

Abstract
PURPOSE
To test the hypothesis that the evaluation of retinal structure can have diagnostic value in differentiating between incomplete congenital stationary night blindness (CSNB2) and retinitis pigmentosa (RP). To compare retinal thickness differences between patients with CSNB2 and myopic controls.
DESIGN
Prospective cross-sectional study.
METHODS
Ten eyes of 5 patients diagnosed with CSNB2 (4 X-linked recessive, 1 autosomal recessive) and 6 eyes of 3 patients with RP (2 autosomal dominant, 1 autosomal recessive) were evaluated with spectral-domain optical coherence tomography (SD OCT) and fundus autofluorescence (FAF). Diagnoses of CSNB2 and RP were confirmed by full-field electroretinography (ERG). Manual segmentation of retinal layers, aided by a computer program, was performed by 2 professional segmenters on SD OCT images of all CSNB2 patients and 4 age-similar, normal myopic controls. Seven patients were screened for mutations with congenital stationary night blindness and RP genotyping arrays.
RESULTS
Patients with CSNB2 had specific findings on SD OCT and FAF that were distinct from those found in RP. CSNB2 patients showed qualitatively normal SD OCT results with preserved photoreceptor inner segment/outer segment junction, whereas this junction was lost in RP patients. In addition, CSNB2 patients had normal FAF images, whereas patients with RP demonstrated a ring of increased autofluorescence around the macula. On SD OCT segmentation, the inner and outer retinal layers of both X-linked recessive and autosomal recessive CSNB2 patients were thinner compared with those of normal myopic controls, with means generally outside of normal 95% confidence intervals. The only layers that demonstrated similar thickness between CSNB2 patients and the controls were the retinal nerve fiber layer and, temporal to the fovea, the combined outer segment layer and retinal pigment epithelium. A proband and his 2 affected brothers from a family segregating X-linked recessive CSNB2 had a mutation, p.R614X, in the gene encoding calcium channel, α 1F subunit.
CONCLUSIONS
CSNB2 patients (X-linked recessive and autosomal recessive) had significantly thinner retinas than myopic controls. However, they demonstrated qualitatively normal SD OCT and FAF images, and therefore can be differentiated from RP patients with these techniques. Although ERG testing remains the gold standard for the diagnosis of these conditions, FAF and SD OCT systems are more widely available to community ophthalmologists, offer shorter acquisition times, and, unlike ERG, can be performed on the same day as the initial clinic visit. Therefore, as a supplement to ERG and genetic testing, we advocate the use of FAF and SD OCT in the examination of patients with CSNB2 and RP.
Congenital stationary night blindness (csnb) refers to a group of disorders characterized by night blindness and nonprogressive retinal dysfunction. Onset of disease occurs in infancy, and patients often have accompanying symptoms of nystagmus and strabismus. Best-corrected visual acuity often is reduced, with myopic and sometimes hyperopic refractive errors, and with astigmatism as well.1,2
CSNB has been classified into complete and incomplete (CSNB2) types, based on electroretinogram (ERG) findings and clinical characteristics.1 In the complete form, full-field ERG testing reveals normal to mildly subnormal cone function and complete absence of rod function. Mutations in the NYX gene have been found to be responsible for the condition.3 In contrast, patients with the incomplete form have greater disturbances of cone function, but they maintain some rod function. X-linked recessive CSNB2 (CSNB2A; Mendelian Inheritance in Man no. 300071) and autosomal recessive CSNB2 (CSNB2B; Mendelian Inheritance in Man no. 610427) inheritance patterns have been reported, with the CSNB2A subtype linked to mutations in the gene encoding a voltage-gated calcium channel, alpha 1F subunit (CACNA1F), and the CSNB2B subtype linked to mutations in the gene encoding calcium-binding protein 4 (CABP4).4–6 Both complete CSNB and CSNB2 demonstrate the classic negative ERG pattern, or Schubert Bornschein type, in which the b-wave is smaller than the a-wave during maximal response.1,7
Retinitis pigmentosa (RP) refers to a diverse group of disorders characterized by night blindness and progressive retinal dysfunction. Visual loss first occurs peripherally and then progresses inward toward the macula. ERG findings demonstrate global photoreceptor dysfunction, with rods more greatly affected than cones.8,9 X-linked recessive, autosomal dominant, and autosomal recessive inheritance patterns have been described previously.9–13 More than 45 genes have been implicated in RP, contributing to the vast heterogeneity of phenotypes in this set of disorders.13,14
Novel imaging techniques and genetic analysis provide greater understanding of ophthalmic diseases. With fundus autofluorescence (FAF) imaging, an abnormal hyperauto-fluorescent ring surrounding the fovea has been demonstrated in 59% to 94% of RP patients.15,16 Spectral-domain optical coherence tomography (SD OCT) has revealed loss of the inner segment/outer segment (IS/OS) junction and generalized decrease in retinal thickness across the width of the hyperautofluorescent ring in RP patients.17 (From our own analysis of more than 100 RP patients, we believe that the incidence of the hyperautofluorescent ring in RP is far closer to 94% than 59% with regard to patients who have at least some preserved IS/OS junction).
The objectives of this study were to describe the qualitative SD OCT features in CSNB2, to compare SD OCT quantitative thickness data between CSNB2 patients and normal myopic controls, to describe FAF features in CSNB2, and to compare the FAF and SD OCT appearances of CSNB2 and RP. We also report the phenotypes of a CSNB2A family carrying a nonsense mutation, p.R614X, in the gene encoding CACNA1F.
METHODS
This prospective, cross-sectional study included 10 eyes of 5 patients diagnosed with CSNB2 (4 X-linked recessive [CSNB2A], 1 autosomal recessive [CSNB2B]), 6 eyes of 3 patients diagnosed with RP (2 autosomal dominant, 1 autosomal recessive), and 8 eyes of 4 age-similar normal myopic controls with spherical equivalents between −2.50 and −5.50 diopters. Three of the 5 patients with CSNB2 were brothers from the same family; all other patients were unrelated. The clinical diagnosis of CSNB2 and RP was established by retina specialists and confirmed by full-field scotopic and photopic ERGs performed according to the International Society for Clinical Electro-physiology of Vision standards.18
FUNDUS AUTOFLUORESCENCE
FAF imaging was performed with a confocal scanning laser ophthalmoscope (OCT-SLO Spectralis/Heidelberg Retina Angiograph 2; Heidelberg Engineering, Heidelberg, Germany) after pupil dilation with topical 0.5% tropicamide and 2.5% phenyl-ephrine. FAF imaging was performed using a 30 × 30-degree field of view at a resolution of 1536 × 1536 pixels. An optically pumped solid-state laser (488 nm) was used for excitation, and a 495-nm barrier filter was used to modulate the blue argon excitation light. Standard procedure was followed for the acquisition of FAF images, including focus of the retinal image in the infrared reflection mode at 820 nm, further focus and sensitivity adjustment at 488 nm, and acquisition of at least 9 single 30 × 30-degree FAF images encompassing the entire macular area with at least a portion of the optic disc. The 9 single images were averaged computationally to produce a single frame with improved signal-to-noise ratio.
SPECTRAL-DOMAIN OPTICAL COHERENCE TOMOGRA-PHY
SD OCT was performed with the OCT-SLO Spectralis (Heidelberg Engineering, Heidelberg, Germany) on CSNB2 and RP patients, as well as on myopic controls. This equipment allows for simultaneous OCT scans and FAF imaging and subsequent image superimposition. OCT imaging was acquired by a broadband 870-nm superluminescent diode that scanned the retina at 40 000 A-scans per second, with an optical axial depth resolution of 7 μm. The standard protocol included at least 25 OCT scans averaged to reduce the signal-to-noise ratio by a factor of 5. The scans included at least 1 9-mm horizontal line scan through the fovea.
SD OCT images also were obtained from patients and controls using the Cirrus Spectral-Domain OCT (Carl Zeiss Meditec, Inc, Dublin, California, USA). The acquisition protocol consisted of a 5-line raster scan and a macular cube 512 × 128 scan pattern in which a 6 × 6-mm region of the retina was scanned (a total of 65 536 sampled points) within a scan time of 2.4 seconds.
To determine retinal layer thicknesses of CSNB2 patients and myopic controls on SD OCT, manual segmentation aided by a computer program was performed by 2 experienced segmenters (M.A.L., R.R.), as previously described.19,20 The results presented below are the averages of these 2 segmenters. All line scans were flattened before thickness analysis to ensure that results were not affected by the curvature of the scans. Six borders were segmented, as shown in Figure 1, and the thicknesses of the following layers were determined: retinal nerve fiber layer (RNFL); retinal ganglion cell layer plus inner plexiform layer (RGC + IPL); inner nuclear layer (INL); total receptor; photoreceptor outer segments plus retinal pigment epithelium (OS + RPE), and total retina. The layers were segmented as follows: RNFL (vitreous/RNFL minus RNFL/RGC), RGC + IPL (RNFL/RGC minus IPL/INL), INL (IPL/INL minus INL/outer plexiform layer), total receptor (INL/outer plexiform layer minus Bruch membrane/choroid), OS + RPE (IS/OS minus Bruch membrane/choroid), and total retina (vitreous/RNFL minus Bruch membrane/choroid border).
FIGURE 1.
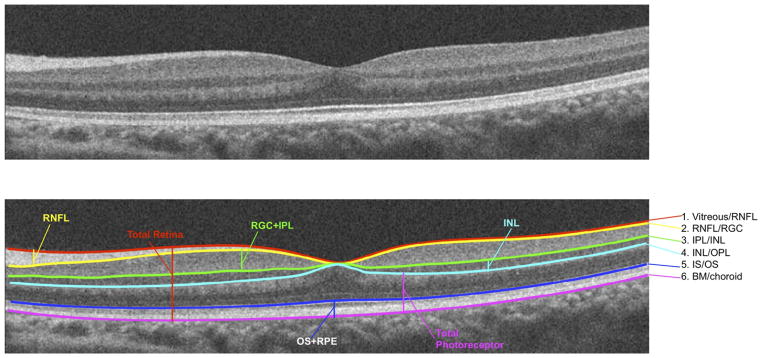
Optical coherence tomography segmentation layers in a normal eye. BM = Bruch membrane; INL = inner nuclear layer; IPL = inner plexiform layer; IS/OS = photoreceptor inner segment/outer segment junction; OPL = outer plexiform layer; RGC = retinal ganglion cell layer; RNFL = retinal nerve fiber layer; RPE = retinal pigment epithelium.
Five CSNB2 patients and 4 age-similar myopic controls imaged with the Cirrus OCT were included in the final segmentation analysis. An additional 10 myopic controls were analyzed during the study; however, because they were imaged only with the Spectralis OCT, these controls were excluded from final analysis for the sake of consistency.
ELECTRORETINOGRAPHY
Full-field ERGs (Diagnosys LLC, Lowell, Massachusetts, USA) were recorded from both eyes with DTL electrodes according to the International Society for Clinical Electrophysiology of Vision standard in both scotopic and photopic states to assess retinal function.18 The amplitudes and implicit times obtained from both eyes of each patient were compared with age-matched normal values, in which lower limits were calculated for 2 standard deviations.
GENETIC ANALYSES
DNA was extracted from whole blood with the QIAamp DNA Blood Maxi Kit 51194 (Qiagen, Inc, Valencia, California, USA). Four CSNB patients were screened with the CSNB APEX array at Asper Biotech, Inc (Tartu, Estonia; www.asperophthalmics.com), and the identified variants were confirmed by direct sequencing. All RP patients were screened with APEX dominant and recessive RP arrays, depending of their mode of inheritance. No RP-associated mutations were detected by the array screening.
RESULTS
The clinical characteristics of the 8 patients are summarized in the Table. Five patients had CSNB2 (4 X-linked recessive [CSNB2A] and 1 autosomal recessive [CSNB2B]), and 3 patients had RP (2 autosomal dominant, 1 autosomal recessive). Snellen best-corrected visual acuity for patients with CSNB2 ranged from 20/40 to 20/70, and patients ranged in age from 6 to 69 years of age. Each of the 3 patients with RP had Snellen best-corrected visual acuity of 20/20 (Table). Fundus photographs from all of these patients looked unremarkable, with the exception of myopic changes (Figure 2). All CSNB2 patients demonstrated an electronegative response on maximal full-field scotopic ERG. All RP eyes exhibited severely diminished full-field scotopic electroretinography responses, and photopic responses were decreased in amplitude and delayed in implicit time compared with those of age-matched normal controls. Representative ERG waveforms of 2 CSNB2 patients, 2 RP patients, and 1 normal control are shown in Figure 3. Compared with controls, all 4 patients demonstrated abnormal rod-specific responses. Patients 3 and 8 had delayed rod-specific implicit peak times, Patient 2 had an extinguished rod-specific response, and Patient 6 showed a characteristic photomyoclonic artifact. On maximal response, Patients 2 and 3 with CSNB2 clearly demonstrated electronegative ERG responses, whereas Patients 6 and 8 with RP had normal maximal b-wave-to-a-wave ratios. Photopic 30-Hz flicker testing revealed a double-peak pattern in Patient 3, characteristic of CSNB2. The first patient (Patient 2, CSNB2B) lost the characteristic double-peak waveform because of narrow band filtering and movement artifact.
TABLE.
Clinical Characteristics of 8 Patients with Incomplete Congenital Stationary Night Blindness or Retinitis Pigmentosa
| Patient No. | Diagnosis | Age (y) | Sex | BCVA
|
|
|---|---|---|---|---|---|
| Right Eye | Left Eye | ||||
| 1 | CSNB2A | 6 | M | 20/70 | 20/60 |
| 2 | CSNB2A | 11 | M | 20/40-2 | 20/40-2 |
| 3 | CSNB2A | 9 | M | 20/40 | 20/40 |
| 4 | CSNB2A | 12 | M | 20/60 | 20/60 |
| 5 | CSNB2B | 69 | M | 20/70 | 20/60 |
| 6 | RP | 7 | M | 20/20 | 20/20 |
| 7 | RP | 10 | M | 20/20 | 20/20 |
| 8 | RP | 30 | M | 20/20 | 20/20 |
BCVA = best-corrected visual acuity; CSNB2A = X-linked recessive congenital stationary night blindness; CSNB2B = autosomal recessive congenital stationary night blindness; M = male; RP = retinitis pigmentosa.
FIGURE 2.

Color fundus photographs of patients with incomplete congenital stationary night blindness (CSNB2) and retinitis pigmentosa (RP). (Top, left to right) Patients 2, 4, and 5, respectively, with CSNB2. (Bottom, left to right) Patients 6, 8, and 7, respectively, with RP. Photographs generally are unremarkable. (Bottom left) White opacity supranasal to foveal center is an image artifact.
FIGURE 3.

Full-field electroretinography (ERG) results from patients with incomplete congenital stationary night blindness (CSNB2), retinitis pigmentosa (RP), and a normal control. (Top row and Second row) Patients 2 and 3 with CSNB2; (Third row and Fourth row) Patients 6 and 8 with RP. (Bottom) Normal control. Rod-specific responses are delayed or abnormal in all patients (dashed line). Scotopic bright flash maximal ERGs for CSNB2 patients typically are electronegative (black arrow, a-wave; red arrow, b-wave) with low b-to-a ratios. Photopic ERGs were mildly delayed and reduced. Patient 3 (CSNB2) demonstrates the characteristic double-peak wave form (black arrow). Photopic responses are scaled in 20-μv increments for CSNB2 patients and 100-μv increments for RP patients and the control.
Three CSNB2 patients were siblings from the same family; they all possessed a nonsense mutation, p.R614X, in the gene encoding CACNA1F. No nystagmus was noted in this family.
On FAF imaging, all 6 study eyes with RP demonstrated a hyperautofluorescent ring that was not evident on fundus biomicroscopy. The autofluorescence appeared normal within the ring for all of these eyes. Other than 1 CSNB2 patient who demonstrated bilateral peripapillary hypoautofluorescence consistent with myopic changes, eyes with CSNB2 demonstrated no abnormal FAF features. Figure 4 compares the autofluorescence findings of CSNB2 patients in the first row with those of RP patients in the second row. No hyperautofluorescent rings are visible in the CSNB2 images, but rings are seen clearly in each of the RP images. The size of this ring varied in the 3 patients—the smallest ring was visible in the parafoveal region (Patient 7, Figure 4), whereas the largest ring was visible just beyond the vascular arcades (Patient 8, Figure 4). The outer boundary of each hyperautofluorescent ring is indicated by a yellow arrow.
FIGURE 4.

Comparison of fundus autofluorescence (FAF) images from patients with incomplete congenital stationary night blindness (CSNB2) and retinitis pigmentosa (RP). (Top, left to right) Normal FAF images of Patients 2, 4, and 5, respectively, with CSNB2. (Top right) An area of hypoautofluorescence around the optic nerve head corresponding to myopic peripapillary atrophy is visible on the color fundus photograph. (Bottom, left to right) Patients 6, 8, and 7, respectively, with RP. Outer boundaries of hyperautofluorescent rings are indicated with a yellow arrow. Autofluorescence appears normal inside the rings.
We compared SD OCT findings in the regions of hyperautofluorescence in RP with SD OCT features in CSNB2 patients. In all eyes with RP, there was loss of the ONL, ELM, and IS/OS junction across the width of the hyperautofluorescent ring. In the zone of normal autofluorescence within the ring, the retinal morphologic features appeared normal in all 6 RP eyes, with intact ONL, ELM, and IS/OS junction (Figure 5). In contrast to RP patients, all CSNB2 patients had normal-appearing retinas on SD OCT and FAF, without qualitative loss of the ONL, ELM, or IS/OS junction (Figure 6).
FIGURE 5.

(Top left) Color fundus photograph, (Top right) fundus autofluorescence (FAF) image, and (Bottom) spectral-domain optical coherence tomography (SD OCT) image of the right eye of Patient 7 with retinitis pigmentosa. Horizontal arrow shows location of Cirrus SD OCT (Carl Zeiss Meditec, Inc, Dublin, California, USA) B scan image. The retina appears thinner temporally in the area that traverses the hyperautofluorescent ring shown in the FAF image. Brackets demonstrate region where there is loss of the outer nuclear layer (ONL), external limiting membrane (ELM), and photoreceptor inner segment/outer segment (IS/OS) junction. Nasal to the area of IS/OS loss and within the ring of normal autofluorescence, the retinal layers appear normal. INL = inner nuclear layer; IPL = inner plexiform layer; NFL = nerve fiber layer; OPL = outer plexiform layer; RPE = retinal pigment epithelium.
FIGURE 6.

(Top left) Color fundus photograph, (Top right) fundus autofluorescence (FAF) image, and (Bottom) spectral-domain optical coherence tomography (SD OCT) of the right eye of Patient 4 with incomplete congenital stationary night blindness. Horizontal arrow shows location of Cirrus SD OCT (Carl Zeiss Meditec, Inc, Dublin, California, USA) B scan image. FAF and SD OCT images appear normal. The photoreceptor inner segment/outer segment (IS/OS) junction can be traced across the length of the scan. ELM = external limiting membrane; INL = inner nuclear layer; IPL = inner plexiform layer; NFL = nerve fiber layer; ONL = outer nuclear layer; OPL = outer plexiform layer; RPE = retinal pigment epithelium.
We performed manual segmentation of both the Spectralis OCT and Cirrus OCT images for all CSNB2 patients and myopic controls. Because not every patient and control eye was imaged with both the Spectralis and Cirrus OCT instruments, we elected to include only patients and controls imaged on the Cirrus OCT instrument for segmentation comparison purposes. However, when we compared segmentation thicknesses for patients imaged with both Cirrus and Spectralis OCT, the results were similar.
Five CSNB2 patients and 4 myopic controls were included in the final segmentation analysis. The 4 controls ranged from 8 to 10 years of age. Figure 7 shows individual patient thickness profiles (colored lines) plotted versus the control mean ± 2 standard deviations (black lines). Figure 8 shows the mean ± 1 standard error of the CSNB2 patients (green and red lines) versus the mean ± 1 standard error of the myopic controls (gray and blue lines). Outside of the foveal center, the RGC + IPL, INL, total photoreceptor, and total retina thickness of CSNB2 patients was consistently below the normal confidence interval. Thicknesses were similar between CSNB2 patients and controls for the RNFL and temporal aspect of the OS + RPE layers. Nasal to the fovea, the OS + RPE thickness appeared thinner compared with myopic controls. Additionally, the thickness profiles of all retinal layers of the 69-year-old CSNB2B patient (Patient 5) fell within the measurements of the 4 younger CSNB2A patients.
FIGURE 7.

Graphs showing individual Cirrus optical coherence tomography (Carl Zeiss Meditec, Inc, Dublin, California, USA) segmentation thicknesses for 5 incomplete congenital stationary night blindness (CSNB2) patients and 4 myopic controls. Thicknesses (in millimeters) of individual CSNB2 patients (colors) are plotted versus the control mean (bold, black) ± 2 standard deviations (black). CSNB2 retinas generally are thinner than those of myopic controls. INL = inner nuclear layer; OS + RPE = outer segment layer and retinal pigment epithelium; RGC + IPL = retinal ganglion cell layer and inner plexiform layer; RNFL = retinal nerve fiber layer.
FIGURE 8.

Graphs showing averaged Cirrus optical coherence tomography (Carl Zeiss Meditec, Inc, Dublin, California, USA) segmentation thicknesses for 5 incomplete congenital stationary night blindness (CSNB2) patients (red traces) and 4 myopic controls (blue traces). Measurements from CSNB2 patients are thinner than those of controls in the following layers: retinal ganglion cell layer and inner plexiform layer (RGC + IPL), inner nuclear layer (INL), nasal outer segment layer and retinal pigment epithelium (OS + RPE), total photoreceptor, and total retina. Thicknesses are similar between CSNB2 patients and myopic controls for retinal nerve fiber layer (RNFL) and temporal OS + RPE.
DISCUSSION
There are several methods that help clinicians differentiate CSNB2 from RP. By history and physical examination, CSNB2 patients usually, but not always, have nystagmus at presentation (the family of 3 CSNB2 patients did not have nystagmus). Unlike RP, CSNB2 can be differentiated by both its nonprogressive nature and the presence of decreased visual acuity from birth (early stage RP patients generally have 20/20 visual acuity). On fundus biomicroscopy, patients with more advanced RP demonstrate RPE mottling, intraretinal pigment migration (i.e., bone spicule pigment), waxy disc pallor, and vessel attenuation, but early stage RP can have an unremarkable fundus appearance, and thus is indistinguishable from CSNB2. Full-field ERG remains the gold standard to differentiate these conditions; however, it can be difficult to perform in young children, which may result in poor traces with significantly decreased informative value. DNA testing is increasingly useful, but the causative mutation is still not found in most RP or CSNB patients,21–24 and screening may take several months. DNA testing also can be costly; therefore, imaging tests that can aid in the accuracy of the initial diagnosis could save the patient and institution from ordering studies aimed at the wrong condition.
In younger, more challenging cases, the ability to make correct diagnoses depends on our analysis of the full constellation of findings: from history and physical examination, electrophysiologic studies, genetic screening, and imaging analysis. Thus, one objective of this study was to identify differences between CSNB2 and RP with FAF and SD OCT imaging in the hope that they will aid in formulating diagnoses.
All study patients had unremarkable fundus biomicroscopic examination results. In all RP eyes, FAF showed a hyperautofluorescent ring in the posterior pole that corresponded to loss of the ONL, ELM, and IS/OS junction on SD OCT imaging. In contrast to RP eyes, all CSNB2 eyes demonstrated normal fundus autofluorescence, and analysis of SD OCT showed preserved outer retinal architecture.
Previous studies have demonstrated that a hyperautofluorescent macular ring is present in most patients with RP.15–17,25,26 The area of this hyperautofluorescent ring demarcates the boundary between normal and severely abnormal retinal function, with normal retinal function within the ring, a transition zone occurring across the ring itself, and severely abnormal retinal function outside the margin of the ring. The size and progressive constriction of the ring correspond to degree of preserved retinal function and visual prognosis.26
The hyperautofluorescent ring represents an abnormal accumulation of lipofuscin in the RPE, likely the result of an increased rate of outer segment degeneration. Lima and associates demonstrated with SD OCT that in the region traversing the hyperautofluorescent ring, total and outer retinal thickness were decreased, and in the area outside the ring, the total and outer retinal thickness were markedly decreased, with loss of the hyperreflective band representing the IS/OS junction.17
With the exception of 1 patient with peripapillary hypoautofluorescence consistent with myopic changes, all of our CSNB2 patients had normal FAF imaging. No CSNB2 patient demonstrated the hyperautofluorescent ring that was present in each of the RP patients. In a previous study of patients with dominantly inherited CSNB, FAF images were obtained from 3 patients. These results were similarly unremarkable, although the oldest patient (58 years of age) did exhibit some small atrophic areas around the central macula as well as larger areas of atrophy in the periphery.27 In light of the other 2 patients in the above study as well as all 5 of our patients having demonstrated normal FAF appearances, we believe that these abnormal features were likely unrelated to CSNB.
Results of SD OCT imaging in all CSNB2 patients were qualitatively normal. When compared with normal myopic controls, however, CSNB2 patients unexpectedly had significantly thinner retinas, despite the apparent preservation of retinal architecture. CSNB2 patients had thinner retinas compared with myopic controls in the RGC + IPL, INL, nasal OS + RPE, and total photoreceptor segments. RNFL thicknesses and temporal OS + RPE segments were not significantly different compared with those of normal myopic controls.
There are 2 clinicopathologic reports of eyes with CSNB.28,29 Vaghefi and associates studied the hyperopic eye of one patient with CSNB, branch retinal vein occlusion, and optic nerve head drusen. With regard to CSNB, the histopathologic analysis demonstrated a normal pattern and proportion of rod and cone cells.28 Watanabe and associates studied the enucleated, myopic left eye of a patient with absolute glaucoma and likely CSNB. The right eye had an electronegative ERG, but the left eye had only a subnormal b-wave response. The authors in this study hypothesized that although the left eye likely was electronegative previously, loss of inhibitory function of optic nerve centrifugal fibers resulting from long-standing glaucoma allowed for a return to subnormal-type ERG. Histologic examination in this left eye revealed a marked loss of ganglion cells, with otherwise normal retinal layers. Electron microscopy showed rod outer segments with a normal arrangement of discs.29
Based on thickness measurements, our OCT segmentation results differed from the above clinicopathologic studies, because the total photoreceptor layer thickness of CSNB2 patients was decreased compared with that of myopic controls. Additionally, the thinning of the segmented inner layers in CSNB2 patients compared with that of controls suggests that there may be structural differences that account for the deficits in inner retinal transmission. The classic electronegative ERG in CSNB, in which the b-wave is smaller in amplitude than the a-wave, indicates defective transmission between photoreceptors and bipolar cells, or between individual bipolar cells. Genetic analyses have revealed mutations in the calcium channel gene CACNA1F and calcium binding protein CABP4 in CSNB2A and CSNB2B, respectively.2,4–6,30,31 Although the thinning of the segmented layers in CSNB2 patients in this study (RGC + IPL, INL, nasal OS + RPE, total photoreceptor) likely is related to mutations affecting calcium channels and calcium binding proteins, there may be other causative factors resulting in the thickness differences between CSNB2 patients and myopic controls.
An additional observation was the fact that the 69-year-old CSNB2B patient displayed thickness profiles of all retinal layers that fell within the measurements of the 4 younger CSNB2A patients. This finding suggests 2 points: first, that the degree of retinal thinning in CSNB2 is similar despite different inheritance patterns, and second, that the natural history of retinal thinning is nonprogressive, as would be expected in this stationary condition.
Our report had several limitations. First, we had a small sample size, with only 5 patients with CSNB2. Second, not all patients and controls were studied with the same imaging instruments (OCT-SLO Spectralis vs Cirrus Spectral Domain OCT). At the beginning of the study, our institution had only the Cirrus OCT, but we acquired the Spectralis OCT toward the end of our study. Therefore, all CSNB2 patients were studied with the Cirrus OCT, but only some also were studied with the Spectralis OCT. Additionally, 10 of the 14 myopic controls were imaged at a separate institution that used only the Spectralis OCT. To simplify the segmentation comparison between CSNB2 eyes and myopic controls, we included only Cirrus OCTs, and therefore excluded 10 of the 14 controls from the final segmentation comparison. Third, although the 4 CSNB2A patients and all myopic controls included in the segmentation comparison were younger than 15 years, the 1 CSNB2B patient was 69 years of age. Despite these limitations, we did segment the 10 excluded myopic controls (imaged on Spectralis OCT; average age, 25 years; range, 15 to 54 years) and found that they had similar thicknesses as the 4 included controls (imaged on Cirrus OCT). Additionally, when we compared the Spectralis controls with the CSNB2 patients imaged on the same instrument, we found similar thickness differences as were found on the Cirrus OCT segmentation comparisons.
In conclusion, we reported the phenotypes of a homozygous disease-causing mutation, p.R614X, in the CACNA1F gene responsible for CSNB2A,31 and we compared FAF and SD OCT findings in RP and CSNB2. Although we do not believe that FAF and SD OCT together can substitute for full-field ERG in the diagnosis of RP or CSNB2, we believe that they offer valuable additional insights, especially in more diagnostically challenging patients with normal funduscopic examinations. In comparison with ERG, these systems offer shorter acquisition times and can be performed on the same day as the initial clinic visit, advantages that may help the clinician communicate more effectively with patients and their families from the outset. Our observation that CSNB2 patients had thinner inner and outer retinas compared with myopic controls suggests that there may be structural differences in CSNB2 eyes that are associated with mutations in the CACNA1F or calcium-binding protein 4 genes. We also suggest that the retinal thinning observed in CSNB2 patients is nonprogressive. We intend to investigate these findings further in a larger cohort of patients and to compare segmentation results of complete versus incomplete CSNB patients.
Acknowledgments
Publication of this article was supported in part by grants EY018213 (S.H.T.), EY021163 and EY13435 (R.A.), and P30EY019007 (Core Support for Vision Research; S.H.T. and R.A.) from the National Eye Institute, National Institutes of Health, Bethesda, Maryland; Foundation Fighting Blindness, Owings Mills, Maryland; Grant TS080017 from the Department of Defense, Washington, DC; and unrestricted funds from Research to Prevent Blindness, Inc, New York, New York. Dr Tsang is a Burroughs-Wellcome Program in Biomedical Sciences Fellow and is also supported by the Charles E. Culpeper Partnership for Cures 07-CS3, the Crowley Research Fund, Schneeweiss Stem Cell Fund, New York State N09G-302, and Joel Hoffmann Scholarship. The authors indicated no financial support or conflicts of interest. Involved in Design and conduct of study (R.W.S.C., J.P.G., J.C.H., S.H.T.); Management, analysis, and interpretation of data (R.W.S.C., J.P.G., M.A.L., R.R., L.H.L., J.C.H., C.S., A.B., R.A., S.H.T.); and Preparation, review, or approval of manuscript (R.W.S.C., J.P.G., M.A.L., R.R., L.H.L., J.C.H., C.S., A.B., R.A., S.H.T.). Institutional Review Board approval no. AAAE5099 was obtained from Columbia University Medical Center/New York Presbyterian Hospital, and all research procedures adhered to the tenets of the Declaration of Helsinki. Informed consent was obtained from all subjects in the study, and Health Insurance Portability and Accountability Act compliance was maintained. The authors thank the medical imaging division at the Edward S. Harkness Eye Institute for their fundus photographs. They also thank Donald C. Hood and R. Theodore Smith, Columbia University, for sharing ideas and equipment.
Biographies

Royce W. S. Chen received his medical degree from Tufts University in 2008 and is currently a second-year resident in ophthalmology at the Edward S. Harkness Eye Institute of Columbia University, New York, New York. After residency, he will pursue a fellowship in vitreoretinal surgery and disease.

Jonathan P. Greenberg received his medical degree in 2008 from the University of the Witwatersrand, South Africa. He is currently a post-doctoral research fellow at the Edward S. Harkness Eye Institute, Columbia University, New York, New York. After his fellowship, he intends tol pursue a residency in Ophthalmology in the United States.
References
- 1.Miyake Y, Yagasaki K, Horiguchi M, Kawase Y, Kanda T. Congenital stationary night blindness with negative electroretinogram: a new classification. Arch Ophthalmol. 1986;104(7):1013–1020. doi: 10.1001/archopht.1986.01050190071042. [DOI] [PubMed] [Google Scholar]
- 2.Nakamura M, Ito S, Terasaki H, Miyake Y. Novel CACNA1F mutations in Japanese patients with incomplete congenital stationary night blindness. Invest Ophthalmol Vis Sci. 2001;42(7):1610–1616. [PubMed] [Google Scholar]
- 3.Bech-Hansen NT, Naylor MJ, Maybaum TA, et al. Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat Genet. 2000;26(3):319–323. doi: 10.1038/81619. [DOI] [PubMed] [Google Scholar]
- 4.Strom TM, Nyakatura G, Apfelstedt-Sylla E, et al. An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat Genet. 1998;19(3):260–263. doi: 10.1038/940. [DOI] [PubMed] [Google Scholar]
- 5.Bech-Hansen NT, Naylor MJ, Maybaum TA, et al. Loss-of-function mutations in a calcium-channel alpha1-subunit gene in Xp11. 23 cause incomplete X-linked congenital stationary night blindness. Nat Genet. 1998;19(3):264–267. doi: 10.1038/947. [DOI] [PubMed] [Google Scholar]
- 6.Zeitz C, Kloeckener-Gruissem B, Forster U, et al. Mutations in CABP4, the gene encoding the Ca(2+)-binding protein 4, cause autosomal recessive night blindness. Am J Hum Genet. 2006;79(4):657–667. doi: 10.1086/508067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schubert G, Bornschein H. Analysis of the human electroretinogram. Ophthalmologica. 1952;123(6):396–413. doi: 10.1159/000301211. [DOI] [PubMed] [Google Scholar]
- 8.Marshall J, Heckenlively JR, Heckenlively JR, editors. Retinitis Pigmentosa. Philadelphia: JB Lippincott; 1988. [Google Scholar]
- 9.Bird AC. Retinal photoreceptor dystrophies LI. Edward Jackson Memorial Lecture. Am J Ophthalmol. 1995;119(5):543–562. doi: 10.1016/s0002-9394(14)70212-0. [DOI] [PubMed] [Google Scholar]
- 10.Bunker CH, Berson EL, Bromley WC, Hayes RP, Roderick TH. Prevalence of retinitis pigmentosa in Maine. Am J Ophthalmol. 1984;97(3):357–365. doi: 10.1016/0002-9394(84)90636-6. [DOI] [PubMed] [Google Scholar]
- 11.Grondahl J. Estimation of prognosis and prevalence of retinitis pigmentosa and Usher syndrome in Norway. Clin Genet. 1987;31(4):255–264. doi: 10.1111/j.1399-0004.1987.tb02804.x. [DOI] [PubMed] [Google Scholar]
- 12.Novak-Lauš K, Suzana Kukulj S, Zoric-Geber M, Bastaic O. Primary tapetoretinal dystrophies as the cause of blindness and impaired vision in the republic of Croatia. Acta Clin Croat. 2002;41(1):23–27. [Google Scholar]
- 13. [Last accessed June 6, 2011];RETNET. Available at: http://www.sph.uth.tmc.edu/Retnet/sumdis.htm#A-geneshttp://www.sph.uth.tmc.edu/Retnet/sum-dis.htm#A-genes.
- 14.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368(9549):1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 15.Murakami T, Akimoto M, Ooto S, et al. Association between abnormal autofluorescence and photoreceptor disorganization in retinitis pigmentosa. Am J Ophthalmol. 2008;145(4):687–694. doi: 10.1016/j.ajo.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 16.Wakabayashi T, Sawa M, Gomi F, Tsujikawa M. Correlation of fundus autofluorescence with photoreceptor morphology and functional changes in eyes with retinitis pigmentosa. Acta Ophthalmol. 2010;88(5):e177–e183. doi: 10.1111/j.1755-3768.2010.01926.x. [DOI] [PubMed] [Google Scholar]
- 17.Lima LH, Cella W, Greenstein VC, et al. Structural assessment of hyperautofluorescent ring in patients with retinitis pigmentosa. Retina. 2009;29(7):1025–1031. doi: 10.1097/IAE.0b013e3181ac2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marmor MF, Fulton AB, Holder GE, Miyake Y, Brigell M, Bach M. ISCEV Standard for full-field clinical electroretinography (2008 update) Doc Ophthalmol. 2009;118(1):69–77. doi: 10.1007/s10633-008-9155-4. [DOI] [PubMed] [Google Scholar]
- 19.Hood DC, Lin CE, Lazow MA, Locke KG, Zhang X, Birch DG. Thickness of receptor and post-receptor retinal layers in patients with retinitis pigmentosa measured with frequency-domain optical coherence tomography. Invest Ophthalmol Vis Sci. 2009;50(5):2328–2336. doi: 10.1167/iovs.08-2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hood DC, Lazow MA, Locke KG, Greenstein VC, Birch DG. The transition zone between healthy and diseased retina in patients with retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2011;52(1):101–108. doi: 10.1167/iovs.10-5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Audo I, Manes G, Mohand-Said S, et al. Spectrum of rhodopsin mutations in French autosomal dominant rod-cone dystrophy patients. Invest Ophthalmol Vis Sci. 2010;51(7):3687–3700. doi: 10.1167/iovs.09-4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Audo I, Sahel JA, Mohand-Said S, et al. EYS is a major gene for rod-cone dystrophies in France. Hum Mutat. 2010;31(5):E1406–E1435. doi: 10.1002/humu.21249. [DOI] [PubMed] [Google Scholar]
- 23.Pelletier V, Jambou M, Delphin N, et al. Comprehensive survey of mutations in RP2 and RPGR in patients affected with distinct retinal dystrophies: genotype-phenotype correlations and impact on genetic counseling. Hum Mutat. 2007;28(1):81–91. doi: 10.1002/humu.20417. [DOI] [PubMed] [Google Scholar]
- 24.Zeitz C, Labs S, Lorenz B, et al. Genotyping microarray for CSNB-associated genes. Invest Ophthalmol Vis Sci. 2009;50(12):5919–5926. doi: 10.1167/iovs.09-3548. [DOI] [PubMed] [Google Scholar]
- 25.Robson AG, El-Amir A, Bailey C, et al. Pattern ERG correlates of abnormal fundus autofluorescence in patients with retinitis pigmentosa and normal visual acuity. Invest Ophthalmol Vis Sci. 2003;44(8):3544–3550. doi: 10.1167/iovs.02-1278. [DOI] [PubMed] [Google Scholar]
- 26.Robson AG, Saihan Z, Jenkins SA, et al. Functional characterisation and serial imaging of abnormal fundus autofluorescence in patients with retinitis pigmentosa and normal visual acuity. Br J Ophthalmol. 2006;90(4):472–479. doi: 10.1136/bjo.2005.082487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kabanarou SA, Holder GE, Fitzke FW, Bird AC, Webster AR. Congenital stationary night blindness and a “Schubert-Bornschein” type electrophysiology in a family with dominant inheritance. Br J Ophthalmol. 2004;88(8):1018–1022. doi: 10.1136/bjo.2003.033555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaghefi HA, Green WR, Kelley JS, Sloan LL, Hoover RE, Patz A. Correlation of clinicopathologic findings in a patient: congenital night blindness, branch retinal vein occlusion, cilioretinal artery, drusen of the optic nerve head, and intraretinal pigmented lesion. Arch Ophthalmol. 1978;96(11):2097–2104. doi: 10.1001/archopht.1978.03910060477019. [DOI] [PubMed] [Google Scholar]
- 29.Watanabe I, Taniguchi Y, Morioka K, Kato M. Congenital stationary night blindness with myopia: a clinico-pathologic study. Doc Ophthalmol. 1986;63(1):55–62. doi: 10.1007/BF00153012. [DOI] [PubMed] [Google Scholar]
- 30.Boycott KM, Pearce WG, Bech-Hansen NT. Clinical variability among patients with incomplete X-linked CSNB and a founder mutation in CACNA1F. Can J Opthalmol. 2000;35(4):204–213. doi: 10.1016/s0008-4182(00)80031-9. [DOI] [PubMed] [Google Scholar]
- 31.Boycott KM, Maybaum TA, Naylor MJ, et al. A summary of 20 CACNA1F mutations identified in 36 families with incomplete X-linked congenital stationary night blindness, and characterization of splice variants. Hum Genet. 2001;108(2):91–97. doi: 10.1007/s004390100461. [DOI] [PubMed] [Google Scholar]