

Abstract
Both the opioid antagonist naltrexone and corticotropin-releasing factor type-1 receptor (CRF-R1) antagonists have been investigated for the treatment of alcoholism. The current study examines the combination of naltrexone and CP154526 to reduce intermittent access ethanol drinking (IAA) in C57BL/6J male mice, and if these compounds reduce drinking via serotonergic mechanisms in the dorsal raphé nucleus (DRN). Systemic injections and chronic icv infusions of naltrexone, CP154526 or CP376395 transiently decreased IAA drinking. Immunohistochemistry revealed CRF-R1 or μ-opioid receptor (MOR) immunoreactivity was co-localized in tryptophan hydroxylase (TPH)-immunoreactive neurons as well as non-TPH neurons in the DRN. Mice with a history of IAA or continuous access to alcohol were microinjected with aCSF, naltrexone, CP154526 or the combination into the DRN or the median raphé nucleus (MRN). Either intra-DRN naltrexone or CP154526 reduced IAA in the initial 2-hours of fluid access, but the combination did not additively suppress IAA, suggesting a common mechanism via which these two compounds affect intermittent drinking. These alcohol-reducing effects were localized to the DRN of IAA drinkers, as intra-MRN injections only significantly suppressed water drinking, and continuous access drinkers were not affected by CRF-R1 antagonism. Extracellular serotonin was measured in the medial prefrontal cortex (mPFC) using in vivo microdialysis after intra-DRN microinjections in another group of mice. Intra-DRN CP154526 increased serotonin impulse flow to the mPFC while naltrexone did not. This suggests the mPFC may not be an essential location to intermittent drinking, as evidenced by different effects on serotonin signaling to the forebrain yet similar behavioral findings.
Keywords: CRF-R1, dorsal raphe nucleus, intermittent alcohol, naltrexone
INTRODUCTION
The neural adaptations during the transition to alcohol dependence remain to be fully characterized, since they may reveal targets for therapeutic intervention. One model of heavy drinking in animals is based on intermittent access to alcohol (IAA), which engenders high-level binge drinking, as observed in alcohol-use disorders. IAA generates high levels of voluntary and preferential ethanol drinking, up to 9 grams/kilogram (g/kg) per day in rats and greater than 20 g/kg/day in C57BL/6J mice which eventually results in convulsive withdrawal symptoms (Wise 1973; Simms et al. 2008; Hwa et al. 2011).
Several pharmacotherapies have been explored to reduce IAA drinking. One U.S. Food and Drug Administration-approved medication for alcoholism, the opioid antagonist naltrexone, decreased 20% ethanol drinking both during intermittent and continuous access schedules (Simms et al. 2008; Sabino et al. 2013). Naltrexone can be contrasted with other compounds that target the negatively reinforcing effects of alcohol withdrawal and stress-induced relapse like corticotropin-releasing factor receptor-type 1 (CRF-R1) antagonists (Heilig and Koob, 2007). Systemic injections of CRF-R1 antagonists like antalarmin or R121919 were not effective in reducing IAA in rats (Cippitelli et al. 2012; Sabino et al. 2013) but other compounds like CP376395 decreased IAA (Simms et al. 2013). Microinjection of CRF-R1 antagonist CP154526 into the dorsal raphé nucleus (DRN) selectively reduced high but not low ethanol drinking (Hwa et al. 2013), which suggests some site-specificity for CRF-R1 in heavy drinkers.
The DRN contains most serotonin (5-HT) neurons that project to the forebrain (Imai et al. 1986) and is a target for neuropeptides such as opioid peptides and CRF. Opioids, like morphine, have excitatory effects on DRN 5-HT neurotransmission indirectly through GABAergic inhibition on 5-HT neurons (Staub et al. 2012). CRF, at low to moderate doses, inhibits 5-HT neuronal activity in the DRN and neuronal release via CRF-R1 on GABA afferents while higher doses stimulate DRN 5-HT neuronal activity (Kirby et al. 2000; Lowry et al. 2000;). These studies imply that opioid and CRF receptors may differentially modulate 5-HT signaling in the DRN. 5-HT has been long implicated in regulating ethanol intake, abuse and dependence (Rolf et al. 1978), so the anti-alcohol effects of neuropeptide antagonists may be caused by modulating 5-HT neurons in the DRN that project to the forebrain. Alcoholics have substantially reduced 5-HT neurotransmission in the prefrontal cortex (PFC; Mantere et al. 2002).
Altogether, this behavioral and neurochemical evidence suggests the hypothesis that both opioid receptors and CRF-R1 may play a role in excessive drinking, possibly through 5-HTergic mechanisms in the DRN. The current studies investigated whether naltrexone and CRF-R1 antagonist CP154526 act dependently or independently in the DRN to reduce IAA drinking in C57BL/6J mice. Co-infusion of naltrexone and CP154526 into the DRN may or may not produce an additive suppression of IAA drinking, revealing common or distinct neural pathways. We also use in vivo microdialysis in a terminal region for the 5-HT DRN system, the PFC, to measure extracellular 5-HT. It is hypothesized that CP154526 may more potently disinhibit 5-HT neurons than naltrexone, increasing 5-HT in the PFC, as its efficacy may be more selective to high-level drinkers rather than moderate-level drinkers. In this, we test if behavioral reductions in alcohol drinking are akin to the effects on 5-HT impulse flow, confirming or refuting another hypothesis that 5-HT transmission to the mPFC is crucial for IAA drinking.
METHODS
Animals
Eight-week old, male C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) were housed in groups of six or more upon arrival. Mice acclimated to a 12-hour reversed light/dark cycle with constant temperature (21 ± 2°C) and humidity (25%). After 5 days of group-housing, mice were singly housed in polycarbonate cages (28 × 17 × 12 cm) with stainless steel wire mesh lids and pine shavings as bedding. Water and standard food (LabDiet 5001 Rodent Diet) were available ad libitum. The Tufts University Institutional Animal Care and Use Committee approved all procedures, which followed guidelines set by the NIH Guide for Care and Use of Laboratory Animals (2011).
Ethanol Drinking Procedures
Mice were given intermittent access to 20% ethanol and water, which has been previously described in detail (Hwa et al. 2011). Briefly, 20% ethanol solution (w/v) was prepared from 95% ethanol (Pharmaco-AAPER, Brookfield, CT) in one bottle accompanied by a bottle of water on Mondays, Wednesdays, and Fridays for 24 hr. Two bottles of water were presented to mice on the remaining days of the week. In experiment 4, a group of mice were given continuous access to 20% ethanol and water for 12 consecutive days, the equivalent number of drinking days to 4 weeks in the IAA schedule. Two, 4, and 24 hr fluid intakes were measured by assessing bottle weights before and after drug manipulations. In order to quantify water and ethanol lost from bottles not due to drinking, full bottles were weighed, then placed on a cage without a mouse, and weighed again at the end of 24 hr. This was done daily, and the average volume lost to “drip” was subtracted from individual values.
Blood ethanol concentrations were measured in a subset of animals given intermittent access (n=11) and continuous access (n=12) to alcohol for 2 hr. Blood was collected from the submandibular vein and centrifuged for 10 min at 1000 G. Plasma was extracted and blood ethanol concentrations were analyzed with the AM1 Analox Analyzer (Analox Instruments Inc., Lunenburg, MA).
Experiment 1: Systemic injections of naltrexone and CRF-R1 antagonists
After 4 weeks of IAA, mice were assessed for ethanol drinking behavior after systemic drug manipulations. Repeated doses of 0.9% saline, 1, and 10 mg/kg naltrexone were administered i.p. 20 min before ethanol and water bottles were given to mice (n=12). In another group of mice (n=12), repeated doses of 0.6% methylcellulose, 10, 17 and 30 mg/kg CP154526 were administered i.p. 20 min before two-bottle choice. In order to test another highly potent, more water-soluble CRF-R1 antagonist, a third group of mice (n=12) was given 0.9% saline, 10, 17 and 30 mg/kg CP376395 20 min before IAA. Injection volumes for systemic drugs were 10 ml/kg. Drug doses were chosen to preserve logarithmically scaled dosing and counterbalanced across 4 two-bottle choice test sessions. Ethanol intake in grams per kilogram of bodyweight (g/kg) ethanol intake in milliliters (ml), and water intake (ml) were measured at 2, 4, and 24 hr after drug injections.
Experiment 2: Chronic i.c.v. infusion of naltrexone and CRF-R1 antagonists
Separate groups of mice (n=6–8/group) were implanted with osmotic minipumps for chronic intracerebroventricular (i.c.v.) drug infusion after 4 weeks of IAA. Mice were anesthetized with a 100 mg/kg ketamine and 10 mg/kg xylazine combination i.p. before surgery and given carprofen (5 mg/kg) subcutaneously (s.c.) for analgesia. Osmotic minipumps (Model 1002, Alzet, Cupertino, CA) connected through a 20 mm-long polyvinylchloride catheter to a 30 gauge cannula (Brain Infusion Kit 3, Alzet). Cannulae were stereotaxically implanted targeting the lateral ventricle at coordinates AP +0.5 mm, ML −0.6 mm from bregma, DV −3.0 mm from dura. Minipumps rested s.c. over the left shoulder blade. Animals recovered for 2 days before tests for ethanol drinking were resumed on day 3.
Groups of mice were given minipumps containing aCSF, 3 μg/μl naltrexone, 30 μg/μl naltrexone, or 0.3 μg/μl CP376395. Other mice were given 4% DMSO or 0.3 μg/μl CP154526. CP154526 did not stay in aCSF for longer than one hr at 37°C (personal observations), so we chose to apply an additional, water soluble CRF-R1 antagonist, CP376395, for the 14-day chronic infusion. All minipumps had flow rates of 0.25 μl/hr. Two-bottle choice drinking behaviors were assessed on minipump infusion days 3, 5, 7, 10, and 12, which corresponded to a normal schedule of IAA. Two, 4, and 24 hr ethanol (g/kg) and water intake (ml) were monitored over the 14-day duration of the minipump.
To verify that naltrexone was chronically administered across the 14-day pump duration, the animals that received naltrexone or aCSF in the minipump were tested in a morphine-sensitive tail withdrawal procedure (adapted from Miczek & Winslow, 1987). After ethanol was removed on minipump day 13, mice were gently restrained so that the distal end of the tail could be submerged in a 54°C water bath. Latency to withdraw the tail (sec) was measured after 0 (saline baseline), 10, 20, 30, and 40 mg/kg i.p. morphine administered through cumulative dosing every 20 min. If the tail withdrawal did not occur within a 20 s cutoff, the test was discontinued, and the mice were not tested with further doses of morphine.
After the drinking tests and/or the tail withdrawal procedure, mice were given an overdose of 100 mg/kg ketamine and 10 mg/kg xylazine combination and perfused with saline and 4% paraformaldehyde. Brains were removed and sliced using a Leica Cryostat (CM1900, Bannockburn, IL). Cannula placement was verified using Nissl staining. Animals with incorrect placements (n=2) into the lateral ventricle were excluded from analysis.
Experiment 3: Immunohistological identification of CRF-R1 or MOR on DRN 5-HT cells
C57BL/6J mice with a history of 4 weeks intermittent alcohol drinking (n=4) or 4 weeks water (n=4) were transcardially perfused with 0.1 M phosphate buffer solution (PBS) and with 4% paraformaldehyde in PBS. Brains were removed and post-fixed overnight at 4°C. They were then placed in a sucrose solution with 0.1% sodium azide and shipped to the Children’s Hospital of Philadelphia for immunohistochemistry. Coronal serial sections (14 μm-thick) were cut onto slides (Fisherbrand ProbeOn Plus; Fisher Scientific). Slides were placed in 4% paraformaldehyde for 1 hour followed by rinses. Sections were incubated with a cocktail of goat anti-CRF1 (C-20, 1:500, Santa Cruz Biotechnology) or rabbit anti-μ-opioid receptor (MOR, 1:1,000, Millipore) and mouse anti-tryptophan hydroxylase (TPH, 1:500, Abcam) for 72 hours at 4°C. Sections were then rinsed and incubated with secondary antibodies for 90 min at room temperature followed by rinses in phosphate buffer. For the CRF1/TPH labeling, AlexaFluor 647-conjugated donkey anti-mouse IgG (1:200, Life Technologies) was used to visualize TPH immunoreactivity and FITC-conjugated donkey anti-goat IgG (1:200, Jackson Immunoresearch) was used to visualize CRF1 immunoreactivity. For MOR/TPH labeling, FITC-conjugated donkey anti-mouse IgG (1:200, Jackson Immunoresearch) was used to visualize TPH immunoreacity and RITC-conjugated donkey anti-rabbit (1:200, Jackson Immunoresarch) was used to visualize MOR. Finally, sections were coverslipped and visualized using a Leica DM5000B. Images were captured using a Hamamatsu ORCA-ER digital camera and Leica Microsystems software.
Experiment 4: Intra-DRN microinjections of naltrexone and CRF-R1 antagonist
Mice with a history of 4-week intermittent ethanol drinking were prepared with intra-DRN cannulae for microinjections of naltrexone and CP154526. In similar surgical methods as Experiment 2, mice were implanted with 26 gauge, 6 mm cannulae (Plastics One, Roanoke, VA) targeting the DRN at the coordinates AP −4.2 mm, ML +1.5 mm from bregma, DV −1.9 mm from dura with 26° angle. To investigate if changes in drinking behavior were site specific to the DRN, another group of mice were implanted with cannulae targeting the median raphé nucleus (MRN) at AP −4.2 mm, ML +1.2 mm, DV −3.0 mm from bregma with 14° angle. Fitted obdurators protruded 0.5 mm beyond the guide cannulae. Mice were allowed 3 days recovery before resuming 2–3 days of ethanol and water drinking to return to pre-surgical drinking behavior. During this period, obdurators were handled daily to habituate animals to microinjection procedures.
Microinjection procedures have been previously described in Hwa et al. (2013). On test days, 33-gauge microinjectors (Plastics One) infused 0.2 μl drug into the DRN at a flow rate of 0.1 μl/min. Injectors were connected through polyethylene tubing to a glass syringe controlled by a microinjection pump (CMA/100, CMA Microdialysis, Sweden). One group of mice (n=8) was injected intra-DRN with aCSF, 6 μg naltrexone, 0.6 μg CP154526, and the combination of 6 μg naltrexone and 0.6 μg CP154526 on 4 intermittent ethanol-water test days. In addition to the group of mice that received the high dose combination, we studied a second group of mice (n=7) that was injected intra-DRN with aCSF, 3 μg naltrexone, 0.3 μg CP154526, and a lower combination of 3 μg naltrexone and 0.3 μg CP154526. To consider if microinjections were specific to the DRN, another group of intermittent access mice (n=9) were microinjected intra-MRN with aCSF, 6 μg naltrexone, 0.6 μg CP154526 and the combination. Finally, to assess the drug effects upon intermittent vs. continuous access drinkers, continuous access mice (n=9) were injected intra-DRN with aCSF, 6 μg naltrexone, 0.6 μg CP154526, and the combination.
During microinjections, mice were allowed to move about freely. Injectors were kept in place for 1 min after microinfusion to minimize capillary backflow up the cannula. Alcohol and water bottles were given to mice 9 min later to measure drug effects on drinking behavior. Two, 4, and 24 hr fluid consumption were assessed after each microinjection. Similar to Experiment 2, animals were perfused after testing procedures, and brains were collected for histological verification of the cannula placement in the DRN or MRN. Mice with missed placements (n=6) were eliminated from analysis.
Experiment 5: 5-HT measurement in the mPFC after intra-DRN naltrexone or CRF-R1 antagonist
To further investigate how opioid receptor and CRF-R1 antagonism in the DRN affect 5-HT impulse flow to the forebrain, IAA mice were dually implanted with 26 gauge, 6 mm cannula for intra-DRN microinjections and a microdialysis probe in the mPFC for 5-HT measurement. Mice that had 4 weeks of intermittent access were surgically prepared with cannulae targeting the DRN, as described in Experiment 4. Additionally, they had microdialysis guide cannulae (CMA 7, Harvard Apparatus, Holliston, MA) targeting the pre- and infralimbic regions of the mPFC (AP +2.0 mm, ML −0.3 mm from bregma, DV −1.0 mm from dura). Animals were allowed 3 days of recovery before resumption of IAA drinking and daily handling of the obdurator.
The night before the test day, mice were anesthetized with isoflurane (Webster Veterinary, Devens, MA) and the dummy probe was replaced with the microdialysis probe with a 1-mm active membrane (CMA 7, Harvard Apparatus). The probe was infused with aCSF at a flow rate of 0.5 μl/min overnight (CMA 400 microinfusion pump).
On test day, the flow rate was increased to 1.5 μl/min. Mice were habituated to the increased flow rate for 1 hr before dialysate sample collection. Samples were collected every 20 min. In similar methods as Experiment 4, 0.2 μl microinjections of either aCSF, 6 μg naltrexone or 0.6 μg CP154526 occurred 10 min before hr 3 of the dark cycle at a flow rate of 0.1 μl/min, which is when access to ethanol and water is normally given. Microinjection and placement verification procedures were identical to those described in Experiment 4. Mice with missed placements in either the DRN or the mPFC (n=7) were excluded from analysis.
5-HT was measured using electrochemical detection equipped with high performance liquid chromatography (HPLC; Takahashi et al. 2010; Shimamoto et al. 2011). A stabilizing agent of 20 mM phosphate buffer with 25 mM EDTA was added to 30 μl dialysate samples. A cation-exchange column (Capcell Pak SCX, 1.5mm × 250 mm, 5 μm I.D., Shiseido, Tokyo, Japan) separated monoamines at a column temperature of 30°C and a flow rate of 0.2 ml/min. The mobile phase consisted of 150 mM ammonium acetate, 50 mM citric acid, 27 μM EDTA, 10% methanol, and 1% acetonitrile with pH adjusted to 4.6. 5-HT concentrations were determined by using standard curves with known amounts of 5-HT in a range of 0.125–0.5 pg. The limit of detection was 2 fg under these conditions with a 10.5% recovery rate.
Drugs
For systemic injections in Experiment 1, naltrexone HCl (NIDA) was dissolved into 0.9% sterile saline at 1 and 10 mg/kg for i.p. delivery. CP154526 is a prototypic CRF-R1 antagonist with high affinity for CRF-R1 (Ki<10; Schulz et al. 1996). CP376395 also has high selectivity for CRF-R1 (IC50=5) with an advantage of being more water-soluble than CP154526 (Chen et al. 2008). CP154526 (Tocris) was freshly suspended in a vehicle of 0.6% methylcellulose, and CP376395 (Tocris) was dissolved in 0.9% sterile saline. For chronic i.c.v. infusion in Experiment 2, 3 μg/μl naltrexone, 30 μg/μl naltrexone and 0.3 μg/μl CP376,395 were dissolved in filtered aCSF. 0.3 μg/μl CP154526 was also chronically delivered in the minipump, though it required a vehicle of 4% DMSO. 4% DMSO was used as a control. Minipump doses were chosen based on a log step reduction from the microinjection doses. Morphine HCl (NIDA), used for the tail withdrawal procedure, was dissolved in sterile saline at 10 mg/kg. For microinjections in Experiments 4, drug doses were chosen based on previous intracerebral studies in C57BL/6J mice with CP154526 in the laboratory (Hwa et al. 2013). Naltrexone and CP154526 were freshly suspended in filtered aCSF on the day of testing. The most effective intra-DRN doses for decreasing IAA, 6 μg naltrexone and 0.6 μg CP154526, were used to evoke changes in 5-HT in Experiment 5 microdialysis.
Statistical Analyses
Blood ethanol concentrations of mice that underwent either IAA or continuous access were compared with a one-way analysis of variance (ANOVA). Ethanol intake (grams/kilogram and milliliter) and water intake (ml) were measured in experiments with drug manipulations. For Experiment 1, one-way repeated measures ANOVAs were run at each time-point after naltrexone, CP154526 or CP376395 injection. For Experiment 2, two-way repeated measures ANOVAs were conducted (Drug × Day) to see if drugs infused i.c.v. via minipump affected fluid intake over 14 days. A two-way repeated measures ANOVA (Drug treatment vs. Morphine dose) was also used to test whether morphine-induced analgesia was effective on minipump day 13 in aCSF and naltrexone-infused animals. For Experiment 4, one-way repeated measures ANOVAs were run for 2, 4, and 24 hr drinking data to assess if microinfusions into the DRN or MRN affected intermittent or continuous access drinking. For Experiment 5, a two-way repeated measures ANOVA (Drug × Time) was conducted to test if the microinjected drugs affected mPFC 5-HT over repeated samples. In all of the statistical tests, post-hoc analyses were Bonferroni t-tests when a main effect of drug was found (p<0.05).
RESULTS
Intermittent access promotes excessive alcohol drinking
At the end of 4 weeks of IAA drinking, male C57BL/6J mice (n=103) consumed, on average, 22.44 ± 0.67 g/kg/day. Blood ethanol concentrations (BECs) in a subset of IAA mice (n=11) were 134 ± 7.53 mg/dl, ranging from 97.9 – 179.4 mg/dl, after 2 hr access to two-bottle choice. In contrast, continuous access mice (n=12) consumed 14.97 g/kg/day after 12 days of continuous two-bottle choice. BECs for that group were 38.4 ± 5.64 mg/dl during the initial 2 hr after bottle measurements. A one-way ANOVA confirmed that BECs from the IAA group were significantly higher than the continuous access group [F(1,21)=106.28, p<.001].
Systemic injections of naltrexone and CRF-R1 antagonists reduce intermittent ethanol drinking
The opioid antagonist naltrexone (0, 1, 10 mg/kg, i.p.) was administered to mice (n=12) to reduce IAA. A one-way repeated measures ANOVA showed that naltrexone decreased 2 hr ethanol drinking (g/kg) [F(2,22)=12.76, p<0.001], at both doses compared to saline vehicle [1 mg/kg, p<0.01, 10 mg/kg, p<0.001]. This suppression of ethanol drinking (g/kg) lasted after 4 hr access [F(2,22)=9.20, p<0.01] at both doses [1 mg/kg, p<0.01; 10 mg/kg, p<0.01], and after 24 hr access [F(2,22)=11.13, p<0.001] also at both naltrexone doses [1 mg/kg, p<0.05; 10 mg/kg, p<0.001]. In accordance with reduced ethanol drinking (g/kg), volume of ethanol intake (ml) was decreased by naltrexone at the 2, 4, and 24 hr time points [2 hr: F(2,22)=12.17, p<0.001; 4 hr: F(2,22)=8.76, p<0.01; 24 hr: F(2,22)=10.52, p<0.001; Table 1], by the same effective doses [2 hr: 1 mg/kg, p<0.01; 10 mg/kg, p<0.001; 4 hr: 1 mg/kg, p<0.01, 10 mg/kg p<0.01; 24 hr: 1 mg/kg, p<0.001, 10 mg/kg, p<0.001]. The fluid-altering effects of naltrexone were not exclusive to ethanol. Naltrexone also increased water intake (ml) at the 4 and 24 hr time points [4 hr: F(2,22)=5.62, p<0.05; 24 hr: F(2,22)=3.90, p<0.05; Table 1] at the 10 mg/kg dose [4 hr: p<0.05; 24 hr: p<0.05].
TABLE 1. SYSTEMIC INJECTIONS OF NALTREXONE AND CRF-R1 ANTAGONISTS ON INTERMITTENT ALCOHOL DRINKING.
Opioid antagonist naltrexone (0, 1, 10 mg/kg, ip, n=12) and CRF-R1 antagonists CP154526 (0, 10, 17, 30 mg/kg, ip, n=9) and CP376395 (0, 10, 17, 30, ip, n=11) reduced intermittent ethanol drinking in male C57BL/6J mice. Shown are mean ethanol (ml) and water (ml) ± SEM consumed over 2, 4, and 24 hour fluid access after drug treatment.
| DRUG | DOSE | HOUR | ETOH (ML) | H2O (ML) |
|---|---|---|---|---|
|
|
||||
| Naltrexone | Saline | 2 | 0.37 ± 0.03 | 0.45 ± 0.05 |
| 4 | 0.93 ± 0.07 | 0.78 ± 0.08 | ||
| 24 | 3.25 ± 0.13 | 2.08 ± 0.19 | ||
|
|
||||
| 1 mg/kg | 2 | 0.23 ± 0.03* | 0.31 ± 0.06 | |
| 4 | 0.65 ± 0.09* | 0.66 ± 0.12 | ||
| 24 | 2.95 ± 0.13 | 2.18 ± 0.24 | ||
|
|
||||
| 10 mg/kg | 2 | 0.18 ± 0.03** | 0.52 ± 0.09 | |
| 4 | 0.65 ± 0.09* | 1.18 ± 0.18* | ||
| 24 | 2.65 ± 0.15** | 2.66 ± 0.24* | ||
|
|
||||
| CP154526 | 0.6% CMC | 2 | 0.29 ± 0.06 | 0.36 ± 0.15 |
| 4 | 0.46 ± 0.07 | 0.83 ± 0.24 | ||
| 24 | 2.37 ± 0.42 | 3.00 ± 0.50 | ||
|
|
||||
| 10 mg/kg | 2 | 0.15 ± 0.06 | 0.44 ± 0.08 | |
| 4 | 0.37 ± 0.10 | 0.71 ± 0.09 | ||
| 24 | 1.95 ± 0.20 | 3.28 ± 0.28 | ||
|
|
||||
| 17 mg/kg | 2 | 0.12 ± 0.02* | 0.14 ± 0.07 | |
| 4 | 0.47 ± 0.12 | 0.40 ± 0.13 | ||
| 24 | 2.39 ± 0.37 | 2.43 ± 0.38 | ||
|
|
||||
| 30 mg/kg | 2 | 0.04 ± 0.01** | 0.23 ± 0.05 | |
| 4 | 0.32 ± 0.08 | 0.34 ± 0.11 | ||
| 24 | 2.19 ± 0.30 | 2.00 ± 0.38 | ||
|
|
||||
| CP376395 | Saline | 2 | 0.21 ± 0.06 | 0.16 ± 0.06 |
| 4 | 0.89 ± 0.07 | 0.24 ± 0.07 | ||
| 24 | 3.45 ± 0.15 | 1.15 ± 0.16 | ||
|
|
||||
| 10 mg/kg | 2 | 0.14 ± 0.04 | 0.34 ± 0.07 | |
| 4 | 0.76 ± 0.12 | 0.50 ± 0.10 | ||
| 24 | 3.31 ± 0.25 | 0.99 ± 0.16 | ||
|
|
||||
| 17 mg/kg | 2 | 0.06 ± 0.03* | 0.90 ± 0.67 | |
| 4 | 0.48 ± 0.11* | 0.99 ± 0.68 | ||
| 24 | 3.07 ± 0.15 | 0.92 ± 0.14 | ||
|
|
||||
| 30 mg/kg | 2 | 0.06 ± 0.04* | 0.58 ± 0.22 | |
| 4 | 0.46 ± 0.12* | 0.67 ± 0.24 | ||
| 24 | 2.94 ± 0.16 | 1.15 ± 0.18 | ||
|
|
||||
Bolded values and *p<0.05 vs. vehicle. **p<0.001 vs. vehicle.
The CRF-R1 antagonist CP154516 (0, 10, 17, 30 mg/kg, i.p.) was given to another group of IAA mice (n=9). A one-way repeated measures ANOVA revealed that CP154526 initially suppressed 2 hr ethanol drinking (g/kg) [F(3,23)=6.03, p<0.01] at the two higher doses compared to 0.6% methylcellulose vehicle [17 mg/kg, p<0.05; 30 mg/kg, p<0.01]. This reduction was also observed in volume of ethanol intake (ml) [F(3,23)=6.40, p<0.01; Table 1] specifically at the higher doses [17 mg/kg, p<0.05, 30 mg/kg, p<0.001]. Systemic CP154526 was not effective at decreasing alcohol after 4 or 24 hr. The transient alcohol-suppressing effects were exclusive to alcohol, as water intake (ml) was not affected [Table 1].
Another CRF-R1 antagonist, CP376395 (0, 10, 17, 30 mg/kg, i.p.), was used to reduce IAA drinking in another group of mice (n=11). One-way repeated measures ANOVAs revealed that CP376395 significantly reduced 2 and 4 hr ethanol drinking (g/kg) [2 hr: F(3,30)=3.74, p<0.05; 4 hr: F(3,30)=4.76, p<0.05]. Post-hoc tests confirm that the 17 mg/kg [p<0.05] and 30 mg/kg [p<0.05] doses were different from saline vehicle. This trend was similar for volume of ethanol intake (ml) during the initial 4 hr of IAA [2 hr: F(3,30)=3.54, p<0.05; 4 hr: F(3,30)=4.42, p<0.05; Table 1] also at the higher doses [2 hr: 17 mg/kg, p<0.05, 30 mg/kg, p<0.05; 4 hr: 17 mg/kg, p<0.05, 30 mg/kg, p<0.05]. Water drinking was not significantly affected by CP376395 at any time point [Table 1].
Chronic i.c.v. infusion of naltrexone and CRF-R1 antagonists transiently reduce IAA
Mice with a history of 4 weeks IAA were implanted with i.c.v. cannulae attached to minipumps that chronically infused aCSF (n=8), 3 μg/μl naltrexone (n=7), 30 μg/μl naltrexone (n=8) or 3 μg/μl CP376395 (n=8) across 14 days in separate groups of mice. These groups were compared statistically, apart from the CP154526 and 4% DMSO groups, as aCSF was vehicle for these groups. A two-way (Drug × Day) repeated measures ANOVA indicated that the only significant interaction for drug effect on ethanol drinking (g/kg) was seen on Day 1 during the initial 2 hr IAA time period [F(3,100)=5.74, p<.01; Figure 1A]. Specifically, 30 μg/μl naltrexone [p<0.01] and 3 μg/μl CP376395 [p<0.05] were different from aCSF infusion within test Day 1. There were no other significant interactions for drug group differences on ethanol drinking (g/kg) or water intake (ml) across the 14 days of chronic infusion at the 2, 4 or 24 hr time points.
FIGURE 1.
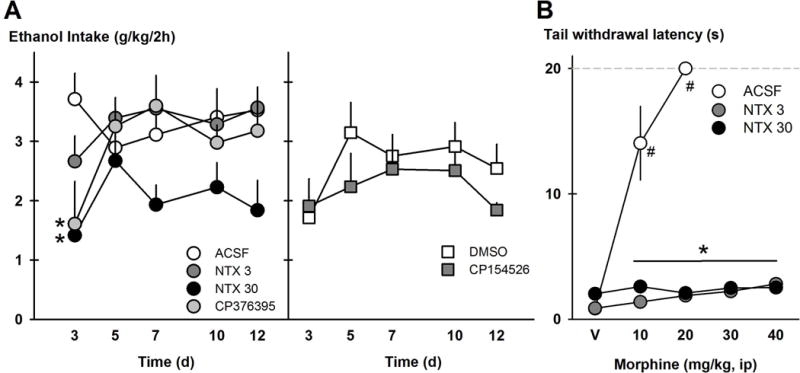
Drugs were chronically administered i.c.v. via osmotic minipumps to affect intermittent access drinking. The left side of panel A shows ethanol drinking (g/kg/2h) over time during naltrexone infusion (3 μg/μl/h, NTX3, n=7 and 30 μg/μl/h, NTX30, n=8), CRF-R1 antagonist CP376395 infusion (0.3 μg/μl/h, n=8) and their vehicle aCSF (n=8). The right side of panel A shows ethanol intake (g/kg/2h) over time during CRF-R1 antagonist CP154526 infusion (0.3 μg/μl/h, n=6) and its vehicle 4% DMSO (n=6). Data points are mean ethanol intake ± SEM. *p<0.05 vs. ACSF vehicle. Panel B shows the tail withdrawal latency in seconds to a hot water bath after cumulative doses of morphine (mg/kg, ip) in mice given ACSF or naltrexone on Day 13 of minipump infusion. Data points are mean latencies ± SEM. *p<0.05 vs. ACSF group. #p<0.05 vs. saline vehicle.
To verify that the minipumps were chronically infusing naltrexone during the 14-day pump duration, the mice given aCSF, 3 μg/μl naltrexone and 30 μg/μl naltrexone were subjected to a test of morphine-induced analgesia on Day 13, after IAA drinking tests. Mouse tails were held in 54°C water, and latency to withdraw the tail was measured after 0, 10, 20, 30, and 40 mg/kg morphine, administered i.p. with cumulative dosing every 20 min. There was a significant Minipump Drug × Morphine dose interaction [F(8,90)=32.94, p<0.001], where during the 10, 20, 30 and 40 mg/kg morphine doses, mice given aCSF had shorter tail withdrawal latencies than mice given 3 μg/μl and 30 μg/μl naltrexone [all p<0.001; Figure 1B]. Mice given aCSF via minipump showed lengthy tail withdrawal latencies of 14.04 ± 2.94 sec with 10 mg/kg morphine, and all aCSF mice met the experimenter-controlled maximum (20 sec) in the hot water with 20 mg/kg morphine, suggesting morphine-induced analgesia. Mice that were given naltrexone via minipump exhibited short tail withdrawal latencies of 2.81 ± 0.20 sec after 40 mg/kg morphine, suggesting blockade of morphine-induced analgesia.
Other groups of IAA mice were implanted with minipumps infusing 4% DMSO (n=6) and 3 μg/μl CP154526 (n=6) for 14 days that were also tested for IAA drinking behavior. These DMSO-based groups were statistically analyzed separately from the aCSF-based vehicle groups. Unlike the other minipump drugs that reduced ethanol drinking on the first day, chronic CP154526 infusion was not different from DMSO infusion on Day 1 [Figure 1A]. A two-way repeated measures (Drug × Day) ANOVA showed that there was a Drug × Day interaction for 4 hr ethanol drinking (g/kg) [F(4,40)=2.93, p<0.05], where Day 5 showed a difference between DMSO and CP154526 treatments (p<0.05). On Day 5, DMSO-treated mice consumed 0.73 ± 0.08 g/kg/4hr while CP154526-treated mice consumed 0.51 ± 0.05 g/kg/4hr. There were no drug effects on water drinking.
Tryptophan hydroxylase immunoreactive DRN neurons co-localize with CRF-R1 or MOR
The brains of some IAA and water-drinking B6 mice (n=4/group) were taken for immunohistochemistry to visualize CRF-R1 and TPH or MOR and TPH in DRN sections. Figure 2 (left panel) shows a representative section through the DRN with dual labeling of CRF-R1 and TPH. Additionally, some cells were CRF-R1 only, which we hypothesize to be GABAergic cells. CRF-R1 immunoreactivity was present within the DRN as well as in the lateral regions. Many TPH-immunoreactive neurons in the DRN were also dual labeled for MOR, although there were occasional single-labeled TPH and single-labeled MOR neurons (Fig. 2, right panel). There were no evident differences between water-drinking and ethanol-drinking mice.
FIGURE 2.
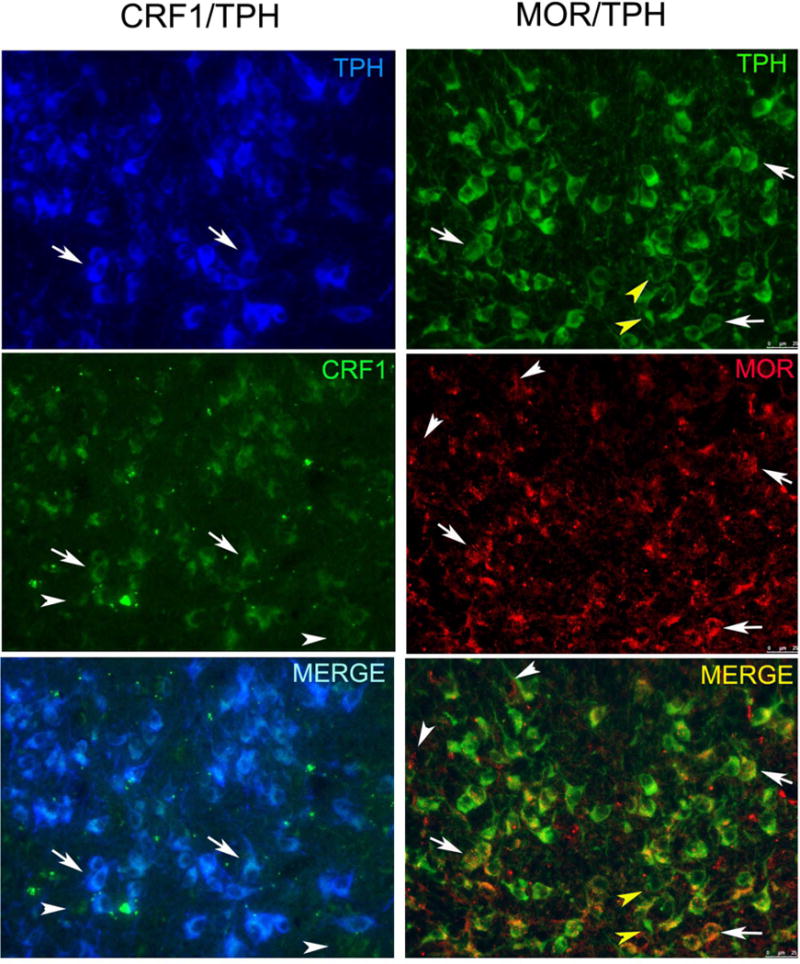
Fluorescent photomicrographs of sections through the DN that were dual labeled to visualize either CRF1 and tryptophan hydroxylase (CRF1/TPH) or MOR and tryptophan hydroxylase (MOR/TPH). The leftmost panel shows TPH cells in blue and CRF1 in green. Dual labeled cells in the merged panel appear whitish blue. Arrows point to examples of dual labeled cells. Arrowheads point to single labeled CRF1 neurons. The rightmost panel shows TPH cells in green and MOR in red. Dual labeled cells in the merged panel appear yellow/orange. Arrows point to examples of dual labeled cells. White arrowheads point to single labeled MOR neurons and yellow arrowheads point to single labeled TPH neurons.
Distinct effects of intra-DRN naltrexone and CRF-R1 antagonist on drinking patterns
A group of mice (n=8) with a history of IAA were outfitted with cannula targeting the DRN and tested for IAA drinking after microinjections of aCSF, 6 μg naltrexone, 0.6 μg CP154526 or the combination of 6 μg naltrexone and 0.6 μg CP154526. Cannula placements in the DRN are shown in Figure 3E. A one-way repeated measures ANOVA revealed that drug treatment affected IAA ethanol drinking (g/kg) at the 2 and 4 hr time points [2 hr: F(3,21)=6.24, p<0.01, Figure 3A; 4hr: F(3,20)=5.28, p<0.01]. Post hoc tests showed that naltrexone, CP154526 and the combination reduced drinking compared to aCSF at both the 2 and 4 hr time points [all p<0.05]. The suppression in IAA drinking caused by the combination of naltrexone and CP154526 was not statistically different from naltrexone or CP154526 administered alone. Water intake (ml) was not affected by any microinjection.
FIGURE 3.
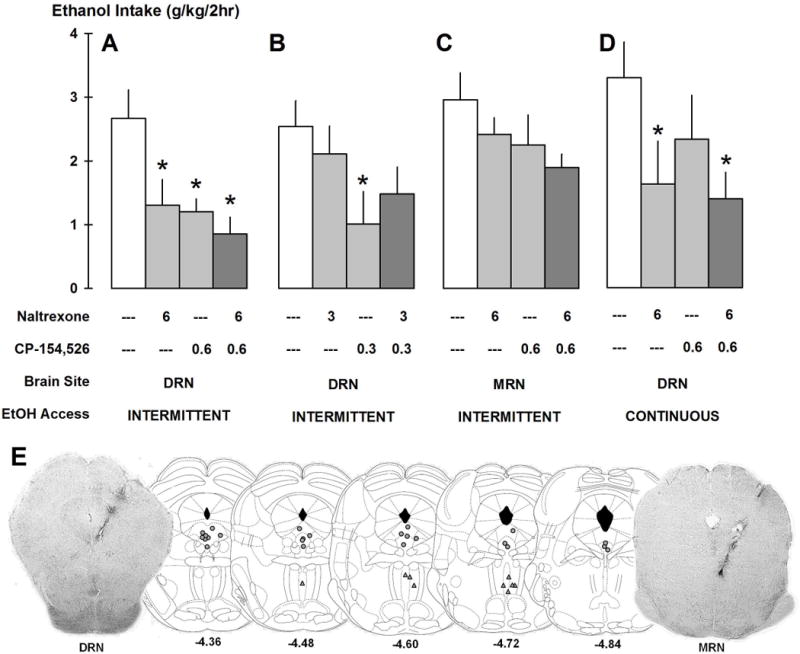
Separate groups of mice were microinjected with aCSF, naltrexone, CP154526 and the combination of naltrexone and CP154526 to reduce ethanol drinking. Panel A shows intra-DRN microinjections of higher doses in intermittent access drinkers (n=8). Panel B shows intra-DRN microinjections of lower doses in intermittent drinkers (n=7). Panel C shows intra-MRN microinjections in intermittent drinkers (n=9). Panel D shows intra-DRN microinjections in continuous access drinkers (n=9). Bars represent mean ethanol intake (g/kg/2h) ± SEM. *p<0.05 vs. aCSF. Panel E shows representative schematics of correct placements of microinjection cannulae in the DRN (grey circles, n=24) or MRN (grey triangles, n=9). Numbers below coronal slices are the distances (mm) from bregma. Photos show correct placement in the brain areas.
Another group of IAA mice (n=7) was microinjected with aCSF and lower doses of naltrexone (3 μg), CP154526 (0.3 μg) and the combination of 3 μg naltrexone and 0.3 μg CP154526 into the DRN. A one-way repeated measures ANOVA showed that 2 hr ethanol drinking (g/kg) was affected by intra-DRN drug [g/kg: F(3,16)=5.45, p<0.01; Figure 3B]. Post hoc tests revealed that only 0.3 μg CP154526 decreased ethanol drinking (g/kg) [p<0.01] compared to aCSF. Lower-dose drug treatment did not affect consumption further than 2 hr. Water drinking (ml) was not changed due to drug infusion.
A group of IAA mice (n=9) were microinjected with aCSF, 6 μg naltrexone, 0.6 μg CP154526 and the combination into the MRN, to contrast drug effects in the DRN. Intra-MRN cannula placements are shown in Figure 3E. A one-way repeated measures ANOVA indicated that water intake (ml) was initially affected by drug treatment in the MRN at 2 hr [F(3,24)=5.67, p<0.01]. Naltrexone [p<0.01], CP154526 [p<0.05] and the combination [p<0.01] reduced 2 hr water intake compared to aCSF. The reduction of water intake (ml) continued until the 4 hr time point [F(3,24)=4.17, p<0.05], caused by all drug doses, again [all p<.05]. Ethanol drinking (g/kg) was only affected at the 4 hr time point [g/kg: F(3,24)=4.00, p<0.05, Figure 3C], but not at the 2 hr time point. Post hoc tests indicated that CP154526 and the combination of naltrexone and CP154526 were different from aCSF for both ethanol drinking (g/kg) at 4 hr [all p<0.05; not shown]. CP154526, naltrexone, and the combination reduced baseline ethanol drinking from 6.64±0.87 after aCSF to 4.29±0.73, 5.43±0.25, and 4.30±0.48 g/kg/4hr, respectively. The drugs suppressed baseline water drinking from 0.92±0.14 after aCSF to 0.48±0.10 after CP154526, 0.51±0.14 after naltrexone, and 0.42±0.13 ml/4hr after the combination.
A final group of mice (n=9) with a history of continuous access to alcohol was tested for drinking behavior after intra-DRN microinjections of aCSF, 6 μg naltrexone, 0.6 μg CP154526 and the combination of naltrexone and CP154526 to reveal if drug effects were exclusive to excessive drinkers compared to moderate-level drinkers. A one-way repeated measures ANOVA indicated that initial 2 hr ethanol drinking (g/kg) was affected by drug treatments [g/kg: F(3,24)=4.70, p<0.05, Figure 3D]. Continuous access to alcohol (g/kg) was specifically decreased by intra-DRN naltrexone [p<0.05] and the combination of naltrexone and CP154526 [p<0.01], but not by CP154526 alone. The naltrexone effects did not last longer than 2 hr. No drug treatments affected water drinking in the continuous access mice.
Intra-DRN CRF-R1 antagonist, but not naltrexone, increased prefrontal cortical 5-HT impulse flow
IAA mice were concurrently implanted with cannulae in the DRN and microdialysis probes in the mPFC for 5-HT measurements after drug microinjections. Correct placements of DRN cannulae and mPFC probes are depicted in Figures 3E and 4B. Eight samples were collected every 20 min, and aCSF (n=6), 6 μg naltrexone (n=7) or 0.6 μg CP154526 (n=7) were infused in different groups of mice 10 min before the 6th sample was collected. A two-way repeated measures ANOVA showed that there was a significant main effect of intra-DRN drug microinjection on 5-HT concentrations [F(2,86)=4.13, p<0.05; Figure 4A], where CP154526, but not naltrexone, was different from aCSF [p<0.05]. There was also a Drug × Time interaction [F(14,86)=1.83, p<0.05]. Post-hoc analysis revealed that 5-HT concentrations 10 and 30 min after the microinjection, the 6th and 7th samples respectively, were significantly increased when CP154526 was injected compared to those when after aCSF [p<0.001] or naltrexone [p<0.001] were injected.
FIGURE 4.
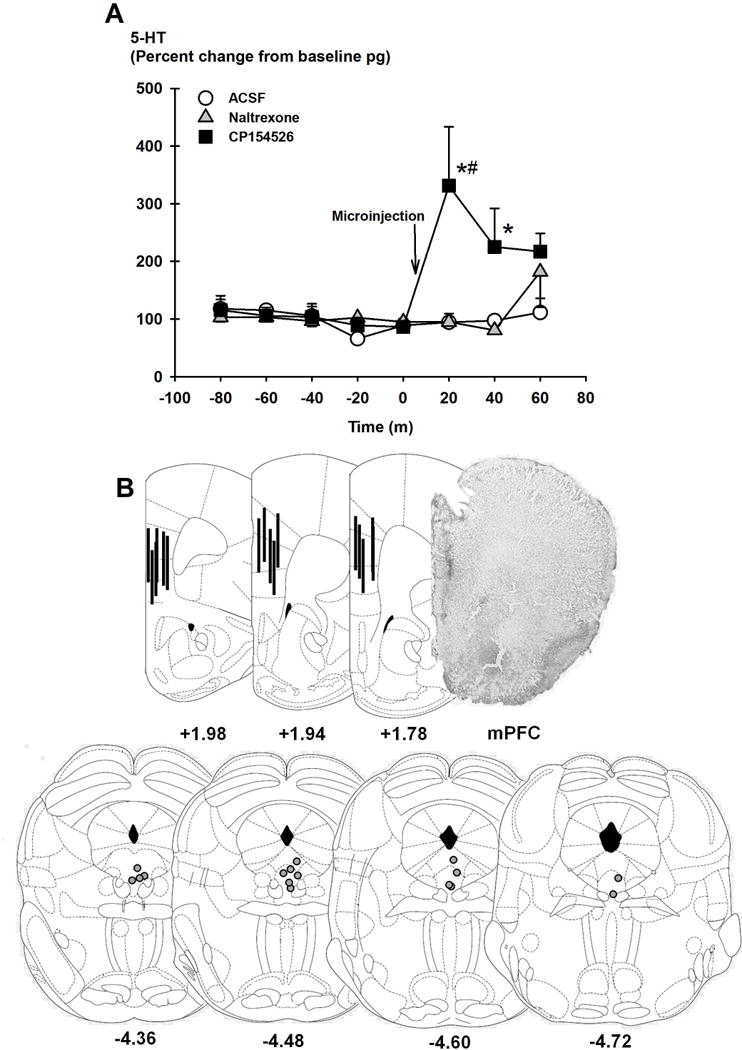
Panel A shows extracellular concentrations of serotonin (5-HT; percent change from baseline pg) were collected from the mPFC after intra-DRN microinjections of aCSF (white circles, n=6), 3 μg naltrexone (grey triangles, n=7), or 0.3 μg CP154526 (black squares, n=7) in mice with a history of intermittent access to alcohol. Data points are mean percent change ± SEM. *p<0.05 vs. aCSF, #p<0.05 vs. baseline time points. Panel B shows representative schematics of correct placements of microinjection cannulae in the DRN and microdialysis probes in the mPFC (n=16). Numbers below coronal sections are the distances (mm) from bregma. The photo shows correct placement in the mPFC.
DISCUSSION
The current studies demonstrate divergent roles of CRF-R1 and opioid receptors in the DRN in volitional excessive ethanol drinking. Persistently high levels of ethanol drinking and BECs were generated using an intermittent schedule of access to two-bottle choice, and systemic and intra-DRN administration of either FDA-approved naltrexone or CP154526 decreased this type of drinking. Intra-DRN naltrexone also reduced moderate-level ethanol drinking, engendered by a continuous schedule of access, but CP154526 did not. These specific anti-ethanol drug effects were limited to the DRN of IAA drinkers, as identical infusions into the MRN had a greater effect on water drinking. Since there is no evidence for an additive effect by CP154526 and naltrexone, this suggests that these compounds accomplished their ethanol-suppressing effects via a common, downstream target. In contrast, in our microdialysis experiments, intra-DRN CP154526 increased extracellular concentrations of 5-HT, presumably reflecting release in the mPFC while naltrexone did not. These differential patterns of impulse flow to the mPFC caused by CP154526 and naltrexone also suggest that the mPFC may not be as relevant for the suppression of IAA drinking.
At the level of the DRN, we co-localized CRF-R1 on 5-HT and non-5-HT cells. This is the first study to confirm CRF-R1 and TPH immunoreactivity in the DRN in the mouse brain, as previous studies have examined co-localization of CRF and TPH in the rat brain (Price et al. 1998; Kirby et al. 2000) and CRF-R1 within the DRN in the rat brain (Waselus et al. 2009). The nearby non-TPH-expressing cells with CRF-R1 immunoreactivity most likely represent GABAergic neurons (Kirby et al. 2008). Our behavioral findings did not reveal any additive effects of co-infusion of both antagonists (e.g. Liu and Weiss 2002), suggesting that the opioid and CRF-R1 systems may be acting on a common mechanism in the DRN to suppress escalated drinking in the current preparation. From a pharmacological perspective, this pattern suggests that they may work through the same mechanism because when one mechanism is saturated, the other drug cannot exert a greater effect to reduce drinking further. We speculate that this putative, common mechanism is located downstream and requires further investigation. At the intracellular level, both naltrexone and CP154526 inhibit adenylate cyclase activity and cyclic AMP (Childers 1991; Schulz et al. 1996). At the receptor level, both morphine and CRF increase GABAergic tone and act on GABAergic and glutamatergic afferents to indirectly influence 5-HT neurons in the DRN (Kirby et al. 2008). Therefore, it is possible that naltrexone and CRF-R1 antagonists similarly influence GABAergic tone in the DRN to decrease excessive ethanol drinking. Alternatively, CP154526 may act through additional interneurons to increase 5-HT flow, as seen in the current study. Future studies measuring GABAergic tone in efferent sites like the PFC would be required to confirm this hypothesis.
In the current preparation, intra-DRN CP154526 significantly increased extracellular 5-HT concentrations in the mPFC in IAA mice, but naltrexone did not. Since we speculate the treatments work via one common neural mechanism to reduce IAA drinking, but they have differential effects on mPFC 5-HT, it suggests that the mPFC may not be a critical site for the suppression of IAA drinking. One limitation of our microdialysis study was that the probes spanned both the infralimbic and prelimbic subregions of the mPFC. We cannot ignore the fact that these regions have discrete projections and are functionally distinct for aspects of drug-seeking behaviors (reviewed by George and Koob, 2010). For example, previous morphological studies have identified high 5-HT1A and 5-HT2A receptor localization and expression throughout both the infralimbic and prelimbic subregions (Amargós-Bosch et al. 2004), which can be interpreted as a collective 5-HT-dense region for extracellular 5-HT measurement. The mPFC, involved in higher order cognitive and motivational functions, is a site that is dysfunctional in alcoholics (Goldstein and Volkow 2002). Alcoholics can have up to 35% loss of 5-HT transporter density in the PFC compared with non-alcoholics, suggesting an association between excessive drinking and dysfunctional 5-HT neurotransmission in the mPFC (Mantere et al. 2002). Connectivity to the PFC becomes disrupted in rats with a history of IAA, which has been shown to be CRF dependent (George et al. 2012). 5-HT in the mPFC appears important for heavy drinking, but our divergent effects of the two compounds on mPFC 5-HT suggest that downstream effects in other brain sites are more important for naltrexone and CP154526.
Neurochemically, CRF, at low to moderate doses inhibits 5-HT DRN activity and local release via CRF-R1 receptors on GABA afferents (Kirby et al. 2000, 2008). Also, stressful events, mediated by endogenous CRF release acting on DRN neurons, lead to inhibition of 5-HT release in the lateral septum (Price et al. 2002). One interpretation of the increased 5-HT is that IAA may contain stressful elements, possibly causing a CRF-R1 antagonist to disinhibit impulse flow from the DRN to the mPFC. Another hypothesis for the increased mPFC 5-HT is that blockade of CRF-R1 may cause relative activation of CRF-R2, stimulating 5-HT flow. A further limitation of the present microdialysis study was that 5-HT was measured only in IAA-experienced mice. It would be interesting to also investigate CRF-R1 influence of mPFC 5-HT in mice with continuous access to ethanol, to confirm if there is a dose-dependent influence of CP154526-induced increased in 5-HT. Although there was not an ethanol-naïve group in the current study, Tanahashi et al. (2012) importantly showed that neither perfusion of CRF-R1 nor CRF-R2 antagonists intra-DRN affect DRN/mPFC 5-HT in ethanol-naïve animals. Paired with the fact that there were no significant differences in CRF-R1 and tryptophan hydroxylase immunoreactivity between the IAA and water-drinking mice in our study, these results suggest that there may not have been a change in 5-HT caused by CP154526 in water drinking mice. However, our experiments with CRF-R1 antagonism in mice with a history of chronic, excessive drinking experience do reveal neuroadaptations in the CRF-R1 system that affect 5-HT.
Systemic morphine increases 5-HT release in the mPFC, which is mediated by the DRN and not MRN (Tao and Auerbach 1995). Like the excitatory effects of acute morphine, MOR agonist DAMGO disinhibits 5-HT neurons in DRN (Tao and Auerbach 2005). Dimatelis et al. (2012) found that naltrexone inhibits striatal 5-HT levels, which is in accordance with MOR activation and disinhibition of 5-HT. Our microdialysis findings did not reveal any changes from baseline 5-HT concentrations caused by intra-DRN naltrexone, suggesting no tonic action of opioids. However, naltrexone may influence DRN function under phasic conditions, where endogenous opioid release is stimulated. Future experiments with morphine microinjections can possibly answer this question. This is the first report to test how naltrexone affects DRN 5-HT impulse flow to the forebrain in excessively drinking mice, so perturbations of 5-HT may be different in more modestly drinking animals, or more heavily influenced by other circuits, like CRF-R1.
We also tested the CRF-R1 antagonist in the MRN, where both CRF-R1 and CRF-R2 are moderately expressed (Van Pett et al. 2000). This suggests CRF-R1 antagonists intra-MRN might also alter ethanol-related behavior. For example, nonspecific CRF receptor antagonist, d-Phe-CRF, into the MRN blocked footshock-induced ethanol reinstatement (Lê et al. 2002). The current results showed intra-MRN CP154526 and the combination of CP154526 and naltrexone reducing ethanol intake after 4 hr, with a trend at the 2 hr time point, confirming its latent involvement in decreasing IAA. However, under the present conditions, it was an issue that intra-MRN CP154526 and naltrexone also decreased water intake in addition to alcohol. 5-HT1A receptors in the MRN are implicated by evidence showing that 8-OH-DPAT microinjections increased locomotor activity, feeding and ethanol drinking (Currie et al. 1994; Tomkins et al. 1994). Since the DRN and MRN have distinct 5-HTergic systems (reviewed by Lechin, van der Dijs and Hernández-Adrián 2006), it is possible that microinjections of a CRF-R1 antagonist suppressed both ethanol and water drinking by indirectly influencing 5-HT regulation in the MRN.
We found that systemic administration of naltrexone decreased intermittent ethanol drinking in B6 mice, confirming our previous reports (Hwa et al. 2014), and intra-DRN naltrexone infusions reduced intermittent and continuous ethanol drinking. Since naltrexone and CP154526 decreased IAA, and naltrexone but not CP154526 reduced continuous access drinking, the data suggest that the mechanism underlying continuous drinking is different from IAA, and it may be opioid-related. These results point to reductions in behavior that are not exclusive to the excessive drinkers. Naltrexone treatment in alcoholics is useful for controlled consumption, but there is a high degree of non-compliance and negative side effects (Croop et al. 1997). Based on these problems, some clinicians do not support the use of naltrexone for the treatment of men with chronic, severe alcohol dependence (Krystal et al. 2001). In preclinical studies, naltrexone has been known to decrease voluntary drinking since the 1980s (Altshuler et al. 1980), but in both dependent and non-dependent animals (Gilpin et al. 2008; Sabino et al. 2006). Under two-bottle choice conditions, systemic naltrexone suppressed 20% ethanol intermittent intake, 20% ethanol continuous intake and 10% ethanol continuous intake in Long-Evans and sP rats (Simms et al. 2008; Sabino et al. 2013). Given these limitations, other medications need to be developed to target excessive drinking.
It has been hypothesized that crhr1 does not have a role in basal alcohol intake or relapse-like drinking situations with a low stress load (Molander et al. 2012). Our studies found that the CRF-R1 antagonist CP154526 intra-DRN decreased IAA, but did not affect continuous access intake. Although IAA and continuous access ethanol drinking (g/kg) values were similar at 2-hr baseline, there were large, significant differences in BECS (three-fold higher in the IAA group), confirming different patterns of overconsumption and the role of CRF-R1 in each group. CRF-R1 antagonists’ effects are most convincing in animals that drink in excess or are dependent on ethanol (see Lowery and Thiele, 2010 for review). For example, CRF-R1 antagonists MPZP and LWH-63 reduced responding for ethanol reinforcement in dependent P and sP rats but not in the non-dependent rats (Sabino et al. 2006; Gilpin et al. 2008). In studies using volitional two-bottle choice, MPZP did not affect binge-like ethanol drinking and R121919 did not affect intermittent intake in rats (Sabino et al. 2013). However, these studies were conducted in non-dependent rats. Comparing treatment efficacies between rats and mice may not be straightforward due to innate differences in ethanol preference and drug metabolism between these species (Martignoni, Groothius and de Kanter 2006). Nevertheless, our current studies using B6 mice demonstrate that CRF-R1 antagonism in the DRN is effective in reducing >20 g/kg daily intake, as this may be more excessive drinking to dependence, replicating previous reports (Hwa et al. 2013).
Despite the efficacy of CRF-R1 antagonists in decreasing alcohol drinking, we only observed transient effects with CP154526 and CP376395 chronic infusion, so future studies of chronic administration need to overcome the short-lasting behavioral effects. To date, this is the first set of experiments to investigate chronic i.c.v. infusions of CRF-R1 antagonists to affect alcohol drinking. Others have seen in voles that had lost their partners, chronic i.c.v. infusions of CP154526 reduced immobility time in the forced swim test (Bosch et al. 2009), but this was a single test, unlike our repeated testing of IAA. We also speculate that the lack of effect of CP154526 was caused by non-specific reductions in drinking due to the vehicle, DMSO. It is also possible that the minipumps did not deliver CP154526 consistently over the 14 days, but results with the water-soluble CP376395 confirm the short-lasting behavioral findings. With repeated testing, it is necessary to avoid compensatory changes with chronic naltrexone. Kaminski, Duke and Weerts (2012) found that naltrexone only reduces alcohol self-administration in the initial drinking bout, but when administered chronically, naltrexone did not decrease progressive ratio break points. To exclude the possibility that the minipumps had failed working, we tested long-lasting opioid receptor antagonism with a morphine-sensitive tail withdrawal test. Mice receiving chronic naltrexone withdrew their tails from the hot water even after 40 mg/kg morphine, suggesting MORs were still potently antagonized on Day 13. We speculate that chronic naltrexone induced specific behavioral neuroadaptations such as the development of tolerance to the ethanol drinking effects of MOR antagonism, but not to other behaviors such as the analgesic effect of opiates. It will be useful to confirm the nature of the neuroadaptations that accompany chronic naltrexone and CRF-R1 antagonist treatment.
In addition to continued exploration of CRF-R1 treatments for excessive drinking, there are apparent future directions with other endogenous opioid receptor systems, like targeting dynorphin/kappa opioid receptor (KOR) systems to treat alcohol abuse and dependence (Walker et al. 2012). Stress and CRF each cause dynorphin-dependent KOR activation in DRN, suggesting KOR antagonists may be therapeutics for stress-related psychiatric disorders like alcoholism (Land et al. 2008). Similarly, naltrexone blocks both MOR and KOR, so it is possible that naltrexone exerts secondary effects on KOR to suppress the escalated component of IAA, in addition to the primary reinforcing effects of ethanol by inhibiting MOR. Altogether, there is an intricate balance between reward-related neuropeptides and stress-related neuropeptides. Perhaps these receptor systems are time-sensitive in the transition to alcohol dependence, first opioid systems, when naltrexone may be more helpful, then CRF-R1 systems, when CRF-R1 antagonists may be more efficacious.
Acknowledgments
The authors would like to acknowledge the research assistance of Jillian Tayeh, Allison Wilens, Danna NiSai and Johana Alvarez. We appreciate Elizabeth Holly for her technical aid with the HPLC and Dr. Lucas Albrechet-Souza for his assistance with histology. The authors declare no competing financial interests. This research was supported by NIH funding R01 AA013983 (KAM), F31 AA021622 (LSH), and R01 MH058250 (RJV).
Footnotes
AUTHORS CONTRIBUTION: LSH, JFD, and KAM were responsible for the experimental design. LSH, TK, and KJN acquired the behavioral data while LSH and AS collected the neurochemical data. RJV performed the immunohistochemistry. LSH drafted the manuscript, and RJV, JFD and KAM provided critical revision and contributed to the interpretation of findings. All authors reviewed the content and approved the final version for publication.
References
- Altshuler HL, Phillips PE, Feinhandler DA. Alteration of ethanol self-administration by naltrexone. Life Sci. 1980;26:679–688. doi: 10.1016/0024-3205(80)90257-x. [DOI] [PubMed] [Google Scholar]
- Amargós-Bosch M, Bortolozzi A, Puig MV, Serrats J, Adell A, Celada P, Toth M, et al. Co-expression and in vivo interaction of serotonin1A and serotonin2A receptors in pyramidal neurons of prefrontal cortex. Cereb cortex. 2004;14:281–299. doi: 10.1093/cercor/bhg128. [DOI] [PubMed] [Google Scholar]
- Bosch O, Nair HP, Ahern TH, Neumann ID, Young LJ. The CRF system mediates increased passive stress-coping behavior following the loss of a bonded partner in a monogamous rodent. Neuropsychopharmacology. 2008;34:1406–1415. doi: 10.1038/npp.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YL, Obach RS, Braselton J, Corman ML, Forman J, Freeman J, Gallaschun RJ, Mansbach R, et al. 2-Aryloxy-4-alkylaminopyridines: Discovery of Novel Corticotropin-Releasing Factor 1 Antagonists. J Med Chem. 2008;51:1385–1392. doi: 10.1021/jm070579c. [DOI] [PubMed] [Google Scholar]
- Childers Opioid receptor-coupled second messengers. Life Sci. 1991;48:1991–2003. doi: 10.1016/0024-3205(91)90154-4. [DOI] [PubMed] [Google Scholar]
- Cippitelli A, Damadzic R, Singley E, Thorsell A, Ciccocioppo R, Eskay RL, Heilig M. Pharmacological blockade of corticotropin-releasing hormone receptor 1 (CRH1R) reduces voluntary consumption of high alcohol concentrations in non-dependent Wistar rats. Pharmacol Biochem Behav. 2012;100:522–529. doi: 10.1016/j.pbb.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croop RS, Faulkner EB, Labriola DF. The safety profile of naltrexone in the treatment of alcoholism: results from a multicenter usage study. Arch Gen Psychiatry. 1997;54:1130. doi: 10.1001/archpsyc.1997.01830240090013. [DOI] [PubMed] [Google Scholar]
- Currie PJ, Fletcher PJ, Coscina DV. Administration of 8-OH-DPAT into the midbrain raphe nuclei: effects on medial hypothalamic NE-induced feeding. Am J Physiol. 1994;266:R1645–R1651. doi: 10.1152/ajpregu.1994.266.5.R1645. [DOI] [PubMed] [Google Scholar]
- Dimatelis JJ, Russell VA, Stein DJ, Daniels WM. The effects of lobeline and naltrexone on methamphetamine-induced place preference and striatal dopamine and serotonin levels in adolescent rats with a history of maternal separation. Metab Brain Dis. 2012;27:351–361. doi: 10.1007/s11011-012-9288-8. [DOI] [PubMed] [Google Scholar]
- George O, Koob GF. Individual differences in prefrontal cortex function and the transition from drug use to drug dependence. Neurosci Biobehav Rev. 2010;35:232–247. doi: 10.1016/j.neubiorev.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George O, Sanders C, Freiling J, Grigoryan E, Vu S, Allen CD, Crawford E, Mandyam CD, Koob GF. Recruitment of medial prefrontal cortex neurons during alcohol withdrawal predicts cognitive impairment and excessive alcohol drinking. Proc Natl Acad Sci U S A. 2012;109:18156–18161. doi: 10.1073/pnas.1116523109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilpin NW, Richardson HN, Koob GF. Effects of CRF1-Receptor and Opioid-Receptor Antagonists on Dependence-Induced Increases in Alcohol Drinking by Alcohol-Preferring (P) Rats. Alcohol Clin Exp Res. 2008;32:1535–1542. doi: 10.1111/j.1530-0277.2008.00745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein RZ, Volkow ND. Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry. 2002;159:1642–1652. doi: 10.1176/appi.ajp.159.10.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig M, Koob GF. A key role for corticotropin-releasing factor in alcohol dependence. TINS. 2007;30:399–406. doi: 10.1016/j.tins.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwa LS, Chu A, Levinson SA, Kayyali TM, DeBold JF, Miczek KA. Persistent escalation of alcohol drinking in C57BL/6J mice with intermittent access to 20% ethanol. Alcohol Clin Exp Res. 2011;35:1938–1947. doi: 10.1111/j.1530-0277.2011.01545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwa LS, DeBold JF, Miczek KA. Alcohol in excess: CRF1 receptors in the rat and mouse VTA and DRN. Psychopharmacology (Berl) 2013;225:313–327. doi: 10.1007/s00213-012-2820-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwa LS, Kalinichev M, Haddouk H, Poli S, Miczek KA. Reduction of excessive alcohol drinking by a novel GABAB receptor positive allosteric modulator ADX71441 in mice. Psychopharmacology (Berl) 2014;231:333–343. doi: 10.1007/s00213-013-3245-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai H, Steindler DA, Kitai ST. The organization of divergent axonal projections from the midbrain raphe nuclei in the rat. J Comp Neurol. 1986;243:363–380. doi: 10.1002/cne.902430307. [DOI] [PubMed] [Google Scholar]
- Kaminski BJ, Duke AN, Weerts EM. Effects of naltrexone on alcohol drinking patterns and extinction of alcohol seeking in baboons. Psychopharmacology (Berl) 2012;223:55–66. doi: 10.1007/s00213-012-2688-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby LG, Freeman-Daniels E, Lemos JC, Nunan JD, Lamy C, Akanwa A, Beck SG. Corticotropin-releasing factor increases GABA synaptic activity and induces inward current in 5-hydoxytryptamine dorsal raphe neurons. J Neurosci. 2008;28:12927–12937. doi: 10.1523/JNEUROSCI.2887-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby LG, Rice KC, Valentino RJ. Effects of corticotropin-releasing factor on neuronal activity in the serotonergic dorsal raphe nucleus. Neuropsychopharmacology. 2000;22:148–162. doi: 10.1016/S0893-133X(99)00093-7. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Cramer JA, Krol WF, Kirk GF, Rosenheck RA. Naltrexone in the treatment of alcohol dependence. N Engl J Med. 2001;345:1734–1739. doi: 10.1056/NEJMoa011127. [DOI] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. The dysphoric component of stress is encoded by activation of the dynorphin kappa-opioid system. J Neurosci. 2008;28:407–414. doi: 10.1523/JNEUROSCI.4458-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lê AD, Harding S, Juzytsch W, Fletcher PJ, Shaham Y. The role of corticotropin-releasing factor in the median raphe nucleus in relapse to alcohol. J Neurosci. 2002;22:7844–7849. doi: 10.1523/JNEUROSCI.22-18-07844.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechin F, van der Dijs B, Hernández-Adrián G. Dorsal raphe vs. median raphe serotonergic antagonism. Anatomical, physiological, behavioral, neuroendocrinological, neuropharmacological and clinical evidences: relevance for neuropharmacological therapy. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30:565–585. doi: 10.1016/j.pnpbp.2005.11.025. [DOI] [PubMed] [Google Scholar]
- Liu X, Weiss F. Additive effect of stress and drug cues on reinstatement of ethanol seeking: exacerbation by history of dependence and role of concurrent activation of corticotropin-releasing factor and opioid mechanisms. J Neurosci. 2002;22:7856–7861. doi: 10.1523/JNEUROSCI.22-18-07856.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery EG, Thiele TE. Pre-clinical evidence that corticotropin-releasing factor (CRF) receptor antagonists are promising targets for pharmacological treatment of alcoholism. CNS Neurol Disord Drug Targets. 2010;9:77–86. doi: 10.2174/187152710790966605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry CA, Rodda JE, Lightman SL, Ingram CD. Corticotropin-Releasing Factor Increases In Vitro Firing Rates of Serotonergic Neurons in the Rat Dorsal Raphe Nucleus: Evidence for Activation of a Topographically Organized Mesolimbocortical Serotonergic System. The Journal of Neuroscience. 2000;20:7728–7736. doi: 10.1523/JNEUROSCI.20-20-07728.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantere T, Tupala E, Hall H, Särkioja T, Räsänen P, Bergström K, Callaway J, Tiihonen J. Serotonin transporter distribution and density in the cerebral cortex of alcoholic and nonalcoholic comparison subjects: a whole-hemisphere autoradiography study. Am J Psychiatry. 2002;159:599–606. doi: 10.1176/appi.ajp.159.4.599. [DOI] [PubMed] [Google Scholar]
- Martignoni M, Groothuis GM, de Kanter R. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opinion on Drug Metabolism and Toxicology. 2006;2:875–894. doi: 10.1517/17425255.2.6.875. [DOI] [PubMed] [Google Scholar]
- Molander A, Vengeliene V, Heilig M, Wurst W, Deussing JM, Spanagel R. Brain-specific inactivation of the Crhr1 gene inhibits post-dependent and stress-induced alcohol intake, but does not affect relapse-like drinking. Neuropsychopharmacology. 2012;37:1047–1056. doi: 10.1038/npp.2011.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price ML, Kirby LG, Valentino RJ, Lucki I. Evidence for corticotropin-releasing factor regulation of serotonin in the lateral septum during acute swim stress: adaptation produced by repeated swimming. Psychopharmacology (Berl) 2002;162:406–414. doi: 10.1007/s00213-002-1114-2. [DOI] [PubMed] [Google Scholar]
- Rolf RH, Matz DR, Brune GG. Serotonin metabolism in chronic alcoholism. Experientia. 1978;34:74–75. doi: 10.1007/BF01921911. [DOI] [PubMed] [Google Scholar]
- Sabino V, Cottone P, Koob GF, Steardo L, Lee MJ, Rice KC, Zorrilla EP. Dissociation between opioid and CRF1 antagonist sensitive drinking in Sardinian alcohol-preferring rats. Psychopharmacology. 2006;189:175–186. doi: 10.1007/s00213-006-0546-5. [DOI] [PubMed] [Google Scholar]
- Sabino V, Kwak J, Rice KC, Cottone P. Pharmacological Characterization of the 20% Alcohol Intermittent Access Model in Sardinian Alcohol-Preferring Rats: A Model of Binge-Like Drinking. Alcohol Clin Exp Res. 2013;37:635–643. doi: 10.1111/acer.12008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz DW, Mansbach RS, Sprouse J, Braselton JP, Collins J, Corman M, Dunaiskis A, Faraci S, et al. CP-154,526: a potent and selective nonpeptide antagonist of corticotropin releasing factor receptors. Proc Natl Acad Sci U S A. 1996;93:10477–10482. doi: 10.1073/pnas.93.19.10477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto A, DeBold JF, Holly EN, Miczek KA. Blunted accumbal dopamine response to cocaine following chronic social stress in female rats: exploring a link between depression and drug abuse. Psychopharmacology (Berl) 2011;218:271–279. doi: 10.1007/s00213-011-2364-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms JA, Nielsen CK, Li R, Bartlett SE. Intermittent access ethanol consumption dysregulates CRF function in the hypothalamus and is attenuated by the CRF-R1 antagonist, CP-376395. Addiction. 2013 doi: 10.1111/adb.12024. [DOI] [PubMed] [Google Scholar]
- Simms JA, Steensland P, Medina B, Abernathy KE, Chandler LJ, Wise R, Bartlett SE. Intermittent access to 20% ethanol induces high ethanol consumption in Long-Evans and Wistar rats. Alcohol Clin Exp Res. 2008;32:1816–1823. doi: 10.1111/j.1530-0277.2008.00753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staub DR, Lunden JW, Cathel AM, Delben EL, Kirby LG. Morphine history sensitizes postsynaptic GABA receptors on dorsal raphe neurons in a stress-induced releapse model in rats. Psychoneuroendocrinology. 2012;37:859–870. doi: 10.1016/j.psyneuen.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi A, Shimamoto A, Boyson CO, DeBold JF, Miczek KA. GABAB receptor modulation of serotonin neurons in the dorsal raphe nucleus and escalation of aggression in mice. J Neurosci. 2010;30:11771–11780. doi: 10.1523/JNEUROSCI.1814-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanahashi S, Yamamura S, Nakagawa M, Motomura E, Okada M. Effect of lamotrigine and carbamazepine on corticotropin-releasing factor-associated serotonergic transmission in rat dorsal raphe nucleus. Psychopharmacology (Berl) 2012;220:599–610. doi: 10.1007/s00213-011-2506-y. [DOI] [PubMed] [Google Scholar]
- Tao R, Auerbach SF. Involvement of the dorsal raphe but not median raphe nucleus in morphine-induced increases in serotonin release in the rat forebrain. Neuroscience. 1995;68:553–561. doi: 10.1016/0306-4522(95)00154-b. [DOI] [PubMed] [Google Scholar]
- Tao R, Auerbach SF. μ-Opioids disinhibit and κ-opioids inhibit serotonin efflux in the dorsal raphe nucleus. Brain res. 2005;104:70–79. doi: 10.1016/j.brainres.2005.04.076. [DOI] [PubMed] [Google Scholar]
- Tomkins DM, Sellers EM, Fletcher PJ. Median and dorsal raphe injections of the 5-HT1A agonist, 8-OH-DPAT, and the GABAA agonist, muscimol, increase voluntary ethanol intake in Wistar rats. Neuropharmacology. 1994;33:349–358. doi: 10.1016/0028-3908(94)90065-5. [DOI] [PubMed] [Google Scholar]
- Van Pett K, Viau V, Bittencourt J, Chan RK, Li HY, Arias C, Prins GS, et al. Distribution of mRNAs encoding CRF receptors in brain and pituitary of rat and mouse. J Comp Neurol. 2000;428:191–212. doi: 10.1002/1096-9861(20001211)428:2<191::aid-cne1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Walker BM, Valdez GR, McLaughlin JP, Bakalkin G. Targeting dynorphin/kappa opioid receptor systems to treat alcohol abuse and dependence. Alcohol. 2012;46:359–370. doi: 10.1016/j.alcohol.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waselus M, Nazzaro C, Valentino RJ, Van Bockstaele EJ. Stress-induced redistribution of corticotropin-releasing factor receptor subtypes in the dorsal raphe nucleus. Biol Psych. 2009;66:76–83. doi: 10.1016/j.biopsych.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise RA. Voluntary ethanol intake in rats following exposure to ethanol on various schedules. Psychopharmacologia. 1973;29:203–210. doi: 10.1007/BF00414034. [DOI] [PubMed] [Google Scholar]