

Abstract
Sphingolipids are a diverse class of signaling molecules implicated in many important aspects of cellular biology, including growth, differentiation, apoptosis, and autophagy. Autophagy and apoptosis are fundamental physiological processes essential for the maintenance of cellular and tissue homeostasis. There is great interest into the investigation of sphingolipids and their roles in regulating these key physiological processes as well as the manifestation of several disease states. With what is known to date, the entire scope of sphingolipid signaling is too broad, and a single review would hardly scratch the surface. Therefore, this review attempts to highlight the significance of sphingolipids in determining cell fate (e.g. apoptosis, autophagy, cell survival) in the context of the healthy lung, as well as various respiratory diseases including acute lung injury, acute respiratory distress syndrome, bronchopulmonary dysplasia, asthma, chronic obstructive pulmonary disease, emphysema, and cystic fibrosis. We present an overview of the latest findings related to sphingolipids and their metabolites, provide a short introduction to autophagy and apoptosis, and then briefly highlight the regulatory roles of sphingolipid metabolites in switching between cell survival and cell death. Finally, we describe functions of sphingolipids in autophagy and apoptosis in lung homeostasis, especially in the context of the aforementioned diseases.
Keywords: Ceramide, Sphingosine, Apoptosis, Caspase, Autophagy, Necroptosis, Lung development, Lung diseases
Introduction
Sphingolipids are a class of lipids that were named after the mythological Sphinx because of their enigmatic nature. They were initially thought to serve strictly as structural components of the membrane bilayer, but have now been implicated in various cell-signaling pathways including cell proliferation, differentiation, and programmed cell death (PCD) [1]. These molecules display amphiphatic properties in which the hydrophobic end is comprised of a sphingoid base that is linked to a fatty acid, while the hydrophilic end varies in structures consisting of hydroxyl groups, phosphates and sugar residues. Different lengths, saturations, and hydroxylations of fatty acids, as well as different head groups result in an immense diversity of the sphingolipids species [2]. All sphingolipids can be generated from a derivation of ceramide. For example, as it is shown in Fig. 1, ceramide can be deacylated by ceramidase to sphingosine that then can be phosphorylated by a sphingosine kinase isoenzyme (SphK1 or SphK2) to form sphingosine-1-phosphate (S1P) [3, 4]. Alternatively, sphingosine can be acylated by ceramide synthase to give rise to ceramide. Both ceramide and sphingosine can act as a second messenger to promote apoptosis, cellular senescence, and growth arrest [5]. Sphingomyelin synthase converts ceramide to sphingomyelin, a structural lipid mainly localized to the outer membrane leaflet, while sphingomyelin can give rise to ceramide by the action of sphingomyelinase (SMase) isoenzymes [6]. Ceramide and S1P have received attention as they appear to play opposing roles in a dynamic relationship known as the “sphingolipid rheostat” [1, 7, 8]. At one end of the scale, ceramide is typically recognized to initiate apoptosis and growth arrest, whereas S1P, at the other end, promotes cell proliferation, survival, mobility, and cell-to-cell adhesion [9–11]. These two sphingolipids have been shown to play a role in cell fate processes such as apoptosis, and more recently, autophagy. A general overview of sphingolipid metabolism and their major functions is shown in Fig. 1. The role of sphingolipids in lung cell fate will be explored in this review.
Fig. 1.
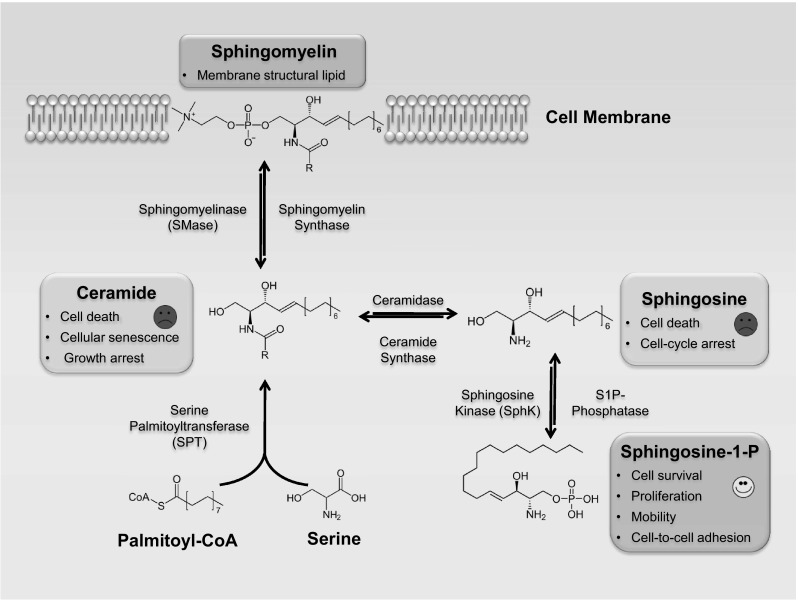
Overview of sphingolipid metabolism and their major functions. Refer to text for further details. CoA coenzyme A, S1P sphingosine-1-phosphate, SMase sphingomyelinase, SphK sphingosine kinase, SPT serine palmitoyltransferase
Ceramide
Ceramide can be generated through three known pathways. De novo synthesis of ceramide is characterized by the rate-limiting step of condensation between serine and palmitoyl-CoA catalyzed by serine palmitoyltransferase (SPT) [12]. Any of the sphingomyelinase isoenzymes, acid sphingomyelinase (aSMase), neutral sphingomyelinase (nSMase), and alkaline sphingomyelinase can use sphingomyelin as a substrate to produce ceramide [13]. Finally, synthesis of ceramide through a recycling loop from sphingosine and glycosphingolipids can also occur by the reverse activity of ceramidase [14].
Ceramide is a well-known critical mediator of various cell death pathways, including apoptosis and necrosis [15, 16]. Increased ceramide levels have been associated with apoptotic cell death in both homeostatic systems as well as pathological settings as a result of cellular insults including oxidative stress, chemotherapeutic agents, ischemia and radiation [5, 17–20]. Studies investigating the mechanism of ceramide-mediated apoptosis have demonstrated that ceramide can act on both the intrinsic (mitochondrial) and extrinsic pathways of apoptosis in a context-dependent manner [21, 22]. Moreover, ceramide is able to induce apoptosis by recruitment of death receptors to lipid rafts and assembly of channels in the outer membrane of the mitochondria promoting the release of cytochrome c, only to mention a few amongst several investigated pathways [22–24]. It has more recently been shown that ceramide has a significant impact on autophagy, influencing cellular fate under stress conditions such as amino acid deprivation, mitochondrial damage, and ER stress [25–30].
Sphingosine-1-phosphate
S1P is well recognized to play critical roles in not only cell proliferation and survival, but also in cell mobility and chemotaxis, cell-to-cell adhesion, angiogenesis, intracellular calcium homeostasis, and cytoskeletal organization [9, 31–33]. Synthesis of S1P can occur by the hydrolysis of sphingolipids from the plasma membrane into ceramide and subsequent N-deacetylation of ceramide to form sphingosine that can be phosphorylated by SphK1 or SphK2 [3, 4] to generate S1P. Signaling actions of S1P can be executed within the intracellular space, as well as the extracellular space when it acts as a ligand of five G-protein coupled receptors (S1P1–S1P5) [34, 35]. These receptors allow S1P to influence signaling processes including angiogenesis, heart development, immunity, and cell movement depending on the receptor that is ligated, cell type, and the cellular context [3]. S1P is currently understood to promote cell survival by inhibition of enzymes involved in ceramide synthesis, as well as activation of the nuclear factor-κB (NF-κB) signaling pathway [36–39]. Furthermore, S1P has also been recently suggested to induce autophagy accordingly with its protective role against apoptosis [7, 40, 41].
The broad effects of second messengers generated through metabolism of sphingolipids (ceramide, S1P) on regulation of cell fate as well as the plasticity of sphingolipid metabolism suggests a variety of possible mechanisms for controlling cell fate. Further study will be required to elucidate the underlying mechanisms regulating cell survival or death decisions by these family members.
Determination of cell fate
Apoptosis
PCD is an essential physiological process involved in development, aging, and tissue homeostasis which maintains normal cellular fate in different organisms [42]. Apoptosis (PCD1) is a widely recognized mode of PCD in which complex molecular signaling systems trigger an orderly, energy-dependent enzymatic breakdown of DNA, lipids, and other macromolecules [43]. Cells undergoing apoptosis show typical, well-defined morphological changes characterized as rounding of the cell, shrinkage of pseudopods, decreased cellular volume, chromatin condensation (pyknosis), nuclear fragmentation (karyorrhexis) along with little or no ultrastructural reformations of organelles in the cytoplasm followed by plasma membrane blebbing, and ingestion by phagocytes [43, 44].
In contrast to necrosis, apoptosis does not induce inflammation since apoptotic cells do not release their cellular contents into the surrounding interstitial tissue and rather are quickly engulfed by macrophages or adjacent normal cells [45, 46]. The mechanism of apoptosis is highly synchronized and is coordinated by an extensively studied group of cysteine proteases known as cysteine aspartate-specific proteases (caspases). Caspases are widely expressed in an inactive proenzyme form (procaspases), localized in the nucleus, cytoplasm, endoplasmic reticulum (ER), the mitochondrial intermediate space, and can be translocated to the plasma membrane [43]. Caspases have proteolytic activity and are able to cleave proteins at an internal aspartic acid site, although different caspases have specificities involving recognition of neighboring amino acids [47, 48]. According to their order of activation, caspases are classified into two groups: (1) the initiator caspases (i.e. caspase-2, -8, -9, and -10), (2) and effector caspases (i.e. caspase-3, -6, and -7) [49, 50]. Once activated, caspases can often activate other procaspases, allowing initiation of a protease cascade that leads to an irreversible commitment towards cell death.
There are at least two major pathways in mammals that are involved in the initiation of apoptosis, namely the extrinsic pathway and the intrinsic pathway (Fig. 2). Initiator caspases are activated upon extrinsic or intrinsic stimuli that lead to the activation of executioner caspases [49, 50]. Extrinsic and intrinsic pathways differ in their induction and regulation, although there is now evidence that the two pathways are linked and intersect at different stages where molecules in one pathway can influence the other [51].
Fig. 2.
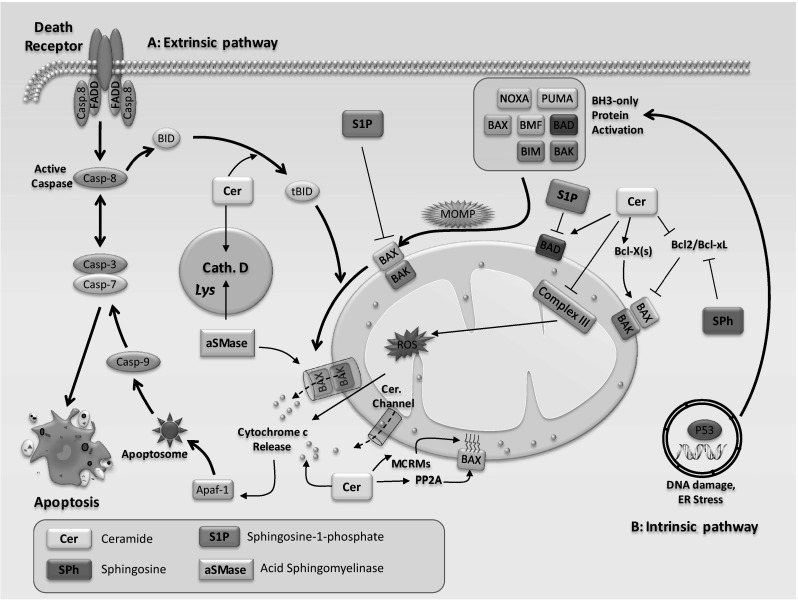
Schematic representation of key events in the apoptotic pathway and regulation of apoptosis by sphingolipids. There are two main apoptotic pathways. A Extrinsic pathway is triggered when cell death ligands (e.g., FasL, APO-2L, TRAIL, TNF) bind to their respective death-receptors (e.g., Fas, DR4, DR5, TNF-R1) and initiates pro-caspase-8 activation by recruiting FADD. Activation of caspase-8 results in cleavage of effector caspases, such as caspase-3,-6,-7, which are involved in the core apoptosis pathway. Furthermore, caspase-8 can truncate BID (tBID), which later induces the intrinsic pathway. B The intrinsic pathway can be directly initiated by a variety of stress signals. Stress signals initiate DNA damage and p53 phosphorylation, which leads to the up-regulation of BH3 only proteins and consequently results in mitochondrial translocation and oligomerization of BAX/BAK, followed by MOMP. Mitochondrial damage leads to cytochrome c release into the cytoplasm. Cytosolic cytochrome c binds to the pro-apoptotic factor Apaf-1 (in the presence of dATP) to form an apoptosome. Apoptosomes then activate caspase-9, which later leads to the activation of caspases-3 and-7, and subsequently to nuclear fragmentation and also chromatin condensation. Sphingolipids have been shown to modulate apoptosis at multiple steps of the process. Sphingolipids may directly affect mitochondria, a strategic center in the control of apoptosis. Ceramide forms channels in mitochondrial outer membranes and promotes the release of cytochrome c for caspase-9 activation. Ceramide channel formation has also shown to be inhibited by dihydroceramide. Furthermore, ceramide generates reactive oxygen species (ROS) via inhibition of mitochondrial complex III. The apoptotic action of ceramide could also be mediated by the recruitment and activation of pro-apoptotic Bax at the mitochondria through the PP2A-dependent dephosphorylation of Bax and formation of mitochondrial ceramide-rich macrodomains (MCRMs). aSMase-released ceramide binds directly to lysosomal protease cathepsin D, leading to cathepsin D activation, resulting in cleavage of the BH3-only protein BID and induction of the mitochondrial pathway of apoptosis. Sphingosine has been shown to downregulate expression of anti-apoptotic proteins, Bcl-2 and Bcl-xL, to enhance apoptosis. While ceramide-mediated activation of pro-apoptotic protein, BAD, promotes apoptosis, S1P suppresses apoptosis via BAD inactivation
The extrinsic pathway, also commonly referred to as the death receptor pathway, is initiated through the ligation of death receptors (Fas, DR4, DR5, TNF-R1) by their specific ligands (e.g., FasL, APO-2L, TRAIL, and TNF) [52]. Interaction of a death ligand to its corresponding receptor leads to activation of initiator caspase-8. Active caspase-8 can affect the mitochondria via truncated BID (tBID) and causes mitochondrial initiator caspase (caspase-9) activation [43, 53]. All of these events culminate to effector caspase activation (caspase-3, -7, -6) [52], resulting in cleavage of different substrates like cytokeratins, PARP, plasma membrane cytoskeletal proteins (alpha fodrin), and subsequently provokes morphological and biochemical aspects of apoptosis.
Sphingolipids have been shown to have direct effects on regulators of the extrinsic pathway of apoptosis (Fig. 2). For instance, ceramide has been reported to activate protein kinase Cζ (PKCζ), which regulates the activation of c-Jun NH2-terminal kinase 1 (JNK1) and inhibition of protein kinase B (PKB or Akt) to induce apoptosis [54–57]. Ceramides are also able to directly bind and activate the lysosomal protease cathepsin D, a direct effector of apoptosis [58, 59]. Upon stimulation with tumor necrosis factor (TNF)-α increased levels of ceramide stimulate cathepsin D-mediated cleavage of BID to activate the apoptotic pathway [58, 59]. Sphingosine acts also as a pro-apoptotic signaling lipid via suppression of the MAPK/ERK signaling pathway [60]. In contrast, S1P is a suppressor of ceramide-mediated activation of JNK1 by activating pro-survival Akt/mTOR complex 1 (mTORC1), MAPK/ERK, and NF-κB signaling pathways [33]. Thus, regulators of the extrinsic pathway of apoptosis are affected in response to various sphingolipids (Fig. 2).
The intrinsic or mitochondrial pathway can be initiated following ER stress, exposure to stresses such as cytotoxic drugs, ultraviolet radiation, and free radicals which cause DNA damage [61–64]. DNA damage and ER stress activates pro-apoptotic members of the Bcl-2 family (Bax/Bak) and induces mitochondrial outer membrane permeabilization (MOMP), ultimately leading to caspase-dependent or independent apoptosis [52, 65]. Anti-apoptotic Bcl-2 proteins (Bcl-2 and Bcl-xL) counteracts pro-apoptotic proteins and can delay or inhibit apoptosis [66]. Following MOMP, release of various polypeptides such as cytochrome c from the mitochondrial intermembrane space promotes caspase activation and apoptosis. Cytochrome c in the cytosol binds with apaf-1 (apoptotic protease-activating factor-1), inducing its oligomerization and thereby forming a structure termed the apoptosome. Formation of the death-inducing signaling complex (DISC) and apoptosome results in activation of initiator caspases 8 and 9, respectively [7, 67]. Each then activates effector caspase-3, which ultimately results in hallmark apoptotic morphological signatures such as loss of cytoplasm, blebbing of the plasma membrane, and fragmentation of DNA in the nucleus [67, 68].
Several studies have highlighted the regulatory roles of sphingolipids on the intrinsic pathway of apoptosis (Fig. 2). Ceramides promote the intrinsic pathway by formation of channels in the outer membrane of the mitochondria to promote the release of cytochrome c resulting in caspase-9 activation [22, 23]. On the other hand, dihydroceramide has been shown to inhibit ceramide channel formation [69]. In addition, the intrinsic pathway can also be stimulated by the inhibitory action of ceramide on mitochondrial complex III to generate reactive oxygen species (ROS) [70, 71]. The apoptotic action of ceramide can also be mediated by the recruitment and activation of pro-apoptotic Bax at the mitochondria through the PP2A-dependent dephosphorylation of Bax and formation of mitochondrial ceramide-rich macrodomains (MCRMs) [72, 73]. Intriguingly, ceramides synthesized in the ER have been shown to transport to the mitochondria where they transiently permeabilize its outer membrane and stimulate the release of cytochrome c [74]. Such exchange may limit the need for ceramide synthesis in the mitochondria to reach ceramide levels required for initiation of the intrinsic apoptotic pathway [74]. It has been shown that aSMase-released ceramide is able to bind directly to lysosomal protease cathepsin D, leading to cathepsin D activation. The activated cathepsin D subsequently cleaves BH3-only protein BID and promotes the mitochondrial apoptotic pathway [58]. Furthermore, while ceramide-mediated activation of pro-apoptotic protein, BAD, promotes apoptosis, S1P suppresses apoptosis via BAD inactivation [75–77]. Sphingosine has been shown to downregulate expression of anti-apoptotic proteins Bcl-2 and Bcl-xL to enhance apoptosis [78, 79].
A far less well understood form of PCD, termed programmed necrosis or necroptosis, has emerged that does not appear to require caspase activity and is morphologically distinct from apoptosis. Necroptosis is an alternative receptor interacting protein (RIP)-mediated form of cell death, which is dependent on TNF receptor, Fas and TNF-related apoptosis-inducing ligand (TRAIL) receptor activation [80–82]. At a biochemical level, while necroptosis displays molecular pathways distinct from those controlling cell deaths by apoptosis or autophagy and represent a different morphology, significant cross-talk between these pathways have been described in different studies [83–85]. In some cases, molecular pathways of necroptosis may involve cellular metabolic alterations associated with mitochondria and the overproduction of ROS [86, 87] via different macromolecules such as ceramide. For instance, Ardestani et al. [87] demonstrated that the mTNF-α isoform is an effective inducer of programmed necrosis through a caspase independent ceramide-induced ROS pathway in mouse fibroblast (L929) cells. In another study with L929 cells, it has been shown that docosahexaenoic acid antagonized TNF-α-induced necroptosis through attenuating ROS generation, ceramide production and lysosomal dysfunction [88]. However, other studies using human intestinal epithelial (HT-29) cells and human monocytic (U937) cells demonstrated that ROS are not required for necroptosis [89, 90]. These disparate findings show the limitation of in vitro bioassays for the evaluation of necroptosis signaling; hence, the exact role of ceramide and whether or not necroptosis requires ROS or mitochondria has yet to be fully determined in vivo.
Autophagy
Autophagy is a tightly regulated catabolic process that supplies energy during development and in response to nutrient stress by carrying out lysosomal degradation of cell contents [91]. Despite its major role as a survival mechanism, autophagy has previously been classified as PCD2 based on morphological grounds, termed “autophagic cell death” to describe a form of caspase-independent necrosis-like cell death associated with the accumulation of autophagosomes in cells [92]. The existence of autophagic cell death as a bona fide death process is still controversial, and the casual relationship between autophagy and cell death remains unproven [93, 94]. Nevertheless, many studies have pointed to intimate relationships between autophagy and cellular death programs, which are not yet fully understood [95].
The autophagy pathway is evolutionarily conserved from early eukaryotes to mammals with as many as 38 Autophagy Related Genes (ATG) identified in yeasts and their human orthologs [96]. Autophagy is divided into three distinct forms: chaperone-mediated autophagy (CMA), microautophagy and macroautophagy [97]. A variety of stress stimuli including long term starvation, exposure to cytotoxic compounds, or oxidative stress can lead to CMA activation which selectively degrades cytosolic proteins in lysosomes [98]. The exact molecular mechanism that triggers microautophagy remains unknown. However, guanosine-5′-triphosphate (GTP) hydrolysis and calcium ions are considered as major initiators of this event in yeast [99]. Macroautophagy (referred to here as autophagy) degrades the bulk of damaged cytoplasmic organelles and proteins. Autophagy includes mitophagy (mitochondrial autophagy), ribophagy (ribosomal autophagy), pexophagy (peroxisome autophagy), ER-phagy (endoplasmic reticulum autophagy), aggrephagy (protein aggregate autophagy) and lipophagy (fat autophagy) [97].
Autophagosomes are the major particles that are formed and processed during the autophagy pathway. An autophagosome includes a double-membrane vesicle destined for degradation of proteins and organelles (cargo) which finally fuse with lysosomes to form autophagolysosomes [100]. Autophagosome formation requires the expression of ATG genes which control levels of Atg proteins [101]. Formation of the structures occurs via three main steps: (1) initiation (induction), (2) elongation, and (3) maturation (closure), with subsequent fusion with lysosomes to form the autolysosome or amphisome [97] (Fig. 3).
Fig. 3.
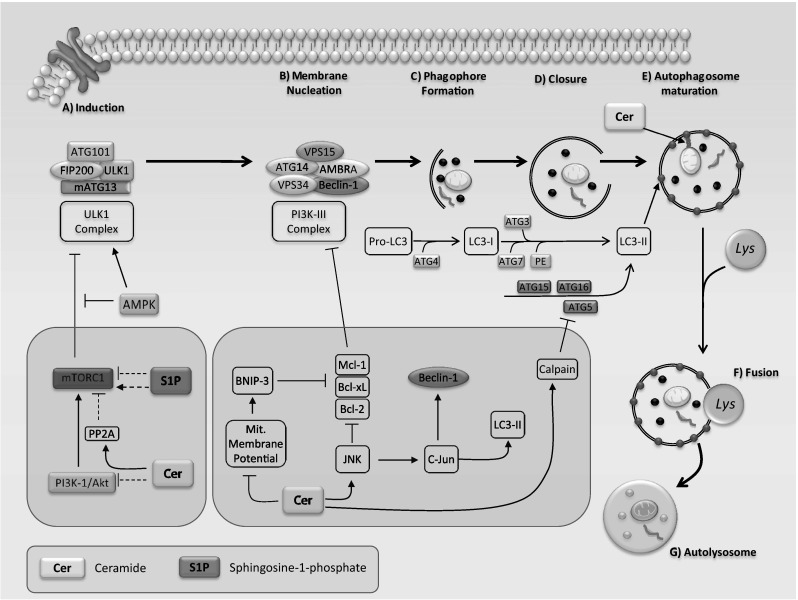
A schematic overview of autophagy machinery and its regulation by sphingolipids. A Autophagy induction and nucleation of phagophore membranes (pre-autophagosomal structures): in nutrient rich conditions (insulin, growth factors and amino acids), the mTORC1 kinase associates with the ULK1 complex to inhibit the initiation of autophagy. However, under growth factor deprivation or nutrient starvation, energy sensor AMPK activates the ULK1 complex by directly phosphorylating ULK1 and by suppression of mTORC1 activity through phosphorylation, and initiates vesicle nucleation. Phosphorylated and active ULK1 also promotes phosphorylation of Atg13 and FIP200, and dissociates from mTORC1. PI3K-III and VPS34 together with ATG14, AMBRA1, VPS15, and Beclin-1 form a protein complex (PI3K-III complex) and initiates phagophore formation. Autophagosome formation and maturation: Two ubiquitin-like proteins, Atg12 and LC3, are involved in double-membrane vesicle (autophagosome) formation, elongation, and closure. Atg12 is conjugated with Atg5 by Atg7 and Atg10, which then form a complex with Atg16. This complex works with Atg7 and Atg3 to conjugate LC3-I with the polar head of phosphatidylethanolamine (PE) to produce LC3-II, which is specifically located on autophagosome structures. Autophagosomes are sequentially fused with lysosomes to form autolysosomes. In the autolysosome, the autophagosomal cargoes are digested by lysosomal hydrolases and the contents are released for metabolic recycling. B Sphingolipids have direct effects on different stages of the autophagy pathway. At the initial step, ceramide may stimulate autophagy via PI3K-1/Akt activation which suppresses the inhibitory effects of mTORC1 on autophagy. Acid sphingomyelinase-derived ceramide also increases autophagy via reducing mTORC1 activity during amino acid deprivation in a PP2A-dependent manner. C2-ceramide-induced decrease of mitochondrial membrane potential upregulates BNIP3 expression which ultimately leads to induction of autophagy through dissociation of Beclin-1 from Bcl-2, Bcl-xL, and Mcl-1 in a competitive manner. Similarly, ceramide-mediated activation of JNK disrupts the inhibitory Beclin-1:Bcl-2 complex through direct phosphorylation of Bcl-2. Furthermore, ceramide-mediated activation of transcription factor c-Jun may increase autophagy activation via upregulation of Beclin-1 and LC3 expression. Ceramide may also activate calpain which subsequently cleaves Atg5 to generate a protein fragment that leads to suppression of autophagy and apoptosis induction. Mitochondrial ceramide has been shown to mediate mitophagy through the direct interaction between ceramide and LC3-II
The mammalian target of rapamycin (mTOR) constitutes a central checkpoint that suppresses autophagy via restraining the kinase activity of UNC-51-like kinase (ULK1) [102, 103]. mTORC1 contains the mTOR catalytic subunit (raptor/GβL/PRAS40/deptor) and phosphorylates ULK1 in the absence of amino acid and growth factor signals [104]. ULK1 Ser/Thr protein kinase, Atg13, and FIP200 (FIP200 is the mammalian homolog of the yeast Atg17) form the ULK1 complex [105–107] and regulates autophagy by phosphorylation of Atg13 and FIP200 [108]. Under stresses such as nutrient starvation, mTORC1 is inhibited by AMP-activated protein kinase (AMPK) resulting in the disassociation of ULK1 and Atg 13. The ULK1 complex is then phosphorylated by AMPK and then initiates vesicle nucleation [105].
Beclin-1 is a platform protein and its complex with class III phosphatidylinositol 3-kinase (PI3K) has a key regulatory role in nucleation and assembly of the initial phagophore membrane [109, 110]. Beclin-1 associates with Vps15, Vps34, and Ambra1 to form a complex that regulates class III PI3K, which forms phosphatidylinositol-3-phosphate (PI3P). Furthermore, Beclin-1 complex can also trigger autophagy via JNK1, and death-associated protein kinase (DAPK) [111]. PI3P is needed for recruitment of other Atg proteins as well as formation of the Ω-shape of initial vesicle nucleation which can be sourced from the outer mitochondrial membrane, ER and/or the plasma membrane [112, 113]. Elongation of the vesicle requires two ubiquitin-like conjugation systems. The first involves the formation of the Atg12–Atg5 conjugate, which appears to act as the E3 ligase of microtubule-associated protein 1 light chain 3 (LC3). The second system initiates autophagosome formation and involves the conjugation of LC3 to the polar head of phosphatidylethanolamine (PE) by release of Atg8 from Atg3 [97]. This conjugation leads to the conversion of the soluble LC3 (LC3-I) to its lipidated form, LC3-II, which is incorporated into the autophagosome membrane [113, 114]. Atg9 has also been shown to be recruited by the Atg1-Atg13 signaling complex and has an important role in expansion of the autophagosome precursor [115]. The conversion of LC3 from LC3-I to LC3-II is regarded as a critical step in autophagosome formation [116] and also represents a hallmark for detecting autophagy [97, 116]. Following elongation, expansion, and closure, the autophagosome with cytoplasmic material finally fuses with the lysosome forming an autophagolysosome, and its contents are subsequently digested by lysosomal enzymes [97].
When the autophagosome proceeds to fuse with a lysosome, its vesicular contents are degraded into macromolecules and are recycled back to the cytosol [7, 91, 112]. This process is conserved in all eukaryotic cells and serves to maintain homeostasis under normal conditions to prevent accumulation of excess/damaged organelles and proteins. However, under conditions of stress such as nutrient starvation, oxidative stress, pathogen infection or hypoxia, autophagy serves as an adaptive cell survival response that could also lead to cell death in situations of defective or excessive autophagy [103, 112].
The regulation of autophagy or apoptosis and the interplay between these two pathways is a complex process involving various key regulators. Among them, sphingolipids have been shown to have significant roles in different organ systems [117–121]. For example, ceramide and S1P have both been demonstrated to induce autophagy by various mechanisms. Because of the paradoxical roles of autophagy, ceramide-dependent autophagy could either promote cell death (by inducing autophagy cell death, or “switching” the cell from autophagy to apoptosis) [27, 122–124] or under certain conditions may induce cytoprotective autophagy [25, 125]. Recently, it has been demonstrated that stimulation of de novo ceramide synthesis results in dissociation of the complex formed between Beclin-1 and Bcl-2 through stimulating the phosphorylation of Bcl-2 by JNK1 leading to autophagy activation [122]. Likewise, inhibition of ceramide synthesis resulted in suppression of autophagy [122]. Ceramide has been reported to suppress the Akt signaling pathway resulting in autophagy activation via negative regulation of mTOR signaling [126], suggesting an upstream regulatory target of the autophagy pathway. Furthermore, ceramide-induced autophagy, either by targeting the mitochondria or by upregulation of Beclin-1, has been reported to be linked to autophagic cell death [27, 126, 127]. The role of ceramide-induced autophagy in the context of the ‘sphingolipid rheostat’ has yet to be fully understood and further investigation is required to determine the mechanisms involved in determination of cell fate when autophagy is activated.
S1P-induced autophagy may be associated with cell survival [41], consistent with the well-established ‘sphingolipid rheostat’. Lavieu et al. [41] reported that SphK1 (S1P)-induced autophagy protects the cell during nutrient starvation and cell death. Other sphingolipids such as gangliosides have been reported to induce the formation of autophagic vacuoles followed by cell death that is inhibited by either knockdown of autophagy genes or by an inhibitor of autophagy [128]. Thus, gangliosides appear to have a similar role in autophagic cell death as ceramides. It has been hypothesized that the structural properties of sphingolipids may mediate autophagosome formation and maturation [129]. In support of this hypothesis, inhibition of sphingolipid synthesis in Saccharomyces cerevisiae reduced autophagic activity, but had no effect on Atg12–Atg5 or Atg8-PE conjugation, or pre-autophagosomal structure formation [130]. This suggests that sphingolipids play a critical role in the formation of the autophagosome. Also, ceramides have been found in the membrane of the autophagosome, in line with the idea of a structural role of sphingolipids in autophagy [131]. This review will further discuss sphingolipid signaling and the consequences of their induced pathways on cell fate and the delicate balance between a healthy and disease state lung.
Sphingolipids in the lung
Significance of sphingolipids in lung development
It is widely understood that sphingolipids play key roles in regulating cellular homeostasis. However, their role in the regulation of lung development has been of particular interest and has received more attention. Ceramide, S1P, and their dynamic relationship have been of particular interest. The de novo pathway of ceramide synthesis has been linked to apoptotic endothelial cell death and decreased pulmonary barrier function [19, 132]. In a study conducted by Medler et al. [132], it was found that de novo ceramide synthesis resulted in apoptotic cell death in lung endothelial cells by both paracellular and TNF-α-stimulated intracellular ceramide signaling. Increased ceramide synthesis has also been linked to the development of emphysema-like disease states in the murine lung, suggesting that the balance between a pro-apoptotic molecule and a pro-survival factor, such as S1P, is essential in maintaining homeostasis in the vasculature of the lung [133]. Furthermore, investigation by Petrache et al. into ceramide-induced effects in the lung have revealed that an excess of ceramide-induced oxidative stress triggered apoptotic cell death which led to alveolar enlargement [134]. Superoxide dismutase has been shown to play a protective role against apoptosis and alveolar enlargement induced by disproportionate ceramide, suggesting that there are protective mechanisms that can be activated to protect or prevent unwarranted cell death in the healthy lung [134].
It is important to emphasize however, that a regulated level of ceramide may be necessary for maintenance of homeostasis in the lung. It is evident now that the preservation of the sphingolipid rheostat is important in the formation of lung structures at all stages of lung development and physiology [119]. A study showed that pigs exposed to fumonisin (FB1) (inhibitor of ceramide synthase) resulted in elevated levels of sphingolipids including sphinganine and sphingosine, which resulted in alveolar endothelial cell damage and fatal pulmonary edema [135]. Chronic exposure to FB1 in rats not only induced renal tumors, but also resulted in metastatic invasion of the lungs [136]. These findings suggest that ceramide synthesis is required for an underlying level of apoptotic cell death in the lungs as an anti-tumorigenic factor [136]. Xu et al. demonstrated that in murine lung epithelia the de novo ceramide synthesis pathway is the major contributing pathway for ceramide production [137]. Longevity assurance homolog 5 (LASS5) was found to be the predominant ceramide synthase in murine primary type II epithelial cells and SV40-transformed murine lung epithelial (MLE) cells. Indeed, inhibition of ceramide synthase activity by FB1 was shown to inhibit the production of ceramide [137]. Furthermore, this study also showed that overexpression of LASS5 reduced phosphatidylcholine (PtdCho) production, suggesting that ceramide may regulate PtdCho metabolism in pulmonary cells [137].
In-depth investigation into the levels of sphingolipids in a developing rat lung revealed that maintenance of sphingolipid levels and their metabolism are highly regulated. In the embryonic rat lung, sphingomyelin and sphingosine levels increase during development and plateau at birth. Interestingly, the activity profiles of SPT, aSMase and nSMase correlate accordingly [138]. In contrast to the rat lung, sphingomyelin levels in the fetal lung of the monkey and lamb decrease during development [139, 140]. These findings suggest that regulation of sphingolipid metabolism is important in proper lung development and any imbalances or misregulations in the sphingolipid rheostat could lead to improper lung structure or disease states (Table 1).
Table 1.
Effects of sphingolipids in pulmonary diseases
| Stimulus | Sphingolipid | Enzyme/pathway | Cell fate | Pathophysiological effect | Disease state | Reference |
|---|---|---|---|---|---|---|
| Sepsis, inhalation of harmful substances | ↑ S1P | NSM pathway, ceramide is converted to S1P | ↓ Neutrophil apoptosis | Expedites inflammation, increase in proteases and ROS resulting in damage to lung tissues | Acute lung injury/ARDS | [142] |
| Mechanical ventilation, high oxygen | ↑ Ceramide | ? (Possibly de novo synthesis pathway) | ↑ Alveolar epithelial cell apoptosis | Halt in alveolarization, fewer and larger alveoli | BPD | [21, 118] |
| Airway response to allergen | ↑ S1P | Mitogenic factor, facilitates G1/S progression | ↑ Airway smooth muscle cell proliferation | Airway wall remodeling | Asthma | [161] |
| Genetic variation at the 17q21 locus (ORMDL3 protein) | ↓ Ceramide | Inhibition of SPT | Increase of S1P, decrease of ceramide results in cell survival and proliferation | Genetic predisposition to play role in pathogenesis of asthma | Asthma | [159, 166, 167] |
| Inhalation of cigarette smoke/pollutants | ↑ Ceramide | ↑ De novo synthesis | ↑ Alveolar epithelial cell apoptosis | Loss of alveolar surface area available for gas exchange | COPD/emphysema | [19] |
| Inhalation of cigarette smoke/pollutants | ↑ Ceramide | ? | ↑ Impaired autophagy | Accumulation of impaired autophagy marker p62 in autophagosomes | COPD/emphysema | [184, 190] |
| P. aeruginosa infection in CFTR-deficient mice | ↑ Ceramide | ? (Possibly by a shift in balance of enzymes involved in ceramide metabolism) | ↑ Bronchial cell apoptosis | Bronchial cell apoptosis and DNA deposition in the upper airways | Cystic fibrosis | [6] |
? means currently unknown
Sphingolipids in pulmonary disease
Acute lung injury/ARDS
Acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) are conditions that are characterized by lung inflammation, increased microvascular permeability, and edema [119, 141]. The contributing role of sphingolipids in attenuating injury to the lung during ALI and ARDS has been recently explored and is now well established that there are a number of mechanisms including the neutral sphingomyelinase (nSMase), ceramide, S1P, and the p38 MAPK pathway, that appear to be involved in the pathological process [2, 142].
The influence that sphingolipids have on neutrophil cell fate in an ALI/ARDS disease state is of particular interest as it is well established that the longevity of neutrophils correlates with the severity of the pathological state [143, 144]. Under homeostatic conditions, neutrophils will undergo unprompted apoptosis [145]. However, neutrophils that are recruited to occupy the alveolar space during lung injury contribute to the pathogenesis of ALI and ARDS by production and secretion of proteases and ROS for the duration of their survival [146]. Thus, neutrophil apoptosis is essential for resolution of inflammation, especially in the context of an injury to the lung.
It has been reported that apoptosis of neutrophils from patients with sepsis was suppressed via a mechanism involving NF-κB, reduced caspase-9 and caspase-3 activity, and maintenance of mitochondrial membrane potential [147]. Another study found that sphingolactone-24 (Sph-24) (nSMase inhibitor) and SKI-II (sphingosine kinase inhibitor) prevented the anti-apoptotic effect of lipopoly saccharides (LPS) on neutrophils, and that LPS stimulates sphingomyelinase activity suggesting that sphingolipids are involved in inhibition of neutrophil apoptosis [142]. When Sph-24 was administered to LPS challenged lungs in mice, counts of leukocytes decreased after a period of 24 h suggesting that nSMase accelerates injury progression during lung injury by deceleration of neutrophil apoptosis [142]. They also demonstrated that LPS challenge of neutrophils that have been recruited to the lung also display an increase in S1P levels. This study also revealed that the increased levels of anti-apoptotic S1P are likely the result of activation of the nSMase-S1P pathway [142]. S1P is generated by SphKs that are stimulated by factors such as histamines and cytokines which could explain the increase of SphK activity in the injured lung [142, 148]. Thus, both nSMase and S1P play key roles in the pathogenic process of ALI/ARDS, particularly in their influence of preventing neutrophil apoptosis. In ALI, increased nSMase activity augments ceramide and subsequent sphingosine formation that then is phosphorylated to S1P by activated sphingosine kinase, leading to phosphorylation of p38 MAPK resulting in inhibition of neutrophil apoptosis [142, 149]. Greater understanding in the pathogenesis of ALI/ARDS and the role sphingolipids play in cell fate will improve detection and treatment of this critical condition.
Bronchopulmonary dysplasia
Bronchopulmonary dysplasia (BPD) is amongst the most common chronic lung diseases of neonates affecting between 52 and 7 % of infants born weighing between 500 and 1500 g, respectively [150]. Very premature infants that are subjected to mechanical ventilation and oxygen supplementation due to respiratory failure are prone to lung injury that may result in chronic lung disease, such as BPD, with lifelong consequences [151]. The lungs of present day BPD patients are characterized by a halt in lung development, with simplification of normal lung complexity as seen with fewer and larger alveoli and vascular abnormalities [152]. It was recently demonstrated that changes in sphingolipid levels may affect proper lung development and function when Tibboel et al. elegantly demonstrated that newborn mice that were exposed to hyperoxic conditions displayed an increase in multiple sphingolipids, including ceramide [118]. This caused abnormal alveolar morphology and obstructive lung function that were only partially recovered in room air [118]. The use of D-sphingosine reduced ceramide levels and partially minimized halt in alveolarization, demonstrating that altered sphingolipid levels may be a factor in hyperoxia-induced lung injury or BPD [118]. Recently, it was shown that levels of vascular endothelial growth factor (VEGF), a critical factor in pulmonary vascular development, were decreased in a rodent animal model of BPD [153], thereby most likely reducing alveolarization [154, 155]. Increased ceramide production has also been shown to decrease VEGF by suppression of hypoxia-inducible factor-1α [156]. These findings suggest that sphingolipids play a role in abnormal lung vascular development through VEGF signaling in BPD. In another study, apoptosis was detected in alveolar epithelial cells in the lungs of preterm infants that were subjected to ventilation and oxygen treatment [157]. Similar results have been reported in mouse models of BPD [158]. Kroon et al. observed that mechanical ventilation of newborn rat resulted in increased number of apoptotic alveolar type II cells [21]. Prolonged maximal cyclic stretch was also associated with increased expression of cleaved caspase-3, -7, and -8, as well as apoptotic mediator Fas ligand (FasL), suggesting that the extrinsic death pathway via the FasL/Fas system is involved in ventilation-induced apoptosis of alveolar type II cells [21].Given these findings, it is worthwhile to investigate the role of ventilation-induced ceramide production in epithelial cell apoptosis as a mechanism responsible for pulmonary apoptosis and inhibition of alveolar development in preterm infants with BPD. The possibility that altered levels of various sphingolipids can change cell fate during mechanical ventilation opens potential areas of therapeutic interventions for BPD to reduce lung damage in premature infants that require respiratory assistance.
Asthma
Asthma is a complex chronic inflammatory lung disease that is characterized by airway wall remodeling, airway smooth muscle contraction and hyperreactivity, increased mucus production, and inflammatory cell gathering [159]. It has increasingly become apparent that sphingolipid metabolites play a regulatory role in the pathogenesis of asthma [160]. S1P is often attributed to act as a pro-survival signal in cells [9], and increased levels in the asthmatic airway appear to play a role in perpetuating the asthmatic response [161]. The role of S1P in the pathogenesis of asthma is well illustrated by the fact that S1P is involved in trafficking and chemotaxis of immune cells, such as mast cells, to the airway. Interestingly, S1P release from the immune cell and binding (in autocrine and paracrine signaling) with S1P1 receptors in low antigen concentrations facilitates mast cell chemotaxis, while activation of S1P2 receptors in high antigen concentrations ceases chemotaxis and initiates degranulation [162]. S1P not only plays an important role in immune cell movement, but also appears to act as a mitogenic factor and a stimulator of proliferation of the airway smooth muscle cells. S1P increases G1/S progression in the cell cycle which results in augmented EGF- and thrombin-induced DNA proliferation [161, 163]. SphK has been shown to play important roles in other inflammatory and hyper proliferative diseases, and has received attention with the prospect of developing specific SphK inhibitors for therapeutic and treatment purposes [164].
Research efforts have recently been focused on the possibility that genetic predisposition of the asthmatic respiratory tract significantly affects the severe reaction to allergic and/or inflammatory stimuli with strong evidence that genetic variation at the 17q21 locus is linked with childhood asthma [165]. The locus gene orosomucoid 1-like 3 (ORMDL3) that belongs to a gene family encoding ER transmembrane proteins has been recently identified to be related to the pathogenesis of asthma [166, 167]. Systematic evaluation of single nucleotide polymorphisms (SNPs) and patients with childhood onset asthma have revealed that genetic variants regulating ORMDL3 expression may alter susceptibility to asthma [167].Orosomucoid-like (ORMDL) proteins act as negative regulators of SPT that are mediated by a feedback response, thus increased levels of the ORMDL proteins result in a decrease in ceramide and higher order sphingolipid biosynthesis as a result of inhibited de novo sphingolipid synthesis [168, 169]. Expression of the ORMDL3 gene is increased in asthmatic airways, which results in decreased de novo ceramide synthesis and also has been associated with airway hyperreactivity in mouse lungs, and human and murine bronchial rings [159]. Worgall et al. [159] investigated the effect of decreased SPT activity in the lung by pharmacologic intervention with myriocin (SPT inhibitor) as well as in SPT heterozygous knockout mice and observed increased bronchial reactivity in the absence of inflammation. They also found that intracellular magnesium homeostasis and bronchial sensitivity to magnesium was affected by decreased SPT activity, providing a mechanistic link between decreased de novo sphingolipid synthesis, altered contractile sensitivity to magnesium, and smooth muscle function in asthma [159]. This emerging evidence that ORMDL proteins affect sphingolipid metabolism in asthmatic airways suggests that it is highly likely that a genetic predisposition to altered sphingolipid metabolism plays a significant role in the pathogenesis of asthma.
COPD and emphysema
Chronic obstructive pulmonary disease (COPD) encompasses a spectrum of pathological conditions, including emphysema, that are characterized by difficulties breathing due to obstructed airflow [170]. Emphysema is found almost exclusively in adults and is often associated with inhalation of cigarette smoke but can also result from exposure to environmental pollutants, typically worsening over time [171]. Patients with emphysema particularly have a loss of alveolar surface area available for gas exchange due to destruction of distal airway spaces [171]. The mechanisms involved in the pathogenesis of COPD include inflammatory responses in the airway, loss of barrier function, oxidative, and ER stress responses. In addition, both apoptosis and autophagy of the cells in the airways are generally accepted to be important events in the pathogenesis of COPD and pulmonary emphysema [172]. However, it is very important to note that in most studies the smoking status of the control population was unknown or was significantly different from that of the COPD subjects [173–176]; thus, it is not fully understood whether the habit of smoking has a role in inducing apoptosis independently of airflow limitation.
It is now recognized that sphingolipids play important roles in the development of COPD with no surprise that ceramide appears to be a crucial mediator in the apoptotic death of alveolar cells [19, 177]. For instance, in rat and mice emphysema models, inhibition of de novo synthesis of ceramide induced by VEGF blockade prevented alveolar cell apoptosis and oxidative stress [19]. Moreover, when ceramide was introduced through intratracheal instillation of mice, apoptosis of lung alveolar cells and total ceramide levels increased [19]. More importantly, lung ceramide levels are markedly higher in individuals with emphysema from chronic cigarette smoking compared to individuals without emphysema [19]. In this study, the authors conclude that de novo ceramide synthesis is essential for the development of murine lung emphysema [19]. At the molecular level, activation of nSMase2 during cigarette smoke-induced oxidative stress of human airway epithelial (A549) cells has been shown to generate ceramide and apoptosis [178]. Both proto-oncogene tyrosine-protein kinase Src and p38 MAPK appear to be involved in regulating nSMase2 phosphorylation and activation [179].
Despite higher expression of ceramide in the lungs of COPD and idiopathic pulmonary fibrosis (IPF) patients, this phenomenon does not appear to be specific and related to COPD severity [180]. Scarpa and colleagues [180] studied 10 subjects with severe COPD, 13 with mild/moderate COPD, 11 with IPF, 12 non-COPD smokers, and 11 nonsmoking controls. They found an increase in ceramide levels in COPD patients compared to control smokers, which correlated to the impairment of gas exchange but not to the degree of airflow limitation. A recent study conducted by Tibboel et al. revealed that the addition of a SPT inhibitor to a rodent model of elastase-induced emphysema diminished the increase in ceramides and improved lung function [177] suggesting that ceramide upregulation in emphysema models may be a critical factor in the development of alveolar destruction in the disease state, and serves as a potential therapeutic target that warrants further investigation.
The functional significance of autophagy in disease states such as COPD has yet to be established since relatively few studies have been done on the lung. Few in vivo and in vitro specimens of COPD lung exhibit increased levels of autophagy markers when exposed to cigarette smoke or cigarette smoke extract, possibly as a response to a source of stress on the lung [73, 128, 181], and there have been reports suggesting that the autophagic process plays an important role in the pathogenesis of COPD perhaps via promoting epithelial cell death [181–183]. Recently, Fujii et al. found autophagy impairment in COPD [184]. Despite high baseline levels of autophagy in primary human bronchial epithelial cells isolated from COPD patients, cigarette smoke extract significantly reduced autophagy [184]. Furthermore, higher levels of p62 and ubiquitinated proteins have been detected in lung homogenates from COPD patients suggesting that insufficient autophagic clearance of damaged proteins may be involved in cell senescence in COPD patients [184]. As mentioned earlier, increased expression of ceramide has been detected in emphysema and may play a critical role in impaired autophagy in the context of COPD. However, the role of ceramide in smoke-induced COPD is not yet well studied and it may be worthwhile to investigate whether the increase of ceramide content in lungs when exposed to cigarette smoke also has an impact on autophagy, and if the pathway plays a pathogenic role in COPD.
Cystic fibrosis
Cystic fibrosis(CF) is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene that codes for a chloride channel that mediates proper movement of chloride ions in epithelial cells [185, 186]. The disease has a median predicted age of survival of 49.7 years [187] and is characterized by accumulation of mucus in the bronchi, and reduced mucociliary clearance ultimately leading to recurrent and/or chronic infections with bacteria including S. aureus and P. aeruginosa [162]. Ceramide has been measured at higher concentrations in CF airways and appears to be an important factor in the hallmark features of CF and cellular fate in the affected lungs [6]. Several studies have shown that CFTR dysfunction leads to an imbalance in sphingolipid homeostasis [6] and that its localization in the lipid raft influences membrane ceramide composition [188, 189]. Furthermore, results of studies using Cftr −/− mice and human lung tissue demonstrate that CFTR expression inversely correlates with ceramide accumulation and severity of emphysema in COPD subjects, demonstrating a critical role of membrane-localized CFTR in ceramide regulation and inflammation in lung injury and emphysema [188, 189]. Ceramide appears to be a critical regulator of P. aeruginosa infection in CFTR-deficient mice [6]. CFTR-deficiency results in alkalinization of acidic prelysosomes and lysosome vesicles which likely alter the balance of enzymes that increase and decrease levels of ceramide in the cells, which ultimately results in an increase of ceramide concentration in the lung epithelial cells of CFTR-deficient mice [6]. Accumulation of ceramide in the cells led to apoptosis and DNA deposition in the upper airways that facilitated P. aeruginosa infection. Treatment of CFTR-deficient mice with amitriptyline (aSMase inhibitor) normalized ceramide levels and prevented pulmonary infection with P. aeruginosa [6]. CFTR-deficient mice also display an upregulation and activation of CD95 (Fas/APO-I receptor) caused by increased ceramide concentrations, further propelling in a feedback cycle [185]. CD95 is also involved in an increase of bronchial cell death, identifying a potential relationship between CD95 and ceramide as an important mechanistic link in CF [185]. It has yet to be fully understood if these findings can be transferred into a human system, however results thus far have suggested that disruption of ceramide formation may serve as a prospective treatment for patients with CF.
Closing remarks
Sphingolipids play an important role in many physiological and pathological processes across many organ systems and diseases. The vast number of sphingolipids is reflected in the spectrum of signaling mechanisms they are involved in. Different sphingolipids have key roles in determining cell fate in many cells throughout nearly all organ systems. Ceramide is known to induce apoptosis in cells while S1P counterbalances ceramide and promotes cell survival in what is referred to as the sphingolipid ‘rheostat’. These effects are of significance in the lungs and its diseases such as ALI/ARDS, BPD, asthma, COPD, emphysema, and CF. Investigation into the lung, its diseases, and the effects of sphingolipids on cell fate using animal and human models have provided great insight into the pathogenesis of the disease states as well as potential interventions that can be used to improve outcomes for patients. In this regard, inhibitors of the sphingolipid pathway show great therapeutic potential and should be investigated and functionally validated for their protective effects and benefit for the patients. This certainly broadens the options of treatment and care for those suffering from devastating lung diseases for which treatments are limited.
Acknowledgments
The authors are supported by the Canadian Institute of Health Research, the Ontario Graduate Scholarship, and The Hospital for Sick Children Restracomp. The authors report no conflicts of interest, financial, or otherwise.
Abbreviations
- ALI
Acute lung injury
- AMPK
AMP-activated protein kinase
- Apaf-1
Apoptotic protease-activating factor-1
- ARDS
Acute respiratory distress syndrome
- aSMase
Acid sphingomyelinase
- ATG
Autophagy related genes
- BPD
Bronchopulmonary dysplasia
- Caspase
Cysteine aspartate-specific protease
- CF
Cystic fibrosis
- CFTR
Cystic fibrosis transmembrane conductance regulator
- CMA
Chaperone-mediated autophagy
- COPD
Chronic obstructive pulmonary disease
- DAPK
Death-associated protein kinase
- DD
Death domains
- DISC
Death-inducing signaling complex
- ER
Endoplasmic reticulum
- FasL
Fas ligand
- FB1
Fumonisin B1
- IPF
Idiopathic pulmonary fibrosis
- JNK1
c-Jun NH2–terminal kinase 1
- LASS5
Longevity assurance homolog 5
- LC3
Microtubule-associated protein 1 light chain 3
- LPS
Lipopolysaccharides
- MCRM
Mitochondrial ceramide-rich macrodomain
- MLE
Murine lung epithelial
- MOMP
Mitochondrial outer membrane permeabilization
- mTOR
Mammalian target of rapamycin
- mTORC1
mTOR complex 1
- NF-κB
Nuclear factor-κB
- nSMase
Neutral sphingomyelinase
- ORMDL
Orosomucoid-Like
- PCD
Programmed cell death
- PE
Phosphatidylethanolamine
- PI3K
Phosphatidylinositol 3-kinase
- PI3P
Phosphatidylinositol-3-phosphate
- PKCζ
Protein kinase C ζ
- PP2A
Protein phosphatase 2
- PtdCho
Phosphatidylcholine
- RIP
Receptor interacting protein
- ROS
Reactive oxygen species
- S1P
Sphingosine-1-phosphate
- SMase
Sphingomyelinase
- SNP
Single nucleotide polymorphism
- Sph-24
Sphingolactone-24
- SphK
Sphingosine kinase
- SPT
Serine palmitoyltransferase
- tBID
Truncated BID
- TNF
Tumor necrosis factor
- TRAIL
TNF-related apoptosis-inducing ligand
- ULK1
UNC-51-like kinase
- UPR
Unfolded protein response
- VEGF
Vascular endothelial growth factor
References
- 1.Spiegel S, Foster D, Kolesnick R. Signal transduction through lipid second messengers. Curr Opin Cell Biol. 1996;8:159–167. doi: 10.1016/s0955-0674(96)80061-5. [DOI] [PubMed] [Google Scholar]
- 2.Yang Y, Uhlig S. The role of sphingolipids in respiratory disease. Ther Adv Respir Dis. 2011;5:325–344. doi: 10.1177/1753465811406772. [DOI] [PubMed] [Google Scholar]
- 3.Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- 4.Liu H, Chakravarty D, Maceyka M, et al. Sphingosine kinases: a novel family of lipid kinases. Prog Nucleic Acid Res Mol Biol. 2002;71:493–511. doi: 10.1016/s0079-6603(02)71049-0. [DOI] [PubMed] [Google Scholar]
- 5.Kolesnick RN, Krönke M. Regulation of ceramide production and apoptosis. Annu Rev Physiol. 1998;60:643–665. doi: 10.1146/annurev.physiol.60.1.643. [DOI] [PubMed] [Google Scholar]
- 6.Teichgräber V, Ulrich M, Endlich N, et al. Ceramide accumulation mediates inflammation, cell death and infection susceptibility in cystic fibrosis. Nat Med. 2008;14:382–391. doi: 10.1038/nm1748. [DOI] [PubMed] [Google Scholar]
- 7.Young MM, Kester M, Wang H-G. Sphingolipids: regulators of crosstalk between apoptosis and autophagy. J Lipid Res. 2013;54:5–19. doi: 10.1194/jlr.R031278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huwiler A, Kolter T, Pfeilschifter J, Sandhoff K. Physiology and pathophysiology of sphingolipid metabolism and signaling. Biochimica et Biophysica Acta (BBA) Mol Cell Biol Lipids. 2000;1485:63–99. doi: 10.1016/s1388-1981(00)00042-1. [DOI] [PubMed] [Google Scholar]
- 9.Pyne S, Pyne NJ. Sphingosine 1-phosphate signalling in mammalian cells. Biochem J. 2000;349:385–402. doi: 10.1042/0264-6021:3490385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Olivera A, Spiegel S. Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature. 1993;365:557–560. doi: 10.1038/365557a0. [DOI] [PubMed] [Google Scholar]
- 11.Hannun YA, Obeid LM. Ceramide: an intracellular signal for apoptosis. Trends Biochem Sci. 1995;20:73–77. doi: 10.1016/s0968-0004(00)88961-6. [DOI] [PubMed] [Google Scholar]
- 12.Dawkins JL, Hulme DJ, Brahmbhatt SB, et al. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat Genet. 2001;27:309–312. doi: 10.1038/85879. [DOI] [PubMed] [Google Scholar]
- 13.Levade T, Jaffrézou J-P. Signalling sphingomyelinases: which, where, how and why? Biochimica et Biophysica Acta (BBA) Mol Cell Biol Lipids. 1999;1438:1–17. doi: 10.1016/s1388-1981(99)00038-4. [DOI] [PubMed] [Google Scholar]
- 14.Okino N, He X, Gatt S, et al. The reverse activity of human acid ceramidase. J Biol Chem. 2003;278:29948–29953. doi: 10.1074/jbc.M303310200. [DOI] [PubMed] [Google Scholar]
- 15.Engedal N, Saatcioglu F. Ceramide-induced cell death in the prostate cancer cell line LNCaP has both necrotic and apoptotic features. Prostate. 2001;46:289–297. doi: 10.1002/1097-0045(20010301)46:4<289::aid-pros1035>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 16.Todaro M, Catalano M, Di Liberto D, et al. High levels of exogenous C2-ceramide promote morphological and biochemical evidences of necrotic features in thyroid follicular cells. J Cell Biochem. 2002;86:162–173. doi: 10.1002/jcb.10203. [DOI] [PubMed] [Google Scholar]
- 17.Verheij M, Bose R, Lin XH, et al. Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature. 1996;380:75–79. doi: 10.1038/380075a0. [DOI] [PubMed] [Google Scholar]
- 18.Hannun YA, Luberto C. Ceramide in the eukaryotic stress response. Trends Cell Biol. 2000;10:73–80. doi: 10.1016/s0962-8924(99)01694-3. [DOI] [PubMed] [Google Scholar]
- 19.Petrache I, Natarajan V, Zhen L, et al. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med. 2005;11:491–498. doi: 10.1038/nm1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hannun YA. Functions of ceramide in coordinating cellular responses to stress. Science. 1996;274:1855–1859. doi: 10.1126/science.274.5294.1855. [DOI] [PubMed] [Google Scholar]
- 21.Kroon AA, DelRiccio V, Tseu I, et al. Mechanical ventilation-induced apoptosis in newborn rat lung is mediated via FasL/Fas pathway. Lung Cell Mol Physiol. 2013;305:L795–L804. doi: 10.1152/ajplung.00048.2013. [DOI] [PubMed] [Google Scholar]
- 22.Siskind LJ, Kolesnick RN, Colombini M. Ceramide channels increase the permeability of the mitochondrial outer membrane to small proteins. J Biol Chem. 2002;277:26796–26803. doi: 10.1074/jbc.M200754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siskind LJ, Colombini M. The lipids C2- and C16-ceramide form large stable channels. Implications for apoptosis. J Biol Chem. 2000;275:38640–38644. doi: 10.1074/jbc.C000587200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schenck M, Carpinteiro A, Grassme H, et al. Ceramide: Physiological and pathophysiological aspects. Arch Biochem Biophys. 2007;462:171–175. doi: 10.1016/j.abb.2007.03.031. [DOI] [PubMed] [Google Scholar]
- 25.Guenther GG, Peralta ER, Rosales KR, et al. Ceramide starves cells to death by downregulating nutrient transporter proteins. PNAS. 2008;105:17402–17407. doi: 10.1073/pnas.0802781105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ponnusamy S, Meyers-Needham M, Senkal CE, et al. Sphingolipids and cancer: ceramide and sphingosine-1-phosphate in the regulation of cell death and drug resistance. Future Oncol. 2010;6:1603–1624. doi: 10.2217/fon.10.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daido S, Kanzawa T, Yamamoto A, et al. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res. 2004;64:4286–4293. doi: 10.1158/0008-5472.CAN-03-3084. [DOI] [PubMed] [Google Scholar]
- 28.Sentelle RD, Senkal CE, Jiang W, et al. Ceramide targets autophagosomes to mitochondria and induces lethal mitophagy. Nat Chem Biol. 2012;8:831–838. doi: 10.1038/nchembio.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Spassieva SD, Mullen TD, Townsend DM, Obeid LM. Disruption of ceramide synthesis by CerS2 down-regulation leads to autophagy and the unfolded protein response. Biochem J. 2009;424:273–283. doi: 10.1042/BJ20090699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taniguchi M, Kitatani K, Kondo T, et al. Regulation of autophagy and its associated cell death by “sphingolipid rheostat”: reciprocal role of ceramide and sphingosine 1-phosphate in the mammalian target of rapamycin pathway. J Biol Chem. 2012;287:39898–39910. doi: 10.1074/jbc.M112.416552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olivera A, Spiegel S. Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature. 1993;365:557–560. doi: 10.1038/365557a0. [DOI] [PubMed] [Google Scholar]
- 32.Zhang H, Desai NN, Olivera A, et al. Sphingosine-1-phosphate, a novel lipid, involved in cellular proliferation. J Cell Biol. 1991;114:155–167. doi: 10.1083/jcb.114.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cuvillier O, Pirianov G, Kleuser B, et al. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature. 1996;381:800–803. doi: 10.1038/381800a0. [DOI] [PubMed] [Google Scholar]
- 34.Kihara Y, Maceyka M, Spiegel S, Chun J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br J Pharmacol. 2014;171:3575–3594. doi: 10.1111/bph.12678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee MJ, Van Brocklyn JR, Thangada S, et al. Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science. 1998;279:1552–1555. doi: 10.1126/science.279.5356.1552. [DOI] [PubMed] [Google Scholar]
- 36.Xia P, Wang L, Gamble JR, Vadas MA. Activation of sphingosine kinase by tumor necrosis factor-alpha inhibits apoptosis in human endothelial cells. J Biol Chem. 1999;274:34499–34505. doi: 10.1074/jbc.274.48.34499. [DOI] [PubMed] [Google Scholar]
- 37.Taha TA, Mullen TD, Obeid LM. A house divided: ceramide, sphingosine, and sphingosine-1-phosphate in programmed cell death. BBA. 2006;1758:2027–2036. doi: 10.1016/j.bbamem.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Le Stunff H, Galve-Roperh I, Peterson C, et al. Sphingosine-1-phosphate phosphohydrolase in regulation of sphingolipid metabolism and apoptosis. J Cell Biol. 2002;158:1039–1049. doi: 10.1083/jcb.200203123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Echten-Deckert G, Zschoche A, Bär T, et al. cis-4-Methylsphingosine decreases sphingolipid biosynthesis by specifically interfering with serine palmitoyltransferase activity in primary cultured neurons. J Biol Chem. 1997;272:15825–15833. doi: 10.1074/jbc.272.25.15825. [DOI] [PubMed] [Google Scholar]
- 40.Lanterman MM, Saba JD. Characterization of sphingosine kinase (SK) activity in Saccharomyces cerevisiae and isolation of SK-deficient mutants. Biochem J. 1998;332:525–531. doi: 10.1042/bj3320525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lavieu G, Scarlatti F, Sala G, et al. Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J Biol Chem. 2006;281:8518–8527. doi: 10.1074/jbc.M506182200. [DOI] [PubMed] [Google Scholar]
- 42.Ameisen JC. On the origin, evolution, and nature of programmed cell death: a timeline of four billion years. Cell Death Differ. 2002;9:367–393. doi: 10.1038/sj.cdd.4400950. [DOI] [PubMed] [Google Scholar]
- 43.Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742–758. doi: 10.1016/j.cell.2011.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature. 2000;407:784–788. doi: 10.1038/35037722. [DOI] [PubMed] [Google Scholar]
- 46.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Path. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stennicke HR, Salvesen GS. Properties of the caspases. Biochimica et Biophysica Acta. 1998;1387:17–31. doi: 10.1016/s0167-4838(98)00133-2. [DOI] [PubMed] [Google Scholar]
- 48.Creagh EM, Conroy H, Martin SJ. Caspase-activation pathways in apoptosis and immunity. Immunol Rev. 2003;193:10–21. doi: 10.1034/j.1600-065x.2003.00048.x. [DOI] [PubMed] [Google Scholar]
- 49.Fischer U, Jänicke RU, Schulze-Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ. 2003;10:76–100. doi: 10.1038/sj.cdd.4401160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yi CH, Sogah DK, Boyce M, et al. A genome-wide RNAi screen reveals multiple regulators of caspase activation. J Cell Biol. 2007;179:619–626. doi: 10.1083/jcb.200708090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Igney FH, Krammer PH. Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer. 2002;2:277–288. doi: 10.1038/nrc776. [DOI] [PubMed] [Google Scholar]
- 52.Ghavami S, Hashemi M, Ande SR, et al. Apoptosis and cancer: mutations within caspase genes. J Med Genet. 2009;46:497–510. doi: 10.1136/jmg.2009.066944. [DOI] [PubMed] [Google Scholar]
- 53.Lakhani SA, Masud A, Kuida K, et al. Caspases 3 and 7: key mediators of mitochondrial events of apoptosis. Science. 2006;311:847–851. doi: 10.1126/science.1115035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bourbon NA, Yun J, Kester M. Ceramide directly activates protein kinase C zeta to regulate a stress-activated protein kinase signaling complex. J Biol Chem. 2000;275:35617–35623. doi: 10.1074/jbc.M007346200. [DOI] [PubMed] [Google Scholar]
- 55.Bourbon NA, Sandirasegarane L, Kester M. Ceramide-induced inhibition of Akt is mediated through protein kinase Czeta: implications for growth arrest. J Biol Chem. 2002;277:3286–3292. doi: 10.1074/jbc.M110541200. [DOI] [PubMed] [Google Scholar]
- 56.Fox TE, Houck KL, O’Neill SM, et al. Ceramide recruits and activates protein kinase C zeta (PKC zeta) within structured membrane microdomains. J Biol Chem. 2007;282:12450–12457. doi: 10.1074/jbc.M700082200. [DOI] [PubMed] [Google Scholar]
- 57.Wang YM, Seibenhener ML, Vandenplas ML, Wooten MW. Atypical PKC zeta is activated by ceramide, resulting in coactivation of NF-kappaB/JNK kinase and cell survival. J Neurosci Res. 1999;55:293–302. doi: 10.1002/(SICI)1097-4547(19990201)55:3<293::AID-JNR4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 58.Heinrich M, Neumeyer J, Jakob M, et al. Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and -3 activation. Cell Death Differ. 2004;11:550–563. doi: 10.1038/sj.cdd.4401382. [DOI] [PubMed] [Google Scholar]
- 59.Dumitru CA, Sandalcioglu IE, Wagner M, et al. Lysosomal ceramide mediates gemcitabine-induced death of glioma cells. J Mol Med. 2009;87:1123–1132. doi: 10.1007/s00109-009-0514-8. [DOI] [PubMed] [Google Scholar]
- 60.Jarvis WD, Fornari FA, Auer KL, et al. Coordinate regulation of stress- and mitogen-activated protein kinases in the apoptotic actions of ceramide and sphingosine. Mol Pharmacol. 1997;52:935–947. doi: 10.1124/mol.52.6.935. [DOI] [PubMed] [Google Scholar]
- 61.Soengas MS, Alarcón RM, Yoshida H, et al. Apaf-1 and caspase-9 in p53-dependent apoptosis and tumor inhibition. Science. 1999;284:156–159. doi: 10.1126/science.284.5411.156. [DOI] [PubMed] [Google Scholar]
- 62.Runyan C, Schaible K, Molyneaux K, et al. Steel factor controls midline cell death of primordial germ cells and is essential for their normal proliferation and migration. Development. 2006;133:4861–4869. doi: 10.1242/dev.02688. [DOI] [PubMed] [Google Scholar]
- 63.Kulik LM, Carr BI, Mulcahy MF, et al. Safety and efficacy of 90Y radiotherapy for hepatocellular carcinoma with and without portal vein thrombosis. Hepatology. 2008;47:71–81. doi: 10.1002/hep.21980. [DOI] [PubMed] [Google Scholar]
- 64.Ahn HJ, Kim KI, Kim G, et al. Atmospheric-pressure plasma jet induces apoptosis involving mitochondria via generation of free radicals. PLoS ONE. 2011;6:e28154. doi: 10.1371/journal.pone.0028154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lorenzo HK, Susin SA, Penninger J, Kroemer G. Apoptosis inducing factor (AIF): a phylogenetically old, caspase-independent effector of cell death. Cell Death Differ. 1999;6:516–524. doi: 10.1038/sj.cdd.4400527. [DOI] [PubMed] [Google Scholar]
- 66.Newmeyer DD, Bossy-Wetzel E, Kluck RM, et al. Bcl-xL does not inhibit the function of Apaf-1. Cell Death Differ. 2000;7:402–407. doi: 10.1038/sj.cdd.4400665. [DOI] [PubMed] [Google Scholar]
- 67.Assuncao Guimaraes C, Linden R. Programmed cell deaths. Apoptosis and alternative deathstyles. Eur J Biochem. 2004;271:1638–1650. doi: 10.1111/j.1432-1033.2004.04084.x. [DOI] [PubMed] [Google Scholar]
- 68.Nagata S. Apoptotic DNA fragmentation. Exp Cell Res. 2000;256:12–18. doi: 10.1006/excr.2000.4834. [DOI] [PubMed] [Google Scholar]
- 69.Stiban J, Fistere D, Colombini M. Dihydroceramide hinders ceramide channel formation: implications on apoptosis. Apoptosis. 2006;11:773–780. doi: 10.1007/s10495-006-5882-8. [DOI] [PubMed] [Google Scholar]
- 70.Gudz TI, Tserng KY, Hoppel CL. Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J Biol Chem. 1997;272:24154–24158. doi: 10.1074/jbc.272.39.24154. [DOI] [PubMed] [Google Scholar]
- 71.García-Ruiz C, Colell A, Marí M, et al. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J Biol Chem. 1997;272:11369–11377. doi: 10.1074/jbc.272.17.11369. [DOI] [PubMed] [Google Scholar]
- 72.von Haefen C, Wieder T, Gillissen B, et al. Ceramide induces mitochondrial activation and apoptosis via a Bax-dependent pathway in human carcinoma cells. Oncogene. 2002;21:4009–4019. doi: 10.1038/sj.onc.1205497. [DOI] [PubMed] [Google Scholar]
- 73.Kim HP, Wang X, Chen Z-H, et al. Autophagic proteins regulate cigarette smoke-induced apoptosis. Autophagy. 2008;4:887–895. doi: 10.4161/auto.6767. [DOI] [PubMed] [Google Scholar]
- 74.Stiban J, Caputo L, Colombini M. Ceramide synthesis in the endoplasmic reticulum can permeabilize mitochondria to proapoptotic proteins. J Lipid Res. 2008;49:625–634. doi: 10.1194/jlr.M700480-JLR200. [DOI] [PubMed] [Google Scholar]
- 75.Basu S, Bayoumy S, Zhang Y, et al. BAD enables ceramide to signal apoptosis via Ras and Raf-1. J Biol Chem. 1998;273:30419–30426. doi: 10.1074/jbc.273.46.30419. [DOI] [PubMed] [Google Scholar]
- 76.Stoica BA, Movsesyan VA, Lea PM, Faden AI. Ceramide-induced neuronal apoptosis is associated with dephosphorylation of Akt, BAD, FKHR, GSK-3beta, and induction of the mitochondrial-dependent intrinsic caspase pathway. Mol Cell Neurosci. 2003;22:365–382. doi: 10.1016/s1044-7431(02)00028-3. [DOI] [PubMed] [Google Scholar]
- 77.Betito S, Cuvillier O. Regulation by sphingosine 1-phosphate of Bax and Bad activities during apoptosis in a MEK-dependent manner. Biochem Biophys Res Commun. 2006;340:1273–1277. doi: 10.1016/j.bbrc.2005.12.138. [DOI] [PubMed] [Google Scholar]
- 78.Sakakura C, Sweeney EA, Shirahama T, et al. Suppression of bcl-2 gene expression by sphingosine in the apoptosis of human leukemic HL-60 cells during phorbol ester-induced terminal differentiation. FEBS Lett. 1996;379:177–180. doi: 10.1016/0014-5793(95)01508-6. [DOI] [PubMed] [Google Scholar]
- 79.Shirahama T, Sakakura C, Sweeney EA, et al. Sphingosine induces apoptosis in androgen-independent human prostatic carcinoma DU-145 cells by suppression of bcl-X(L) gene expression. FEBS Lett. 1997;407:97–100. doi: 10.1016/s0014-5793(97)00304-9. [DOI] [PubMed] [Google Scholar]
- 80.Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 81.Chan FK-M, Shisler J, Bixby JG, et al. A role for tumor necrosis factor receptor-2 and receptor-interacting protein in programmed necrosis and antiviral responses. J Biol Chem. 2003;278:51613–51621. doi: 10.1074/jbc.M305633200. [DOI] [PubMed] [Google Scholar]
- 82.Kawahara A, Ohsawa Y, Matsumura H, et al. Caspase-independent cell killing by Fas-associated protein with death domain. The Journal of Cell Biology. 1998;143:1353–1360. doi: 10.1083/jcb.143.5.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Declercq W, Vanden Berghe T, Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell. 2009;138:229–232. doi: 10.1016/j.cell.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 84.Lin Y, Devin A, Rodriguez Y, Liu ZG. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999;13:2514–2526. doi: 10.1101/gad.13.19.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Feng S, Yang Y, Mei Y, et al. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal. 2007;19:2056–2067. doi: 10.1016/j.cellsig.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 86.Zhang D-W, Shao J, Lin J, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 87.Ardestani S, Deskins DL, Young PP. Membrane TNF-alpha-activated programmed necrosis is mediated by Ceramide-induced reactive oxygen species. J Mol Signal. 2013;8:12. doi: 10.1186/1750-2187-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pacheco FJ, Almaguel FG, Evans W, et al. Docosahexanoic acid antagonizes TNF-α-induced necroptosis by attenuating oxidative stress, ceramide production, lysosomal dysfunction, and autophagic features. Inflamm Res. 2014;63:859–871. doi: 10.1007/s00011-014-0760-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 90.He S, Wang L, Miao L, et al. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 91.Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 92.Shimizu S, Kanaseki T, Mizushima N, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 93.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Publ Group. 2008;9:1004–1010. doi: 10.1038/nrm2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shen H-M, Codogno P. Autophagic cell death: Loch Ness monster or endangered species? Autophagy. 2011;7:457–465. doi: 10.4161/auto.7.5.14226. [DOI] [PubMed] [Google Scholar]
- 95.Jain MV, Paczulla AM, Klonisch T, et al. Interconnections between apoptotic, autophagic and necrotic pathways: implications for cancer therapy development. J Cell Mol Med. 2013;17:12–29. doi: 10.1111/jcmm.12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Araki Y, Ku W-C, Akioka M, et al. Atg38 is required for autophagy-specific phosphatidylinositol 3-kinase complex integrity. J Cell Biol. 2013;203:299–313. doi: 10.1083/jcb.201304123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 2012;22:407–417. doi: 10.1016/j.tcb.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Uttenweiler A, Mayer A. Microautophagy in the yeast Saccharomyces cerevisiae. Methods Mol Biol. 2008;445:245–259. doi: 10.1007/978-1-59745-157-4_16. [DOI] [PubMed] [Google Scholar]
- 100.Chen H-Y, White E. Role of autophagy in cancer prevention. Cancer Prev Res (Phila) 2011;4:973–983. doi: 10.1158/1940-6207.CAPR-10-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–1109. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- 102.Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death Differ. 2005;12:1509–1518. doi: 10.1038/sj.cdd.4401751. [DOI] [PubMed] [Google Scholar]
- 104.Mehrpour M, Esclatine A, Beau I, Codogno P. Autophagy in health and disease. 1. Regulation and significance of autophagy: an overview. Am J Physiol Cell Physiol. 2010;298:C776–C785. doi: 10.1152/ajpcell.00507.2009. [DOI] [PubMed] [Google Scholar]
- 105.Ganley IG, Lam DH, Wang J, et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–12305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jung CH, Jun CB, Ro S-H, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chan EYW, Kir S, Tooze SA. siRNA screening of the kinome identifies ULK1 as a multidomain modulator of autophagy. J Biol Chem. 2007;282:25464–25474. doi: 10.1074/jbc.M703663200. [DOI] [PubMed] [Google Scholar]
- 109.Zhong Y, Wang QJ, Yue Z. Atg14L and Rubicon: yin and yang of Beclin 1-mediated autophagy control. Autophagy. 2009;5:890–891. doi: 10.4161/auto.9162. [DOI] [PubMed] [Google Scholar]
- 110.Zhong Y, Wang QJ, Li X, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11:468–476. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Pattingre S, Espert L, Biard-Piechaczyk M, Codogno P. Regulation of macroautophagy by mTOR and Beclin 1 complexes. Biochimie. 2008;90:313–323. doi: 10.1016/j.biochi.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 112.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kroemer G, Mariño G, Levine B. Autophagy and the Integrated Stress Response. Mol Cell. 2010;40:280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hanada T, Noda NN, Satomi Y, et al. The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem. 2007;282:37298–37302. doi: 10.1074/jbc.C700195200. [DOI] [PubMed] [Google Scholar]
- 115.Orsi A, Razi M, Dooley HC, et al. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell. 2012;23:1860–1873. doi: 10.1091/mbc.E11-09-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cervia D, Perrotta C, Moscheni C, et al. Nitric oxide and sphingolipids control apoptosis and autophagy with a significant impact on Alzheimer’s disease. J Biol Regul Homeost Agents. 2013;27:11–22. [PubMed] [Google Scholar]
- 118.Tibboel J, Joza S, Reiss I, et al. Amelioration of hyperoxia-induced lung injury using a sphingolipid-based intervention. Eur Respir J. 2013;42:776–784. doi: 10.1183/09031936.00092212. [DOI] [PubMed] [Google Scholar]
- 119.Tibboel J, Reiss I, de Jongste JC, Post M. Sphingolipids in lung growth and repair. Chest. 2014;145:120. doi: 10.1378/chest.13-0967. [DOI] [PubMed] [Google Scholar]
- 120.Pullmannová P, Staňková K, Pospíšilová M, et al. Effects of sphingomyelin/ceramide ratio on the permeability and microstructure of model stratum corneum lipid membranes. Biochim Biophys Acta. 2014;1838:2115–2126. doi: 10.1016/j.bbamem.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 121.Awojoodu AO, Keegan PM, Lane AR, et al. Acid sphingomyelinase is activated in sickle cell erythrocytes and contributes to inflammatory microparticle generation in SCD. Blood. 2014;124:1941–1950. doi: 10.1182/blood-2014-01-543652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pattingre S, Bauvy C, Carpentier S, et al. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J Biol Chem. 2009;284:2719–2728. doi: 10.1074/jbc.M805920200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Young MM, Takahashi Y, Khan O, et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem. 2012;287:12455–12468. doi: 10.1074/jbc.M111.309104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lépine S, Allegood JC, Edmonds Y, et al. Autophagy induced by deficiency of sphingosine-1-phosphate phosphohydrolase 1 is switched to apoptosis by calpain-mediated autophagy-related gene 5 (Atg5) cleavage. J Biol Chem. 2011;286:44380–44390. doi: 10.1074/jbc.M111.257519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Park MA, Zhang G, Martin AP, et al. Vorinostat and sorafenib increase ER stress, autophagy and apoptosis via ceramide-dependent CD95 and PERK activation. Cancer Biol Ther. 2008;7:1648–1662. doi: 10.4161/cbt.7.10.6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Scarlatti F, Bauvy C, Ventruti A, et al. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J Biol Chem. 2004;279:18384–18391. doi: 10.1074/jbc.M313561200. [DOI] [PubMed] [Google Scholar]
- 127.Li D-D, Wang L-L, Deng R, et al. The pivotal role of c-Jun NH2-terminal kinase-mediated Beclin 1 expression during anticancer agents-induced autophagy in cancer cells. Oncogene. 2009;28:886–898. doi: 10.1038/onc.2008.441. [DOI] [PubMed] [Google Scholar]
- 128.Hwang J, Lee S, Lee JT, et al. Gangliosides induce autophagic cell death in astrocytes. Br J Pharmacol. 2010;159:586–603. doi: 10.1111/j.1476-5381.2009.00563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Goñi FM, Alonso A. Biophysics of sphingolipids I. Membrane properties of sphingosine, ceramides and other simple sphingolipids. Biochim Biophys Acta. 2006;1758:1902–1921. doi: 10.1016/j.bbamem.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 130.Yamagata M, Obara K, Kihara A. Sphingolipid synthesis is involved in autophagy in Saccharomyces cerevisiae. Biochemical and Biophysical Research Communications. 2011;410:786–791. doi: 10.1016/j.bbrc.2011.06.061. [DOI] [PubMed] [Google Scholar]
- 131.Sims K, Haynes CA, Kelly S, et al. Kdo2-lipid A, a TLR4-specific agonist, induces de novo sphingolipid biosynthesis in RAW264.7 macrophages, which is essential for induction of autophagy. J Biol Chem. 2010;285:38568–38579. doi: 10.1074/jbc.M110.170621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Medler TR, Petrusca DN, Lee PJ, et al. Apoptotic Sphingolipid Signaling by Ceramides in Lung Endothelial Cells. Am J Respir Cell Mol Biol. 2008;38:639–646. doi: 10.1165/rcmb.2007-0274OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Petrache I, Petrusca DN, Bowler RP, Kamocki K. Involvement of ceramide in cell death responses in the pulmonary circulation. Proc Am Thorac Soc. 2011;8:492–496. doi: 10.1513/pats.201104-034MW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Petrache I, Medler TR, Richter AT, et al. Superoxide dismutase protects against apoptosis and alveolar enlargement induced by ceramide. Lung Cell Mol Physiol. 2008;295:L44–L53. doi: 10.1152/ajplung.00448.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gumprecht LA, Beasley VR, Weigel RM, et al. Development of fumonisin-induced hepatotoxicity and pulmonary edema in orally dosed swine: morphological and biochemical alterations. Toxicol Pathol. 1998;26:777–788. doi: 10.1177/019262339802600610. [DOI] [PubMed] [Google Scholar]
- 136.Hard GC, Howard PC, Kovatch RM, Bucci TJ. Rat kidney pathology induced by chronic exposure to fumonisin B1 includes rare variants of renal tubule tumor. Toxicol Pathol. 2001;29:379–386. doi: 10.1080/019262301316905345. [DOI] [PubMed] [Google Scholar]
- 137.Xu Z, Zhou J, McCoy DM, Mallampalli RK. LASS5 is the predominant ceramide synthase isoform involved in de novo sphingolipid synthesis in lung epithelia. J Lipid Res. 2005;46:1229–1238. doi: 10.1194/jlr.M500001-JLR200. [DOI] [PubMed] [Google Scholar]
- 138.Longo CA, Tyler D, Mallampalli RK. Sphingomyelin metabolism is developmentally regulated in rat lung. Am J Respir Cell Mol Biol. 1997;16:605–612. doi: 10.1165/ajrcmb.16.5.9160843. [DOI] [PubMed] [Google Scholar]
- 139.Adams FH, Fujiwara T, Latta H. “Alveolar” and whole lung phospholipids of premature newborn lambs. Correlations with surface tension, respiratory distress and pathology. Biol Neonate. 1971;17:198–218. doi: 10.1159/000240314. [DOI] [PubMed] [Google Scholar]
- 140.Perelman RH, Engle MJ, Kemnitz JW, et al. Biochemical and physiological development of fetal rhesus lung. J Appl Physiol Respir Environ Exerc Physiol. 1982;53:230–235. doi: 10.1152/jappl.1982.53.1.230. [DOI] [PubMed] [Google Scholar]
- 141.Rubenfeld GD. Epidemiology and outcomes of acute lung injury. Chest. 2007;131:554–562. doi: 10.1378/chest.06-1976. [DOI] [PubMed] [Google Scholar]
- 142.Lin W-C, Lin C-F, Chen C-L, et al. Inhibition of neutrophil apoptosis via sphingolipid signaling in acute lung injury. J Pharmacol Exp Ther. 2011;339:45–53. doi: 10.1124/jpet.111.181560. [DOI] [PubMed] [Google Scholar]
- 143.Zemans RL, Colgan SP, Downey GP. Transepithelial Migration of Neutrophils. Am J Respir Cell Mol Biol. 2009;40:519–535. doi: 10.1165/rcmb.2008-0348TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Uhlig S, Yang Y. Sphingolipids in Acute Lung Injury. In: Gulbins E, Petrache I, editors. Sphingolipids in Disease. Vienna: Springer; 2013. pp. 227–246. [DOI] [PubMed] [Google Scholar]
- 145.Fox S, Leitch AE, Duffin R, et al. Neutrophil apoptosis: relevance to the innate immune response and inflammatory disease. J Innate Immun. 2010;2:216–227. doi: 10.1159/000284367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Grommes J, Soehnlein O. Contribution of neutrophils to acute lung injury. Mol Med. 2011;17:293–307. doi: 10.2119/molmed.2010.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Taneja R, Parodo J, Jia SH, et al. Delayed neutrophil apoptosis in sepsis is associated with maintenance of mitochondrial transmembrane potential and reduced caspase-9 activity. Crit Care Med. 2004;32:1460–1469. doi: 10.1097/01.ccm.0000129975.26905.77. [DOI] [PubMed] [Google Scholar]
- 148.Mastrandrea LD, Sessanna SM, Laychock SG. Sphingosine kinase activity and sphingosine-1 phosphate production in rat pancreatic islets and INS-1 cells: response to cytokines. Diabetes. 2005;54:1429–1436. doi: 10.2337/diabetes.54.5.1429. [DOI] [PubMed] [Google Scholar]
- 149.Chihab R, Pörn-Ares MI, Alvarado-Kristensson M, Andersson T. Sphingosine 1-phosphate antagonizes human neutrophil apoptosis via p38 mitogen-activated protein kinase. Cell Mol Life Sci. 2003;60:776–785. doi: 10.1007/s00018-003-2357-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ehrenkranz RA, Walsh MC, Vohr BR, et al. Validation of the National Institutes of Health consensus definition of bronchopulmonary dysplasia. Pediatrics. 2005;116:1353–1360. doi: 10.1542/peds.2005-0249. [DOI] [PubMed] [Google Scholar]
- 151.Baraldi E, Filippone M. Chronic Lung Disease after Premature Birth. The New England Journal of Medicine. 2007;357:1946–1955. doi: 10.1056/NEJMra067279. [DOI] [PubMed] [Google Scholar]
- 152.Jobe AH. Lung development and lung injury—the new BPD. Chin J Contemp Pediatr. 2001;3:433–437. [Google Scholar]
- 153.Hosford GE, Olson DM. Effects of hyperoxia on VEGF, its receptors, and HIF-2alpha in the newborn rat lung. Lung Cell Mol Physiol. 2003;285:L161–L168. doi: 10.1152/ajplung.00285.2002. [DOI] [PubMed] [Google Scholar]
- 154.Jakkula M, Le Cras TD, Gebb S, et al. Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Lung Cell Mol Physiol. 2000;279:L600–L607. doi: 10.1152/ajplung.2000.279.3.L600. [DOI] [PubMed] [Google Scholar]
- 155.Le Cras TD, Markham NE, Tuder RM, et al. Treatment of newborn rats with a VEGF receptor inhibitor causes pulmonary hypertension and abnormal lung structure. Lung Cell Mol Physiol. 2002;283:L555–L562. doi: 10.1152/ajplung.00408.2001. [DOI] [PubMed] [Google Scholar]
- 156.Yasuo M, Mizuno S, Allegood J, et al. Fenretinide causes emphysema, which is prevented by sphingosine 1-phoshate. PLoS One. 2013;8:e53927. doi: 10.1371/journal.pone.0053927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.May M, Strobel P, Preisshofen T, et al. Apoptosis and proliferation in lungs of ventilated and oxygen-treated preterm infants. Eur Respir J. 2004;23:113–121. doi: 10.1183/09031936.03.00038403. [DOI] [PubMed] [Google Scholar]
- 158.Dieperink HI, Blackwell TS, Prince LS. Hyperoxia and apoptosis in developing mouse lung mesenchyme. Pediatr Res. 2006;59:185–190. doi: 10.1203/01.pdr.0000196371.85945.3a. [DOI] [PubMed] [Google Scholar]
- 159.Worgall TS, Veerappan A, Sung B, et al. (2013) Impaired sphingolipid synthesis in the respiratory tract induces airway hyperreactivity. Sci Trans Med 5:186ra67–186ra67. doi: 10.1126/scitranslmed.3005765 [DOI] [PubMed]
- 160.Maceyka M, Spiegel S. Sphingolipid metabolites in inflammatory disease. Nature. 2014;510:58–67. doi: 10.1038/nature13475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ammit AJ, Hastie AT, Edsall LC, et al. Sphingosine 1-phosphate modulates human airway smooth muscle cell functions that promote inflammation and airway remodeling in asthma. FASEB J. 2001;15:1212–1214. doi: 10.1096/fj.00-0742fje. [DOI] [PubMed] [Google Scholar]
- 162.Uhlig S, Gulbins E. Sphingolipids in the Lungs. Am J Respir Crit Care Med. 2008;178:1100–1114. doi: 10.1164/rccm.200804-595SO. [DOI] [PubMed] [Google Scholar]
- 163.Ediger TL, Toews ML. Synergistic stimulation of airway smooth muscle cell mitogenesis. JPET. 2000;294:1076–1082. [PubMed] [Google Scholar]
- 164.Plano D, Amin S, Sharma AK. Importance of sphingosine kinase (SphK) as a target in developing cancer therapeutics and recent developments in the synthesis of novel SphK inhibitors. J Med Chem. 2014;57:5509–5524. doi: 10.1021/jm4011687. [DOI] [PubMed] [Google Scholar]
- 165.Ono JG, Worgall TS, Worgall S. 17q21 locus and ORMDL3: an increased risk for childhood asthma. Pediatr Res. 2013;75:165–170. doi: 10.1038/pr.2013.186. [DOI] [PubMed] [Google Scholar]
- 166.Galanter J, Choudhry S, Eng C, et al. ORMDL3 gene is associated with asthma in three ethnically diverse populations. Am J Respir Crit Care Med. 2008;177:1194–1200. doi: 10.1164/rccm.200711-1644OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Moffatt MF, Kabesch M, Liang L, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470–473. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- 168.Breslow DK, Collins SR, Bodenmiller B, et al. Orm family proteins mediate sphingolipid homeostasis. Nature. 2010;463:1048–1053. doi: 10.1038/nature08787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Siow DL, Wattenberg BW. Mammalian ORMDL proteins mediate the feedback response in ceramide biosynthesis. J Biol Chem. 2012;287:40198–40204. doi: 10.1074/jbc.C112.404012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Md DDMM, Md ASB. ReviewGlobal burden of COPD: risk factors, prevalence, and future trends. Lancet. 2007;370:765–773. doi: 10.1016/S0140-6736(07)61380-4. [DOI] [PubMed] [Google Scholar]
- 171.Yoshida T, Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev. 2007;87:1047–1082. doi: 10.1152/physrev.00048.2006. [DOI] [PubMed] [Google Scholar]
- 172.Gulbins E, Petrache I. Sphingolipids Dis. 2013;216:1–473. [Google Scholar]
- 173.Segura-Valdez L, Pardo A, Gaxiola M, et al. Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest. 2000;117:684–694. doi: 10.1378/chest.117.3.684. [DOI] [PubMed] [Google Scholar]
- 174.Yokohori N, Aoshiba K, Nagai A, Respiratory Failure Research Group in Japan Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysema. Chest. 2004;125:626–632. doi: 10.1378/chest.125.2.626. [DOI] [PubMed] [Google Scholar]
- 175.Calabrese F, Giacometti C, Beghe B, et al. Marked alveolar apoptosis/proliferation imbalance in end-stage emphysema. Respir Res. 2005;6:14. doi: 10.1186/1465-9921-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Imai K, Mercer BA, Schulman LL, et al. Correlation of lung surface area to apoptosis and proliferation in human emphysema. Eur Respir J. 2005;25:250–258. doi: 10.1183/09031936.05.00023704. [DOI] [PubMed] [Google Scholar]
- 177.Tibboel J, Reiss I, de Jongste JC, Post M. Ceramides: a potential therapeutic target in pulmonary emphysema. Respir Res. 2013;14(1):96. doi: 10.1186/1465-9921-14-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ravid T, Tsaba A, Gee P, et al. Ceramide accumulation precedes caspase-3 activation during apoptosis of A549 human lung adenocarcinoma cells. Lung Cell Mol Physiol. 2003;284:L1082–L1092. doi: 10.1152/ajplung.00172.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chung S, Vu S, Filosto S, Goldkorn T. Src regulates cigarette smoke-induced ceramide generation via nsmase2 in the airway epithelium. Am J Respir Cell Mol Biol. 2014 doi: 10.1165/rcmb.2014-0122OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Scarpa MC, Baraldo S, Marian E, et al. Ceramide expression and cell homeostasis in chronic obstructive pulmonary disease. Respiration. 2013;85:342–349. doi: 10.1159/000341185. [DOI] [PubMed] [Google Scholar]
- 181.Chen Z-H, Kim HP, Sciurba FC, et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One. 2008;3:e3316. doi: 10.1371/journal.pone.0003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Chen Z-H, Lam HC, Jin Y, et al. Autophagy protein microtubule-associated protein 1 light chain-3B (LC3B) activates extrinsic apoptosis during cigarette smoke-induced emphysema. Proc Natl Acad Sci USA. 2010;107:18880–18885. doi: 10.1073/pnas.1005574107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ryter SW, Chen Z-H, Kim HP, Choi AMK. Autophagy in chronic obstructive pulmonary disease: homeostatic or pathogenic mechanism? Autophagy. 2009;5:235–237. doi: 10.4161/auto.5.2.7495. [DOI] [PubMed] [Google Scholar]
- 184.Fujii S, Hara H, Araya J, et al. Insufficient autophagy promotes bronchial epithelial cell senescence in chronic obstructive pulmonary disease. Oncoimmunology. 2012;1:630–641. doi: 10.4161/onci.20297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73:1251–1254. doi: 10.1016/0092-8674(93)90353-r. [DOI] [PubMed] [Google Scholar]
- 186.Rommens JM, Iannuzzi MC, Kerem B-S, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- 187.Stephenson AL, Tom M, Berthiaume Y, et al. A contemporary survival analysis of individuals with cystic fibrosis: a cohort study. Eur Respir J. 2014 doi: 10.1183/09031936.00119714. [DOI] [PubMed] [Google Scholar]
- 188.Hamai H, Keyserman F, Quittell LM, Worgall TS. Defective CFTR increases synthesis and mass of sphingolipids that modulate membrane composition and lipid signaling. J Lipid Res. 2009;50:1101–1108. doi: 10.1194/jlr.M800427-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Bodas M, Min T, Mazur S, Vij N. Critical modifier role of membrane-cystic fibrosis transmembrane conductance regulator-dependent ceramide signaling in lung injury and emphysema. J Immunol. 2011;186:602–613. doi: 10.4049/jimmunol.1002850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ryter SW, Lee S-J, Choi AM. Autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. Expert Rev Respir Med. 2010;4:573–584. doi: 10.1586/ers.10.61. [DOI] [PMC free article] [PubMed] [Google Scholar]