

Abstract
Scope
Eicosapentaenoic acid (EPA), abundant in oily fish, is reported to reduce skin inflammation and provide photoprotection, potential mechanisms include competition with arachidonic acid (AA) for metabolism by cyclooxygenases/lipoxygenases to less pro-inflammatory mediators. We thus examine impact of EPA intake on levels of AA, EPA and their resulting eicosanoids in human skin with or without ultraviolet radiation (UVR) challenge.
Methods and results
In a double-blind randomised controlled study, 79 females took 5 g EPA-rich or control lipid for 12 wk. Pre- and post-supplementation, red blood cell and skin polyunsaturated fatty acids were assessed by GC, and eicosanoids from unexposed and UVR-exposed skin by LC-MS/MS. Active supplementation increased red blood cell and dermal EPA versus control (both p < 0.001), lowering relative AA:EPA content (4:1 versus 15:1 and 5:1 versus 11:1, respectively; both p < 0.001). Pre-supplementation, UVR increased PGE2, 12-hydroxyeicosatetraenoic acids, 12-HEPE (all p < 0.001) and PGE3 (p < 0.05). Post-EPA, PGE2 was reduced in unchallenged skin (p < 0.05) while EPA-derived PGE3 (non-sign) and 12-HEPE (p < 0.01) were elevated post-UVR. Thus, post-EPA, PGE2:PGE3 was lower in unchallenged (12:1 versus 28:1; p < 0.05) and UVR exposed (12:1 versus 54:1; p < 0.01) skin; 12-hydroxyeicosatetraenoic acids:12-HEPE was lower in UVR-exposed skin (3:1 versus 11:1; p < 0.001).
Conclusion
Dietary EPA augments skin EPA:AA content, shifting eicosanoid synthesis towards less pro-inflammatory species, and promoting a regulatory milieu under basal conditions and in response to inflammatory insult.
Keywords: EPA, 12-HETE, Inflammation, PGE2, UVR
1. Introduction
Exposure of the skin to ultraviolet radiation (UVR) induces acute inflammation characterised clinically by erythema, which becomes visible after 4–6 h and peaks in intensity at 24–48 h post-exposure. This vasodilatory response is accompanied by a dermal leukocytic infiltrate comprising neutrophils, macrophage, mast cells and T-lymphocytes 1,2 and, when severe, can be associated with oedema and pain. Prostaglandin (PG)E2, nitric oxide and pro-inflammatory cytokines are established mediators of UVR-induced skin inflammation, but more recently it has been demonstrated that a wider range of eicosanoids are implicated in the sunburn response 3. Ultraviolet radiation stimulates the membrane release of omega-6 (n-6) polyunsaturated fatty acid, arachidonic acid (AA) and its metabolism by cyclooxygenase (COX) and lipoxygenase (LOX) enzymes 4,5, augmenting the synthesis of PG and hydroxyeicosatetraenoic acids (HETE), respectively. While at low concentrations, these eicosanoids regulate tissue homeostasis, at higher concentrations they mediate inflammatory processes. In UVR-exposed skin, PGE2 levels are elevated and peak in the first 24 h of the sunburn response, stimulating vasodilatation and epidermal cell proliferation 3,5–7. In contrast, 12-HETE, the most abundant HETE expressed in UVR-inflamed skin, continues to rise at 72-h post-UVR exposure 3. This eicosanoid has powerful leukocyte chemoattractant properties 8–10 and is believed to promote the infiltration of leukocytes into the skin following UVR exposure 3.
Eicosapentaenoic acid (EPA), a long chain omega-3 (n-3) PUFA, competes with AA for release from membrane phospholipids and for metabolism to eicosanoids by COX and LOX 11. Synthesis of eicosanoids from EPA is reported to occur with reduced efficiency compared to AA 12 and many EPA-derived metabolites exhibit less potent inflammatory properties than AA-derived species in vitro 13–15. Additionally, it is reported that EPA can reduce expression of COX-2 in some in vitro models 16,17. The photoprotective potential of n-3 PUFA supplements is supported by in vivo studies when administered topically 18,19 or systemically 20–25, and small studies in humans indicate that oral EPA reduces the sunburn response in association with reduced PGE2. However, the potential effects of EPA through an impact on LOX-produced eicosanoids, and perturbation in the relative profiles of AA- and EPA-derived eicosanoids in human skin require further exploration.
We hypothesise that increased EPA ingestion may, through altered skin AA/EPA content, result in modification of the profiles of their respective COX- and LOX-derived eicosanoids that could counter inflammation in human skin. Accordingly, this double-blind randomised controlled study in 79 women examined the influence of 12-wk dietary EPA supplementation on skin levels of AA and EPA and the consequent impact on eicosanoid levels, in both basal skin and skin following a standardised pro-inflammatory UVR challenge. Attention was focused on the most abundant skin AA-derived pro-inflammatory eicosanoids, i.e. PGE2 and 12-HETE, and their EPA-derived counterparts PGE3 and 12-HEPE.
2. Materials and methods
2. 1. Participants
Seventy-nine healthy female volunteers were recruited to the study. All volunteers had a history of nickel contact allergy as this study was performed as part of a trial assessing the effects of systemic EPA-rich lipid on skin immunity. Recruitment was from the contact dermatitis investigation unit at Salford Royal Hospital, Manchester, UK and by open advertisement. Volunteers were eligible to take part if they were aged between 18 and 60 years and of sun reactive skin type I or II (i.e. sunburns easily, with no or minimal ability to tan). Volunteers were excluded from the study if they were taking n-3 PUFA-rich supplements, were pregnant or breastfeeding, had sunbathed or used a tanning bed in 3 months prior to the study and if they were taking photoactive medication, had a history of photosensitivity, skin cancer, or atopy. They did not have active contact dermatitis at the time of the study. Ethical approval was granted by North Manchester local research ethics committee (08/H1006/30) and the study was performed in accordance with the Declaration of Helsinki principles (revised Seoul 2008). Written informed consent was provided by all volunteers before inclusion in the study.
2. 2. Study design and intervention
The study took place in the Photobiology Unit, Dermatology Centre, Salford Royal Hospital (Manchester, UK) between 2008 and 2010. It was a double-blind randomised (1:1) controlled parallel group study. The treatment allocation sequence was by permuted block design (mixed blocks of four to six) and was produced by the study biostatistician using statistical software (v2.7.7; StatsDirect, Altrincham, UK). Active and control lipid supplements, identical in appearance, were packaged and labeled with sequential numbers according to the allocation sequence by GP Solutions (Manchester, UK), and the code was held securely by the study biostatistician until study completion. Volunteers were assigned the intervention on enrolment to the study and in view of ethical restrictions on the number of samples taken per volunteer, were concurrently randomised to have either suction blister fluid sampled for analysis of skin lipid mediators or have skin punch biopsies taken for assessment of dermal EPA and AA levels. All volunteers provided blood samples pre- and post-supplementation to assess their compliance, through measurement of red blood cell (RBC) EPA and AA levels. A UVR dose series was applied to volunteers’ buttock skin pre- and post-supplementation for assessment and quantification of their sunburn response. A standardised pro-inflammatory UVR challenge was given to small areas of buttock skin pre- and post-supplementation in order that skin biopsy and blister fluid samples could be taken from both unexposed and UVR-exposed skin. The EPA supplements were 1 g gelatine capsules containing Incromega E7010 SR ethyl ester, which comprises ∼70% EPA and 10% DHA (Croda Chemicals Leek, Staffordshire, UK). The control supplements comprised 1 g gelatine capsules of identical appearance containing glyceryl tricoprylate coprate (Croda Chemicals Leek), a medium chain triglyceride containing four fatty acids: caproic, caprylic, capric and lauric acid, in the ratio of 2:55:42:126–28. Both supplements were taken as five capsules daily with breakfast for 12 wk.
2. 3. UVR exposure
The lamps used to administer the UVR doses were a TL 20W/12 fluorescent broadband UVR lamp (270–400 nm, peak 310 nm; Philips, Hamburg, Germany) housed in a black plastic casing which contains five apertures, each 10 mm in diameter and incorporating a series of neutral density filters (Medical Physics Department, Dryburn Hospital, Durham, UK) 29 and a Waldman UV800 canopy fitted with 10 Waldmann 20W UV21 fluorescent broadband UVR lamps (270–400 nm; Waldmann, VS-Schwenningen, Germany). The two lamps emit exactly the same spectrum of radiation, i.e. 44% UVB, 56% UVA and 1% UVC, with the difference in designation solely due to different manufacturers. The irradiance was monitored during each exposure using radiometers (Medical Physics Department, Dryburn Hospital and Waldmann IL 730A, International Light, Newburyport, MA, USA) calibrated for the two sources, respectively, and with standards ultimately traceable to the UK National Physical Laboratory. Pre-supplementation, a geometric series of ten erythemally weighted UVR doses was applied to the upper buttock of each volunteer, and the individual's minimal erythemal dose (MED; see below) was determined at 24-h post-exposure. Sites on the upper buttock were then exposed to four times the individual's MED of UVR prior to blister fluid sampling, in order to provide a pro-inflammatory challenge. Following the supplement course, the individualised dose of UVR (4XMED) and the MED test were repeated.
2. 4. Erythema assessment
The MED was defined as the lowest dose of UVR (mJ/cm2) causing a visually perceptible erythema at 24-h post-UVR exposure. Erythemal intensity was also measured using a reflectance instrument (Dia-Stron; Andover, UK), providing an erythema index. Readings were taken in triplicate at each of the ten UVR-exposed sites and at two adjacent unexposed sites. The mean of the latter readings was subtracted from readings taken at the UVR-exposed sites to give delta E values; these were used to construct UVR-erythema dose response curves with non-linear regression using designated software (Copyright © Regional Medical Physics Department, Gateshead and South Tyneside Health Authority, 1999) as previously described 30. The MED and dose response measurements allowed for comparison of UVR-induced erythema between supplement groups.
2. 5. Tissue sampling
All tissue sampling was performed both pre- and post-supplement course.
Blood sampling: Blood samples (7 mL) were taken from the antecubital fossa of all participants and collected in 3.4 mL EDTA monovette tubes (Sarsstedt, Newton, UK). Samples were centrifuged for 15 min at 1500 rpm and the RBC fractions were collected and stored at −70°C until analysis.
Suction blister fluid sampling: Suction blisters were raised using commercially available hand-held vacuum pumps (Mityvac, St. Louis, MO, USA) and perspex suction cups containing a central 1 cm diameter aperture (Medical Physics Department, Salford Royal Hospital, Manchester, UK) as described 3. In brief, suction cups were placed on unexposed buttock skin and buttock skin exposed 24 h earlier to four times the MED of UVR. Pumps were maintained at a constant pressure of 250 mm Hg for approximately 90 min until blisters formed. Blister fluid was aspirated using a syringe, snap frozen in liquid nitrogen and stored at −70°C until analysis.
Skin sampling: Skin biopsies were performed after intradermal injection of Lidocaine (2%) local anesthetic (Antigen Pharmaceuticals, Tipperary, Ireland). Five millimetres diameter punch biopsies were taken using Militex biopsy punches (Militex, York, PA, USA) and placed into 1.8 mL cryotubes (Thermo Fisher Scientific, Roskilde, Denmark) prior to snap freezing in liquid nitrogen. Samples were stored at −70°C until analysis.
All samples were analysed at completion of the human study.
2. 6. Assessment of red blood cell (RBC) and dermal AA and EPA
Skin samples were defrosted on ice. Dermal tissue sections were available for this analysis and these were incubated in chloroform:methanol (4 mL) (2:1, v/v) containing butylated hydroxy toluene (BHT) 0.01% w/v overnight at 4°C and homogenised using a blade homogeniser. Lipids were extracted using a further two volumes of chloroform:methanol:BHT as described 31. RBC samples (1 mL) were defrosted on ice and lipids were extracted with chloroform:methanol:BHT (4 mL). The solvent was removed under nitrogen and the lipid extract was stored at −20°C prior to analysis.
Fatty acid methyl esters were prepared using BF3 in methanol, and analysed by gas chromatography on a BPX70 GC capillary column (SGE Europe, Milton Keynes, UK), using helium as the carrier gas and a flame ionisation detector. Heneicosaenoic acid (C21:0) was used as the internal standard (ACS reagent, Sigma Aldrich, UK) with a 37 fatty acid methyl esters mixed standard (Supelco, Belleforte, PA, USA) the reference for identification of fatty acids. The principal PUFA measured were EPA and AA, expressed as percentage of total fatty acids in each sample.
2. 7. Assessment of eicosanoids
Lipidomic analyses were performed as described previously 32,33. In summary, lipid mediators were extracted by diluting blister fluid samples (50–200 μL) in methanol-water (15% w/w). Internal standards PGB2-d4 (40 ng) and 12-HETE-d8 (80 ng) (Cayman Chemicals, Ann Arbor, MI, USA) were added to each sample. Solutions were then acidified to pH 3.0 and applied to preconditioned SPE cartridges (C18-E 500 mg, 6 mL) (Phenomenex, Macclesfield, UK) and lipid mediators eluted with methyl formate. Chromatographic analysis was performed on a C18 column (Luna, 5 μm, 2.0 mm, Phenomenex, Macclesfield, UK) using HPLC (Alliance 2695, Waters, Elstree, Hertfordshire, UK) coupled to a triple quadrupole mass spectrometer with ESI (Quattro Ultima, Waters). The following multiple reaction monitoring transitions were used to assay for the presence of prostanoids and hydroxy fatty acids in the blister fluid extracts: PGE1 m/z 353 >317, PGE2 m/z 351>271, PGE3 m/z 349>269, 13,14 dihydro-15-keto PGE2 m/z 351>333, 11-HETE m/z 319>167, 12-HETE m/z 319>179, 15-HETE m/z 319>175, 11-HEPE m/z 317>167, 12-HEPE m/z 317>179, 15-HEPE m/z 317>175, 15-HETrE m/z 321>221, 9-HODE m/z 295>171 and 13-HODE m/z 295>195. Results are expressed as picogram of eicosanoid per microlitre of blister fluid, based on calibration lines constructed from commercially available standards (Cayman Chemicals).
2. 8. Statistical analysis
The study was powered to detect a difference in aspects of skin immunity between active and control groups 34. Previous studies support that the subject number exceeded that for detection of an impact of n-3 PUFA on skin UVR-induced inflammatory endpoints, for example it is estimated that to detect a 30% difference in the sunburn threshold between treatment groups at the 5% significance level and with 90% power ≥14 subjects per group are required 22. Statistical analysis was performed in SPSS 16.0 and the normality of the data was determined using the Kolmogorov–Smirnov test. RBC and skin PUFA and eicosanoid data were transformed using natural log (ln) to obtain normally distributed data. ANCOVA analyses were performed to compare differences in UVR-induced erythema, tissue PUFA content and skin eicosanoid levels between EPA and control groups post-supplementation with baseline data as the covariate. Paired t-tests were performed to compare eicosanoids levels in unexposed and UVR-exposed skin within groups. A p value of <0.05 was considered statistically significant.
3. Results
3. 1. Volunteers and compliance
Seventy-nine volunteers were recruited but six did not complete due to personal reasons unrelated to the study (Fig.1). Three volunteers in the EPA group showed no increase in RBC EPA levels post-supplementation and were thus excluded from analyses for poor compliance (all three were in the suction blister subgroup). Of the remaining 70 volunteers (median age (range) 43 years (22–60)) who completed the study, 33 were in the control group and 37 in the EPA supplementation group. No adverse effects were reported for either of the oral supplements. Baseline dietary EPA intake in the study population was found to be 23 mg/day as assessed by food frequency questionnaire 35. This average intake is below the current recommendations for the UK according to the Food Standards Agency and Scientific Advisory Committee on Nutrition 36.
Figure 1.
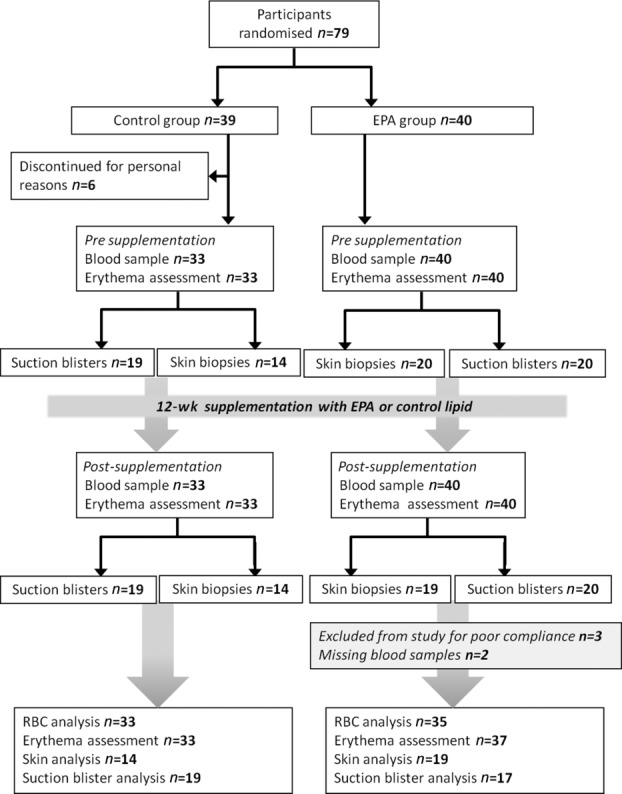
Flow diagram of study design and volunteer participation.
3. 2. Tissue AA and EPA content and AA:EPA ratio
3. 2. 1. RBC
Due to technical reasons, there was no RBC PUFA data for one individual and no post-supplementation data for another; both were in the n-3 PUFA group. Therefore, RBC PUFA data was available for 35 individuals in the EPA group and 33 individuals in the control group. Pre-supplementation (baseline) levels of EPA and AA did not differ between the two groups, therefore the groups were combined. Baseline EPA levels were less than 0.9% of total fatty acids and AA accounted for 13% of the total fatty acids (Table1). After supplementation, the mean percent EPA was elevated in the EPA group where levels were fourfold higher than in the control group (p < 0.001; Table1). There was no significant change in the AA content of RBC after supplementation. At baseline, the mean AA:EPA ratio of the pooled control and EPA group was 18:1 (percent of total fatty acids; Table1). Following 3 months of supplementation, the AA:EPA ratio in the EPA group was significantly lower than in the control group (4:1 versus 15:1, p < 0.001; Table1).
Table 1.
Tissue AA and EPA content at baseline and following 12-wk supplementation with 5 g/day of EPA-rich or control lipid
| Mean (SEM) (Percent of total fatty acids) | ||||
|---|---|---|---|---|
| Baselinea | Control | EPA | ||
| RBCb | AA | 12.9 (0.4) | 12.8 (0.4) | 11.8 (0.3) |
| EPA | 0.9 (0.05) | 0.9 (0.06) | 3.6 (0.2)*** | |
| AA:EPA | 18:1 | 15:1 | 4:1*** | |
| Dermisc | AA | 0.7 (0.05) | 0.7 (0.09) | 0.6 (0.04) |
| EPA | 0.07 (0.005) | 0.06 (0.009) | 0.12 (0.008)*** | |
| AA:EPA | 11:1 | 11:1 | 5:1*** | |
Baseline: combined presupplementation group data.
RBC: baseline n = 68, control n = 33, EPA n = 35.
Dermis: baseline n = 33, control n = 14, EPA n = 19
***p < 0.001 compared to the control group postsupplementation.
3. 2. 2. Dermis
One volunteer declined to have skin sampling post-supplementation therefore dermal PUFA data was available for 19 volunteers in the EPA group and 14 in the control group. At baseline, both treatment groups had the same dermal content of EPA and AA, thus the baseline data was combined. Prior to supplementation, the percent EPA was 0.07% and AA content was 0.7% (Table1). At 12 wk, the mean percent EPA in the dermis was increased following EPA and was twofold higher than in the control group (p < 0.001; Table1). At baseline, the mean AA:EPA ratio of the pooled groups was 11:1 (Table1). Post-supplementation, the dermal AA:EPA ratio in the EPA group was significantly lower than in the control group (5:1 versus 11:1, p < 0.001; Table1).
3. 3. COX-mediated eicosanoid production: PGE2 and PGE3
PGE2 and 12-HETE and their EPA-derived counterparts PGE3 and 12-HEPE were the eicosanoids of interest in this study due to their abundance and pro-inflammatory properties. Results for a complete range of eicosanoids quantified in the blister fluid are shown in Supporting Information Table 1; no significant differences between active and control groups were seen other than those presented below.
3. 3. 1. PGE2
At baseline, PGE2 (mean (SEM)) was elevated twofold in skin blister fluid following UVR exposure (p < 0.001; Table2). Following 3 months of supplementation, the mean level of PGE2 in unexposed skin was ∼30% lower in the EPA group compared to the control group (p < 0.05; Table2), while there was no significant difference in UVR-exposed skin.
Table 2.
EPA- and AA-derived eicosanoids in skin blister fluid at baseline and following 12-wk supplementation with 5 g/day of EPA-rich or control lipid
| Mean (SEM) (pg/μL)a | ||||||
|---|---|---|---|---|---|---|
| Baseline (n = 36)b | Control (n = 19) | EPA (n = 17) | ||||
| Unexposed | UVR exposed | Unexposed | UVR exposed | Unexposed | UVR exposed | |
| PGE2 | 10.2 (1.5) | 20.8 (2.4)††† | 10.7 (2.2) | 28.1 (5.4)†† | 6.0 (1.1)* | 19.9 (3.4)††† |
| PGE3 | 0.6 (0.1) | 1.1 (0.2)† | 0.6 (0.2) | 1.2 (0.3)† | 0.8 (0.2) | 3.1 (1.0)† |
| 12-HETE | 12.2 (1.3) | 35.4 (4.0)††† | 13.1 (2.9) | 51.4 (8.6)††† | 13.4 (3.8) | 50.3 (8.0)††† |
| 12-HEPE | 2.8 (0.3) | 4.5 (0.4)††† | 3.0 (0.6) | 6.4 (1.9)† | 5.9 (1.6) | 18.2 (3.4)†††,** |
Results are presented as picogram of eicosanoid per microlitre of blister fluid.
Baseline: combined treatment groups pre-supplementation.
†p < 0.05, ††p < 0.01, †††p < 0.001 compared to unexposed skin in the same group, *p < 0.05, **p < 0.01 compared to the corresponding skin (i.e. unexposed or UVR exposed) in the control group post-supplementation.
3. 3. 2. PGE3
At baseline, the mean level of PGE3 was increased ∼twofold in skin blister fluid following UVR exposure (p < 0.05; Table2). Post-supplementation, the mean level of PGE3 in unexposed skin in the EPA group was similar to that in the control group. In UVR-exposed skin, PGE3 levels were 2.6-fold higher in the EPA group than the control group (Table2), although this difference did not show statistical significance. Post-supplementation, the UVR-induced rise in PGE2 and PGE3 was maintained in both the control and EPA group (all p < 0.05).
3. 3. 3. PGE2:PGE3 ratio
At baseline, the mean ratio of blister fluid PGE2:PGE3 in all volunteers combined was elevated by UVR, from 27:1 in unexposed skin to 47:1 (p = 0.054). After 3 months of EPA supplementation, the ratio in unexposed skin was ∼50% lower than in the control group (12:1 versus 28:1, p < 0.05), and in UVR-exposed skin, the ratio was ∼75% lower than in the control group (12:1 versus 54:1, p < 0.01; Fig.2). Notably, the pre-supplementation rise in PGE2:PGE3 ratio induced by UVR was abolished post-EPA supplementation.
Figure 2.
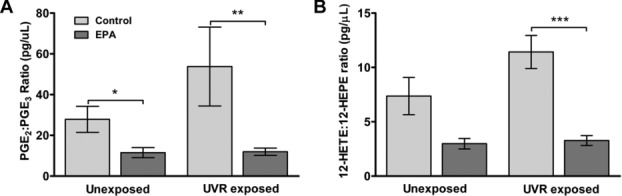
(A) PGE2:PGE3 ratio (B) and 12-HETE:12-HEPE ratio postsupplementation with 5 g/day of EPA-rich or control lipid (control n = 19; EPA n = 17). Results are expressed as mean ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001 compared to control group).
3. 4. LOX-mediated eicosanoid production: 12-HETE and 12-HEPE
3. 4. 1. 12-HETE
In all volunteers combined pre-supplementation the mean level of 12-HETE was raised threefold in UVR-exposed skin compared to unexposed skin (p < 0.001; Table2). Supplementation with n-3 PUFA had no effect on the absolute level of 12-HETE in human blister fluid.
3. 4. 2. 12-HEPE
Baseline levels of 12-HEPE were elevated 1.6-fold in skin blister fluid in response to UVR exposure (p < 0.001; Table2). Post-supplementation 12-HEPE levels were elevated ∼twofold in unexposed skin in the EPA group compared to the control group, although this did not show statistical significance. In UVR-exposed skin, levels of 12-HEPE were threefold higher in the EPA group than in the control group (p < 0.01; Table2). Post-supplementation, the UVR-induced rise in 12-HETE and 12-HEPE was maintained in both the control and EPA group (all p < 0.05).
3.4.3. 12-HETE:12-HEPE ratio
At baseline, the mean 12-HETE:12-HEPE ratio rose from 7:1 in unexposed skin to 9:1 in UVR-exposed skin (p < 0.01). Post-supplementation, in the EPA group 12-HETE:12-HEPE ratio was ∼60% lower than that of the control group in unexposed skin (3:1 versus 7:1); this was not statistically significant. In UVR-exposed skin, the 12-HETE:12-HEPE ratio was ∼70% lower in the EPA group than the control group (3:1 versus 11:1, p < 0.001; Fig.2). As with the PGE2:PGE3 ratio, the UVR-induced increase in 12-HETE:12-HEPE seen at baseline was abolished following 12-wk EPA supplementation.
3. 5. Erythemal response to UVR in human skin
There was no difference in the mean (SEM) MED in the EPA group versus the control group post-supplementation (29 (1.3) mJ/cm2 versus 28.5 (2.2) mJ/cm2). In order to determine the impact of oral EPA on the slope of the UVR–erythemal dose responses, i.e. the size of the response per log unit change in dose, slope analysis was performed on the dose response curves. The mean (SEM) Hill slope value for the baseline UVR–erythema dose response was 0.055 (0.004). There was no difference in the slope of the dose response curve post-supplementation between control and EPA groups (0.051 (0.004) versus 0.054 (0.005).
4. Discussion
This double-blind randomised controlled nutritional study has demonstrated how enhanced oral EPA intake can profoundly alter tissue PUFA composition, with consequent impact on the eicosanoid profile in human skin. The AA-derived mediators PGE2 and 12-HETE are highly expressed, potent eicosanoids in basal and UVR-exposed skin 3; hence they and their EPA-derived counterparts PGE3 and 12-HEPE were the focus of this assessment. The higher availability of EPA in the active group post-supplementation resulted in a doubling of the relative dermal EPA:AA content, which was accompanied by a doubling of the basal PGE3:PGE2 level, while following UVR-exposure, skin PGE3:PGE2 increased threefold and the 12-HEPE:12-HETE was fourfold higher than on control lipid (Fig. 3). Lower absolute levels of PGE2 were seen in the unexposed skin of the EPA-supplemented group, and following a pro-inflammatory UVR challenge, there were increases in the levels of EPA-derived PGE3 and 12-HEPE. Prostaglandin E2 is known to be an important regulator of numerous cellular responses during homeostasis, including keratinocyte proliferation and apoptosis 37,38 and is a key molecule mediating the vasodilatation of UVR-induced inflammation in humans 7, while 12-HETE has powerful chemoattractant properties believed to be important in the dermal leukocytic influx in sunburn 3 and some inflammatory skin conditions 39,40. Less is known regarding the actions of PGE3 and 12-HEPE in skin, but both have been reported to show a partial agonist effect to their AA-derived counterparts 13,41. Thus, the significant reduction in PGE2 level in unchallenged skin, plus the increases in PGE3 and 12-HEPE levels following UVR exposure, support that increased dietary EPA promotes a milieu that may assist regulation of cutaneous inflammatory responses.
Figure 3.
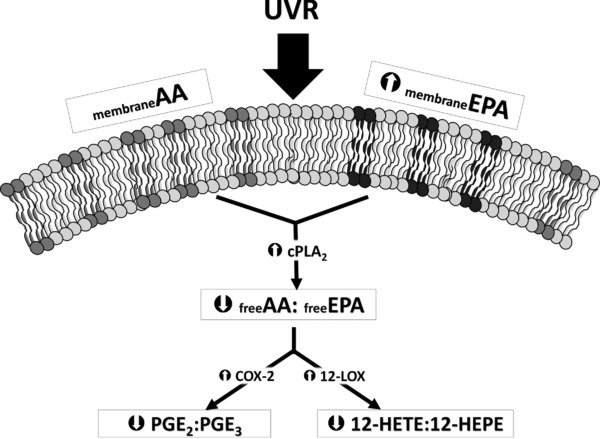
Schematic illustrating the reduction in AA:EPA-derived eicosanoid ratios following EPA supplementation.
Compliance with the oral n-3 PUFA ingestion was demonstrated by significantly elevated RBC EPA content in the active group at 12 wk. The change observed is in line with that reported in other studies assessing changes in RBC EPA content after n-3 PUFA supplementation 42,43. The dermal EPA content was also increased although by a smaller magnitude. Previous studies have shown skin EPA content to reach levels from 0.6 to 9% total fatty acids following oral n-3 PUFA 21,22, while in the current study, the EPA group showed a mean of 0.12% of total fatty acids post-supplementation, versus 0.06% in the control. Importantly, baseline EPA intake was assessed in the study population using a validated food questionnaire; this was low at 23 mg/day, and substantially less than the recommended daily amount 35. This low dietary level was reflected in the skin EPA content at baseline (0.07% of total fatty acids) while in a previous study by Rhodes et al., baseline levels were considerably higher 21. In addition to diet, differences in the tissue samples analysed (i.e. dermal in the current study versus whole skin/epidermal shave biopsies in others) may have contributed to the differing EPA content. The lower skin EPA uptake may also underlie the absence of an effect on the clinical erythemal response. Nevertheless, after n-3 PUFA the AA:EPA ratio was significantly reduced in both RBC and dermis. No absolute changes in the levels of AA were observed in either RBC or dermis, indicating that the reduced ratios were largely due to increased incorporation of EPA into membrane phospholipids as a result of systemic supplementation. Although several studies report a reduction in AA concomitant with an increase in EPA in RBCs after n-3 PUFA supplementation, this is not consistently found 42 and the size of the change can vary 44–47. Possible explanations include differences in protocols and baseline dietary status, or other regulatory mechanisms 48. In the skin, the effect of EPA on lipid content is less described. The modest uptake in this study may account for the lack of impact on AA content, although previously, Rhodes et al. found no impact of n-3 supplementation on total n-6 PUFA levels in skin despite an eightfold increase in skin EPA content 21, and rectal mucosal tissue increases in EPA uptake (∼threefold) are reported in the absence of a reduction in AA 26.
At 3 months post-supplementation, mean PGE2 levels were significantly lower in the EPA group in unexposed skin but not in UVR-exposed skin. The mean PGE3 levels were similar between groups in unexposed skin, but in UVR-exposed skin levels were twofold higher in the EPA group. It has been reported from in vitro studies that EPA inhibits COX-1 activity, and is itself metabolised with only 10% of the efficiency of AA by this isoform 12, which may explain our finding of a lowered PGE2 in basal skin, unaccompanied by any evident increase in PGE3 levels. The inducible isoform, COX-2, is reported to metabolise EPA more efficiently (with 30% of the efficiency of AA in an in vitro model) 12. Even a modest change in PGE3 could be relevant to skin physiology, in view of its reported partial agonist activity to PGE2. For example, in colorectal cancer cells PGE3 binding at the prostaglandin EP4 receptor reduced resistance to apoptosis, and in a fibroblast cell line PGE3 was less efficient in inducing COX-2 mRNA expression than PGE2, while in a murine macrophage cell line PGE3 proved less effective in stimulating secretion of pro-inflammatory IL-6 13,49. To date, related studies have not been reported in skin cells, but it is anticipated that the lower PGE2 levels observed in unexposed skin after EPA could help promote a less inflammatory basal state. In vitro studies support that EPA can inhibit COX-2 expression, and while smaller human trials found that n-3 PUFA supplementation significantly reduced the absolute levels of UVR-induced skin PGE2 20,25, this was not seen in the current double-blind randomised study, potentially attributable to the lower EPA uptake as discussed above.
Notably, this is the first report that UVR exposure increases the levels of 12-HEPE in human skin. The large UVR-induced rise in 12-HEPE may indicate that free EPA is preferentially directed through the 12-LOX pathway or that 12-LOX expression/activity is considerably greater than other LOX and COX enzymes in UV-irradiated skin. It is conceivable that the UVR-upregulated 12-HEPE may act as a partial agonist to 12-HETE, analogous to the effect of PGE3 to PGE2; currently few studies have examined the synthesis and activity of EPA-derived eicosanoids in human skin. Rose et al. reported that 12-HEPE derived from exogenous EPA suppressed 12-HETE synthesis in alveolar macrophages in vitro, while von Schacky et al. showed that 12-HEPE competed with AA for 5-LOX metabolism in stimulated neutrophils in vitro, producing biologically inactive 5, 12-diHEPE, which was associated with a decrease in chemoattractant LTB4 50. Moreover, Cunningham et al. demonstrated that 12-HEPE was less efficient at inducing erythema compared to 12-HETE, following its topical application to human skin 41. A decrease in the 12-HETE:12-HEPE ratio might potentially also act to reduce leukocytic infiltration into the skin during UVR inflammation, as 12-HETE is known to be a powerful chemoattractant. Studies are thus indicated to assess the relative chemoattractant properties of 12-HEPE and for any impact of the altered 12-HETE:12-HEPE ratio on dermal leukocytic infiltration.
In contrast to previous reports in smaller studies by our group and Orengo et al. 20–22,24, no alteration in the clinical erythemal response to UVR, i.e. the sunburn threshold, was seen. This was confirmed by quantitative analysis using the UVR–erythema dose response. Reports of a fall in UVR-induced PGE2 levels following EPA supplementation have previously been associated with a greater uptake of skin EPA than was measured in this study 20,25. Thus, the relatively low uptake of EPA could have contributed to the lack of a significant effect on absolute PGE2 levels and consequently, the erythemal response in UV-irradiated skin. Nonetheless, the observed significant alteration in PGE2:PGE3 ratio may influence a range of subclinical effects including cell proliferation and apoptosis. Since UVR-induced PGE2 has potent immunosuppressive properties, the altered cutaneous PGE2:PGE3 expression potentially contributed to the protection against UVR-induced immunosuppression that we reported in the volunteers ingesting EPA 34.
Strengths of this study include the double-blind, randomised controlled design and the large subject number; this is one of the biggest study groups in the assessment of n-3 PUFA impact on UVR inflammation. Limitations include that the study population was comprised entirely of females with a history of nickel allergy. The volunteers did not have active dermatitis at the time of the study and we do not anticipate the study population to be different to the general population with regard to their lipid metabolism or baseline inflammatory status. However, further research could include a broader population sample.
In summary, we have demonstrated that systemic EPA supplementation can significantly reduce the levels of key pro-inflammatory mediators PGE2 and 12-HETE relative to their EPA-derived counterparts PGE3 and 12-HEPE, which may have important consequences for skin physiology and regulation of responses to inflammatory stimuli. These findings support the evidence base, including their cardioprotective properties and potential benefit in conditions such as inflammatory bowel disease and rheumatoid arthritis 51, for tissue protective effects during an inflammatory response. As PGE2 and 12-HETE are reported to be tumour promoters when expressed at high concentrations 52,53, it is possible the supplement-induced shift in eicosanoid ratios could protect against carcinogenesis in the longer term, consistent with evidence of n-3 PUFA chemopreventive effect in skin 54–58. Further exploration is indicated to examine the impact of EPA on eicosanoid profile during the resolution phase of UV inflammation, and on COX and LOX expression/activity in human skin.
Acknowledgments
This research was supported by a BBSRC/Croda funded CASE Ph.D. studentship (SMP), the Association for International Cancer Research, AICR (LER, AN) (08-0131), and Yorkshire Cancer Research (KAM). The lipid supplements were provided by Croda, and packaged by GP Solutions (UK). We thank Andy Vail (University of Manchester) for statistical input, and Susan P. Bennett (University of Manchester) and Andrew Healey (University of Bradford, Analytical Centre) for excellent technical support. We would also like to thank all the volunteers who took part in the study. This study was registered at http://www.clinicaltrials.gov as NCT01032343.
Glossary
- AA
arachidonic acid
- BHT
butylated hydroxy toluene
- COX
cyclooxygenase
- EI
erythema index
- EPA
eicosa-pentaenoic acid
- HETE
hydroxyeicosatetraenoic acid
- LOX
lipoxygenase
- MED
minimal erythemal dose
- PG
prostaglandin
- RBC
red blood cell
- UVR
ultraviolet radiation
The authors have declared no conflicts of interest.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Table S1. Mean eicosanoid levels in suction blister fluid from unexposed and UVR-exposed skin at baseline and after 12 wk supplementation with 5 g/day of EPA-rich or control lipid.
5 References
- Di Nuzzo S, Sylva-Steenland RM, de Rie MA, Das PK, et al. UVB radiation preferentially induces recruitment of memory CD4+ T cells in normal human skin: long-term effect after a single exposure. J. Invest. Dermatol. 1998;110:978–981. doi: 10.1046/j.1523-1747.1998.00220.x. [DOI] [PubMed] [Google Scholar]
- Teunissen MB, Piskin G, di Nuzzo S, Sylva-Steenland RM, et al. Ultraviolet B radiation induces a transient appearance of IL-4+ neutrophils, which support the development of Th2 responses. J. Immunol. 2002;168:3732–3739. doi: 10.4049/jimmunol.168.8.3732. [DOI] [PubMed] [Google Scholar]
- Rhodes LE, Gledhill K, Masoodi M, Haylett AK, et al. The sunburn response in human skin is characterized by sequential eicosanoid profiles that may mediate its early and late phases. FASEB J. 2009;23:3947–3956. doi: 10.1096/fj.09-136077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckman SY, Gresham A, Hale P, Hruza G, et al. COX-2 expression is induced by UVB exposure in human skin: implications for the development of skin cancer. Carcinogenesis. 1998;19:723–729. doi: 10.1093/carcin/19.5.723. [DOI] [PubMed] [Google Scholar]
- Black AK, Fincham N, Greaves MW, Hensby CN. Time course changes in levels of arachidonic acid and prostaglandins D2, E2, F2 alpha in human skin following ultraviolet B irradiation. Br. J. Clin. Pharmacol. 1980;10:453–457. doi: 10.1111/j.1365-2125.1980.tb01788.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black AK, Greaves MW, Hensby CN, Plummer NA. Increased prostaglandins E2 and F2alpha in human skin at 6 and 24 h after ultraviolet B irradiation (290–320 nm) Br. J. Clin. Pharmacol. 1978;5:431–436. doi: 10.1111/j.1365-2125.1978.tb01650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes LE, Belgi G, Parslew R, McLoughlin L, et al. Ultraviolet-B-induced erythema is mediated by nitric oxide and prostaglandin E2 in combination. J. Invest. Dermatol. 2001;117:880–885. doi: 10.1046/j.0022-202x.2001.01514.x. [DOI] [PubMed] [Google Scholar]
- Cunningham FM, Wong E, Woollard PM, Greaves MW. The chemokinetic response of psoriatic and normal polymorphonuclear leukocytes to arachidonic acid lipoxygenase products. Arch. Dermatol. Res. 1986;278:270–273. doi: 10.1007/BF00407736. [DOI] [PubMed] [Google Scholar]
- Ruzicka T, Burg G. Effects of chronic intracutaneous administration of arachidonic acid and its metabolites. Induction of leukocytoclastic vasculitis by leukotriene B4 and 12-hydroxyeicosatetraenoic acid and its prevention by prostaglandin E2. J. Invest. Dermatol. 1987;88:120–123. doi: 10.1111/1523-1747.ep12525265. [DOI] [PubMed] [Google Scholar]
- Dowd PM, Kobza Black A, Woollard PM, Camp RD, et al. Cutaneous responses to 12-hydroxy-5,8,10,14-eicosatetraenoic acid (12-HETE) J. Invest. Dermatol. 1985;84:537–541. doi: 10.1111/1523-1747.ep12273537. [DOI] [PubMed] [Google Scholar]
- Lands WE. Biochemistry and physiology of n-3 fatty acids. FASEB J. 1992;6:2530–2536. doi: 10.1096/fasebj.6.8.1592205. [DOI] [PubMed] [Google Scholar]
- Wada M, DeLong CJ, Hong YH, Rieke CJ, et al. Enzymes and receptors of prostaglandin pathways with arachidonic acid-derived versus eicosapentaenoic acid-derived substrates and products. J. Biol. Chem. 2007;282:22254–22266. doi: 10.1074/jbc.M703169200. [DOI] [PubMed] [Google Scholar]
- Bagga D, Wang L, Farias-Eisner R, Glaspy JA, et al. Differential effects of prostaglandin derived from omega -6 and omega -3 polyunsaturated fatty acids on COX-2 expression and IL-6 secretion. Proc. Natl. Acad. Sci. USA. 2003;100:1751–1756. doi: 10.1073/pnas.0334211100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman DW, Pickett WC, Goetzl EJ. Human neutrophil chemotactic and degranulating activities of leukotriene B5 (LTB5) derived from eicosapentaenoic acid. Biochem. Biophys. Res. Commun. 1983;117:282–288. doi: 10.1016/0006-291x(83)91572-3. [DOI] [PubMed] [Google Scholar]
- Yang P, Chan D, Felix E, Cartwright C, et al. Formation and antiproliferative effect of prostaglandin E(3) from eicosapentaenoic acid in human lung cancer cells. J. Lipid Res. 2004;45:1030–1039. doi: 10.1194/jlr.M300455-JLR200. [DOI] [PubMed] [Google Scholar]
- Obata T, Nagakura T, Masaki T, Maekawa K, et al. Eicosapentaenoic acid inhibits prostaglandin D2 generation by inhibiting cyclo-oxygenase-2 in cultured human mast cells. Clin. Exp. Allergy. 1999;29:1129–1135. doi: 10.1046/j.1365-2222.1999.00604.x. [DOI] [PubMed] [Google Scholar]
- Calviello G, Di Nicuolo F, Gragnoli S, Piccioni E, et al. n-3 PUFAs reduce VEGF expression in human colon cancer cells modulating the COX-2/PGE2 induced ERK-1 and -2 and HIF-1alpha induction pathway. Carcinogenesis. 2004;25:2303–2310. doi: 10.1093/carcin/bgh265. [DOI] [PubMed] [Google Scholar]
- Puglia C, Tropea S, Rizza L, Santagati NA, et al. In vitro percutaneous absorption studies and in vivo evaluation of anti-inflammatory activity of essential fatty acids (EFA) from fish oil extracts. Internat. J. Pharmaceut. 2005;299:41–48. doi: 10.1016/j.ijpharm.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Kim HH, Cho S, Lee S, Kim KH, et al. Photoprotective and anti-skin-aging effects of eicosapentaenoic acid in human skin in vivo. J. Lipid Res. 2006;47:921–930. doi: 10.1194/jlr.M500420-JLR200. [DOI] [PubMed] [Google Scholar]
- Rhodes LE, Durham BH, Fraser WD, Friedmann PS. Dietary fish oil reduces basal and ultraviolet B-generated PGE2 levels in skin and increases the threshold to provocation of polymorphic light eruption. J. Invest. Dermatol. 1995;105:532–535. doi: 10.1111/1523-1747.ep12323389. [DOI] [PubMed] [Google Scholar]
- Rhodes LE, O'Farrell S, Jackson MJ, Friedmann PS. Dietary fish-oil supplementation in humans reduces UVB-erythemal sensitivity but increases epidermal lipid peroxidation. J. Invest. Dermatol. 1994;103:151–154. doi: 10.1111/1523-1747.ep12392604. [DOI] [PubMed] [Google Scholar]
- Rhodes LE, Shahbakhti H, Azurdia RM, Moison RM, et al. Effect of eicosapentaenoic acid, an omega-3 polyunsaturated fatty acid, on UVR-related cancer risk in humans. An assessment of early genotoxic markers. Carcinogenesis. 2003;24:919–925. doi: 10.1093/carcin/bgg038. [DOI] [PubMed] [Google Scholar]
- Rhodes LE, White SI. Dietary fish oil as a photoprotective agent in hydroa vacciniforme. Br. J. Dermatol. 1998;138:173–178. doi: 10.1046/j.1365-2133.1998.02047.x. [DOI] [PubMed] [Google Scholar]
- Orengo IF, Black HS, Wolf JE. Influence of fish oil supplementation on the minimal erythema dose in humans. Arch. Dermatol. Res. 1992;284:219–221. doi: 10.1007/BF00375797. [DOI] [PubMed] [Google Scholar]
- Shahbakhti H, Watson RE, Azurdia RM, Ferreira CZ, et al. Influence of eicosapentaenoic acid, an omega-3 fatty acid, on ultraviolet-B generation of prostaglandin-E2 and proinflammatory cytokines interleukin-1 beta, tumor necrosis factor-alpha, interleukin-6 and interleukin-8 in human skin in vivo. Photochem. Photobiol. 2004;80:231–235. doi: 10.1562/2004-01-27-RA-066. [DOI] [PubMed] [Google Scholar]
- West NJ, Clark SK, Phillips RK, Hutchinson JM, et al. Eicosapentaenoic acid reduces rectal polyp number and size in familial adenomatous polyposis. Gut. 2010;59:918–925. doi: 10.1136/gut.2009.200642. [DOI] [PubMed] [Google Scholar]
- Belluzzi A, Brignola C, Campieri M, Pera A, et al. Effect of an enteric-coated fish-oil preparation on relapses in Crohn's disease. N. Engl. J. Med. 1996;334:1557–1560. doi: 10.1056/NEJM199606133342401. [DOI] [PubMed] [Google Scholar]
- Henz BM, Jablonska S, van de Kerkhof PC, Stingl G, et al. Double-blind, multicentre analysis of the efficacy of borage oil in patients with atopic eczema. Br. J. Dermatol. 1999;140:685–688. doi: 10.1046/j.1365-2133.1999.02771.x. [DOI] [PubMed] [Google Scholar]
- Diffey BL, Oliver RJ. An inexpensive luminaire for diagnostic phototesting to UVB radiation. Photodermatology. 1985;2:260–262. [PubMed] [Google Scholar]
- Diffey BL, Farr PM. Quantitative aspects of ultraviolet erythema. Clin. Phys. Physiol. Meas. 1991;12:311–325. doi: 10.1088/0143-0815/12/4/001. [DOI] [PubMed] [Google Scholar]
- Green P, Anyakoha N, Yadid G, Gispan-Herman I, et al. Arachidonic acid-containing phosphatidylcholine species are increased in selected brain regions of a depressive animal model: implications for pathophysiology. Prostagl. Leukot. Essent. Fatty Acids. 2009;80:213–220. doi: 10.1016/j.plefa.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Masoodi M, Nicolaou A. Lipidomic analysis of twenty-seven prostanoids and isoprostanes by liquid chromatography/electrospray tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2006;20:3023–3029. doi: 10.1002/rcm.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masoodi M, Mir AA, Petasis NA, Serhan CN, et al. Simultaneous lipidomic analysis of three families of bioactive lipid mediators leukotrienes, resolvins, protectins and related hydroxy-fatty acids by liquid chromatography/electrospray ionisation tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2008;22:75–83. doi: 10.1002/rcm.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilkington SM, Massey KA, Bennett SP, Al-Aasswad NM, et al. Randomized controlled trial of oral omega-3 PUFA in solar-simulated radiation-induced suppression of human cutaneous immune responses. Am. J. Clin. Nutr. 2013;97:646–652. doi: 10.3945/ajcn.112.049494. [DOI] [PubMed] [Google Scholar]
- Wallingford SC, Pilkington SM, Massey KA, Al-Asswaad NMI, et al. Three-way assessment of long chain omega-3 polyunsaturated fatty acid nutrition: by questionnaire and matched blood and skin samples. Br. J. Nutr. 2012;23:1–8. doi: 10.1017/S0007114512001997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scientific Advisory Committee on Nutrition (SACN) Advice on Fish Consumption: Benefits and Risks. London: The Stationary Office; 2004. [Google Scholar]
- Chun KS, Akunda JK, Langenbach R. Cyclooxygenase-2 inhibits UVB-induced apoptosis in mouse skin by activating the prostaglandin E2 receptors, EP2 and EP4. Cancer Res. 2007;67:2015–2021. doi: 10.1158/0008-5472.CAN-06-3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun KS, Lao HC, Langenbach R. The prostaglandin E2 receptor, EP2, stimulates keratinocyte proliferation in mouse skin by G protein-dependent and {beta}-arrestin1-dependent signaling pathways. J. Biol. Chem. 2010;285:39672–39681. doi: 10.1074/jbc.M110.117689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr RM, Wong E, Mallet AI, Olins LA, et al. The analysis of arachidonic acid metabolites in normal, uninvolved and lesional psoriatic skin. Prostaglandins. 1984;28:57–65. doi: 10.1016/0090-6980(84)90113-8. [DOI] [PubMed] [Google Scholar]
- Black AK, Barr RM, Wong E, Brain S, et al. Lipoxygenase products of arachidonic acid in human inflamed skin. Br. J. Clin. Pharmacol. 1985;20:185–190. doi: 10.1111/j.1365-2125.1985.tb05059.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham FM, Woollard PM, Camp RDR. Proinflammatory properties of unsaturated fatty acids and their monohydroxy metabolites. Prostaglandins. 1985;30:497–509. doi: 10.1016/0090-6980(85)90122-4. [DOI] [PubMed] [Google Scholar]
- van Rensburg S, Smuts C, Hon D, Kidd M, et al. Changes in erythrocyte membrane fatty acids during a clinical trial of eicosapentaenoic acid (EPA) supplementation in schizophrenia. Metabolic Brain Dis. 2009;24:659–672. doi: 10.1007/s11011-009-9160-7. [DOI] [PubMed] [Google Scholar]
- Palozza P, Sgarlata E, Luberto C, Piccioni E, et al. n-3 fatty acids induce oxidative modifications in human erythrocytes depending on dose and duration of dietary supplementation. Am. J. Clin. Nutr. 1996;64:297–304. doi: 10.1093/ajcn/64.3.297. [DOI] [PubMed] [Google Scholar]
- Rizzo AM, Corsetto PA, Montorfano G, Opizzi A, et al. Comparison between the AA/EPA ratio in depressed and non depressed elderly females: omega-3 fatty acid supplementation correlates with improved symptoms but does not change immunological parameters. Nutr. J. 2012;11:82–92. doi: 10.1186/1475-2891-11-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siener R, Alteheld B, Terjung B, Junghans B, et al. Change in the fatty acid pattern of erythrocyte membrane phospholipids after oral supplementation of specific fatty acids in patients with gastrointestinal diseases. Eur. J. Clin. Nutr. 2010;64:410–418. doi: 10.1038/ejcn.2009.151. [DOI] [PubMed] [Google Scholar]
- Dawczynski C, Hackermeier U, Viehweger M, Stange R, et al. Incorporation of n-3 PUFA and gamma-linolenic acid in blood lipids and red blood cell lipids together with their influence on disease activity in patients with chronic inflammatory arthritis–a randomized controlled human intervention trial. Lipids Health Dis. 2011;10:130–141. doi: 10.1186/1476-511X-10-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marangoni F, Angeli MT, Colli S, Eligini S, et al. Changes of n-3 and n-6 fatty acids in plasma and circulating cells of normal subjects, after prolonged administration of 20:5 (EPA) and 22:6 (DHA) ethyl esters and prolonged washout. Biochim. Biophys. Acta. 1993;1210:55–62. doi: 10.1016/0005-2760(93)90049-f. [DOI] [PubMed] [Google Scholar]
- von Schacky C, Fischer S, Weber PC. Long-term effects of dietary marine omega-3 fatty acids upon plasma and cellular lipids, platelet function, and eicosanoid formation in humans. J. Clin. Invest. 1985;76:1626–1631. doi: 10.1172/JCI112147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawcroft G, Loadman PM, Belluzzi A, Hull MA. Effect of eicosapentaenoic acid on E-type prostaglandin synthesis and EP4 receptor signaling in human colorectal cancer cells. Neoplasia. 2010;12:618–627. doi: 10.1593/neo.10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schacky C, Marcus AJ, Safier LB, Ullman HL, et al. Platelet-neutrophil interactions. 12S,20- and 5S,12S-dihydroxyeicosapentaenoic acids: two novel neutrophil metabolites from platelet-derived 12S-hydroxyeicosapentaenoic acid. J. Lipid Res. 1990;31:801–810. [PubMed] [Google Scholar]
- Calder PC. n-3 polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am. J. Clin. Nutr. 2006;83:1505S–1519S. doi: 10.1093/ajcn/83.6.1505S. [DOI] [PubMed] [Google Scholar]
- Ansari KM, Rundhaug JE, Fischer SM. Multiple signaling pathways are responsible for prostaglandin E2-induced murine keratinocyte proliferation. Mol. Cancer Res. 2008;6:1003–1016. doi: 10.1158/1541-7786.MCR-07-2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun K-S, Langenbach R. A proposed COX-2 and PGE2 receptor interaction in UV-exposed mouse skin. Mol. Carcinogenesis. 2007;46:699–704. doi: 10.1002/mc.20354. [DOI] [PubMed] [Google Scholar]
- Hakim IA, Harris RB, Ritenbaugh C. Fat intake and risk of squamous cell carcinoma of the skin. Nutr. Cancer. 2000;36:155–162. doi: 10.1207/S15327914NC3602_3. [DOI] [PubMed] [Google Scholar]
- Kune GA, Bannerman S, Field B, Watson LF, et al. Diet, alcohol, smoking, serum beta-carotene, and vitamin A in male nonmelanocytic skin cancer patients and controls. Nutr. Cancer. 1992;18:237–244. doi: 10.1080/01635589209514224. [DOI] [PubMed] [Google Scholar]
- Black HS, Thornby JI, Gerguis J, Lenger W. Influence of dietary omega-6, -3 fatty acid sources on the initiation and promotion stages of photocarcinogenesis. Photochem. Photobiol. 1992;56:195–199. doi: 10.1111/j.1751-1097.1992.tb02147.x. [DOI] [PubMed] [Google Scholar]
- Orengo IF, Black HS, Kettler AH, Wolf JE., Jr Influence of dietary menhaden oil upon carcinogenesis and various cutaneous responses to ultraviolet radiation. Photochem. Photobiol. 1989;49:71–77. doi: 10.1111/j.1751-1097.1989.tb04080.x. [DOI] [PubMed] [Google Scholar]
- Lou Y-R, Peng Q-Y, Li T, Medvecky CM, et al. Effects of high-fat diets rich in either omega-3 or omega-6 fatty acids on UVB-induced skin carcinogenesis in SKH-1 mice. Carcinogenesis. 2011;32:1078–1084. doi: 10.1093/carcin/bgr074. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Mean eicosanoid levels in suction blister fluid from unexposed and UVR-exposed skin at baseline and after 12 wk supplementation with 5 g/day of EPA-rich or control lipid.