pb2+ from that in the presence of Zn2+ or Mg2+ (Scheme 1).
Interestingly, apparent folding was observed upon simultaneous addition of both Pb2+ and Mg2+ with the reaction rate much closer to that of Pb2+. Thus, the Pb2+–dependent reaction did occur from the Mg2+–induced folded state. The two–fold reduction in the reaction rate compared to that of Pb2+ only may arise due to competitive binding of Mg2+ to a Pb2+ binding site or due to the Mg2+–induced distortion of the conformations most active for Pb2+–dependent reaction. These results imply that there are multiple reaction pathways depending on the metal cofactors, similar to the proposals made for the hammerhead ribozyme35. The 8–17 DNAzyme in the presence of Zn2+ or Mg2+ ions behave similarly to many proteins and ribozymes in that it requires a metal–dependent global folding step before it can be functional. However, the DNAzyme may be prearranged to accept Pb2+ and may not require global conformational changes for the catalytic reaction. A similar feature has been reported for the GlmS ribozyme36,37, whose ligand binding pocket is pre–organized for the ligand to participate in the chemistry directly. Our results imply that DNAzymes can also use all modes of activation that other metalloprotein enzymes and some ribozymes use.
In conclusion, divalent metal–dependent folding, cleavage, and release of the product of a DNAzyme were probed at the single molecule, providing a unique opportunity to dissect the reaction pathways and to determine the rates of both cleavage reaction and product release unambiguously. Our study suggests that the catalytically active DNAzyme does not undergo global folding before cleavage if Pb2+ is used as the cofactor. The different reaction pathways between Pb2+–dependent and Zn2+– and Mg2+–dependent reactions may contribute to the high activity of the Pb2+–dependent 8–17 DNAzyme, which is among the fastest nucleic acid enzymes.
Methods
Bulk activity assays
HPLC–purified oligonucleotides were purchased from Integrated DNA Technology Inc. The enzyme–substrate complex (Fig. 1a) with excess enzyme (3 µM substrate + 5 µM enzyme) was annealed in 50 mM Tris (USB Corporation)–HCl (Alfa Aesar) (pH 7.3) and 50 mM NaCl (Alfa Aesar) by immersing the samples in 85 °C water bath, then cooling down to 4 °C over 3 hours. The reaction was initiated by adding 10 times concentrated metal ion solutions into the DNA solution at room temperature and the reaction was quenched at various time points by adding an aliquot of the reaction mixture into a stop-buffer containing 50 mM EDTA (Fisher Scientific) and 8 M urea (USB Corporation). The reaction products and the uncleaved substrates were separated by electrophoresis on a 20% denaturing polyacrylamide gel and analyzed with a fluorescence imager (FLA-3000, Fujifilm). The fraction of the cleaved product was plotted against time and observed rate constant (kobs) was obtained by nonlinear curve fitting using SigmaPlot 6.0 based on the equation, y = y0 + a(1-e−kt), where y is the cleaved fraction at time t, y0 is the background cleavage at t = 0, a is the fraction reacted at t = ∞, and k is the observed rate constant, kobs.
smFRET study
Single molecule fluorescence measurements were performed using a wide–field prism–type TIR fluorescence microscope system. Molecules were excited by TIR of a 532 nm laser (and a 633 nm laser for alternate excitation) and fluorescence was measured with 100 ms time resolution. Annealed biotin modified enzyme–substrate complex (1 µM Enzyme + 2 µM substrate) was diluted and immobilized on a BSA–biotin–Neutravidin (Pierce) coated quartz slide via Neutravidin–biotin interactions38. The DNA concentration was adjusted until a sufficient number of molecules were observed (usually 100–200 pM).
We characterized the DNAzyme activity in the presence of individual components required for prolonging photobleaching lifetimes of fluorophores. We found that the commonly used β–mercaptoethanol chelates Zn2+, inhibiting folding of the DNAzyme, and therefore used Trolox (Sigma) instead. Trolox did not chelate Zn2+ nor interfere with the DNAzyme activity, and turned out to eliminate acceptor blinking and to enhance fluorophores’ photostability39. In addition, the standard concentration of glucose oxidase (165 U ml−1, Sigma) decreased the Pb2+–dependent activities (Supplementary Fig. 1 online). Thus, glucose oxidase concentrations of 33 U ml−1 and 16 U ml−1 were used for the wild type and the mutant enzymes, respectively, as a compromise between slowing photobleaching and maintaining the cleavage activity. No other reagents affected the activity of the enzyme (Supplementary Fig. 1 Online).
Single molecule fluorescence measurements were performed in an imaging buffer of 50 mM Tris–HCl (pH 7.3), 50 mM NaCl in saturated (~ 2 mM) Trolox (Sigma) with an oxygen scavenging system (0.4 % β–D–glucose (Sigma), 16 U ml−1 or 33 U ml−1 glucose oxidase (Sigma), and 2170 U ml−1 catalase (Roche)). Reactions were initiated by injecting 80 µL of metal ion solution at a flow rate of 0.5 ml min−1 using a flow system coupled to a syringe pump during fluorescence measurement. In order to distinguish EFRET decrease due to cleavage from acceptor photobleaching, the acceptor’s presence was checked with a briefly turned on red laser (633 nm). For data analysis, only molecules with functional acceptors at the end of the reaction were included. Cleavage and product release rates were determined by fitting the dwell time histograms to a single exponential decay curve. EFRET is approximated by the acceptor intensity divided by the sum of the donor and acceptor intensities after correcting for background and cross–talks between the two detection channels (~ 15%). Data were analyzed with Matlab 7.0 student version and Microcal Origin 6.0.
Supplementary Material
Acknowledgement
We would like to thank Chirlmin Joo and Rahul Roy for generous help with performing experiments and data analysis and Jung Heon Lee for help with drawing the graphical abstract. This material is based upon work supported by the U.S. Department of Energy (DEFG02–01–ER63179), the National Institutes of Health (GM065367), and the National Science Foundation (CTS–0120978 and DMI- 0328162). T.H. is a Howard Hughes Medical Institute Investigator.
Footnotes
Author Contributions
H.K. designed and carried out all the experiments and wrote the manuscript; I.R. designed and carried out single molecule FRET experiments; J.L. designed single molecule FRET experiments; T.H. designed single molecule FRET experiments and wrote the manuscript; Y.L. designed all the experiments and wrote the manuscript.
Competing Financial Interests Statement
The authors declare there are no competing financial interests.
References
- 1.Bren KL, Pecoraro VL, Gray HB. Metalloprotein Folding. Inorg. Chem. 2004;43:7894–7896. doi: 10.1021/ic040121q. [DOI] [PubMed] [Google Scholar]
- 2.Kruger K, et al. Self-splicing RNA: autoexcision and autocyclization of the ribosomal RNA intervening sequence of Tetrahymena. Cell. 1982;31:147–157. doi: 10.1016/0092-8674(82)90414-7. [DOI] [PubMed] [Google Scholar]
- 3.Guerrier-Takada C, Gardiner K, Marsh T, Pace N, Altman S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell. 1983;35:849–857. doi: 10.1016/0092-8674(83)90117-4. [DOI] [PubMed] [Google Scholar]
- 4.Breaker RR, Joyce GF. A DNA enzyme that cleaves RNA. Chem. Biol. 1994;1:223–229. doi: 10.1016/1074-5521(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 5.Sen D, Geyer CR. DNA enzymes. Curr. Opin. Chem. Biol. 1998;2:680–687. doi: 10.1016/s1367-5931(98)80103-8. [DOI] [PubMed] [Google Scholar]
- 6.Li Y, Breaker RR. Deoxyribozymes: new players in the ancient game of biocatalysis. Curr. Opin. Struct. Biol. 1999;9:315–323. doi: 10.1016/S0959-440X(99)80042-6. [DOI] [PubMed] [Google Scholar]
- 7.Lu Y. New Transition Metal-Dependent DNAzymes as Efficient Endonucleases and as Selective Metal Biosensors. Chem. Eur. J. 2002;8:4588–4596. doi: 10.1002/1521-3765(20021018)8:20<4588::AID-CHEM4588>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 8.Santoro SW, Joyce GF. A general purpose RNA-cleaving DNA enzyme. Proc. Natl. Acad. Sci. USA. 1997;94:4262–4266. doi: 10.1073/pnas.94.9.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J, Lu Y. A highly sensitive and selective catalytic DNA biosensor for lead ions. J. Am. Chem. Soc. 2000;122:10466–10467. [Google Scholar]
- 10.Liu J, Lu Y. A Colorimetric Lead Biosensor Using DNAzyme-Directed Assembly of Gold Nanoparticles. J. Am. Chem. Soc. 2003;125:6642–6643. doi: 10.1021/ja034775u. [DOI] [PubMed] [Google Scholar]
- 11.Liu J, Lu Y. Improving Fluorescent DNAzyme Biosensors by Combining Inter- and Intramolecular Quenchers. Anal. Chem. 2003;75:6666–6672. doi: 10.1021/ac034924r. [DOI] [PubMed] [Google Scholar]
- 12.Lu Y, Liu J. Functional DNA nanotechnology: emerging applications of DNAzymes and aptamers. Curr. Opin. Biotechnol. 2006;17:580–588. doi: 10.1016/j.copbio.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 13.Lu Y, Liu J. Smart Nanomaterials Inspired by Biology: Dynamic Assembly of Error-Free Nanomaterials in Response to Multiple Chemical and Biological Stimuli. Inorg.Chem. 2007;40:315–323. doi: 10.1021/ar600053g. [DOI] [PubMed] [Google Scholar]
- 14.Faulhammer D, Famulok M. The Ca2+ ion as a cofactor for a novel RNA-cleaving deoxyribozyme. Angew. Chem. Int. Ed. 1996;35:2837–2841. [Google Scholar]
- 15.Li J, Zheng W, Kwon AH, Lu Y. In vitro selection and characterization of a highly efficient Zn(II)-dependent RNA-cleaving deoxyribozyme. Nucleic Acids Res. 2000;28:481–488. doi: 10.1093/nar/28.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cruz RPG, Withers JB, Li Y. Dinucleotide Junction Cleavage Versatility of 8–17 Deoxyribozyme. Chem. Biol. 2004;11:57–67. doi: 10.1016/j.chembiol.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 17.Schlosser K, Li Y. Tracing Sequence Diversity Change of RNA-Cleaving Deoxyribozymes under Increasing Selection Pressure during in Vitro Selection. Biochemistry. 2004;43:9695–9707. doi: 10.1021/bi049757j. [DOI] [PubMed] [Google Scholar]
- 18.Peracchi A. Preferential activation of the 8–17 deoxyribozyme by Ca2+ ions. Evidence for the identity of 8–17 with the catalytic domain of the MG5 deoxyribozyme. J. Biol. Chem. 2000;275:11693–11697. doi: 10.1074/jbc.275.16.11693. [DOI] [PubMed] [Google Scholar]
- 19.Brown AK, Li J, Pavot CMB, Lu Y. A Lead-Dependent DNAzyme with a Two-Step Mechanism. Biochemistry. 2003;42:7152–7161. doi: 10.1021/bi027332w. [DOI] [PubMed] [Google Scholar]
- 20.Xiao Y, Rowe AA, Plaxco KW. Electrochemical Detection of Parts-Per-Billion Lead via an Electrode-Bound DNAzyme Assembly. J. Am. Chem. Soc. 2007;129:262–263. doi: 10.1021/ja067278x. [DOI] [PubMed] [Google Scholar]
- 21.Clegg RM. Fluorescence resonance energy transfer and nucleic acids. Meth. Enzymol. 1992;211:353–388. doi: 10.1016/0076-6879(92)11020-j. [DOI] [PubMed] [Google Scholar]
- 22.Lilley DMJ, Wilson TJ. Fluorescence resonance energy transfer as a structural tool for nucleic acids. Curr. Opin. Chem. Biol. 2000;4:507–517. doi: 10.1016/s1367-5931(00)00124-1. [DOI] [PubMed] [Google Scholar]
- 23.Bassi GS, Murchie AIH, Walter F, Clegg RM, Lilley DMJ. Ion-induced folding of the hammerhead ribozyme: a fluorescence resonance energy transfer study. EMBO J. 1997;16:7481–7489. doi: 10.1093/emboj/16.24.7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walter NG, Burke JM, Millar DP. Stability of hairpin ribozyme tertiary structure is governed by the interdomain junction. Nat. Struct. Biol. 1999;6:544–549. doi: 10.1038/9316. [DOI] [PubMed] [Google Scholar]
- 25.Fang X-w, Pan T, Sosnick TR. Mg2+-dependent folding of a large ribozyme without kinetic traps. Nat. Struct. Biol. 1999;6:1091–1095. doi: 10.1038/70016. [DOI] [PubMed] [Google Scholar]
- 26.Wilson TJ, Lilley DMJ. Metal ion binding and the folding of the hairpin ribozyme. RNA. 2002;8:587–600. doi: 10.1017/s1355838202020514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim H-K, et al. Metal-Dependent Global Folding and Activity of the 8–17 DNAzyme Studied by Fluorescence Resonance Energy Transfer. J. Am. Chem. Soc. 2007;129:6896–6902. doi: 10.1021/ja0712625. [DOI] [PubMed] [Google Scholar]
- 28.Nahas MK, et al. Observation of internal cleavage and ligation reactions of a ribozyme. Nat. Struct. Mol. Biol. 2004;11:1107–1113. doi: 10.1038/nsmb842. [DOI] [PubMed] [Google Scholar]
- 29.Ha T, et al. Probing the interaction between two single molecules: fluorescence resonance energy transfer between a single donor and a single acceptor. Proc. Natl. Acad. Sci. USA. 1996;93:6264–6268. doi: 10.1073/pnas.93.13.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Myong S, Stevens BC, Ha T. Bridging Conformational Dynamics and Function Using Single-Molecule Spectroscopy. Structure. 2006;14:633–643. doi: 10.1016/j.str.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 31.Zhuang X, et al. Correlating structural dynamics and function in single ribozyme molecules. Science. 2002;296:1473–1476. doi: 10.1126/science.1069013. [DOI] [PubMed] [Google Scholar]
- 32.Tan E, et al. A four-way junction accelerates hairpin ribozyme folding via a discrete intermediate. Proc. Natl. Acad. Sci. USA. 2003;100:9308–9313. doi: 10.1073/pnas.1233536100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bokinsky G, et al. Single-molecule transition-state analysis of RNA folding. Proc. Natl. Acad. Sci. USA. 2003;100:9302–9307. doi: 10.1073/pnas.1133280100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhuang X, et al. A single-molecule study of RNA catalysis and folding. Science. 2000;288:2048–2051. doi: 10.1126/science.288.5473.2048. [DOI] [PubMed] [Google Scholar]
- 35.Roychowdhury-Saha M, Burke DH. Distinct reaction pathway promoted by non-divalent-metal cations in a tertiary stabilized hammerhead ribozyme. RNA. 2007;13:841–848. doi: 10.1261/rna.339207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hampel KJ, Tinsley MM. Evidence for Preorganization of the glmS Ribozyme Ligand Binding Pocket. Biochemistry. 2006;45:7861–7871. doi: 10.1021/bi060337z. [DOI] [PubMed] [Google Scholar]
- 37.Cochrane JC, Lipchock SV, Strobel SA. Structural Investigation of the GlmS Ribozyme Bound to Its Catalytic Cofactor. Chem. Biol. 2007;14:97–105. doi: 10.1016/j.chembiol.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hohng S, et al. Conformational Flexibility of Four-way Junctions in RNA. J. Mol. Biol. 2004;336:69–79. doi: 10.1016/j.jmb.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 39.Rasnik I, McKinney SA, Ha T. Nonblinking and long-lasting single-molecule fluorescence imaging. Nature Methods. 2006;3:891–893. doi: 10.1038/nmeth934. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.