

Abstract
Protease activated receptors (PARs) populate neurons and astrocytes in the brain. The serine protease thrombin, which activates PAR-1 during the first hours after stroke, appears to be associated with the cytotoxicity. Thrombin antagonists and PAR-1 inhibitors have been correlated with reduced cell death and behavioral protection after stroke, but no data yet supports a mechanistic link between PAR-1 action and benefit. We sought to establish the essential role of PAR-1 in mediating ischemic damage. Using a short hairpin mRNA packaged with green fluorescent protein in a lentivirus vector, we knocked downPAR-1 in the medial caudate nucleus prior to rat middle cerebral artery occlusion (MCAo) and in rat neurons prior to oxygen-glucose deprivation. We also compared aged PAR-1 knockout mice with aged PAR-3, PAR-4 mice and young wild-type mice in a standard MCAo model. Silencing PAR-1 significantly reduced neurological deficits, reduced endothelial barrier leakage, and decreased neuronal degeneration in vivo during MCAo. PAR-1 knock-down in the ischemic medial caudate allowed cells to survive the ischemic injury; infected cells were negative for TUNEL and c-Fos injury markers. Primary cultured neurons infected with PAR-1 shRNA showed increased neuroprotection during hypoxic/aglycemic conditions with or without added thrombin. The aged PAR-1 knockout mice showed decreased infarction and vascular disruption compared to aged controls or young wild types. We demonstrated an essential role for PAR-1 during ischemia. Silencing or removing PAR-1 significantly protected neurons and astrocytes. Further development of agents that act at PAR-1or its downstream pathways could yield powerful stroke therapy.
Keywords: Protease Activated Receptor-1, Thrombin, cerebral ischemia, Lentivirus knock-down, oxygen –glucose deprivation
1.0 Introduction
Thrombolysis remains the mainstay of stroke therapy, while effective neuroprotection remains under development. In addition to neuroprotection and thrombolysis, however, early investigators pursued stroke therapies designed to limit microvascular thrombosis downstream of an occluded cerebral artery (Ames et al., 1968, Adams et al., 2008). When several large clinical trials of anti-thrombotic therapy for acute stroke failed, interest waned in developing these drugs (Biller et al., 1989, Gordon et al., 1990). A new chapter in anti-thrombotic therapy opened with the discovery of cell-membrane receptors that could be activated by proteases, named the protease activated receptors or PARs (Vu et al., 1991). Thrombin, normally present in the serum as prothrombin until activated, is a serine protease that plays a central mediating role in the coagulation cascade. Accumulating data from in vitro studies with brain cells indicate that thrombin induces cell death in glia and neurons (Donovan et al., 1997, Striggow et al., 2000). Furthermore, thrombin seems to be particularly toxic to stressed neuronal cells (Weinstein et al., 1998). We showed significant benefit after acute ischemia with direct thrombin inhibitors (Lyden et al., 2014). Thrombin induces protection at low doses (thrombin preconditioning) but acts as a neurotoxin at high doses, killing cells via the PARs (Xi et al., 2003).
PARs belong to a G-protein coupled receptor family including PAR-1 to PAR-4. PAR-1 is also known as thrombin receptor and is considered to be the main receptor subtype activated by thrombin and plasmin (Macfarlane et al., 2001, Hollenberg and Compton, 2002). PAR-1 is activated by proteolytic cleavage of an N-terminus extra-cellular blocking segment, unmasking an amino acid sequence that acts as an auto-ligand (Coughlin, 2000, Macfarlane et al., 2001). Blood derived thrombin and other PAR-1 activators enter brain tissue during traumatic brain injury, hemorrhagic stroke, ruptured aneurysms and therapeutic treatments with rt-PA (Gingrich et al., 2000, Gingrich and Traynelis, 2000, Davalos et al., 2014). During ischemia, thrombin enters brain via a permeable blood brain barrier (Chen et al., 2012, Davalos et al., 2014). Thrombin injected in rat brain causes edema, neuronal cell death and precipitates seizures (Lee et al., 1996, Lee et al., 1997). PAR-1 is up regulated during ischemic conditions and has been associated with neuronal cell death, neurite retraction, and glial proliferation (Gurwitz and Cunningham, 1988, Cavanaugh et al., 1990, Grabham and Cunningham, 1995, Donovan et al., 1997). PAR-1 is expressed in most brain regions and is associated with brain injury during hemorrhagic and ischemic stroke (Donovan et al., 1997, Sarker et al., 1999, Rohatgi et al., 2004).
Agents acting at the PAR-1 receptor to either block thrombin or induce cell-survival signaling have been associated with beneficial effects after ischemia, as measured with behavioral or histological outcomes. For example, activated protein-C (APC) and mutant analogs that cleave the PAR-1 tail at a distinct site, causing cytoprotective signaling, have been correlated with benefit in animal stroke models (Wang et al., 2012). Previous studies have shown that mice lacking PAR-1 tolerate ischemia better compared to wild type mice (Junge et al., 2003). A recent study using PAR-1 siRNA infected animals showed decrease in lesion area and improved behavior in animals after stroke and correlated with HSP-70 expression (Zhang et al., 2012). To date, however, data confirming a required role for presence of PAR-1 on neurons and astrocytes (cells mainly affected during ischemia) in mediating cytotoxicity has been lacking; there remains a possibility that previous treatment correlations could be due to off-target effects. We sought to demonstrate obligate involvement of the PAR-1 receptor in mediating cell death in the brain using genomic modification. We also sought to confirm in aged animals the previous findings that PAR-1 knock out provided protection to young animals suffering ischemia. We used in vivo and in vitro knockdown of PAR-1 in neurons and astrocytes to protect the ischemic lesion. We subjected aged PAR-1 knockout mice to focal cerebral ischemia.
2.0 Materials and Methods
2.1 Reagents
All analytical grade reagents were purchased from Sigma-Aldrich. Cell culture media were purchased from Life Technologies (formerly Invitrogen). Other reagents were purchased as noted.
2.2 Animal Surgery
Institutional Animal Care and Use Committee (IACUC) at Cedars-Sinai Medical Center approved all animal handling and surgery protocols, as per the national guideline for the care of experimental animals. Procedures recommended by the STAIR and RIGOR guidelines were followed (Fisher et al., 2009, Lapchak et al., 2013).
2.3 shRNA Lentivirus
A potential siRNA sequence of 20 nucleotide duplexes against the PAR-1 receptor consensus coding (GenBank Accession No. NM_012950) was designed to create a small hairpin-blocking segment (Liu et al., 2004, 2006, Villares et al., 2008). The sequence used was specific to rat and mouse PAR-1 and was cloned in pLKO.1-puro-UbC-TurboGFPTM vector. The sequence for shRNA of PAR-1 knockdown used was “AATCCCAGTGAAGATACATTT”. We used a negative control siRNA that mismatches the sequence of PAR-1mRNA (misRNA PAR-1) for comparison. Lentiviruses were packaged in the vector by Sigma-Aldrich labs with the sequence provided. The vectors included the code for green fluorescent protein (GFP). The vectors were titrated using flow cytometry and PCR analysis. The titers of the vectors used in the study were in the range of 1.0 × 109 – 3.9 × 109 TU per ml.
2.4 In vivo PAR-1 knockdown
To demonstrate cell survival after PAR-1 knockdown, male Sprague Dawley rats weighing 290–310g were divided at random into two groups and injected with GFP tagged shRNA PAR-1 or misPAR-1 using a lentivirus delivery vector. Animals were anesthetized with 4% isoflurane mixed in oxygen and nitrous oxide (30:70) and maintained on1.5–2% isoflurane during the surgery. A volume of 2 μl of lentivirus was stereotaxically injected into the medial striatum using a 10μl Hamilton syringe at a rate of 0.2 μl/min. The coordinates for striatal injections were −0.3 mm rostral, 3.0 mm lateral and 6.5 mm ventral to bregma. Following injections animals survived for 1 week and were then subjected to MCAo. The MCAo procedure was performed by a surgeon blind to treatment assignment, as described previously in our laboratory (Chen et al., 2012, Van Winkle et al., 2012). Briefly, a midline neck incision was made exposing the left common carotid artery. The external carotid and occipital arteries were ligated with 4–0 silk suture and an incision was made in the wall of the external carotid artery close to the bifurcation point of the external and internal carotid arteries. A 4–0 heat-blunted nylon suture (Ethicon) was inserted and advanced 17.5mm from the bifurcation point into the internal carotid arteries for 2h. After the 2h occlusion duration, the nylon suture was removed from carotid artery to allow the reperfusion of blood flow into the MCA. Neurological function was tested during reperfusion and 24 h after onset of ischemia using rodent neurological grading system. Animals were killed with an overdose of ketamine and xylazine, and then intracardially perfused with saline and 4% paraformaldehyde.
2.5 Effect of PAR knockout
To evaluate the role of PAR in mediating ischemic injury, PAR-1, PAR-3 and PAR-4 age-matched mice from 12–14 months old were used. Young wild-type C57/bl6-J mice were purchased from Jackson Labs (Bar Harbor, Maine). PAR-1, PAR-3 and PAR-4 knockout animals were kindly provided by Dr. Berislav Zlokovic (University of Southern California, Los Angeles) and Dr. Sean Coughlin (University of California, San Francisco). Animals were anesthetized with 4% isoflurane mixed in oxygen and nitrous oxide (30:70) and received 2MDa dextran conjugated with FITC in PBS (Sigma-Aldrich, FD2000S). The MCAo model as described earlier was adapted for mice (Chen et al., 2012, Van Winkle et al., 2012). Cerebral blood flow was monitored using laser Doppler flowmetry (LDF) by fixing a probe on the skull, 4-mm lateral to the bregma (Moor Instruments, Devon, United Kingdom). Nylon 6-0 silicon-coated suture was inserted and advanced 10 mm from the bifurcation into the internal carotid artery. After 2 hours the suture was removed and reperfusion confirmed with LDF. Animals survived for 24 hours after stroke and neurological functions were examined during reperfusion and 24 hours after onset of ischemia. Brains were removed and divided coronally into two halves: the anterior one-half was sectioned into 2 or 3 1-mm blocks and incubated for 30min in 2,3,5-triphenyltetrazolium chloride (TTC) at 37°C. The posterior half was post-fixed, frozen, and 3 evenly spaced 50μm thick sections were cut and imaged for FITC fluorescence as described previously (Chen et al., 2009, Chen et al., 2012). The surgeon and the behavioral examiners were blinded to the genomic status of the mice.
2.6 Cell target characterization and cell injury
Brain sections were used for immunofluorescence studies for characterization of infected cells using NeuN, IBA1, GFAP antibodies and Neurotrace (Nissl Staining) (details provided in Table). Briefly, rats were anesthetized with halothane and perfused with cold saline and 4% paraformaldehyde and post-fixed overnight in 4% paraformaldehyde, followed by cryopreservation in 30% sucrose solution. Using a sliding microtome, 50-μm-thick brain sections were cut and free-floating sections were collected in phosphate-buffered saline (PBS). Brain sections were incubated in 0.2% Triton X-100 in PBS for 15 min and followed by three subsequent washes with PBS. Sections were then incubated in 5% normal goat serum for 1 h at room temperature and followed by overnight incubation with mouse anti-NeuN, GFAP and rabbit anti c-Fos and IBA1 (1:1000) antibodies. Following three subsequent washes in PBS; brain sections were incubated with Alexa 594 (red) secondary antibodies for 1 h at room temperature. For Nissl staining with Neurotrace (NeuroTrace 640/660, Life Technologies) the sections were permeablised with 0.2% Triton X-100, following three washes with PBS the sections were incubated with Neurotrace for 15 min (1:3000). The sections were washed, mounted on slides and viewed under Leica and Olympus (FV10i) microscopes. Images showing co-localization were generated using NIH ImageJ software; Adobe Photoshop (San Jose, CA, USA) was used to make the composites from smaller micrographs. Separate sections were used for Fluoro-Jade-C (Histo-Chem Inc. Jefferson, Arkansas) and In Situ Cell Death Kit, TMR red (TUNEL kit, Roche Applied Science, Germany catalogue number 12156792910) to determine the neuronal cell death as per the manufacturer’s instructions as described previously (Chen et al., 2010, Chen et al., 2012).
Table 1.
Antibody list for immunohistochemistry
| Primary Antibody | Species | Vendor/Catalogue# | Secondary Antibody | Vendor/Catalogue# |
|---|---|---|---|---|
| NeuN | MS-monoclonal | Millipore/MAB377 | A594 GtαMs | Invitrogen/A11032 |
| GFAP | MS-monoclonal | Millipore/MAB360 | A594 GtαMs | Invitrogen/A11032 |
| C-Fos | Rb-polyclonal | Abcam/ab7963 | A594 GtαRb | Invitrogen/A11037 |
| Iba1 | Rb-polyclonal | Wako/019-19741 | A594 GtαRb | Invitrogen/A11037 |
| PAR-1 receptor | Rb-polyclonal | Santa Cruz Biotechnology/H-111 | Goat anti-rabbit IgG-HRP | Santa Cruz/SC-2030 |
| NeuroTrace-Red 530/615 | N/A | Invitrogen/N-21482 | N/A | N/A |
2.7 Image analysis
The investigator conducting all image analysis was unaware of group assignment or genomic status and all sections were masked and coded. Sections were decoded and assigned to proper groups only after all image data was collected and finalized. All sections were imaged at low power using epifluorescence microscopy with a highly sensitive air-cooled CCD camera (Apogee, Alta U32) (Chen et al., 2010, Chen et al., 2012). For quantification of Fluoro-Jade-C positive neurons and TUNEL positive cells NIH ImageJ software was used. The images were converted to 8-bit grey scale format; the Image J pseudo flat field plugin was used to control variation in the fluorescence across all sections. Using the Image J nucleus counter particle analysis plugin, the numbers of neurons from each brain section were automatically counted (Wang et al., 2013).
2.8 Primary Neuronal Culture
Cortices from E14 to E16 embryos were removed and isolated, cleaned of meninges, and finely minced. The cortical tissue was then digested in 0.25% Trypsin for 5 minutes in a 37 °C water bath with occasional gentle shaking. DNAse I was added to the cells and the mixture was returned to the 37 °C water bath for another 5 minutes. The cells were then removed and brought up to 50mL with warmed DMEM and centrifuged for 5 minutes at 1000g. The supernatant was aspirated and the cell pellet was resuspended in pre-warmed complete media (DMEM containing 5% HS, 2 mM glutamine and 1% penicillin/streptomycin). The cells were then filtered through a 70μM diameter membrane, removed and resuspended, and counted for total cell concentration for plating. After two days survival, cells were infected with PAR-1 shRNA lentivirus or misRNA (1μl/ml. of media) for 24 hours and medium was replaced with fresh medium. Successful infection was confirmed using the GFP reporter; plates were not used unless more than about 80% of cells expressed the GFP.
2.9 Oxygen-Glucose Deprivation
Neurons for oxygen-glucose deprivation (OGD) were grown on 96-well plates at a density of 1×105 cells per well. OGD media was made with glucose-free DMEM (5% HS, 2 mM glutamine and 1% penicillin/streptomycin) and subsequently bubbled with 95% nitrogen, 5% CO2 gas mixture for 15 minutes. The cells were then washed twice with OGD media and placed in an anoxic incubator for 2 hours (95% N2 and 5% CO2). Wells were randomly divided into three groups: Control, Thrombin 50 U/ml treated during 2 hours of OGD or Thrombin 50 U beginning and the end of OGD and continuing during 24 hours of reoxygenation. Wells not subjected to OGD were washed twice with normal media and randomly divided into similar groups: control, Thrombin 50U/ml for 2 hours or 24 hours and placed in a normal incubator for an equivalent amount of time. The medium from each treatment cohort was removed after 2 hours of OGD and replaced with normal medium. After 24 hours medium was removed and used for lactate dehydrogenase (LDH) assay (Cytotoxicity Detection KitPLUS (LDH) Roche Applied Science, Germany catalogue number -04744934001). To determine a maximum LDH value for each well, after removing the media, the remaining cells in each well were lysed using a provided lysis reagent according to manufacturer’s instructions. Each measurement was normalized to the total of the LDH found after 2 hours, 24 hours and lysis.
2.10 Statistical Analysis
All continuous data were analyzed with parametric statistics and reported as mean ± SE. For cell counting, data are presented as number of neurons that stained positive for Fluoro-Jade-C or using TUNEL. Briefly, from 3–4 sections per animal, the numbers of positive neurons were averaged over 4 high power fields per section. For c-Fos immunoreactivity the area was measured using Image J analysis and represented as mm2. Quantitative neurological scores were compared using an independent sample student t-test. Two-way ANOVA with Bonferroni post-hoc test was used to compare the results of OGD experiment (LDH assay) performed in triplicates. Areal measurements (c-Fos, TTC, and vascular disruption) were compared using 1-way ANOVA with Dunnett’s multiple comparison post-hoc test. GraphPad Prism 5.0 (San Diego, CA) was used to perform all the statistical analysis.
3.0 Results
3.1 Effect of in vivo PAR-1 Knock-down
We confirmed PAR-1 knockdown and protein expression after vector infection in brain sections (Fig 1A) and cultured cells (Fig 1B and Fig 1C) using immunohistochemistry and western blot analysis. As shown in Figure 1A brain sections from animals injected with misRNA PAR-1 (control) and shRNA PAR-1 were stained for PAR-1 antibody. The misRNA PAR-1 panel shows colocalization with infected cells in green whereas; the shRNA PAR-1 panel shows no colocalization with GFP labeled lentivirus infected cells. We also confirmed the knockdown by western blot analysis using protein lysates from cultured neuronal cells and see significant decrease in PAR-1 protein expression in cells infected with PAR1 shRNA in comparison to cells infected with misRNA and uninfected cells Fig (1B and 1C).
Figure 1. Effective knockdown of PAR-1 with PAR-1 shRNA lentivirus in brain sections and transfected neuronal cells.

(A) Lentivirus was injected into rat striatum. After one week the animals were sacrificed and brain sections were stained to determine the effective knockdown of PAR-1 mediated by lentivirus. Sections were stained with rabbit polyclonal antibody against PAR-1 (Alexa 594 goat anti-rabbit secondary antibody.) Pictomicrograph shows cells infected with lentivirus expressing a mis-sense RNA (misRNA) PAR-1 colocalize with PAR-1 protein indicated with arrows, demonstrating no knock down. Cells infected with shRNA PAR-1 lentivirus show no colocalization with PAR-1 receptor antibody, indicated with arrow heads. (B) Cell lysate taken from infected cells was used to determine the protein levels of PAR-1 with western blot analysis. Decreased expression of PAR-1 was observed in cells infected with PAR-1 shRNA when compared to control cells and PAR-1 misRNA infected cells. (C) Densitometry analysis shows a significant reduction in PAR-1 expression upon infection with PAR-1 shRNA lentivirus *** p<0.001.
Brain sections taken from lesioned rats one week after lentivirus injection were used for characterization of the infected cells. Cells infected in the medial caudate—the medial MCA perfusion bed—avidly took up the vector and expressed vector-delivered proteins (Fig. 2A and 2B). The infected cells were further characterized using specific antibodies to label neurons, astrocytes or microglia. Of the entire lentivirus infected cell population, about 42% also stained for NeuN, about 28% stained for GFAP, and about 30% stained for neither (Fig. 2 C, D and E). Of considerable importance, infected cells showed no positive immunoreactivity when stained with IBA1—a microglial marker (Fig. 2E)—suggesting viability, or at least the absence of phagocytosis around the infected cells. For further confirmation of neuronal staining we used NeuroTrace Red Nissl staining to co-localize with lentivirus infected GFP positive cells. Using CytoFx© software supplied by Dr. Kolja Wawrowsky, we created a 3D contour image from a z-stack showing a clear co-localization of GFP positive cells with NeuroTrace Red (Fig. 2F).
Figure 2. Stereotaxic injection of PAR-1 shRNA lentivirus infects neurons and astrocytes in the ischemic zone.

(A) Lentivirus was injected into rat striatum. One week later the animals were subjected to MCAo and survived for 24 hours. A schematic representation shows TTC-negative areas in rat coronal brain sections at the level of Bregma −0.3mm from 6 different animals. The absent TTC staining concentrated mostly in the striatum indicated by darker color to demonstrate the consistency of the ischemia area after 2 hr occlusion in our laboratory. The medial striatum was lesioned in 100% of the subjects and thus the lentivirus injections targeted this area. (B) High power photomicrograph from the medial striatum of one animal. GFP positive cells were detected one week following injection. (C, D, F) Confocal microscopy for several cell-type specific antigens co-localized with GFP labeled cells: NeuN (neuronal marker), GFAP (astrocytes) and Nissl body (neurons). (E) GFP positive cells did not co-localize with microglia marker IBA1. (G) Quantification of the proportion of GFP positive cells co-localizing with GFAP+ or NeuN+ profiles. At least 1000–1500 cells were counted for the quantification from 7 different animals. Scale bars (B) 100μm; (C, D, E) 50μm; (F) 20μm. In all figures, white arrows indicate GFP positive cells showing co-localization and in (E) the white arrowhead indicates GFP positive cells showing no co-localization with IBA1.
3.2 Post-ischemic cell injury
To identify areas of cellular injury after ischemia, we labeled for c-Fos, an immediate-early proto-oncogene whose expression is increased after stimulation (Hu et al., 1994, Sharp and Sagar, 1994). We observed increased expression of c-Fos in ischemic areas of animals subjected at 24 hours with 2 hours of MCAo. We noted c-Fos expression was significantly decreased in the PAR-1 shRNA-injected animals compared to misRNA injection (Fig. 3). To assure that this finding was not due to random variation in the sizes of the ischemic zones induced in our model, we confirmed in a separate series that the medial caudate is involved in every animal subject to MCAo, even though the sizes of the infarctions may vary (Fig. 2A). A signification reduction in c-Fos positive cells in PAR-1 shRNA injected animals was regularly observed in the ischemic zone (Fig. 3D). Upon high-power magnification, there was a clear distinction (i.e. absent co-localization) between infected cells and c-Fos positive cells, even juxtaposed in the same ischemic bed (Fig 3E). That is, in a single ischemic zone—in which all factors related to ischemia (temperature, cerebral blood flow, acidosis) affected all cells equally—dead and viable cells differing only by the presence or absence of PAR-1 could be identified next to each other, (Fig. 3E). On sections taken 24 hours after stroke, we observed a significantly reduced number of TUNEL positive cells in animals with PAR-1 knock-down (Fig. 4). Interestingly, the cells positive for GFP (indicating presence of shRNA) showed no co-localization with TUNEL positivity; these viable cells were found in the ischemic zone, and were surrounded by degenerating cells. In other words, in one ischemic milieu, there was a clear distinction (absent co-localization) between infected cells and degenerating cells. We used Fluoro-Jade-C, a marker for degenerating neurons, to demonstrate a neuroprotective effect of PAR-1 shRNA knockdown. Similar to what was observed with TUNEL staining, increased numbers of Fluoro-Jade-C positive cells were observed in the misRNA injected animals (Fig. 5A) and this was markedly reduced in shRNA injected animals (Fig. 5B). The quantification of Fluoro-Jade-C positive cells demonstrated that degenerating neurons were significantly fewer in MCAo induced PAR-1 knockdown animals (Figs. 5C and 5D).
Figure 3. Distribution of c-Fos positive neurons in the ischemic striatum.

(A, B) Animals subjected to 2-hour MCAo and 24 hours reperfusion showed marked increased expression of c-Fos, an immediate early gene associated with neuronal stress or injury. Animals pre-injected with mismatch RNA showed greater expression of c-Fos compared to animals pre-injected with PAR-1 shRNA. (C) Par-1 knockdown significantly reduced the total area of c-Fos expression in ischemic animals, measured with planimetry from stained sections, Mean ± SE (n=7). ***P<0.001, Students t-test for independent samples. (D, E) Cells counted in 20× magnification high power fields showed essentially no co-localization of PAR-1 shRNA positive GFP cells with c-Fos positive cells. Scale bars (A, B) 200μm (inset: 100μm); (E) 20μm.
Figure 4. Representative TUNEL staining of brain sections after MCAo.

(A, B) Photomicrographs showing TUNEL positive cells in PAR-1 mismatch and shRNA injected animals 24 hours after 2hr. of MCAo. (C) PAR-1 shRNA positive GFP cells show no co-localization with TUNEL positive cells. Animals with PAR-1 knockdown show significant reduction in dying cells. Scale bars (A, B) 200μm (inset: 100μm); (C) 20μm. (D) Data presented as Mean ± SE (n=7). ***P<0.001, Students t-test for Independent samples.
Figure 5. PAR-1 knockdown induces cytoprotection after MCAo.

(A, B) Representative images showing Fluoro-Jade-C positive cells indicating neuronal damage in mismatch and shRNA PAR-1 lentivirus injected animals. (C) Fluoro-Jade-C positive cells in a selected high power field. (D) PAR-1 shRNA injected animals show significantly reduced number of Fluoro-Jade-C positive cells. Scale bars (A, B) 200μm; (C) 100μm. (D) Data presented as mean ± SE (n=7). ***P<0.001, Students t-test for independent samples.
3.3 Neurological behavior outcomes
A rodentneurological scale was used to asses the changes in the behavior of animals after MCAo (Bederson et al., 1986). Figure 6 illustrates the effect of PAR-1 knockdown by shRNA on neurolgical behavior 24 hours after MCAo. We observed no difference between the groups 2 hours after stroke, suggesting that the level of acute injury was similar between the groups. The scores were significantly lower (better) 24 hours after stroke in the animals injected with PAR-1 shRNA lentivirus compared to misRNA injected animals (p< 0.001). Thus, the data suggest that PAR-1 knockdown ameliorates the behavioral deficits after ischemic stroke.
Figure 6. Effect of PAR-1 knockdown on neurological function.
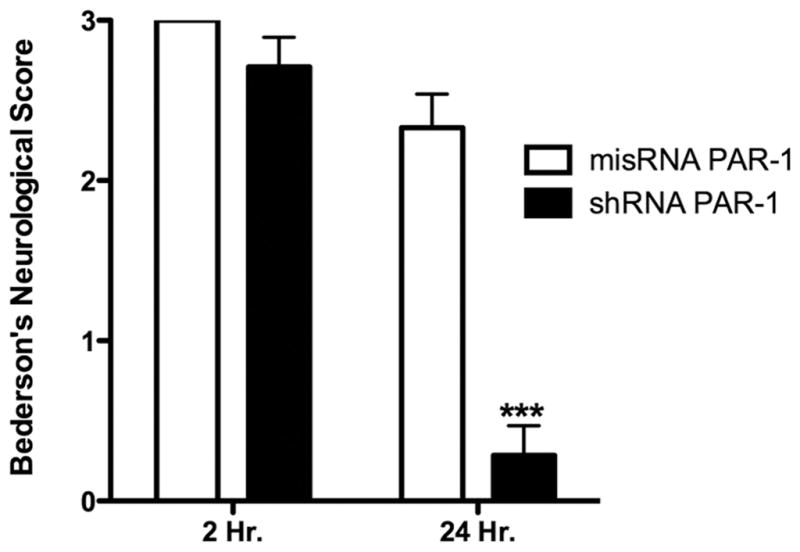
Stereotaxic injection of PAR-1shRNA in animals 1 week before MCAo mediated significant recovery of neurological functions compared to PAR-1 misRNA injection. Animals were tested for forelimb withdrawal when suspended by tail, twisting of animal toward contralateral side and circling behavior based on a standard rodent grading system. Each neurological deficit accounted for 1 point for a total score of 3 points. Data presented as mean ± SE (n=7), ***P<0.001, Students t-test for independent samples.
3.4 Oxygen-Glucose Deprivation in PAR-1 knockdown primary neuronal culture
Primary neuronal cultures were infected with misRNA or PAR-1 shRNA 1 week before OGD. Visualizing the cells with green fluorescence microscopy and counting the GFP positive cells we estimated the infection efficiency to be 80% (data not shown). Thrombin concentration for the OGD experiment was first determined from a separate dose escalation cell death assay (Fig. 7 inset). Under normoxic conditions 10U, 20U 50U and 100U per ml thrombin treatment resulted in approximately 50% cell death during 2-hour treatment in misRNA-infected cells while no effect was observed in shRNA infected cells (Fig. 7). After 24 hours, cell death increased in both treatment groups, but less so in the PAR-1 knockdown cells. However, during 2 hours of OGD the PAR-1 knockdown cells showed significantly decreased cell death with or without thrombin treatment when compared to misRNA infected cells without thrombin p<0.05 or with thrombin p<0.01. Upon treatment with thrombin during 24 hours reoxygenation after OGD, the PAR-1 knockdown cultured cells showed a significant reduction in cell death as compared to misRNA-infected cells (Fig. 7, p<0.01).
Figure 7. PAR-1 knockdown mediates neuroprotection in primary cultured neurons subjected to oxygen/glucose deprivation.

Primary cultured neurons infected with PAR-1 misRNA or shRNA for 24 hours and then grown for 5 or 6 days until at least 80% of cells were infected. The cells were divided into OGD (2 hours) or non-OGD groups at random. During OGD or non-OGD 50U/ml thrombin was added to the wells. The media was removed 2 hr and 24 hr after OGD followed by LDH assay to estimate cell death using spectrophotometric analysis at 492 nm. Data is presented as percentage mean±SE from three different experiments, each carried out in triplicate. Cells infected with PAR- 1 shRNA showed decreased neuronal cell death after OGD or without OGD with thrombin treatment in comparison to PAR-1 misRNA infected cells. PAR-1 knockdown significantly rescued the cells exposed to OGD with or without thrombin treatment compared to misRNA. Inset: Primary cultured cells were treated with increasing concentration of thrombin (1 U/ml to 100U/ml) for 2 hours and replaced with fresh medium after treatment. Medium withdrawn at 2 hours and 24 hours was used for LDH assay to determine percentage LDH release. Thrombin dose above 10U/ml showed a significant increase in cell death at 24 hours. Mean±SE. In both experiments the final results were analyzed using two-way ANOVA and the Bonferroni test for post-hoc comparisons. Significant difference misRNA compared to shRNA: * p<0.05, **p<0.01. Significant difference thrombin compared to control: # p<0.05, ### p<0.01, *** p<0.001
3.5 Transient ischemia in PAR-1−/− mice
Young PAR-1 knockout mice have been shown to resist ischemia-induced injury compared to young wild type mice (Junge et al., 2003). Since stroke typically occurs in aged subjects, we compared aged PAR-1−/− mice with aged PAR-3−/− and PAR-4−/− mice and compared them to young wild type mice. Animals were excluded if they showed less than a minimum 75% LDF drop in blood flow during MCAo. Ischemic lesion volume determined using TTC was significantly reduced in PAR-1−/− and PAR-3−/− mice compared with wild type (p<0.05) or PAR-4−/−mice (<0.01) (Fig 8A). We also compared the area of vascular disruption measured by high molecular weight dextran-FITC leakage in the penumbra. Vascular disruption was reduced inPAR-1−/− and PAR-3−/− mice compared to young wild type and PAR-4−/− mice (Fig. 8B, p<0.05).
Figure 8. PAR-1 knockout animals show reduction in neurovascular damage after acute ischemia.
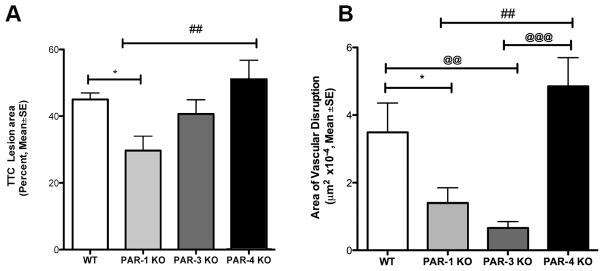
Animals were subjected to 2 hr MCAo and 48 hr of reperfusion. (A) Brains were processed for TTC staining for cellular metabolism. Using calibrated planimetry, the area of TTC pallor was measured and a blinded examiner compared the areas between different groups of animals. The percent TTC corresponds to percent of the whole brain section. PAR-1−/− animals showed a significant reduction in comparison with young wild types (* p<0.05) and PAR-4−/− mice (##p<0.01). (B) Severe vascular disruption was quantified by measuring the accumulation and leakage of high-molecular weight dextran-FITC fluorescence in coronal sections. PAR-1−/− animals showed significant reduction in vascular damage compared to wild type (* p<0.05) and age matched PAR-4 −/− (##p<0.01) mice. The PAR-3−/− animals also showed significant reduction in vascular disruption in comparison to young wild type and aged PAR-4−/− mice (@@@ p<.01). N=15–17 for each group; One-way ANOVA followed by a post hoc Bonferroni adjustment for multiple comparisons.
4.0 Discussion
In this study, we found that PAR-1 plays an essential role mediating damage to the neurovascular unit during cerebral ischemia. The selective silencing of PAR-1 using RNA interference techniques resulted in improved neurological function induced by MCAo ischemia. PAR-1 knockdown selectively decreased the ischemia-induced expression of c-Fos. PAR-1 shRNA positive cells showed no co-localization with TUNEL positive cells. PAR-1 knockdown results in 70% reduction in the number of Fluoro-Jade-C positive degenerating neurons counted in the ischemic striatum.
4.1 PAR-1 mediates cytotoxicity
Our data confirm an essential role for PAR-1 in mediating ischemic injury using 3 different models. After intracerebral injection of PAR-1 silencing mRNA, we showed significant cytoprotection in the infected cells in the ischemic area. The effect of the PAR-1 knock down was widespread, as evidenced by resistance to injury (c-Fos expression) throughout the ischemic bed (Fig. 3). Benefit was also expressed as improved behavioral outcome (Fig. 6) and as evidenced by several histological markers of cell injury (Figs 4, 5 and 7). Using an in vitro model of oxygen and glucose deprivation, cells with pre-injury PAR-1 knockdown significantly resisted injury (Fig 7). Finally, aged PAR-1 knock out animals showed significant protection from focal ischemia (Fig. 8).
Previously, a role for PAR-1 mediated neuroprotection has been suggested; our data show for the first time a direct link between PAR-1 and neurovascular injury. For example, in the ischemic zone, we identified degenerating cells immediately juxtaposed to viable cells lacking the PAR-1 receptor (Fig. 4c). Non-specific effects of the virus injection cannot explain the protective effect because the cells are residing next to each other, subject to the identical ischemic milieu. Similarly, injured cells expressing c-Fos could be identified next to healthy cells that lacked PAR-1 (Fig. 3E). Given the identical milieu in which the viable and dying cells are found, the only explanation for the survival is the absence of PAR-1. In cultured neuronal and astrocyte cells, resistance to both thrombin toxicity and OGD in the presence of thrombin was reduced by PAR-1 knockdown. In the controlled in vitro environment, there are no other factors that could explain the protective effect.
4.2 PAR-1 knockout correlated with protection in aged animals
Previous authors have shown that young PAR-1−/− animals resist ischemia (Junge et al., 2003). We replicated these findings in aged animals to assure the consistency of the result during advanced age (Fig 8A). This replication is important because knockout animals live without the PAR-1 receptor their entire lifetime, and could develop unknown compensatory mechanisms. Thus, compensatory effects such as upregulation of other PAR receptors could mediate the observed resistance to ischemia. In fact, recently one group reported that young PAR-4−/− animals showed ischemia resistance, an effect we did not see in aged animals (Mao et al., 2010). Those authors attributed the neuroprotective effect of PAR-4 knock out to an anti-platelet effect since the PAR-4 receptor in mice mediates platelet response to injury. We used the identical strain of animals, so we suspect that the different results indicate either a chance variation in experimental response, or a confounding effect of aging in our experiment. Previous authors have confirmed significant neuroprotection from agents that act on the PAR-1 receptor using an embolic thrombus model (Wang et al., 2012).
4.3 Astrocytes may participate in cytoprotection
Cells infected with PAR-1 shRNA lentivirus showed co-localization with neuronal markers as well as astrocytes (Fig. 2). In 30% of infected cells, neither neuronal nor astrocytic markers were detected, but neither NeuN nor GFAP reliably label 100% of their respective cell types. Infected cells showed no co-localization with microglial markers, consistent with the viability of the cells and resistance to inflammatory attack. In previous work, we have shown that PAR-1 mediated ischemic cell injury occurs primarily in neurons and endothelial cells, but to a lesser extent in astrocytes. The present data suggest that there may be a protective effect mediated via astrocytes as well. Further study will be needed to identify the specific roles played by each component of the neurovascular unit during PAR-1 dependent cell death. While our data clearly establish a direct link between the PAR-1 receptor and cell death (Figs. 3, 4, 5), the widespread zone of protection seen around the injection site (Fig. 3) raises the possibility of additional effects. Significant improvement in neurological deficit withPAR-1 knockdown indicates a critical role of PAR-1 in mediating ischemia injury (Fig 6). Recently, using siRNA injected into the lateral ventricle, behavioral and lesion volume results similar to ours were obtained (Zhang et al., 2012).
4.4 Limitations
There are some limitations to our methods that suggest a cautious interpretation. Although we used LDF to assure ischemia, we could not perform regional cerebral blood flow studies in each subject. Therefore, there is the remote possibility that the subjects receiving the shRNA somehow all suffered less ischemia than the misRNA injected subjects. We view this possibility as extremely unlikely, given the very high reproducibility of the model—as shown in Fig. 2A—in which the medial striatum is involved in 100% of subjects, a finding we have published consistently over the course of several years (Chen et al., 2009). To minimize possible bias, all animals were randomly assigned to one treatment group or the other; all surgeries and injections were done without knowledge of the treatment group assignment; and all behavioral and imaging assessments were done by an investigator blinded to treatment group (Lapchak et al., 2013). Stroke modeling with the nylon filament technique, while standard in this field, is not physiologically representative; our results could be confirmed using an embolic thrombus approach, but no data exists to suggest the findings would be significantly different between the 2 approaches.
4.5 Clinical Translational Implications
These data have significant implications for stroke victims. In the past, experimental findings in pre-clinical animal models have not translated well to human stroke trials. We and others have shown powerful benefits of drugs that block the main activator for PAR-1, the serine protease thrombin, and we showed that a thrombin inhibitor could be effective even if delayed to a clinically relevant 3 hours after ischemia onset (Lyden et al., 2014). Agents that act on the PAR-1 receptor directly also show powerful neuroprotective effects in multiple laboratories, using different models, at clinical relevant delay times (Shibata et al., 2001). One such agent is in early phase clinical trials (Lyden et al., 2013). Our present data suggest that the protective effect of these drugs is mediated directly by PAR-1, not by off-target effects, thus significantly increasing the likelihood that such stroke trials could be successful. Our data also suggest that future strategies, perhaps aimed at downstream transduction events triggered by PAR-1 actions, could similarly be successful. Future studies will be needed to explore these downstream actions of PAR-1 agonists, biased agonists, and antagonists. Even though the shRNA injections occurred before the MCAo, the results provided here indicate direct mediation of PAR-1 in several post-ischemia pathophysiological pathways. PAR-1 is present on astrocytes and neurons along with other cell types hence engineering various cellular targets will be critical for developing a therapeutic for ischemic injury.
5.0 Conclusion
Our data confirms the role of thrombin-activated PAR-1 in mediating ischemic cell death. Further, we have demonstrated for the first time that PAR-1 is both necessary and sufficient to mediate cell death during ischemia. Treatment strategies targeting PAR-1 are expected to protect the neurovascular unit during ischemic injury.
PAR-1 knockdown neuronal and astrocytes cells are negative for TUNEL positive cells.
PAR-1 knockdown results 70% reduction of dying neurons with improved neurological deficits.
Knockdown or inhibition of PAR-1 would provide neuroprotection in brain from ischemic injury.
Acknowledgments
This work was supported by the National Institute of Neurological Disorders and Stroke R01 NS075930 (Lyden). PAR knock out mouse breeding pairs were kindly provided by Dr. Berislav Zlokovic and Dr. Sean Coughlin. Dr. Kolja Wawrowsky kindly supplied the CytoFx© software.
Abbreviations
- ANOVA
analysis of variance
- APC
Activated Protein-C
- CCD
charge-coupled Device
- DMEM
Dulbecco’s modified eagle’s medium
- DNAse
deoxyribonuclease
- E14
embryonic day 14
- G-protein
guanosine nucleotide-binding protein
- GFP
green fluorescent protein
- GFAP
glial fibrillary acidic protein
- IACUC
Institutional animal care and use committee
- IBA1
Ionized calcium-binding adapter molecule 1
- LDF
laser Doppler flowmetry
- LDH
lactate dehydrogenase
- MCA
middle cerebral artery
- MCAo
middle cerebral artery occlusion
- NeuN
neuronal nuclei
- OGD
oxygen glucose deprivation
- PARs
protease activated receptors
- PBS
phosphate buffered saline
- PCR
polymerase chain reaction
- rtPA
recombinant tissue plasminogen activator
- STAIR
stroke therapy academic industry roundtable
- shRNA
short hairpin ribonucleic acid
- siRNA
small interfering ribonucleic acid
- TTC
2,3,5-triphenyl-2H-tetrazolium chloride
- TU
Titer Unit
- TUNEL
terminal deoxynucleotidyltransferase mediated dUTP Nick End Labeling
- U
Unit
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams HP, Jr, Effron MB, Torner J, Davalos A, Frayne J, Teal P, Leclerc J, Oemar B, Padgett L, Barnathan ES, Hacke W. Emergency administration of abciximab for treatment of patients with acute ischemic stroke: results of an international phase III trial: Abciximab in Emergency Treatment of Stroke Trial (AbESTT-II) Stroke. 2008;39:87–99. doi: 10.1161/STROKEAHA.106.476648. [DOI] [PubMed] [Google Scholar]
- Ames A, III, Wright RL, Kowada M, Thurston JM, Majno G. Cerebral ischemia. II. The no-reflow phenomenon. Am J Pathol. 1968;52:437. [PMC free article] [PubMed] [Google Scholar]
- Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- Biller J, Massey EW, Marler JR, Adams HP., Jr A dose escalation study of ORG 10172 (low molecular weight heparinoid) in stroke. Neurology. 1989;39:262. doi: 10.1212/wnl.39.2.262. [DOI] [PubMed] [Google Scholar]
- Cavanaugh KP, Gurwitz D, Cunningham DD, Bradshaw RA. Reciprocal modulation of astrocyte stellation by thrombin and protease nexin-1. J Neurochem. 1990;54:1735–1743. doi: 10.1111/j.1471-4159.1990.tb01228.x. [DOI] [PubMed] [Google Scholar]
- Chen B, Cheng Q, Yang K, Lyden PD. Thrombin mediates severe neurovascular injury during ischemia. Stroke. 2010;41:2348–2352. doi: 10.1161/STROKEAHA.110.584920. [DOI] [PubMed] [Google Scholar]
- Chen B, Friedman B, Cheng Q, Tsai P, Schim E, Kleinfeld D, Lyden PD. Severe blood-brain barrier disruption and surrounding tissue injury. Stroke. 2009;40:e666–674. doi: 10.1161/STROKEAHA.109.551341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Friedman B, Whitney MA, Winkle JA, Lei IF, Olson ES, Cheng Q, Pereira B, Zhao L, Tsien RY, Lyden PD. Thrombin activity associated with neuronal damage during acute focal ischemia. J Neurosci. 2012;32:7622–7631. doi: 10.1523/JNEUROSCI.0369-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- Davalos D, Baeten KM, Whitney MA, Mullins ES, Friedman B, Olson ES, Ryu JK, Smirnoff DS, Petersen MA, Bedard C, Degen JL, Tsien RY, Akassoglou K. Early detection of thrombin activity in neuroinflammatory disease. Annals of Neurology. 2014;75:303–308. doi: 10.1002/ana.24078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donovan FM, Pike CJ, Cotman CW, Cunningham DD. Thrombin induces apoptosis in cultured neurons and astrocytes via a pathway requiring tyrosine kinase and RhoA activities. J Neurosci. 1997;17:5316–5326. doi: 10.1523/JNEUROSCI.17-14-05316.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher M, Feuerstein G, Howells DW, Hurn PD, Kent TA, Savitz SI, Lo EH. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40:2244–2250. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingrich MB, Junge CE, Lyuboslavsky P, Traynelis SF. Potentiation of NMDA receptor function by the serine protease thrombin. J Neurosci. 2000;20:4582–4595. doi: 10.1523/JNEUROSCI.20-12-04582.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingrich MB, Traynelis SF. Serine proteases and brain damage - is there a link? Trends Neurosci. 2000;23:399–407. doi: 10.1016/s0166-2236(00)01617-9. [DOI] [PubMed] [Google Scholar]
- Gordon DL, Linhardt R, Adams HP., Jr Low-molecular-weight heparins and heparinoids and their use in acute or progressing ischemic stroke. Clin Neuropharmacology. 1990;13:522. doi: 10.1097/00002826-199012000-00005. [DOI] [PubMed] [Google Scholar]
- Grabham P, Cunningham DD. Thrombin receptor activation stimulates astrocyte proliferation and reversal of stellation by distinct pathways: involvement of tyrosine phosphorylation. J Neurochem. 1995;64:583–591. doi: 10.1046/j.1471-4159.1995.64020583.x. [DOI] [PubMed] [Google Scholar]
- Gurwitz D, Cunningham DD. Thrombin modulates and reverses neuroblastoma neurite outgrowth. Proc Natl Acad Sci U S A. 1988;85:3440–3444. doi: 10.1073/pnas.85.10.3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenberg MD, Compton SJ. International Union of Pharmacology. XXVIII. Proteinase-activated receptors. Pharmacol Rev. 2002;54:203–217. doi: 10.1124/pr.54.2.203. [DOI] [PubMed] [Google Scholar]
- Hu E, Mueller E, Oliviero S, Papaioannou VE, Johnson R, Spiegelman BM. Targeted disruption of the c-fos gene demonstrates c-fos-dependent and - independent pathways for gene expression stimulated by growth factors or oncogenes. The EMBO journal. 1994;13:3094–3103. doi: 10.1002/j.1460-2075.1994.tb06608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junge CE, Sugawara T, Mannaioni G, Alagarsamy S, Conn PJ, Brat DJ, Chan PH, Traynelis SF. The contribution of protease-activated receptor 1 to neuronal damage caused by transient focal cerebral ischemia. Proc Natl Acad Sci U S A. 2003;100:13019–13024. doi: 10.1073/pnas.2235594100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapchak PA, Zhang JH, Noble-Haeusslein LJ. RIGOR Guidelines: Escalating STAIR and STEPS for Effective Translational Research. Trans Stroke Res. 2013;4:279–285. doi: 10.1007/s12975-012-0209-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KR, Colon GP, Betz AL, Keep RF, Kim S, Hoff JT. Edema from intracerebral hemorrhage: the role of thrombin. J Neurosur. 1996;84:91–96. doi: 10.3171/jns.1996.84.1.0091. [DOI] [PubMed] [Google Scholar]
- Lee KR, Drury I, Vitarbo E, Hoff JT. Seizures induced by intracerebral injection of thrombin: a model of intracerebral hemorrhage. J Neurosur. 1997;87:73–78. doi: 10.3171/jns.1997.87.1.0073. [DOI] [PubMed] [Google Scholar]
- Liu J, Schuff-Werner P, Steiner M. Double transfection improves small interfering RNA-induced thrombin receptor (PAR-1) gene silencing in DU 145 prostate cancer cells. FEBS Lett. 2004;577:175–180. doi: 10.1016/j.febslet.2004.09.079. [DOI] [PubMed] [Google Scholar]
- Liu J, Schuff-Werner P, Steiner M. Thrombin/thrombin receptor (PAR-1)-mediated induction of IL-8 and VEGF expression in prostate cancer cells. Biochem Biophys Res Commun. 2006;343:183–189. doi: 10.1016/j.bbrc.2006.02.136. [DOI] [PubMed] [Google Scholar]
- Lyden P, Levy H, Weymer S, Pryor K, Kramer W, Griffin JH, Davis TP, Zlokovic B. Phase 1 safety, tolerability and pharmacokinetics of 3K3A-APC in healthy adult volunteers. Current Pharma Design. 2013;19:7479–7485. doi: 10.2174/1381612819666131230131454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyden P, Pereira B, Chen B, Zhao L, Lamb J, Lei IF, Rajput P. Direct thrombin inhibitor argatroban reduces stroke damage in 2 different models. Stroke. 2014;45:896–899. doi: 10.1161/STROKEAHA.113.004488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlane SR, Seatter MJ, Kanke T, Hunter GD, Plevin R. Proteinase-activated receptors. Pharmacol Rev. 2001;53:245–282. [PubMed] [Google Scholar]
- Mao Y, Zhang M, Tuma RF, Kunapuli SP. Deficiency of PAR4 attenuates cerebral ischemia/reperfusion injury in mice. J Cereb Blood Flow Metab. 2010;30:1044–1052. doi: 10.1038/jcbfm.2009.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi T, Sedehizade F, Reymann KG, Reiser G. Protease-activated receptors in neuronal development, neurodegeneration, and neuroprotection: thrombin as signaling molecule in the brain. Neuroscientist. 2004;10:501–512. doi: 10.1177/1073858404269955. [DOI] [PubMed] [Google Scholar]
- Sarker KP, Abeyama K, Nishi J, Nakata M, Tokioka T, Nakajima T, Kitajima I, Maruyama I. Inhibition of thrombin-induced neuronal cell death by recombinant thrombomodulin and E5510, a synthetic thrombin receptor signaling inhibitor. Thromb Haemost. 1999;82:1071–1077. [PubMed] [Google Scholar]
- Sharp FR, Sagar SM. Alterations in gene expression as an index of neuronal injury: heat shock and the immediate early gene response. Neurotoxicology. 1994;15:51–59. [PubMed] [Google Scholar]
- Shibata M, Kumar SR, Amar A, Fernandez JA, Hofman F, Griffin JH, Zlokovic BV. Anti-inflammatory, antithrombotic, and neuroprotective effects of activated protein C in a murine model of focal ischemic stroke. Circulation. 2001;103:1799–1805. doi: 10.1161/01.cir.103.13.1799. [DOI] [PubMed] [Google Scholar]
- Striggow F, Riek M, Breder J, Henrich-Noack P, Reymann KG, Reiser G. The protease thrombin is an endogenous mediator of hippocampal neuroprotection against ischemia at low concentrations but causes degeneration at high concentrations. Proc Natl Acad Sci U S A. 2000;97:2264–2269. doi: 10.1073/pnas.040552897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Winkle JA, Chen B, Lei IF, Pereira B, Rajput PS, Lyden PD. Concurrent middle cerebral artery occlusion and intra-arterial drug infusion via ipsilateral common carotid artery catheter in the rat. J Neurosci Meth. 2012;213:63–69. doi: 10.1016/j.jneumeth.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villares GJ, Zigler M, Wang H, Melnikova VO, Wu H, Friedman R, Leslie MC, Vivas-Mejia PE, Lopez-Berestein G, Sood AK, Bar-Eli M. Targeting melanoma growth and metastasis with systemic delivery of liposome-incorporated protease-activated receptor-1 small interfering RNA. Cancer Res. 2008;68:9078–9086. doi: 10.1158/0008-5472.CAN-08-2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang Z, Chow N, Davis TP, Griffin JH, Chopp M, Zlokovic BV. An activated protein C analog with reduced anticoagulant activity extends the therapeutic window of tissue plasminogen activator for ischemic stroke in rodents. Stroke. 2012;43:2444–2449. doi: 10.1161/STROKEAHA.112.658997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhao Z, Chow N, Rajput PS, Griffin JH, Lyden PD, Zlokovic BV. Activated protein C analog protects from ischemic stroke and extends the therapeutic window of tissue-type plasminogen activator in aged female mice and hypertensive rats. Stroke. 2013;44:3529–3536. doi: 10.1161/STROKEAHA.113.003350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein JR, Lau AL, Brass LF, Cunningham DD. Injury-related factors and conditions down-regulate the thrombin receptor (PAR-1) in a human neuronal cell line. J Neurochem. 1998;71:1034–1050. doi: 10.1046/j.1471-4159.1998.71031034.x. [DOI] [PubMed] [Google Scholar]
- Xi G, Reiser G, Keep RF. The role of thrombin and thrombin receptors in ischemic, hemorrhagic and traumatic brain injury: deleterious or protective? J Neurochem. 2003;84:3–9. doi: 10.1046/j.1471-4159.2003.01268.x. [DOI] [PubMed] [Google Scholar]
- Zhang J, Wang Y, Zhu P, Wang X, Lv M, Feng H. siRNA-mediated silence of protease-activated receptor-1 minimizes ischemic injury of cerebral cortex through HSP70 and MAP2. J Neurological Sci. 2012;320:6–11. doi: 10.1016/j.jns.2012.05.040. [DOI] [PubMed] [Google Scholar]