

Abstract
Background
Rapeseed (B. napus, AACC, 2n = 38) is one of the most important oil seed crops in the world, it is also one of the most common oil for production of biodiesel. Its oil is a mixture of various fatty acids and dissection of the genetic network for fatty acids biosynthesis is of great importance for improving seed quality.
Results
The genetic basis of fatty acid biosynthesis in B. napus was investigated via quantitative trail locus (QTL) analysis using a doubled haploid (DH) population with 202 lines. A total of 72 individual QTLs and a large number pairs of epistatic interactions associated with the content of 10 different fatty acids were detected. A total of 234 homologous genes of Arabidopsis thaliana that are involved in fatty acid metabolism were found within the confidence intervals (CIs) of 47 QTLs. Among them, 47 and 15 genes homologous to those of B. rapa and B. oleracea were detected, respectively. After the QTL mapping, the epistatic and the candidate gene interaction analysis, a potential regulatory pathway controlling fatty acid biosynthesis in B. napus was constructed, including 50 enzymes encoded genes and five regulatory factors (LEC1, LEC2, FUS3, WRI1 and ABI3). Subsequently, the interaction between these five regulatory factors and the genes involved in fatty acid metabolism were analyzed.
Conclusions
In this study, a potential regulatory pathway controlling the fatty acid was constructed by QTL analysis and in silico mapping analysis. These results enriched our knowledge of QTLs for fatty acids metabolism and provided a new clue for genetic engineering fatty acids composition in B. napus.
Electronic supplementary material
The online version of this article (doi:10.1186/s12870-015-0475-8) contains supplementary material, which is available to authorized users.
Keywords: Brassica napus, Fatty acid composition, QTL, Epistatic interaction, Regulatory pathway
Background
Oilseed rape (Brassica napus L., AACC, 2n = 38) is one of the most important oil crops producing multi-purpose oil for food and biofuel in many parts of the world. In 2007, biodiesel production accounted for 7% of the global vegetable oil supplies, in which 68% were used for biofuels in the EU [1]. As the global requirements for rapeseed oil are growing rapidly, increasing the oil content and improving the oil composition are important ways to meet the demands of agricultural feed stocks.
The fatty acid composition of rapeseed oil is considered to be genetically more variable than any other major vegetable oils [2]. Rapeseed oil is a mixture of seven main fatty acids [3]. Fatty acid biosynthetic pathways are generally controlled by multiple genes and considered as quantitative traits regulated by QTLs. So far, a number of QTLs controlling oil composition were identified in B. napus. Ecke et al. identified two QTLs for erucic acid distributed on chromosomes A6 and C2 [4]. Four QTLs for erucic acid distributed across chromosomes A1, A2, A8 and C3 were reported and three of these coincided with QTLs for the accumulation of oil content [5]. Burns et al. observed 13 QTLs affecting composition of 10 fatty acids, and seven also affected oil content [6]. Hu et al. identified two QTLs for oleic (on A1 and A5) and linolenic acids (on A4 and C4), respectively [7]. One to eight QTLs were detected for seven individual fatty acids by Zhao et al., and eight of these also affected oil content [8]. Recently, Smooker et al. identified 34 QTLs for five major fatty acids [9], and Yan et al. detected a total of 40 QTLs for six fatty acids, which were most clustered on chromosomes A8, A9 and C3 [10].
The allotetraploid B. napus has two progenitor species, B. rapa and B. oleracea, which shared their last common ancestor with A. thaliana about 20 million years ago [11,12]. Both a high degree of sequence similarities and chromosomal colinearities between Brassica species and Arabidopsis were reported [13-15]. Parkin et al. reported 21 conserved blocks within the Arabidopsis genome shared with B. napus [16], and Schranz et al. proposed a set of 24 conserved chromosomal blocks in B. napus [17]. Furthermore, all the genome sequence of B. rapa, B. oleracea and B. napus have been released [18-20]. It is feasible to predict the Arabidopsis orthologous genes for specific agronomic traits within the Brassica genome. For example, a number of candidate genes were mapped to CIs of QTLs for flowering time by in silico mapping [21], and a total of 14 lipid-related candidate gene loci were located in the CIs of six QTLs for seed oil content [22]. In fact, many important genes involved in fatty acid metabolism were identified in Arabidopsis, such as FAB2, FAD2, FAD3 and FAE1 [23-26], and the orthologs of these genes in B. napus were also reported and mapped. BnaFAD2 was mapped on A1, A5, C1 and C5 chromosomes [27-29], and one major QTL BnaA.FAD2.a located on A5 was responsible for high C18:1 [27]. BnaFAD3 was mapped on A3, A4, A5, C3 and C4 [9,27], and two major QTLs BnaA.FAD3.b and BnaC.FAD3.b were both responsible for low C18:3 [27]. BnaFAE1 was mapped on both A8 and C3 [9], and two FAE1 homologous genes on A8 and C3 linkage groups were also found by Qiu et al. [5] and Fourmann et al. [30]. Collectively, although genes or QTLs for fatty acid biosynthesis have been identified, the genetic network for all these metabolic pathways in B. napus needs to be elucidated.
In Arabidopsis, more than 120 enzymatic reactions and at least 600 genes are involved in acyl-lipid metabolism [31]. Li-Beisson et al. gave metabolic pathways associated with the biosynthesis and degradation of acyl-lipids in Arabidopsis [31]. The genome of polyploid B. napus may typically contain six distinct alleles for each gene present in Arabidopsis [32], the fatty acid biosynthesis and the gene regulation in B. napus might have a more complex pathway than that in Arabidopsis. Though much attention was given to genes and regulatory factors involved in acyl-lipid metabolism in Arabidopsis [31,33-37], similar questions concerning the genetic basis of fatty acid biosynthesis in B. napus remain open, mainly due to the lack of integrative studies at a population scale. Moreover, the interaction of genes involved in acyl-lipid metabolism has not yet been studied based on co-location of mapped candidate genes with QTLs in B. napus. To determine these key steps in relevant complex metabolic pathways of acyl-lipids in B. napus, it is first necessary to identify QTLs or genes associated with fatty acids composition.
In this paper, we describe the genetic bases of seed fatty acid composition through QTL mapping in B. napus. The aims of this study were as follows: (1) to add knowledge concerning QTL mapping of the fatty acid composition in B. napus; (2) to predict candidate genes of major QTLs for different fatty acids’ biosynthesis by comparative genome analysis; and (3) to construct a regulatory pathway for fatty acids metabolism in B. napus.
Results
Variation and single QTL analysis of fatty acid composition in the ‘Tapidor’ × ‘Ningyou7’ cross (TN) DH population
Means of all traits measured from the TN DH population over six environments were close to the mid-parent values (Table 1). There was a wide range of variations and transgressive segregations for the concentration of each fatty acid (Figure 1). The population appeared to have a normal or near-normal distribution for C16:0, C18:0, C18:2, C18:3, C20:0, C22:0 and FAS (Saturated Fatty Acid), suggesting complexity of their genetic networks. However, C18:1, C20:1 and C22:1 showed bi-modal distributions, indicating that they might be controlled by few major genes with a relatively large effect. The distribution patterns of 10 fatty acids’ compositions showed that they were genetically stable but also affected by environment. The correlation between different fatty acid compositions showed great differences (Table 2). Erucic acid (C22:1) content was highly and positively correlated with the level of C20:0, C20:1 and C22:0 (Coefficients 0.30–0.63), but was negatively correlated with other fatty acids (−0.92 to −0.03), especially C18:1 (−0.92) (Table 2). C18:1 showed a high positive correlation with C16:0 and C18:0 (0.70–0.75) and moderate positive correlation with C18:2 and C18:3 (Table 2), but showed a high negative correlation with other fatty acids (−0.92 to −0.33).
Table 1.
Means and ranges for seed fatty acids of TN DH population evaluated in six environments
| Traits | 16:0 | C18:0 | C18:1 | C18:2 | C18:3 | C20:0 | C20:1 | C22:0 | C22:1 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Tapidor | Meana | 4.82 ± 0.03 | 1.89 ± 0.1 | 57.43 ± 2.46 | 17.5 ± 0.84 | 7.76 ± 0.52 | 0.67 ± 0.01 | 2.41 ± 1.24 | 0.35 ± 0.01 | 2.83 ± 0.91 |
| Ningyou7 | Mean | 3.15 ± 0.01 | 1.11 ± 0.04 | 17.45 ± 2.85 | 12.92 ± 1.45 | 8.54 ± 0.48 | 0.85 ± 0.04 | 8.82 ± 0.08 | 0.73 ± 0.01 | 45.55 ± 0.27 |
| DH | Mean | 3.94 ± 0.53 | 1.63 ± 0.41 | 34.12 ± 15.63 | 15.44 ± 3.3 | 8.22 ± 0.97 | 0.77 ± 0.3 | 10.88 ± 5.62 | 0.4 ± 0.16 | 24.3 ± 15.56 |
| Max | 5.87 | 4.92 | 73.54 | 27.24 | 12.02 | 6.88 | 19.66 | 1.86 | 52.22 | |
| Min | 2.84 | 0.89 | 9.4 | 8.61 | 1.31 | 0.14 | 1.08 | 0 | 0 |
aMean value ± SE.
Figure 1.
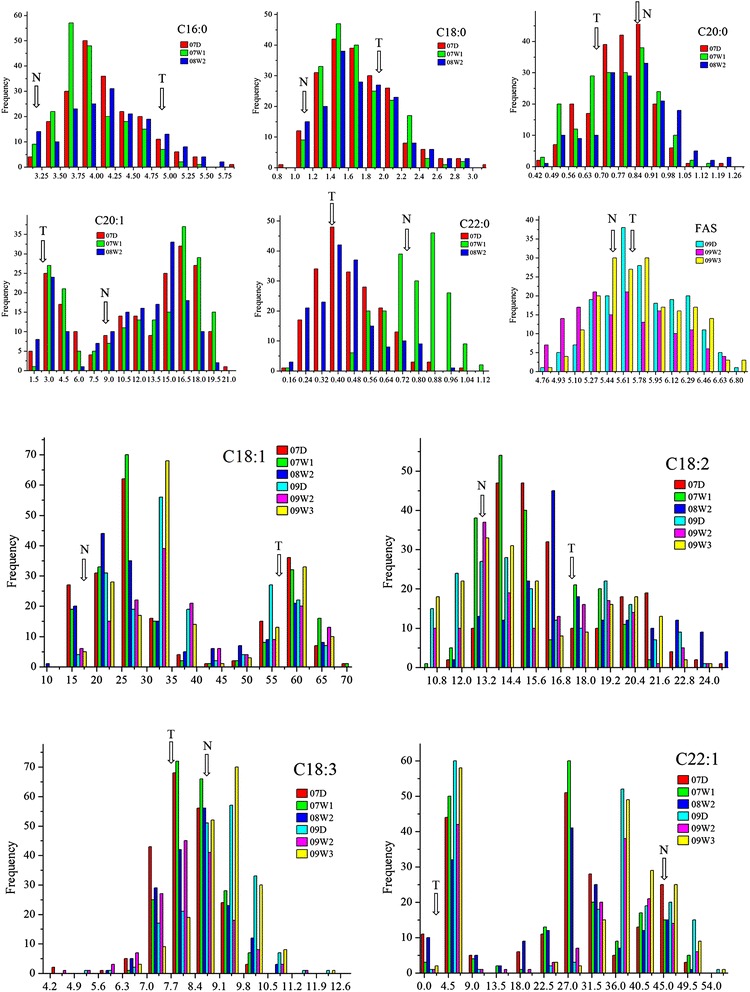
Distribution of fatty acid concentrations of TN DH population in multiple environments. The unit of x-axis means percentage of the specific fatty acid composition in the sum of all fatty acids. The unit of y-axis means the number of lines. N represents the parent “Ningyou7” and T the parent “Tapidor” of TN population.
Table 2.
Pearson correlation coefficients for trait pairs affecting fatty acid compositions in the DH population
| C16:0 | C18:0 | C18:1 | C18:2 | C18:3 | C20:0 | C20:1 | C22:0 | C22:1 | |
|---|---|---|---|---|---|---|---|---|---|
| C16:0 | 1 | ||||||||
| C18:0 | 0.62** | 1 | |||||||
| C18:1 | 0.70** | 0.75** | 1 | ||||||
| C18:2 | 0.06 | 0.05 | 0.03 | 1 | |||||
| C18:3 | 0.26** | 0.06 | 0.03 | −0.01 | 1 | ||||
| C20:0 | −0.23** | −0.12** | −0.33** | −0.03 | −0.13** | 1 | |||
| C20:1 | −0.58** | −0.52** | −0.77** | −0.03 | −0.12** | 0.31** | 1 | ||
| C22:0 | −0.37** | −0.41** | −0.51** | −0.04 | −0.16** | 0.27** | 0.11** | 1 | |
| C22:1 | −0.77** | −0.78** | −0.92** | −0.04 | −0.03 | 0.30** | 0.63** | 0.60** | 1 |
**Significant at P = 0.001.
For QTL mapping analysis, Wincart_2.5 detected a total of 139 QTLs distributed across 15 chromosomes (except for C1, C2, C4 and C7) and individual QTL for any given trait explained 1.27–47.56% of phenotypic variance (PV) (Additional file 1). QTLNetwork_2.0 detected a total of 44 QTLs across 15 chromosomes (Additional file 2). After combining QTLs for different traits clustered in the same regions indicated by the same close-linked molecular markers, a total of 72 QTLs controlling fatty acid composition were identified (Figure 2, Table 3), and 10, 44 and 18 QTLs were detected by using QTLNetwork_2.0 only, Wincart_2.5 only and both QTLNetwork_2.0 and Wincart_2.5, respectively (Table 3).
Figure 2.

QTL distribution of fatty acid concentrations on linkage groups in B. napus . Whole linkage groups are shown with black lines labeled with molecular markers (short vertical bars) on the bottom, and the Arabic numerals listed on the right side show the length of linkage groups containing QTLs. The names of traits are listed on the left side of the linkage groups. The black lines on the linkage groups show the QTL confidence interval and the circles indicate the peak position. The pseudo-chromosomes of Arabidopsis are aligned under each linkage group of B. napus.
Table 3.
The combined QTLs for fatty acid contents detected by WinQTLCart_2.5 and QTLNetwork_2.0
| QTL | Chr. a | Marker interval | LOD | Additive | PV(%) | range | S b | Env. c | traits |
|---|---|---|---|---|---|---|---|---|---|
| qA1-1 | A1 | znS13M26-100-CB10081 | 3.1 | −1.28 | 4.87 | 0.3-7.2 | W | 07D | C20:1 |
| qA1-2 | A1 | CB10097-JICB0313 | 3.6 | 0.19 | 7.15 | 26.1-28.8 | W | 07 W1 | C18:3 |
| qA1-3 | A1 | HBr006-ZAAS156a | 7.6 | 0.29 | 14.64 | 35.9-37 | W | 07 W1 | C18:3 |
| qA1-4 | A1 | JICB0455-znS08M15-320 | 4.8 | 0.24 | 11.42 | 39.8-43.1 | W | 07 W1 | C18:3 |
| qA1-5 | A1 | em09me21-70-ZAAS165 | 4.0 | 0.07 | 3.43 | 80.4-84.6 | W&Q | 07 W1 | C18:0 |
| qA2-1 | A2 | CB10355-Ol10F04 | 3.1 | 0.23 | 6.64 | 0-18.2 | W&Q | 09 W3 | C18:3 |
| qA2-2 | A2 | HR-Sp1-210-pX154 | 3.3 | 0.31 | 7.85 | 56.2-61.5 | W | 09 W2 | C18:3 |
| qA2-3 | A2 | BRMS-082-HG-FT-A2 | 3.5 | 0.32 | 8.43 | 61.5-67.3 | W | 09 W2 | C18:3 |
| qA3-1 | A3 | ZNS13M26-360-HBR160 | 0.05 | 2.02 | 15.7-21.4 | Q | C16:0 | ||
| qA3-2 | A3 | RA2E11-BRMS-303 | 3.8 | −0.03 | 4.02 | 35.9-37.7 | W | 07 W1 | C20:0 |
| qA3-3 | A3 | CB10271-CNU250 | 3.3-4.0 | −0.52-0.02 | 3.01-5.66 | 37.8-43.0 | W&Q | 07D/07 W1 | C16:0/C18:2/C20:0/C22:0 |
| qA3-4 | A3 | HBr124-KBrB043L02-12 | 3.1 | −0.10 | 3.22 | 61.5-63.7 | W | 07D | C16:0 |
| qA3-5 | A3 | HBr137-CNU270 | 3.0-3.5 | −0.1-3.2 | 3.52-3.56 | 67.4-76.1 | W&Q | 07D/08 W2 | C16:0/C18:1 |
| qA3-6 | A3 | HR-S2-130-HR-Tp4-165 | 3.8 | −0.03 | 4.78 | 79.0-86.5 | W | 07D | C22:0 |
| qA4-1 | A4 | OL11H02C-SR12307I | −1.02 | 0.39 | 100.1-104.1 | Q | C22:1 | ||
| qA4-2 | A4 | znS13M26-220-sN3514f | 4.4 | 0.18 | 8.86 | 14.0-23.2 | W | 08 W2 | C16:0 |
| qA4-3 | A4 | IGF3365B-SN13034 | −0.09 | 5.43 | 25.7-28.5 | Q | C16:0 | ||
| qA4-4 | A4 | BRMS-276-HR-C001-A4 | 4.6 | 0.06-0.17 | 1.04-8.02 | 29.5-35.2 | W&Q | 08 W2 | C16:0/C18:0 |
| qA4-5 | A4 | niab048-JICB0134 | 3.4-5.4 | −0.03-0.18 | 5.45-6.26 | 45.6-60.5 | W | 07D/07 W1 | C16:0/C18:3/C20:0 |
| qA4-6 | A4 | JICB0134-HBr091 | 5.22 | −0.05_-0.04 | 0.37-5.44 | 60.5-67.2 | W&Q | 07 W1 | C18:3/C20:0 |
| qA4-7 | A4 | HBr202-HS-k02-2 | 3.2 | 0.24 | 6.98 | 70.4-82.3 | W | 07D | C18:3 |
| qA5-1 | A5 | BRMS-034-niab017 | 3.3 | 0.03 | 4.18 | 22.9-29.3 | W | 07D | C22:0 |
| qA5-2 | A5 | niab013-pX129a | 3.8 | 0.09 | 4.40 | 57.9-59.9 | W | 09D | FAS |
| qA5-3 | A5 | CNU206-PIE1-6 | 3.2-3.7 | 0.09-0.26 | 4.38-8.17 | 65.3-79.0 | W&Q | 09 W3/09D | C18:3/FAS |
| qA5-4 | A5 | CNU268-HR-Tp4-200 | 3.1-3.3 | −2.3-0.22 | 1.88-5.96 | 82.3-89.7 | W | 07D | C18:1/C18:3 |
| qA5-5 | A5 | IGF3134a-CNU362 | 3.8 | 0.23 | 6.76 | 95.5-109.3 | W | 07D | C18:3 |
| qA6-1 | A6 | Ol11F12a-ZAAS92a | 4.8 | 0.29 | 10.56 | 0-4.6 | W&Q | 07D | C18:3 |
| qA6-2 | A6 | HBr201-BRMS-030 | 3.2 | −0.07 | 3.05 | 40.7-51.1 | W&Q | 07D/07 W1 | C18:0 |
| qA6-3 | A6 | HR-Tp3-320-pW217 | 4.1 | 0.25 | 7.51 | 9.1-14.2 | W | 07D | C18:3 |
| qA7-1 | A7 | IGF1226l-CNU053b | 3 | −1.70 | 1.27 | 100.2-105.5 | W | 07D | C22:1 |
| qA7-2 | A7 | HBr030-znS06M34-50 | 5.4 | −0.10 | 4.36 | 64.0-70.7 | W | 07 W1 | C16:0 |
| qA7-3 | A7 | CNU167-CNU044 | 4.4 | −0.09 | 3.62 | 71.2-74.8 | W | 07 W1 | C16:0 |
| qA7-4 | A7 | BRMS-036-HBr021 | 4.5 | −0.10 | 4.10 | 74.8-79.8 | W | 07 W1 | C16:0 |
| qA8-1 | A8 | em10me26-120-JICB0335 | −0.04-0.64 | 4.39-6.42 | 18.6-26.8 | Q | C18:1/C20:0 | ||
| qA8-2 | A8 | HBr107-HBr017 | 8.1 | 0.07 | 15.24 | 27.4-34.0 | W | 08 W2 | C20:0 |
| qA8-3 | A8 | HBr104-HBr010 | 4.7 | 0.05 | 9.30 | 36.8-41.9 | W | 08 W2 | C20:0 |
| qA8-4 | A8 | Na12B05a-HBr031 | 3.4 | −0.22 | 6.16 | 48.0-53.3 | W | 07D | C18:3 |
| qA8-5 | A8 | IGF1108c-sR7178 | 3.1-59.9 | −10.4-9.14 | 5.7-47.55 | 57.4-74.6 | W&Q | 07D/07 W1/08 W2/09D/09 W2/09 W3 | C16:0/C18:0/C18:1/C18:2/C18:3/C20:0/C20:1/C22:0/C22:1/FAS |
| qA8-6 | A8 | HBr015-HBr026 | 3.9-33.2 | −9.1-8.9 | 7.7-38.7 | 75.3-90.4 | W | 07D/07 W1/08 W2/09 W2 | C18:1/C18:2/C20:0/C20:1/C22:0/C22:1/FAS |
| qA9-1 | A9 | pW123aH-HBr178 | 3.6 | 0.08 | 2.74 | 18.3-25.3 | W&Q | 07 W1 | C16:0 |
| qA9-2 | A9 | CNU296-KBrB073D09-12 | 3.2 | −3.20 | 4.64 | 45.5-52.0 | W | 09 W2 | C18:1 |
| qA9-3 | A9 | HBr197-HBr205 | 4.5 | 0.04 | 5.59 | 63.5-66.9 | W | 07D | C20:1 |
| qA9-4 | A9 | HBr205-niab003 | 4.3 | −1.50 | 6.47 | 66.9-70.0 | W&Q | 07 W1 | C20:1 |
| qA9-5 | A9 | IGF1087g-niab127 | 3.3 | 0.52 | 3.32 | 70.9-71.5 | W | 07D | C18:2 |
| qA9-6 | A9 | em18me23-380-HBr196 | 3.9 | 0.03 | 4.97 | 73.2-75.1 | W | 07D | C22:0 |
| qA9-7 | A9 | pW203b-HBr053 | 3.4 | −0.08 | 3.96 | 86.1-95.5 | W | 09D | FAS |
| qA10-1 | A10 | RA2E03-HS-b14-1 | 3.5 | 0.24 | 6.29 | 14.6-18.9 | W | 07D | C18:3 |
| qA10-2 | A10 | HS-j90-HG4-HG-CO-A10 | 3.3-4.6 | −2.59-0.42 | 2.0-12.9 | 18.9-34.2 | W&Q | 07D/07 W1/09 W2 | C18:0/C18:1/C18:3 |
| qA10-3 | A10 | JICB0551-JICB0573 | −0.05 | 0.28 | 60.8-71.3 | Q | C18:0 | ||
| qC1-1 | C1 | EM18ME23-300-CB10258 | 0.58 | 3.85 | 33.0-35.9 | Q | C20:1 | ||
| qC3-1 | C3 | IGF5376b-HBr014 | 3.4-15.1 | −8.20-8.63 | 5.0-30.0 | 118.4-126.1 | W | 07D/09D | C18:1/C22:0/C22:1 |
| qC3-2 | C3 | HBr014-Ol13C12 | 4.7-38.6 | −9.83-8.86 | 6.3-37.7 | 126.1-131.5 | W | 07D/07 W1/08 W2/09D/09 W2/09 W3 | C16:0/C18:0/C18:1/C18:2/C20:0/C20:1/C22:0/C22:1/FAS |
| qC3-3 | C3 | IGF0235b-BRMS-093 | 3.8-64.9 | −10.96-9.78 | 3.0-45.8 | 133.8-152.3 | W&Q | 07D/07 W1/08 W2/09D/09 W2/09 W3 | C16:0/C18:0/C18:1/C18:2/C20:0/C20:1/C22:0/C22:1/FAS |
| qC3-4 | C3 | pX141bE-HBr032 | 4.9 | 0.10 | 4.80 | 15.7-26.0 | W&Q | 07D | C18:0 |
| qC3-5 | C3 | pW221-pX141aE | 4.4 | 0.07 | 3.68 | 26.5-34.2 | W | 07D/07 W1 | C18:0 |
| qC3-6 | C3 | HBr211-HBr152 | 4.2 | 1.52 | 6.46 | 51.7-59.0 | W | 07 W1 | C20:1 |
| qC3-7 | C3 | Ol11G11b-pW143 | −0.05 | 4.98 | 80.3-91.5 | Q | C20:0 | ||
| qC3-8 | C3 | HBr139-CNU099 | 4.7 | −3.10 | 2.91 | 94.1-98.8 | W | 07D | C18:1 |
| qC5-3 | C5 | Ol10B02-JICB0509 | 3.2 | 0.10 | 5.01 | 30-62.6 | W | 09 W3 | FAS |
| qC5-1 | C5 | IGF3112a-em12me21-150 | 3.5-7.2 | −0.06-0.55 | 3.7-13.9 | 63.8-82.9 | W | 07D/08 W2 | C18:2/C20:0 |
| qC5-2 | C5 | em12me21-150-IGF0193C | 4.2-4.6 | 0.28-0.56 | 3.9-9.8 | 82.9-88.3 | W | 07D/09 W3 | C18:2/C18:3 |
| qC6-1 | C6 | HBr025-Na12E01a | 5.5 | 0.24 | 11.4 | 74.3-80.8 | W | 09 W2 | FAS |
| qC6-2 | C6 | HBr057-HBr047 | 4.0 | −0.21 | 8.64 | 91.0-95.4 | W | 09 W2 | FAS |
| qC6-3 | C6 | SR12387-EM14ME28-200 | −0.57 | 0.10 | 41.2-45.3 | Q | C22:1 | ||
| qC7-1 | C7 | SNRH63-CNU400 | −0.13 | 1.68 | 18.8-37.2 | Q | C18:3 | ||
| qC8-1 | C8 | CB10504-sN11670a | 3.4 | 0.36-0.54 | 3.0-3.65 | 32.4-46.8 | W&Q | 07D/09 W3 | C18:2/C20:1 |
| qC9-1 | C9 | CB10064-em20me27-230 | 6.1 | −0.12 | 6.28 | 26.3-42.9 | W | 07 W1 | C16:0 |
| qC9-2 | C9 | EM20ME27-230-HR-TP3-360 | 0.13 | 3.83 | 46.2-50.3 | Q | C18:3 | ||
| qC9-3 | C9 | BRMS-154-HBr144 | 5 | −0.12 | 5.31 | 52.7-61.2 | W&Q | 07D/07 W1 | C16:0 |
| qC9-4 | C9 | HBr186-SA30 | 3.1-4.1 | −0.14-0.08 | 3.97-5.4 | 62.5-74.2 | W | 07D/08 W2 | C16:0/C18:0 |
| qC9-5 | C9 | S16071-3F3R-sNRG42 | 3.4 | 0.07-0.90 | 0.03-3.3 | 76.2-82.8 | W&Q | 07D | C18:0/C22:1 |
| qC9-6 | C9 | HR-Sp1-170-sR12384I | 3.6 | 2.71 | 3.36 | 92.7-98.4 | W | 09 W3 | C18:1 |
aChromosome.
bThe software used to detect QTL. W, WinQTLCart_2.5; Q, QTLNetwork_2.0.
cThe environment in which the QTLs are detected.
The QTL with bold indicates that no candidate genes are located in the confidence interval of this QTL in the present study.
Individual saturated fatty acids were only analyzed in three environments (07D, 07 W1 and 08 W2). For C16:0, 18 QTLs were detected across seven chromosomes (Table 3). Among these QTLs, seven (39%) were detected by two types of software. The additive effect ranged from −0.29 to 1.72, and explained 2.74–37.99% of PV (Additional files 1 and 2). For C18:0, 12 QTLs distributed across seven chromosomes had −0.24 to 0.10 additive effect and explained 3.05–37.73% of PV (Table 3, Additional files 1 and 2). Three of them (on A1, A4 and C3) were environment-specific QTLs detected only in one environment. A total of 13 QTLs controlling C20:0 explained 4.02–47.55% of PV (Table 3, Additional files 1 and 2). Nine QTLs on five chromosomes for C22:0 were detected, and three were environment-specific, their additive effect ranged from −0.06 to 0.11, and explained 4.02–47.56% of PV (Table 3, Additional files 1 and 2). In 09D, 09 W2 and 09 W3, all saturated fatty acid compositions were considered as one trait named FAS, and a total of 10 QTLs were detected with additive effect ranged from −0.28 to 0.24 and explaining 3.96–31.33% of PV (Table 3, Additional files 1 and 2).
For the other four unsaturated fatty acid components, phenotypic data were obtained from six different environments, except for C20:1 from three environments (07D, 07 W1 and 08 W2). Twelve QTLs for C18:1 were distributed across seven chromosomes and their additive effect ranged from −10.96 to 3.18 and explained 1.88–42.43% of PV (Table 3, Additional files 1 and 2). Nine QTLs on six chromosomes were associated with C18:2, with the additive effect ranging from −1.69 to 0.56 and explaining 3.10–36.26% of PV (Table 3, Additional files 1 and 2). Twenty-one QTLs on 10 chromosomes were significantly associated with C18:3, which had −1.64 to 0.42 additive effect and explained 5.70–22.29% of PV (Table 3, Additional files 1 and 2). For C20:1, 10 QTLs were detected across A1, A8, A9, C1, C3 and C8 in three environments (Table 3). Six of them were found in two or more environments, and the remaining four were only in one environment. Nine QTLs were observed for C22:1 distributed across A4, A7, A8, C3, C6 and C9, and singly explained 1.27–45.79% of PV (Table 3, Additional files 1 and 2).
Co-localization of mapped candidate genes of B. rapa and B. oleracea with single-locus QTL
A total of 932 molecular markers were mapped to the new version of the TN map. This map covered a total length of 2116.73 cM with an average marker interval of 2.27 cM. The length of the 19 linkage groups varied from 65.83 (C1) to 154.53 (C3) cM (Additional file 3). Thirty-four synteny blocks (28 for A genome, 6 for C genome) and 149 insertion fragment islands (95 for A genome, 54 for C genome) were identified between Arabidopsis pseudochromosomes and TN DH genetic linkage groups by the in silico mapping approach (Additional file 3). More than twenty-nine thousand homologous genes were found to underline the CIs of 61 QTLs associated with the concentrations of 10 different fatty acids, by comparative analysis between the linkage map of TN DH and the genome of Arabidopsis (Additional file 4). Among them, a total of 111 key genes involving fatty acid metabolism were used as candidate genes in the present study (Additional file 4). A number of important genes were found, such as FAB2, FAD2, FAD3 and FAE1, but also five regulatory factors, LEC1, LEC2, FUS3, WRI1 and ABI3. As the polyploid B. napus genome may typically contain six loci for each gene present in Arabidopsis, these 111 genes were found to have 824 homologous genes mapped on the TN DH linkage map in total, including 97 key genes of 234 homologs underlying the CIs of 47 QTLs (Additional file 4). All of the QTLs with CIs containing homologous genes were separately compared to the physical genomic regions of B. rapa (A genome) and B. oleracea (C genome). To compare with the B. rapa genome, 32 QTLs containing candidate gene(s) distributed on the A genome of the TN DH linkage map were used for analysis, accounting for 65.3% of the total QTLs on the A genome. In total, 47 genes in B. rapa matched those in Arabidopsis underlying 24 QTL CIs (Additional file 4). For the C genome of the TN DH linkage map, 23 QTLs were detected and 15 (65.2%) of them contained candidate gene(s) in Arabidopsis. Comparison of the candidate genes showed 15 genes in B. oleracea matched those in Arabidopsis underlying 7 QTL CIs (Additional file 4). For example, on the C3 linkage group, 33 candidate genes (40 homologous genes) of Arabidopsis were located in the CIs of eight QTLs, and 10 candidate genes of B. oleracea were found based on the candidate genes of Arabidopsis, including the regulatory factor ABI3 of previous studies (Figure 3).
Figure 3.
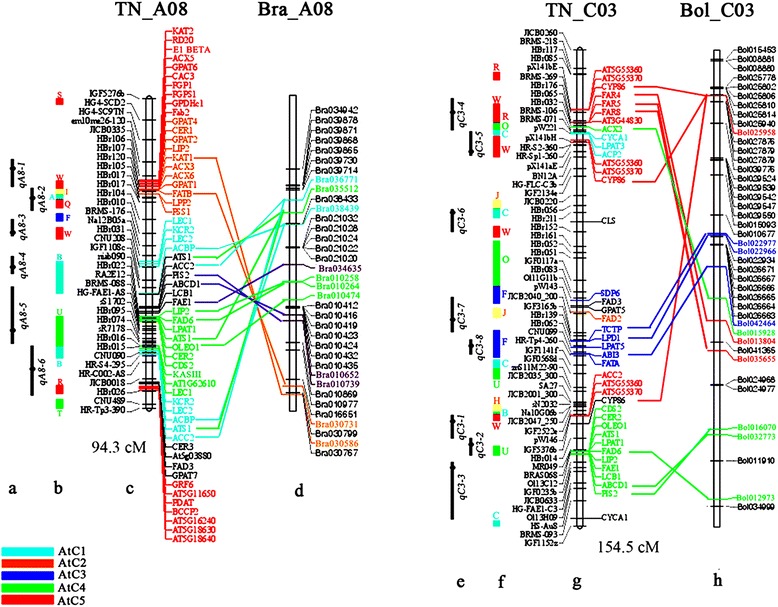
Comparative mapping of homologous linkage groups between B. napus and B. rapa/B. oleracea . (a, e) QTLs associated with 10 traits in multiple experiments, and the ring point in the bar indicates the QTL peak position. (b, f) The building blocks of Brassica genomes are identified on TN genetic maps. (c, g) On the left side are the markers on TN chromosome, and on the right side are candidate genes of Arabidopsis underlying the QTL confidence interval. (d, h) Genes with colored font are the candidate genes in B. rapa/B. oleracea of Arabidopsis, and genes with black font are the candidate genes in B. rapa/B. oleracea identified by each informative marker. The homologous genes in A/C genomes of B. rapa/B. oleracea and B. napus are connected with colored lines using the homologous genes as anchors. The genetic length of TN chromosome is annotated at the bottom of chromosome. The color-coding of the blocks is based on the report of Schranz et al. (2006) [66] and the candidate genes with the same color as the block.
Epistatic QTLs and interaction analysis of candidate genes for fatty acid compositions
QTLNetwork_2.0 and Genotype Matrix Mapping ver2.1 (GMM) software were used to identify the epistatic interactions for fatty acid compositions. Twenty-four pairs of epistatic QTLs involving 28 loci were identified by QTLNetwork_2.0 for 10 measured traits (Additional files 5 and 6), with 1–5 epistatic QTL pairs for each trait. The proportion of total PV explained by all epistatic QTLs was 1.03–38.26% for each trait. Among 24 epistatic QTLs, two, three and 19 were NN, AN and AA interactions, respectively. A total of 395 loci interactions were identified by GMM, including 34 pairs of digenic and 361 pairs of trigenic interactions (Additional file 7). By comparing the epistatic interactions by GMM and single QTL based on common markers, 312 pairs of epistatic interactions were associated with QTLs. Some of the epistatic QTLs affected the level of more than one fatty acid composition. For example, the interactions of two loci associated with QTLs qA8-5 and qC3-3 were detected by both types of software, which controlled different fatty acid compositions (C16:0, C18:0, C18:1, C20:1, C22:0 and FAS by QTLNerwork_2.0 and nine traits except FAS by GMM; Additional files 6 and 7). This indicated that epistatic interactions were very important for fatty acid metabolism in B. napus.
For C16:0, four pairs of epistatic QTLs were detected by QTLNetwork_2.0, explaining 0.66–4.99% of PV. The interaction between QTL qA8-5 and qC9-3 explained 3.39% of PV (Additional file 6). The genes OLEO1, ACC2, FAE1, LPAT1 and ATS1 were underlying the QTL CI of qA8-5, while LEC1, LEC2, ACC2, TAG1, KASIII and ATS1 were underlying the CI of qC9-3 (Additional file 4). A total of 34 significant loci interactions were identified for C16:0 by GMM, containing one pair with digenic and 33 pairs with trigenic interactions, most of these trigenic interactions have loci underlying three QTLs of CIs, including QTLs qA8-5, qA8-6 and qC3-3 (Additional file 7). A number of homologous genes were mapped to the CI of qA8-6, including LEC1, LEC2, ACC2, KASIII, FAD3, GPAT7, BCCP2, ACBP, PDAT, and ATS1, but only one homologous gene CYCA1 for qC3-3 was found (Additional file 7). All the results above suggested that the potential of these gene interactions increased the level of C16:0. Similar to C16:0, a series of pairs of epistatic QTLs and loci interactions for another nine traits were identified by QTLNetwork_2.0 (Additional file 6) and GMM (Additional file 7), and numerous important genes underlying QTL CIs were identified (Additional file 4). For example, for C18:0, C18:1 and C18:2, besides the trigenic interaction of QTLs qA8-5, qA8-6 and qC3-3, the interactions between QTLs qC3-2 and qA8-5 were also identified, and FAE1 was underlying the CI of qC3-2.
Cytoscape_V2.6.3 software was used to investigate the interaction of candidate genes that were observed from the single and epistatic QTL results and the gene interaction network was constructed. The results revealed that the whole network incorporated 167 nodes and 416 edges, that could be divided into three sub-clusters: the five regulatory factors (FUS3, ABI3, WRI1, LEC1 and LEC2) and the genes that were directly affected by only one regulatory factor (A cluster, Figure 4a), the genes affected by two or more regulatory factors (B cluster, Figure 4b) and the genes indirectly affected by regulatory factors (C cluster, Figure 4c). The A cluster consisted of 32 nodes and 105 edges in total. The five regulatory factors formed a tightly intra-linked group, with each regulatory factor under the influence of at least two other regulatory factors, and LEC1 and LEC2 were especially affected by all other four regulatory factors. In addition to the five regulatory factors, BCCP2 and CAC2 were underlying the QTL CIs. BCCP2 was associated with QTLs qA3-1 and qA8-6, which affected C16:0, C18:2 and C20:0; CAC2 was associated with QTL qC9-4 and affected C18:0. These two genes were both regulated by WRI1. The B cluster was composed of seven nodes and 40 edges, in which the genes were affected by at least two regulatory factors. SEP2 and LFY in this group were directly affected by all five regulatory factors. LFY was linked to other eight genes (Figure 4), and two of them (FAD2 and FAE1 in cluster C) were well-known important genes involved in fatty acid synthesis. The C cluster included 128 nodes and 319 edges, and 27 genes were detected to underline the QTL CIs. FATB associated with QTL qA8-2 was notably affected by both FAE1 and FAD2, and FATB also affected another eight genes (ACP1, FAD3, FATA, ACT1, TES, TAG1, CYP86A2 and GPAT5) that were underlying the QTL CIs. The network mentioned above indicated that the genes involved in fatty acid synthesis were directly or indirectly affected by regulatory factors.
Figure 4.

Gene interaction pathway. Network visualization for interaction of the 93 candidate genes and regulatory factors observed from the QTL results using Cytoscape_V2.6.3 software. Genes are presented as nodes and gene interactions are presented as edges. (a) The five regulatory factors and genes that are directly affected by only one regulatory factor: the five regulatory factors indicated by round rectangle with different color (yellow, LEC2; pink, LEC1; green, FUS3; light blue, ABI3; red, WRI1), while the genes are depicted as hexagon with the same color of the regulatory factors which linked to them. (b) The genes affected by two or more regulatory factors, denoting as diamond-shaped nodes, and the two diamond-shaped nodes with light blue color are LFY and SEP2, separately. (c) The genes indirectly affected by regulatory factors. In the middle of the cluster are three very important genes in fatty acid synthesis denoted as rectangle nodes with different color (pink, FAD2; green, FAE1; light yellow, FATB), and other genes are depicted as ellipse pink-filled nodes.
Construct of a potential controlling pathway of fatty acids in B. napus
Among the 97 genes of Arabidopsis located in the CIs of 47 QTLs in B. napus, 57 were shown to have roles in 32 different pathways (Additional file 8). Among them, 31 genes were involved in at least one of the five pathways, including biosynthesis of unsaturated fatty acids, fatty acid metabolism, glycerophospholipid metabolism, fatty acid biosynthesis and alpha-linolenic acid metabolism, with 13, nine, nine, eight and six candidate genes, respectively (Additional file 9). The potential pathway for fatty acid synthesis in B. napus, which included 50 genes and the five regulatory factors mentioned above, was constructed by combining the main pathways that involved interaction of candidate genes as well as knowledge of fatty acid regulatory pathways in Arabidopsis (Figure 5). The candidate genes in the deduced pathways could be divided into three types: genes associated with the fatty acid biosynthetic pathway (Figure 5a), the phospholipid synthesis and other pathways (Figure 5b) and the triacylglycerol and fatty acid degradation (Figure 5c).
Figure 5.
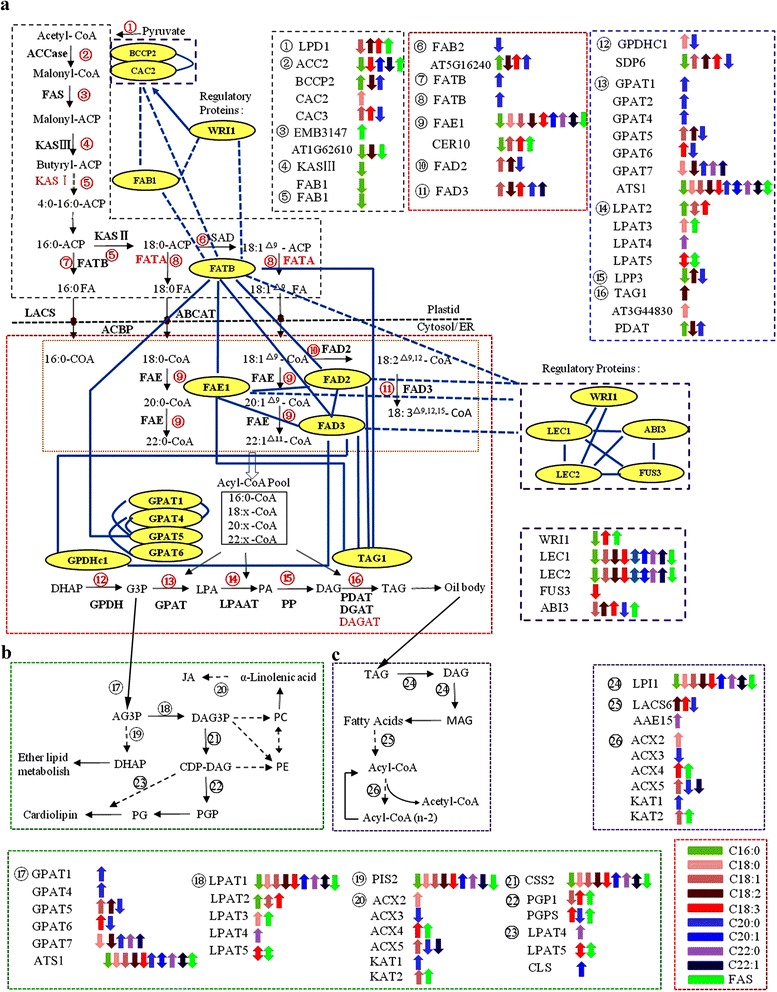
Potential regulatory pathways and candidate genes associated with fatty acid synthesis in B. napus . a, the fatty acid biosynthetic pathway; b, the phospholipid synthesis and other pathways; c, the triacylglycerol and fatty acid degradation. The black characters indicate genes detected in previous studies and also in this study, the red characters indicate genes not found in this study but in previous studies. The colored arrows indicate genes underlying a QTL which affects traits with additive effect, up-arrows indicate QTLs with positive additive and down-arrows a negative additive effect. The double-headed arrows indicate genes underlying different QTLs which affect the same trait with opposite additive effect at the same time based on the WinQTLcart2.5 and QTLNetwork_2.0 results (not combined results of the two). Different colors denote different traits as the bar shown at the lower right corner of the picture. The abbreviations of gene name are same with the following reference papers [30,37,38].
The diagrams of the fatty acid biosynthetic pathway in the plastid and the cytosol/endoplasmic reticulum could be divided into four categories: genes participating in early stages of fatty acid synthesis, fatty acid elongation and desaturation, triacylglycerol biosynthesis and regulators of fatty acid biosynthesis. At least 24 enzymes were involved in the plastidial fatty acid synthetic pathway [38]. ACCase was a major control point of the pathway among these enzymes [39,40], and four subunits of ACCase were detected (Figure 5a) and assigned to 13 QTLs: ACC2 was assigned to seven QTLs: qA3-3, qA5-2, qA8-5, qA8-6, qA9-4, qC3-1 and qC9-3; BCCP2 to QTLs qA3-1 and qA8-6; CAC2 to QTL qC9-4; and CAC3 to QTLs qA4-7, qA8-1 and qC6-1 (Additional file 4). Downstream from ACCase, fatty acids were constructed and elongated by each cycle with four reactions (steps 3–5). In step 3, two genes EMB3147 and AT1G62610 were found underlying the CIs of one (qA5-3) and four QTLs (qA8-6, qA9-7, qC9-3 and qC9-4), respectively. Candidate genes of KASIII (qA8-6, qA9-7, qC9-3 and qC9-4) and FAB1 (qA7-2) in step 4 and FAB1 in step 5 were also observed. This suggested that all these candidate genes in QTL CIs could affect the traits through their action in early stages of fatty acid synthesis.
Most C18:0-ACP produced by elongation was desaturated by the stearoyl-ACP desaturase in step 6, and FAB2 (qA8-1 and qC6-1) and AT5G16240 (qA3-1, qA8-6 and qA10-2) were found. Some important candidate genes were found to be associated with fatty acid elongation and desaturation; for example, FAE1 and CER10 appeared in step 9. FAE1 was associated with QTLs qA1-4 detected by C18:3 and qA8-5 with all 10 traits, and qC3-2 with nine traits but not C18:3. CER10 was assigned to qA4-3, qA5-2, qA10-1 and qC9-6, which were detected with C16:0, FAS, C18:3 and C18:1, respectively.
Triacylglycerol synthesis could be divided into four main steps [38,41]. DHAP could be reduced to G3P catalyzed by GPDH, and GPDHc1 (qA8-1, qC6-1 and qC9-4) and SDP6 (qA3-5, qA5-4, qC3-7 and qC5-1) were revealed in this step. From G3P to PA of steps 13 and 14, fatty acids were sequentially transferred from CoA to positions 1 and 2 of G3P catalyzed by GPAT and LPAT, respectively. GPAT was composed of 10 subunits [38], seven of which except for GPAT3, GPAT8 and GPAT9 were observed in this step. Among them, GPAT1, GPAT2 and GPAT4 were associated with QTL qA8-2; GPAT5 with QTLs qA3-5, qC3-7 and qC5-1; GPAT6 with QTLs qA4-7, qA8-1 and qC6-1; GPAT7 with QTLs qA8-6 and qA10-3; and ATS1 with QTLs qA3-3, qA5-2, qA8-5, qA8-6, qA9-4 and qC9-3 (Additional file 4). LPAT was a main enzyme in step 14 with five subunits [38], and LPAT2, LPAT3, LPAT4 and LPAT5 were associated with four (qA4-2, qA9-2, qA10-1 and qC9-6), two (qA5-2 and qC3-5), one (qA9-6) and four (qA3-6, qA5-3, qC3-8 and qC5-1) QTLs, respectively. LPP3 was associated with QTLs qA3-4 and qC5-1 that catalyze dephosphorylation of PA to release DAG in step 15. Step 16 was the final step of TAG synthesis and TAG1 (qC8-1 and qC9-3), AT3G44830 (qC3-4) and PDA (qA3-1 and qA8-6) were observed in this step. Many genes or QTLs involved in one step indicated that the fatty acid synthesis in B. napus was very complicated.
Besides those genes that had direct activities in catalyzing fatty acid synthesis, some regulators (LEC1, LEC2, FUS3, WRI1 and ABI3) underlying QTL CIs were also found (Figure 5). The interaction analysis indicated that WRI1 directly affected BCCP2 and CAC2, and indirectly affected FAB1 and FATB in early stages of fatty acid synthesis regulation, while WRI1, LEC1/2, FUS3 and ABI3 all indirectly affected FAE1, FAD2 and FAD3, which played important roles in later modifying and transporting processes. LEC1 and LEC2 were underlying the CIs of five QTLs (qA3-3, qA8-4, qA8-6, qA9-6 and qC9-3) and six QTLs (qA3-3, qA5-2, qA8-4, qA8-6, qA9-4 and qC9-3), respectively. Both LEC1 and LEC2 affected nine fatty acid compositions, except for C18:0, in different environments. FUS3 located in the CI of the QTL qC7-1 that was only associated with C18:3. WRI1 located in the CIs of QTLs qA4-3, qA5-2 and qA10-1 was associated with C16:0, FAS and C18:3, respectively. ABI3 was associated with three QTLs (qA5-3, qC3-8 and qC5-1) and affected the levels of C18:3, FAS, C18:1, C18:2 and C20:0 (Figure 5). Moreover, the epistatic effects of major gene–gene interactions were also identified in fatty acid synthesis (Figure 5). For example, the major genes FATB, FAE1, FAD2 and FAD3 were all under the influence of each other and all could affect the gene TAG1 in the final step of TAG synthesis, and then affected the level of different fatty acids (Figure 5). All these findings indicated that the regulatory pathway of fatty acid synthesis was a complex network, controlled by a large number of genes and also affected by gene–gene interactions.
Potential phospholipid synthesis (Figure 5b), and triacylglycerol and fatty acid degradation (Figure 5c), were also predicted in B. napus based on knowledge of acyl-lipid metabolism in Arabidopsis [31]. G3P was converted to DAG3P by sequential acylation reactions in steps 17 and 18, which was regulated by GPAT and LPAT, and the homologous genes involved in the two steps were very similar to the genes in steps 13 and 14, respectively. Then DAG3P was converted to PC or PE by a series of enzyme reactions. In the pathway of triacylglycerol and fatty acid degradation, a number of homologous genes were also found, and the six genes in step 26 were the same as the genes in step 20. Moreover, gene LPI1, which functioned in degradation of TAG to MAG, affected the level of nine traits but not C20:0.
In addition to the 47 QTLs with candidate genes mapped in the CIs, the remaining 25 QTLs also affected the concentrations of one or two fatty acids. QTL qA4-4 on chromosome A4 with additive effect on traits C16:0 and C18:0 might be assigned to step 5 (Figure 5) with potential ketoacyl ACP synthetase activity. QTL qC5-2 might be assigned to step 11 with a similar function to FAD3, as the QTL had a consistent additive effect on traits C18:2 and C18:3. QTL qC9-5 might be a pleiotropic locus with a similar function to FAE1 as it had an additive effect on traits C18:0 and C22:1. The remaining 22 QTLs only had additive effects on one trait. Based on the premise that plastidic synthesis and modification should result in consistent QTL effects on products in any of the individual sets of saturated fatty acids [42], qA7-3, qA7-4 and qC9-1 affected C16:0, and qA1-5 and qA6-2 affected C18:0, which allows us to assign these QTLs tentatively to early stages of synthesis. The other 17 QTLs may be assigned to desaturation and elongase steps, as they affected C18:3, C20:0, C20:1, C22:0, C22:1 and FAS. Generally, it is important to take into account all the preceding synthetic and modification steps where a QTL could be identified for a particular fatty acid [42]; however, assigning a specific enzyme activity to these QTLs is relatively complex.
Discussion
A total of 72 QTLs and a large number of pairs of epistatic loci for 10 fatty acid compositions were identified using QTL analysis. Then 234 homologous genes were mapped in the CIs of 47 QTLs. QTL mapping and interaction analysis of candidate genes enabled the construction of a regulatory pathway that controlled the different concentrations of fatty acids in B. napus.
Single QTL mapping of each fatty acid concentration showed that QTLs on A8 and C3 were highly correlated. QTL qA8-5 on A8 affected concentrations of all 10 types of fatty acids, and explained PV of individual fatty acids ranging from 15.89% for C20:1 to 47.55% for C20:0, and QTL qA8-6 affected seven fatty acids; meanwhile, both QTLs qC3-2 and qC3-3 on C3 affected nine fatty acids but not C18:3. All these results revealed that these QTLs were major QTLs, consistent with previous studies [6,9,10]. For the two QTLs on C3, the alleles from ‘Ningyou 7’ conferred higher levels of C20:0, C20:1, C22:0 and C22:1, but had negative effects on levels of C16:0, C18:0, C18:1 and C18:2. The group C16:0, C18:0, C18:1 and C18:2 were positively correlated with each other, and likewise for the group C20:0, C20:1, C22:0 and C22:1; however, the two groups showed a negative correlation (Table 2). The fact that QTLs clustered in certain linkage groups and controlled different traits with opposite effects, corroborated strongly with the significant positive and negative correlations among the fatty acids analyzed. Besides the two major QTLs on A8 and C3, another 15 QTLs also affected more than one fatty acid. Moreover, the number of QTLs associated with a single fatty acid in the present study ranged from nine (C18:2, C20:0 and C22:1) to 21 (C18:3), and a total of 72 QTLs for all 10 traits detected were more than that from previous studies [6-10], with a number of new QTLs for different fatty acids. No QTLs for C16:0 were found on A4, A7 and C9 previously [6,8,10], suggesting that the 10 QTLs on these chromosomes in the TN population were all new QTLs. The unsaturated fatty acids were synthesized as a result of the fatty acid desaturation pathway, starting from C18:0; however, only a few QTLs for C18:0 were detected in previous studies. Zhao et al. identified two QTLs for C18:0 on A6 and A7 [8], Smooker et al. identified one QTL on A9 [9], and Burns et al. detected four QTLs respectively on A1, A6, A7 and A8 [6]. Herein, 12 QTLs across seven chromosomes were detected, and the QTLs on A4 (one QTL), A10 (two QTLs), C3 (four QTLs) and C9 (two QTLs) were new. QTLs for C18:1 were identified on 14 chromosomes except A4, A6, C2, C5 and C7 [6-10]. In the present study, there were 12 QTLs associated with C18:1 across seven chromosomes, nine of which also affected other fatty acids. QTLs for C18:2 were identified on 15 chromosomes except A4, A10, C5 and C9 [6,8-10], indicating that the two QTLs (qC5-1 and qC5-2) for C18:2 on C5 were new. There were 21 QTLs for C18:3 identified here, and three QTLs (qA2-1, qA2-2 and qA2-3) mapped on A2 and one QTL (qC7-1) on C7 were new QTLs, but the former 18 QTLs were distributed across 14 chromosomes excluding A2, A3, A9, C6 and C7 [6-10]. Many QTLs for C22:1 were identified on 14 chromosomes excluding A4, A5, A7, C1 and C5 [4-6,8-10,43]. Two major QTLs associated with C22:1 on A8 and C3 chromosomes were observed in the present study, which was consistent with previous studies [5,9], and two new QTLs (qA4-1 and qA7-1) were mapped on A4 and A7 chromosomes (Table 3).
Generally, additive effects are considered as the main factors contributing to variations in quantitative traits [44,45]. However, epistatic effects can also play an important role in complex traits in B. napus [46,47]. Epistatic QTLs was found to explain PV ranging from 0.01% for C18:2 to 25.47% for C20:1 (Additional file 6), showing similar trends to previous studies [48]. Moreover, interactions among 395 loci were identified by GMM, including 34 pairs of digenic and 361 pairs of trigenic interactions (Additional file 7). A number of candidate genes were mapped to the CIs of these QTLs. These results showed that epistasis may substantially contribute to variation in oil concentration in different crops.
The regulatory pathway constructed in this study was based on QTL information and the interaction of candidate genes associated with the single and epistatic QTLs of 10 fatty acids, as well as previous knowledge concerning Brassica species and Arabidopsis [31,42]. A large number of important genes were found to be candidate genes for the processes of fatty acid metabolism. Among the 50 genes and five regulatory factors detected, 31 genes (62.0%) affected more than one trait, and FAE1 and ATS1 affected nine (but not C20:0) and 10 traits, respectively. Previous studies found that FAE1 affected oil composition in several steps of seed fatty acid synthesis and modification pathways in B. oleracea [42] and B. napus [30,49]. In B. napus, the orthologs of FAD2 and FAD3 were mapped in different linkage groups of the genome. BnaFAD2 loci were mapped in linkage groups A1, A5, C1 and C5 [28], and BnaFAD2 located in different linkage groups was found to be responsible for the high C18:1 level [29] and erucic acid content [9]. It seemed clear that a major functional variant of FAD2 orthologs in the A genome was located on linkage group A5, but unclear whether FAD2 loci were present in the Brassica C genome [8,29]. The ortholog of FAD3 was identified in the control of C18:3 level in B. napus, and a specific marker of fad3 was located in the CI of the QTL for C18:3 in linkage group A1 [50]. In the present study, FAD2 was located in three QTLs CIs: qA3-5 by C16:0 and C18:1, qC3-7 by C20:0, and qC5-1 by C18:2 and C20:0 using in silico mapping. The homologous genes of FAD3 were mapped in the CIs of seven QTLs: qA3-5 by C16:0 and C18:1; qA5-3 by C18:3 and FAS; qA5-4 by C18:1 and C18:3; qA8-6 by C18:1, C18:2, C20:0, C20:1, C22:0, C22:1 and FAS; qA10-3 by C18:0; qC3-7 by C20:0; and qC5-1 by C18:2 and C20:0. Taken together, these findings increased the understanding of the important genes affecting the processes of fatty acid metabolism in B. napus.
Despite the multiple studies on the function of several important genes in fatty acid biosynthesis [23-26], the regulatory mechanism of the pathway is still not well understood in B. napus. Five regulatory factors (LEC1, LEC2, WRI1, FUS3 and ABI3) were involved in the process concerning the fatty acid biosynthetic pathways, which might correspond to new important findings in this study. Both LEC1 and LEC2 could act as positive regulators upstream of ABI3 and FUS3 in the control of seed maturation [51-53], and several studies revealed that these four regulatory factors might have a function in regulation of fatty acid metabolism [37,51-54]. WRI1, which acted downstream of LEC2, controlled expression of a subset of genes involved in fatty acid biosynthesis [33], and overexpression of WRI1 caused an increased TAG level in both seeds and leaves of Arabidopsis [34]. In this study, each of the five regulatory factors was under the influence of at least two other regulatory factors, and LEC1 and LEC2 were especially affected by all four other regulatory factors (Figures 4 and 5). Moreover, all five regulatory factors were associated with C18:3, but none affected C18:0. Conversion of acetyl-CoA to malonyl-CoA by acetyl carboxylase (ACCase) is one of the most committed steps in fatty acid biosynthesis [39,40], and reduction of ACCase activity caused a decrease of fatty acid content in the range of 1.5–16.0% in transgenic seeds of oilseed crops [55]. Most of the enzymes involved in de novo fatty acid synthesis seemed to be affected by WRI1 [33], and only WRI1 of the five regulatory factors had a direct impact on transcription levels of genes in the plastidial fatty acid synthetic pathway [38]. As a result of the interaction analysis of candidate genes, two genes CAC2 and BCCP2, which encoded subunits of ACCase, were found to be under the influence of WRI1. In the process of fatty acid elongation and desaturation, no candidate genes were shown to be under the influence of the five regulatory factors in this study (Figure 5). Among the targets of the five regulatory factors, LFY might represent a key factor mediating the regulatory action of elongation and desaturation processes, as it was directly affected by all five regulatory factors and also regulated important downstream genes, including FAD2, FAE1 and FATB, and these three genes regulated a series of downstream genes (Figure 4). LFY was arguably the most important floral meristem identity gene, which encoded a plant-specific transcription factor and controlled multiple aspects of floral morphogenesis [56,57]. It was striking that LFY played an important role in the regulatory pathway of fatty acid metabolism, and is worthy of future study.
Overall, these results provided important new insights into the regulatory model for the control of oil synthesis in B. napus and enhanced our understanding of the fatty acid synthesis pathways.
Conclusions
In this study, 72 individual QTLs and a large number pairs of epistatic interactions contributing to fatty acid biosynthesis were identified. By using in silico mapping analysis, 234 homologous genes of A. thaliana that could be involved in fatty acid metabolism within the CIs of 47 QTLs were found. After QTL mapping and candidate gene analysis, a potential regulatory network controlling fatty acid metabolism was revealed in B. napus. Our results provided new insights into the regulatory model for the control of oil synthesis in B. napus.
Methods
Plant materials and field experiments
The segregating DH population (the TN DH population), with 202 DH lines derived from the cross ‘Tapidor’ × ‘Ningyou7’ was constructed by Qiu et al. [5]. The DH lines as well as their parents were grown in six independent environments in China. The population was grown at Wuhan (W1) for one year (September 2007 to May 2008, 07 W1), Huanggang (W2) for two years (September 2008 to May 2009, 08 W2; September 2009 to May 2010, 09 W2) and Qichun (W3) for one year (September 2009 to May 2010, 09 W3) in Hubei Province; and Dali for two years (September 2007 to May 2008, 07D; September 2009 to May 2010, 09D) in Shaanxi Province. Wuhan, Qichun and Huanggang were the experiment bases of Huazhong University of Science and Technology, and Dali was the experiment base of Hybrid Rapeseed Research Center of Shaanxi Province. No specific permissions were required for the field trials. For each field trial, all lines were planted in a randomized complete-block designed with three replicates, and each plot contained 30 plants per genotype [58].
Measurements of fatty acid composition
Bulked seed samples (200 mg) from each of replicates in the three environments (07D, 07 W1 and 08 W2) were analyzed for their fatty acid compositions by gas liquid chromatography according to Rücker and Röbbelen [59]. In three other environments (09D, 09 W2 and 09 W3), fatty acid compositions were determined by near-infrared reflectance spectroscopy (NIRS) using standard methods [60] and all saturated fatty acid compositions in these three environments were considered as one trait named FAS.
Linkage map construction and map alignment with Arabidopsis, B. rapa and B. oleracea
The TN genetic linkage map of B. napus, which was introduced by Jiang et al. was updated in the present study [61]. A total of 932 markers were mapped to the new linkage map generated with the TN DH population using JoinMap_4.0 [62]. Detailed information of TN genetic linkage map was summarized in Additional file 3. Of the 932 linked markers, 429 with known sequence information or corresponding to Arabidopsis genes were used as anchored markers to carry out map alignment between B. napus and Arabidopsis according to the method of Long et al. [21]. If more than three homologous sequence-informative markers in the TN DH population were closely linked within one conserved block of Arabidopsis as described by Schranz et al. [17], a synteny block was considered to exist. If there were only one or two sequence-informative marker(s), it was recognized as an insertion segment. The homologous genes underlying the block or island were considered to be candidate genes if they were just underlying the QTL CI. There were 111 key genes (824 homologous genes) (Additional file 4) involved in fatty acid metabolism of Arabidopsis collected from the TAIR website [63] and in silico mapping in the genetic map as described by Long et al. [21]. The comparative mapping analysis between B. napus and B. rapa/B. oleracea was done using BLAST analysis on the Brassica Database [64] or B. oleracea Database [65]. The sequence of markers in the TN DH linkage map was used as a query sequence. All available sequence-informative markers on B. napus were subjected to BLASTn analysis to identify their physical positions on Arabidopsis, B. rapa and B. oleracea chromosomes. Then the Brassicaceae building blocks that corresponded to each informative marker were identified. The candidate gene(s) in B. napus, B. rapa and B. oleracea for each informative marker were also identified.
QTL and epistasis analysis
QTL analysis was done using the QTLNetwork_2.0 software described by Yang et al. [66,67] and WinQTLCart_2.5 [68], and epistasis QTL mapping analyses were performed using the QTLNetwork_2.0 and GMM [69]. For QTLNetwork_2.0, a mixed linear model was used to identify QTLs at 2-cM intervals with a window size of 10 cM. Two-dimensional (2D) genome scans were used to search for multiple interacting QTLs. For each trait, a genome-wide threshold value of the F-statistic (P = 0.05) for declaring the presence of a QTL was estimated by 1000 random permutations [70]. A Bayesian method with Gibbs sampling was used to estimate QTL effects [71]. The sum of individual PV explained by each QTL was calculated as the total PV explained by all QTLs for each trait. Based on genetic effects, mapped epistatic QTLs comprised three types: interactions between two QTLs with additive effects (AA), interactions between a QTL with additive effect and a locus without significant additive effect (AN or NA), and interactions between two loci within non-QTL (NN) [45]. For WinQTLCart_2.5, composite interval mapping (CIM) was used with a significance threshold of P = 0.05 using the permutation test method [68], based on 1000 runs of randomly shuffling the trait values [72]. QTL CIs were determined by 2-LOD intervals surrounding the QTL peak. QTLs for different traits that mapped to the same region with overlapping CIs were assumed to be the same [58,73]. For GMM, two and three loci interactions were detected with default parameters. For the QTL comparison between different populations, QTLs detected in this study were preliminarily compared with those in other publications, because few common markers were available between TN and other populations. If one or more QTLs were identified on a chromosome in this study but not in previous studies, these were considered as new QTLs.
The genetic interaction analysis candidate genes
The network was constructed and analyzed using Cytoscape_V2.6.3 with the Agilent Literature Search Plug-in [74]. The degree of connectivity, the clustering coefficient, the network density and the diameter were designed according to Toubiana et al. [75]. Search controls option by using the default parameter settings and extraction controls option with appropriate choice and choosing Arabidopsis in concept lexicon, the network was laid out by using group attributes layout.
Construction of potential controlling pathways of fatty acids in B. napus
To identify candidate genes underlying the QTL CIs involved in the fatty acid regulatory pathways, the Kyoto Encyclopedia of Genes and Genomes (KEGG) database was applied to identify pathways in which these genes were involved [76,77]. Potential controlling pathways of fatty acids in B. napus were inferred based on the pathways of fatty acid synthesis within Arabidopsis [31,38] and other plants [42,78] and also using gene interaction analysis and QTL epistasis analysis.
Acknowledgments
The authors are very grateful to Professor Xingwang Deng of Yale University for his critical reading of the manuscript. The work was supported by National Natural Science Foundation of China (31171582, 31071447), the High Technology Program of China (2011AA10A104) and the New Century Talents Support Program of the Ministry of Education of China (NCET110172).
Abbreviations
- ABCAT
ABC acyl transporter
- ACBP
Acyl CoA binding protein
- ACCase
Acetyl-CoA carboxylase
- CI
Confidence interval
- DAG
Diacylglycerol
- DAGAT
Diacylglycerol transacylase
- DGAT
Acyl-CoA,diacylglycerol acyltransferase
- DH
Doubled haploid
- FAE
Fatty acid elongase
- FAS
Fatty acid synthase
- FAT
Fatty ACP thioesterase
- G3P
Glycerol-3-phosphate
- GMM
Genotype Matrix Mapping ver2.1software
- GPAT
Glycerol-3-phosphate acyltransferase
- GPDH
NAD-dependent glycerol-3-phosphate dehydrogenase
- JA
Jasmonate
- KAS
Ketoacyl-ACP synthase
- LACS
Long-chain acyl-CoA synthetase
- LPA (AG3P)
Lysophosphatidic acid
- LPAAT
1-acylglycerol-3-phosphate acyltransferase
- MAG
Monoacylglycerol
- PA (DAG3P)
Phasphatidic acid
- PC
Phosphatidylcholine
- PDAT
Phospholipid,diacylglycerol acyltransferase
- PE
Phosphatidylethanolamine
- PG
Phosphatidylglycerol
- PGP
Phosphatidyglycerophosphate
- PP
Phosphatidate phosphatase
- PV
Phenotypic variance
- QTL
Quantitative trait loci
- SAD
Stearoyl-ACP desaturase
- TAG
Triacylglycerol
Additional files
Single QTL detection by WinQTLCart_2.5 in TN DH population.
Single QTL detection by QTLNetwork_2.0 in TN DH population.
The TN DH genetic linkage map and its syntenic segmental alignment with the Arabidopsis genome.
Positions of homologous genes of Arabidopsis mapped on TN DH linkage map by in silico mapping and orthologous genes between B. napus and B. rapa/B. oleracea .
Demonstration of a complex epistatic network for fatty acids in B. napus .
Epistatic QTLs for fatty acid compositions detected by QTLNetwork_2.0 in TN DH population.
Digenic and trigenic interactions for fatty acid compositions detected by genotype matrix mapping software ver2.1 in TN DH population.
The distribution of 57 candidate genes in 32 different pathways by the KEGG analysis.
Five KEGG pathway maps of candidate genes in Arabidopsis.
Footnotes
Xiaodong Wang, Yan Long and Yongtai Yin contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XW and YL participated in field experiment and the fatty acid compositions measurement, carried out the QTL and epistasis analysis, inferred potential controlling pathway of fatty acids in B. napus and co-wrote the manuscript. YY, LG and CZ participated in the interaction analysis of candidate genes and mapped alignment with A. thaliana, B. rapa and B. oleracea. LL, LY and JM made helpful suggestions to the manuscript. ML designed, led and coordinated the overall study. All authors read and approved the final manuscript.
Contributor Information
Xiaodong Wang, Email: xdwang120@163.com.
Yan Long, Email: longren0807@gmail.com.
Yongtai Yin, Email: yinyongtai@163.com.
Chunyu Zhang, Email: zhchy@mail.hzau.edu.cn.
Lu Gan, Email: ganluhust@gmail.com.
Liezhao Liu, Email: liezhao2003@126.com.
Longjiang Yu, Email: yulongjaing@hust.edu.cn.
Jinling Meng, Email: jmeng@mail.hzau.edu.cn.
Maoteng Li, Email: limaoteng426@mail.hust.edu.cn.
References
- 1.Hoekman B. Global Economic Prospects 2008: Technology Diffusion in the Developing World. Washington, DC: World Bank Publications; 2007. pp. 40–1. [Google Scholar]
- 2.Sovero M. Rapeseed, a new oilseed crop for the United States. New York: Wiley; 1993. pp. 302–7. [Google Scholar]
- 3.Velasco L, Becker HC. Estimating the fatty acid composition of the oil in intact-seed rapeseed (Brassica napus L.) by near-infrared reflectance spectroscopy. Euphytica. 1998;101(2):221–30. doi: 10.1023/A:1018358707847. [DOI] [Google Scholar]
- 4.Ecke W, Uzunova M, Weissleder K. Mapping the genome of rapeseed (Brassica napus L.). II. Localization of genes controlling erucic acid synthesis and seed oil content. Theor Appl Genet. 1995;91(6–7):972–7. doi: 10.1007/BF00223908. [DOI] [PubMed] [Google Scholar]
- 5.Qiu D, Morgan C, Shi J, Long Y, Liu J, Li R, et al. A comparative linkage map of oilseed rape and its use for QTL analysis of seed oil and erucic acid content. Theor Appl Genet. 2006;114(1):67–80. doi: 10.1007/s00122-006-0411-2. [DOI] [PubMed] [Google Scholar]
- 6.Burns MJ, Barnes SR, Bowman JG, Clarke MH, Werner CP, Kearsey MJ. QTL analysis of an intervarietal set of substitution lines in Brassica napus: (i) Seed oil content and fatty acid composition. Heredity. 2003;90(1):39–48. doi: 10.1038/sj.hdy.6800176. [DOI] [PubMed] [Google Scholar]
- 7.Hu X, Sullivan-Gilbert M, Gupta M, Thompson SA. Mapping of the loci controlling oleic and linolenic acid contents and development of fad2 and fad3 allele-specific markers in canola (Brassica napus L.) Theor Appl Genet. 2006;113(3):497–507. doi: 10.1007/s00122-006-0315-1. [DOI] [PubMed] [Google Scholar]
- 8.Zhao J, Dimov Z, Becker HC, Ecke W, Möllers C. Mapping QTL controlling fatty acid composition in a doubled haploid rapeseed population segregating for oil content. Mol Breed. 2008;21(1):115–25. doi: 10.1007/s11032-007-9113-y. [DOI] [Google Scholar]
- 9.Smooker AM, Wells R, Morgan C, Beaudoin F, Cho K, Fraser F, et al. The identification and mapping of candidate genes and QTL involved in the fatty acid desaturation pathway in Brassica napus. Theor Appl Genet. 2011;122(6):1075–90. doi: 10.1007/s00122-010-1512-5. [DOI] [PubMed] [Google Scholar]
- 10.Yan XY, Li JN, Wang R, Jin MY, Chen L, Qian W, et al. Mapping of QTLs controlling content of fatty acid composition in rapeseed (Brassica napus) Genes Genomics. 2011;33(4):365–71. doi: 10.1007/s13258-010-0149-8. [DOI] [Google Scholar]
- 11.Koch MA, Haubold B, Mitchell-Olds T. Comparative evolutionary analysis of chalcone synthase and alcohol dehydrogenase loci in Arabidopsis, Arabis, and related genera (Brassicaceae) Mol Biol Evol. 2000;17(10):1483–98. doi: 10.1093/oxfordjournals.molbev.a026248. [DOI] [PubMed] [Google Scholar]
- 12.Yang YW, Lai KN, Tai PY, Li WH. Rates of nucleotide substitution in angiosperm mitochondrial DNA sequences and dates of divergence between Brassica and other angiosperm lineages. J Mol Evol. 1999;48(5):597–604. doi: 10.1007/PL00006502. [DOI] [PubMed] [Google Scholar]
- 13.Lan TH, DelMonte TA, Reischmann KP, Hyman J, Kowalski SP, McFerson J, et al. An EST-enriched comparative map of Brassica oleracea and Arabidopsis thaliana. Genome Res. 2000;10:776–88. doi: 10.1101/gr.10.6.776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parkin I, Sharpe AG, Lydiate DJ. Patterns of genome duplication within the Brassica napus genome. Genome. 2003;46:291–303. doi: 10.1139/g03-006. [DOI] [PubMed] [Google Scholar]
- 15.Babula D, Kaczmarek M, Barakat A, Delseny M, Quiros CF, Sadowski J. Chromosomal mapping of Brassica oleracea based on ESTs from Arabidopsis thaliana: complexity of the comparative map. Mol Genet Genomics. 2003;268(5):656–65. doi: 10.1007/s00438-002-0782-2. [DOI] [PubMed] [Google Scholar]
- 16.Parkin IA, Gulden SM, Sharpe AG, Lukens L, Trick M, Osborn TC, et al. Segmental structure of the Brassica napus genome based on comparative analysis with Arabidopsis thaliana. Genetics. 2005;171(2):765–81. doi: 10.1534/genetics.105.042093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schranz ME, Lysak MA, Mitchell-Olds T. The ABC’s of comparative genomics in the Brassicaceae: building blocks of crucifer genomes. Trends Plant Sci. 2006;11(11):535–42. doi: 10.1016/j.tplants.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 18.Wang X, Wang H, Wang J, Sun R, Wu J, Liu S, et al. The genome of the mesopolyploid crop species Brassica rapa. Nat Genet. 2011;43(10):1035–9. doi: 10.1038/ng.919. [DOI] [PubMed] [Google Scholar]
- 19.Chalhoub B, Denoeud F, Liu S, Parkin IA, Tang H, Wang X, et al. Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science. 2014;345(6199):950–3. doi: 10.1126/science.1253435. [DOI] [PubMed] [Google Scholar]
- 20.Liu S, Liu Y, Yang X, Tong C, Edwards D, Parkin IA, et al. The Brassica oleracea genome reveals the asymmetrical evolution of polyploid genomes. Nat Commun. 2014;5:3930. doi: 10.1038/ncomms4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Long Y, Shi J, Qiu D, Li R, Zhang C, Wang J, et al. Flowering time quantitative trait loci analysis of oilseed Brassica in multiple environments and genomewide alignment with Arabidopsis. Genetics. 2007;177(4):2433–44. doi: 10.1534/genetics.107.080705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao JY, Huang JX, Chen F, Xu F, Ni XY, Xu HM, et al. Molecular mapping of Arabidopsis thaliana lipid-related orthologous genes in Brassica napus. Theor Appl Genet. 2012;124(2):407–21. doi: 10.1007/s00122-011-1716-3. [DOI] [PubMed] [Google Scholar]
- 23.Kachroo A, Shanklin J, Whittle E, Lapchyk L, Hildebrand D, Kachroo P. The Arabidopsis stearoyl-acyl carrier protein-desaturase family and the contribution of leaf isoforms to oleic acid synthesis. Plant Mol Biol. 2007;63(2):257–71. doi: 10.1007/s11103-006-9086-y. [DOI] [PubMed] [Google Scholar]
- 24.Browse J, McConn M, James DJ, Miquel M. Mutants of Arabidopsis deficient in the synthesis of alpha-linolenate. Biochemical and genetic characterization of the endoplasmic reticulum linoleoyl desaturase. J Biol Chem. 1993;268(22):16345–51. [PubMed] [Google Scholar]
- 25.Miquel M, Browse J. Arabidopsis mutants deficient in polyunsaturated fatty acid synthesis. Biochemical and genetic characterization of a plant oleoyl-phosphatidylcholine desaturase. J Biol Chem. 1992;267(3):1502–09. [PubMed] [Google Scholar]
- 26.Kunst L, Taylor DC, Underhill EW. Fatty acid elongation in developing seeds of Arabidopsis thaliana. Plant Physiol Biochem. 1992;30(4):425–34. [Google Scholar]
- 27.Yang Q, Fan C, Guo Z, Qin J, Wu J, Li Q, et al. Identification of FAD2 and FAD3 genes in Brassica napus genome and development of allele-specific markers for high oleic and low linolenic acid contents. Theor Appl Genet. 2012;125(4):715–29. doi: 10.1007/s00122-012-1863-1. [DOI] [PubMed] [Google Scholar]
- 28.Scheffler JA, Sharpe AG, Schmidt H, Sperling P, Parkin I, Lühs W, et al. Desaturase multigene families of Brassica napus arose through genome duplication. Theor Appl Genet. 1997;94(5):583–91. doi: 10.1007/s001220050454. [DOI] [Google Scholar]
- 29.Schierholt A, Becker HC, Ecke W. Mapping a high oleic acid mutation in winter oilseed rape (Brassica napus L.) Theor Appl Genet. 2000;101(5–6):897–901. doi: 10.1007/s001220051559. [DOI] [Google Scholar]
- 30.Fourmann M, Barret P, Renard M, Pelletier G, Delourme R, Brunel D. The two genes homologous to Arabidopsis FAE1 co-segregate with the two loci governing erucic acid content in Brassica napus. Theor Appl Genet. 1998;96(6–7):852–8. doi: 10.1007/s001220050812. [DOI] [Google Scholar]
- 31.Li-Beisson Y, Shorrosh B, Beisson F, Andersson MX, Arondel V, Bates PD, et al. Acyl-lipid metabolism. The Arabidopsis book/American Society of Plant Biologists. 2010;8:e0133. doi: 10.1199/tab.0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lysak MA, Koch MA, Pecinka A, Schubert I. Chromosome triplication found across the tribe Brassiceae. Genome Res. 2005;15(4):516–25. doi: 10.1101/gr.3531105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baud S, Mendoza MS, To A, Harscoet E, Lepiniec L, Dubreucq B. WRINKLED1 specifies the regulatory action of LEAFY COTYLEDON2 towards fatty acid metabolism during seed maturation in Arabidopsis. Plant J. 2007;50(5):825–38. doi: 10.1111/j.1365-313X.2007.03092.x. [DOI] [PubMed] [Google Scholar]
- 34.Cernac A, Benning C. WRINKLED1 encodes an AP2/EREB domain protein involved in the control of storage compound biosynthesis in Arabidopsis. Plant J. 2004;40(4):575–85. doi: 10.1111/j.1365-313X.2004.02235.x. [DOI] [PubMed] [Google Scholar]
- 35.Stone SL, Kwong LW, Yee KM, Pelletier J, Lepiniec L, Fischer RL, et al. LEAFY COTYLEDON2 encodes a B3 domain transcription factor that induces embryo development. Proc Natl Acad Sci U S A. 2001;98(20):11806–11. doi: 10.1073/pnas.201413498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luerßen H, Kirik V, Herrmann P, Miséra S. FUSCA3 encodes a protein with a conserved VP1/ABI3-like B3 domain which is of functional importance for the regulation of seed maturation in Arabidopsis thaliana. Plant J. 1998;15(6):755–64. doi: 10.1046/j.1365-313X.1998.00259.x. [DOI] [PubMed] [Google Scholar]
- 37.Lotan T, Ohto M, Yee KM, West MA, Lo R, Kwong RW, et al. Arabidopsis LEAFY COTYLEDON1 is sufficient to induce embryo development in vegetative cells. Cell. 1998;93(7):1195–205. doi: 10.1016/S0092-8674(00)81463-4. [DOI] [PubMed] [Google Scholar]
- 38.Beisson F, Koo AJ, Ruuska S, Schwender J, Pollard M, Thelen JJ, et al. Arabidopsis genes involved in acyl lipid metabolism. A 2003 census of the candidates, a study of the distribution of expressed sequence tags in organs, and a web-based database. Plant Physiol. 2003;132(2):681–97. doi: 10.1104/pp.103.022988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sasaki Y, Nagano Y. Plant acetyl-CoA carboxylase: structure, biosynthesis, regulation, and gene manipulation for plant breeding. Biosci Biotechnol Biochem. 2004;68(6):1175–84. doi: 10.1271/bbb.68.1175. [DOI] [PubMed] [Google Scholar]
- 40.Baud S, Guyon V, Kronenberger J, Wuilleme S, Miquel M, Caboche M, et al. Multifunctional acetyl-CoA carboxylase 1 is essential for very long chain fatty acid elongation and embryo development in Arabidopsis. Plant J. 2003;33(1):75–86. doi: 10.1046/j.1365-313X.2003.016010.x. [DOI] [PubMed] [Google Scholar]
- 41.Ohlrogge J, Browse J. Lipid biosynthesis. Plant Cell. 1995;7(7):957–70. doi: 10.1105/tpc.7.7.957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barker GC, Larson TR, Graham IA, Lynn JR, King GJ. Novel insights into seed fatty acid synthesis and modification pathways from genetic diversity and quantitative trait loci analysis of the Brassica C genome. Plant Physiol. 2007;144(4):1827–42. doi: 10.1104/pp.107.096172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jourdren C, Barret P, Horvais R, Foisset N, Delourme R, Renard M. Identification of RAPD markers linked to the loci controlling erucic acid level in rapeseed. Mol Breed. 1996;2(1):61–71. doi: 10.1007/BF00171352. [DOI] [Google Scholar]
- 44.Delourme R, Falentin C, Huteau V, Clouet V, Horvais R, Gandon B, et al. Genetic control of oil content in oilseed rape (Brassica napus L.) Theor Appl Genet. 2006;113(7):1331–45. doi: 10.1007/s00122-006-0386-z. [DOI] [PubMed] [Google Scholar]
- 45.Zhao JY, Becker HC, Zhang DQ, Zhang YF, Ecke W. Oil content in a European × Chinese rapeseed population: QTL with additive and epistatic effects and their genotype-environment interactions. Crop Sci. 2005;45(1):51–9. [Google Scholar]
- 46.Carlborg OR, Haley CS. Epistasis: too often neglected in complex trait studies? Nat Rev Genet. 2004;5(8):618–25. doi: 10.1038/nrg1407. [DOI] [PubMed] [Google Scholar]
- 47.Lark KG, Chase K, Adler F, Mansur LM, Orf JH. Interactions between quantitative trait loci in soybean in which trait variation at one locus is conditional upon a specific allele at another. Proc Natl Acad Sci U S A. 1995;92(10):4656–60. doi: 10.1073/pnas.92.10.4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao J, Becker HC, Zhang D, Zhang Y, Ecke W. Conditional QTL mapping of oil content in rapeseed with respect to protein content and traits related to plant development and grain yield. Theor Appl Genet. 2006;113(1):33–8. doi: 10.1007/s00122-006-0267-5. [DOI] [PubMed] [Google Scholar]
- 49.Barret P, Delourme R, Renard M, Domergue F, Lessire R, Delseny M, et al. A rapeseed FAE1 gene is linked to the E1 locus associated with variation in the content of erucic acid. Theor Appl Genet. 1998;96(2):177–86. doi: 10.1007/s001220050725. [DOI] [Google Scholar]
- 50.Jourdren C, Barret P, Brunel D, Delourme R, Renard M. Specific molecular marker of the genes controlling linolenic acid content in rapeseed. Theor Appl Genet. 1996;93(4):512–8. doi: 10.1007/BF00417942. [DOI] [PubMed] [Google Scholar]
- 51.To A, Valon C, Savino G, Guilleminot J, Devic M, Giraudat J, et al. A network of local and redundant gene regulation governs Arabidopsis seed maturation. Plant Cell. 2006;18(7):1642–51. doi: 10.1105/tpc.105.039925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kagaya Y, Okuda R, Ban A, Toyoshima R, Tsutsumida K, Usui H, et al. Indirect ABA-dependent regulation of seed storage protein genes by FUSCA3 transcription factor in Arabidopsis. Plant Cell Physiol. 2005;46(2):300–11. doi: 10.1093/pcp/pci031. [DOI] [PubMed] [Google Scholar]
- 53.Kroj T, Savino G, Valon C, Giraudat J, Parcy F. Regulation of storage protein gene expression in Arabidopsis. Development. 2003;130(24):6065–73. doi: 10.1242/dev.00814. [DOI] [PubMed] [Google Scholar]
- 54.Mu J, Tan H, Zheng Q, Fu F, Liang Y, Zhang J, et al. LEAFY COTYLEDON1 is a key regulator of fatty acid biosynthesis in Arabidopsis. Plant Physiol. 2008;148(2):1042–54. doi: 10.1104/pp.108.126342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thelen JJ, Ohlrogge JB. Metabolic engineering of fatty acid biosynthesis in plants. Metab Eng. 2002;4(1):12–21. doi: 10.1006/mben.2001.0204. [DOI] [PubMed] [Google Scholar]
- 56.Siriwardana NS, Lamb RS. The poetry of reproduction: the role of LEAFY in Arabidopsis thaliana flower formation. Int J Dev Biol. 2012;56(4):207–21. doi: 10.1387/ijdb.113450ns. [DOI] [PubMed] [Google Scholar]
- 57.Grandi V, Gregis V, Kater MM. Uncovering genetic and molecular interactions among floral meristem identity genes in Arabidopsis thaliana. Plant J. 2012;69(5):881–93. doi: 10.1111/j.1365-313X.2011.04840.x. [DOI] [PubMed] [Google Scholar]
- 58.Shi J, Li R, Qiu D, Jiang C, Long Y, Morgan C, et al. Unraveling the complex trait of crop yield with quantitative trait loci mapping in Brassica napus. Genetics. 2009;182(3):851–61. doi: 10.1534/genetics.109.101642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rücker B, Röbbelen G. Impact of low linolenic acid content on seed yield of winter oilseed rape (Brassica napus L.) Plant Breed. 1996;115(4):226–30. doi: 10.1111/j.1439-0523.1996.tb00908.x. [DOI] [Google Scholar]
- 60.Mika V, Nerusil P, Koprna R, Kucera V. Fast prediction of quality parameters in whole seeds of oilseed rape (Brassica napus) Plant Soil Environ. 2003;49(4):141–5. [Google Scholar]
- 61.Jiang C, Ramchiary N, Ma Y, Jin M, Feng J, Li R, et al. Structural and functional comparative mapping between the Brassica A genomes in allotetraploid Brassica napus and diploid Brassica rapa. Theor Appl Genet. 2011;123(6):927–41. doi: 10.1007/s00122-011-1637-1. [DOI] [PubMed] [Google Scholar]
- 62.Van Ooijen JW. JoinMap 4. Software for the calculation of genetic linkage maps in experimental populations. Wageningen, Netherlands: Kyazma BV; 2006. [Google Scholar]
- 63.The Arabidopsis Information Resource. [http://www.arabidopsis.org/]
- 64.Brassica Database. [http://brassicadb.org/brad/]
- 65.Brassica oleracea Database. [http://www.ocri-genomics.org/bolbase/]
- 66.Yang J, Zhu J, Williams RW. Mapping the genetic architecture of complex traits in experimental populations. Bioinformatics. 2007;23(12):1527–36. doi: 10.1093/bioinformatics/btm143. [DOI] [PubMed] [Google Scholar]
- 67.Yang J, Hu C, Hu H, Yu R, Xia Z, Ye X, et al. QTLNetwork: mapping and visualizing genetic architecture of complex traits in experimental populations. Bioinformatics. 2008;24(5):721–3. doi: 10.1093/bioinformatics/btm494. [DOI] [PubMed] [Google Scholar]
- 68.Wang S, Basten CJ, Zeng ZB. Windows QTL cartographer 2.5. Raleigh, NC: Department of Statistics, North Carolina State University; 2007. [Google Scholar]
- 69.Isobe S, Nakaya A, Tabata S. Genotype matrix mapping: searching for quantitative trait loci interactions in genetic variation in complex traits. DNA Res. 2007;14(5):217–25. doi: 10.1093/dnares/dsm020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Doerge RW, Churchill GA. Permutation tests for multiple loci affecting a quantitative character. Genetics. 1996;142(1):285–94. doi: 10.1093/genetics/142.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang CS, Rutledge JJ, Gianola D. Bayesian analysis of mixed linear models via Gibbs sampling with an application to litter size in Iberian pigs. Genet Sel Evol. 1994;26(2):91–115. doi: 10.1186/1297-9686-26-2-91. [DOI] [Google Scholar]
- 72.Churchill GA, Doerge RW. Empirical threshold values for quantitative trait mapping. Genetics. 1994;138(3):963–71. doi: 10.1093/genetics/138.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Arcade A, Labourdette A, Falque M, Mangin B, Chardon F, Charcosset A, et al. BioMercator: integrating genetic maps and QTL towards discovery of candidate genes. Bioinformatics. 2004;20(14):2324–6. doi: 10.1093/bioinformatics/bth230. [DOI] [PubMed] [Google Scholar]
- 74.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Toubiana D, Semel Y, Tohge T, Beleggia R, Cattivelli L, Rosental L, et al. Metabolic profiling of a mapping population exposes new insights in the regulation of seed metabolism and seed, fruit, and plant relations. PLoS Genet. 2012;8(3):e1002612. doi: 10.1371/journal.pgen.1002612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ogata H, Goto S, Fujibuchi W, Kanehisa M. Computation with the KEGG pathway database. Biosystems. 1998;47(1–2):119–28. doi: 10.1016/S0303-2647(98)00017-3. [DOI] [PubMed] [Google Scholar]
- 77.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rawsthorne S. Carbon flux and fatty acid synthesis in plants. Prog Lipid Res. 2002;41(2):182–96. doi: 10.1016/S0163-7827(01)00023-6. [DOI] [PubMed] [Google Scholar]