

Abstract
A major challenge in neuronal stem cell biology lies in characterization of lineage-specific reprogrammed human neuronal cells, a process that necessitates the use of an assay sensitive to the single-cell level. Single-cell gene profiling can provide definitive evidence regarding the conversion of one cell type into another at a high level of resolution. The protocol we describe employs Fluidigm Biomark dynamic arrays for high-throughput expression profiling from single neuronal cells, assaying up to 96 independent samples with up to 96 qPCR probes (equivalent to 9216 reactions) in a single experiment, which can be completed within 2–3 days. The protocol enables simple and cost-effective profiling of several hundred transcripts from a single cell, and could have numerous utilities.
INTRODUCTION
The human central nervous system (CNS) contains 10 to 100 billion neurons and glia, which mediate cognitive functions and regulate behavior. CNS neurons have been segregated into hundreds of different subytpes depending on their location, connectivity, neurotransmitter identity, passive and active electrophysiological properties and molecular markers. Dysfunction of specific neuronal subtypes and perhaps glia underlies all neuropsychiatric disorders such as Parkinson’s, Alzheimer diseases and autism spectrum disorders (ASD). Autologous de-novo generation of neurons useful for transplantation-based therapy of CNS disorders has been a central aim of regenerative medicine and, thanks to recent advances in stem cell biology, is fast becoming a reality. This development has the capacity to revolutionize understanding and treatment of CNS diseases.
Like diverse neuronal types in the brain, neurons formed through cellular reprogramming are heterogeneous in the acquisition of lineage-specificity. Thus, a major challenge in the neuronal stem cell field is the characterization of lineage-specific reprogrammed human neuronal cells, a process that necessitates the use of an assay sensitive to the single-cell level1. Traditional immunocytochemical analysis is limited both by the availability of specific antibodies and the limited number of proteins that can be assayed per cell. In contrast, single-cell gene profiling can provide definitive evidence regarding the identity of generated neurons, such as their expression of specific lineage markers and neurotransmitter identities. Together with functional analysis, the molecular characterization of single cells is crucial for characterizing the conversion of cells from various sources into neurons. While detection of small numbers of transcripts from single cells has been performed for over 20 years2,3–13, a significant limitation of these analyses has been the ability to comprehensively profile expression of multiple genes from a single cell and the low throughput of comparisons between cells. Furthermore, whole-genome single-cell gene profiling14,15 is still in its infancy and is prohibitively expensive for analysis of multiple cells.
A number of applications that enable high-throughput qPCR (higher than 384 reactions in an experiment) are in advanced development, such as emulsion-based digital PCR (dPCR) technologies (e.g. Quantalife's "Droplet Digital"; Raindance Technologies' "Rainstorm"), that could potentially enable absolute quantification of the number of transcripts in a sample, or microfluidic platforms for parallel analysis of multiple inputs16 (e.g. Stokes Bio's high-throughput diagnostic system). Additionally, direct quantitation of target transcripts can be performed by multiplexed profiling (e.g. Nanostring's "nCounter"). To our knowledge, the only platform currently commercially available for comprehensive analysis of the limiting material found in single cells is Fluidigm's "Biomark" high throughput qPCR chip. Therefore, this protocol is tailored to use of the Fluidigm Biomark system for comprehensive qPCR profiling of single cells. This system utilizes a pressure-regulated microfluidic circuit in order to perform mixing of nanoliter volumes of samples and probes within individual chambers on the microfluidic chip. After loading and mixing is complete, thermal cycling is performed, coupled to imaging of the chip at the end of each cycle17.
Recently, we successfully converted human skin fibroblasts into functional neurons and addressed the challenge of identifying the subtype-specificity of the newly converted neurons, by employing comprehensive qPCR-based single-cell gene profiling18. The protocol we developed uses microfluidic qPCR chips (Fluidigm Biomark), which enable high-throughput qPCR-based parallel analysis of multiple genes from a single cell sample. The system is compatible with commercially-available Taqman probes as well as experimenter-designed primers in conjunction with DNA-binding dyes such as Evagreen. Evagreen-based qPCR is significantly more cost-effective and maintains the capacity for enhanced flexibility and quality assurance. The method we describe is based upon aspiration of single cells into a fine glass pipette, followed by target-specific amplification using a mix of primers to the sequences of interest. Fluidigm BioMark dynamic arrays (48.48 or 96.96) are then utilized for high-throughput qPCR on a large number of independent samples (up to 48/96) across a large number of qPCR probes (up to 48/96), equivalent to 2304 or 9216 independent reactions on a single chip. Utilizing this system, we explored the variation in neuronal gene expression patterns of individual iN cells18. A protocol describing the derivation of iN cells is currently in preparation (Vierbuchen et al., 2011). Similar approaches have also been utilized recently for the investigation of the variability in the acquisition of neuronal phenotypes following microRNA-induced conversion of human fibroblasts19, as well as for addressing the heterogeneity amongst human induced pluripotent stem cells20.
APPLICATIONS OF THE METHOD
The protocol we described here, utilizing microfluidic qPCR for single-cell gene profiling, enables the investigation of up to several hundred transcripts from single neuronal cells. While the version of the protocol described below is specific to cells in culture, further developments of this protocol could be applied to profiling of single cells collected directly from tissue samples10 or following cell sorting3. For example, in the context of investigation of neuronal function in brain slice preparations following standard whole-cell patch-clamp electrophysiology, cytosolic material containing cellular mRNA can be aspirated into the patch pipette. Single-cell gene profiling, using the protocol described here, will enable correlation of gene profiling with cellular function. This approach has been utilized in the past for correlation of gene expression with cellular function, using traditional PCR3,6,21,22, but has been limited by the number of transcripts profiled from a single neuron. As neurons within the brain are very heterogeneous, both in their function and in their incorporation into independent neuronal circuits, an assay enabling comprehensive single-cell profiling could be applied to enabling a higher resolution of experimentation. For example: 1) Experience-dependent plasticity relies on de-novo gene transcription for the encoding of memories23, but only neuronal sub-circuits are involved in encoding the experience24. More so, neurons encoding opposing functions may sit adjacent to each other within the tissue, and when assaying gene expression within a brain region25, this cell- and circuit-specific information is lost. Comprehensive single cell gene profiling will enable association of specific transcripts with specific cells, increasing the resolution of investigation. 2) Many neuronal genes, including ion channels and cell adhesion molecules display complex splicing patterns26,27 which are expected to vary in a cell-type specific manner. The method we describe may significantly simplify analysis of the cell-type specific expression patterns of splice isoforms. Potentially, this could lead to novel insight into mechanisms underlying CNS pathologies; 3) Obviously, applications of this technique can be extended to any context in which comprehensive information on gene expression at the single cell level is of interest. This is true for all branches of stem cell investigation, as well as diverse fields such as development, cancer and microbiology.
CONSIDERATIONS FOR CHOICE OF METHODOLOGY
This method is applicable in situations where parallel analysis of a number of different cellular markers is necessary for characterization of the variation among single cells in a population. Given the limitations of the throughput and availability of reagents for techniques based on immunohistochemistry and in-situ hybridization, single-cell qPCR array is our method of choice. Additional protocols describing single cell PCR have been described elsewhere4,28–31
COMPARISON WITH OTHER METHODS
Expression profiling of single cells can be performed either at the level of RNA or of protein. Obviously, it is more informative to assay protein levels within a cell, since a gap may exist between RNA levels and protein levels. Still, the techniques for parallel analysis of multiple proteins within a single cell, such as immunohistochemistry-based techniques, are extremely limited both by the availability of good antibodies, as well as by the availability of fluorophores enabling multiplexed analysis of a single sample. Recent advances enable parallel analysis of up to a 100 different proteins in a single immune cell32. While this suggests exciting prospects for the future, the wide implementation of the technique may not be trivial.
A number of methods exist for analysis of RNA levels within single cells. One approach is based on in-situ hybridization, which offers high sensitivity and the potential to probe different splicing variant of specific gene. However, the throughput of the readout is very limited, in that only one or two genes can be probed in a single cell. At the other end of the spectrum, single-cell deep sequencing14,15 is a recent development, which enables comprehensive profiling of transcripts from a single cell. However, the technology is still in its infancy and is costly and labor intensive, rendering it impractical for standard lab use at this stage.
In contrast, the use of microfluidic based qPCR chips, utilizing regular qPCR primers (see Box 1) with a DNA-binding dye, is highly cost effective, and enables parallel analysis of a large number of samples across many probes, with relatively low effort. For the collection of cells, while laborious, we utilize manual aspiration of single cells, as this allows the unambiguous collection of specific cells, based on their morphology, to be performed in a minimal volume, while minimally disrupting the complex morphology of the neuronal branches (in contrast to the enzymatic treatment necessary for fluorescence-aided cell sorting).
BOX 1: CHOOSING GOOD PRIMERS.
Excellent PCR primers and simple quality assurance measures are crucial to establish the validity of single-cell qPCR analysis. Optimal PCR primers should provide specific and efficient amplification of the target, minimizing potential contamination33. Our preference is for simple nonspecific detection of qPCR products using fluorescent dyes like EvaGreen. The principle of the reaction is that the binding of the fluorophore to double-stranded DNA causes it to emit a strong fluorescent signal. The result is an increase in fluorescent intensity which is directly proportional to the increase in dsDNA. This type of detection system is the simplest and most economical choice for qPCR but is not selective, due to the potential for nonspecific amplification. Therefore, careful planning of qPCR primers, followed by vigilant quality assurance, is crucial for the acquisition of valid data. Importantly, use of fluorescent dyes to detect double-stranded DNA enables simple quality assurance thanks to the possibility to measure the efficiency of the primer pair (by performing dilution analysis of a reference sample; Box 1a,b) as well as the specificity of the amplification (by addressing the melting curve of the amplicon; Box 1c). Choosing primers: Optimal primers should be 16–30 bases long and exhibit a melting temperature (Tm) of 56°C–68°C (optimally 60°–64°C). The Tm of primers within a primer pair should be within 1°C of each other. Product size should ideally be between 80 and 150 bp. If comparisons are to be made between products, the products should be of similar length. Nucleotide repeats should be avoided as they promote mispriming.
An optimal primer pair for qPCR of mRNA transcripts will be one in which at least one of the primers covers an exon-exon junction, or in which the amplicon contains an intron (intron-spanning), so as to avoid amplification of genomic DNA contaminants. A point often ignored is that of alternative splicing of the target sequence. It is important to note that when planning primers, attention should be given to alternative splice isoforms, and extra care should be made when choosing primers, noting the splice variants that will be identified, and those that will be missed.
In our experience, reliable resources for calculation of primers are software based on Primer334 (such as Roche Probefinder (http://qpcr.probefinder.com/organism.jsp) and NCBI primer-blast (http://www.ncbi.nlm.nih.gov/tools/primer-blast/)). Public repositories for qPCR primers are becoming increasingly prevalent, with popular ones being rtprimerdb (http://www.rtprimerdb.org/) and primerbank (http://pga.mgh.harvard.edu/primerbank/). Detailed, step-by-step descriptions of how to choose primers have been described previously35.
Testing primers:
In order to ensure the quality of the primers, control reactions should be performed, in which the specificity and efficiency of the primers is addressed. In these reactions an abundant reference RNA is utilized (in the case of human iN cells, we utilized a commercial source of human brain RNA). A comprehensive titration of this RNA is prepared (e.g. 8 4-fold dilutions from a concentration of ~1 µg/µl). This dilution curve should be subject to identical treatment to the samples (target-specific pre-amplification, ExoSAP-IT etc’), preferably in duplicate parallel reactions, and analyzed in the same experiment as the single-cell samples. Due to the high-throughput nature of the Fluidigm Biomark PCR chips, it is preferable to include the control reactions in the first experiment in which they are introduced, so that a direct comparison of the results from the control reactions and the experimental cells may be performed. An additional control reaction important to include is the non-template control, which controls for contamination within the solutions used for the reaction.
Primer efficiency is a crucial aspect of qPCR analysis of limiting samples, since reduced efficiency would result in non-linear amplification of the targets, which would render the results uninterpretable. Efficiency is calculated by plotting the results of the titration of the reference RNA (Ct vs. log[dilution]; Figure 1B) within the range of linear amplification. An optimally efficient PCR reaction will result in doubling of the quantity of the target RNA in each amplification cycle. Therefore, efficiency is defined as 100*(10^(−1/slope)/2), such that the curve of the optimal slope is −3.3 (10^(−1/3.3))=2.
The specificity of the amplicon is addressed by visually inspecting the melting-curve of the amplification product (Box 1c). Specific amplification will be demonstrated by a single prominent peak, common to all samples, while off-target amplification should be clearly evident (Box 1c). It should be noted that aside from sequencing the amplicons, there is no way of definitively ensuring that no off-target amplification occurred. Performing such analysis will entail setting up a parallel assay in a traditional PCR tube (since the amplified targets are not accessible from within the Fluidigm Biomark chip). However, a more practical (albeit less definitive) approach to gain additional confidence in the specificity of the reaction may be to analyze a particular gene of interest with multiple primer pairs.
Other options for PCR primers include FRET-based hydrolysis probes (such as Taqman), in which beyond the PCR primer pair, an additional probe is included in the reaction, which improves the specificity of the reaction. These options are currently significantly more expensive than the Evagreen-based approach, and another limitation is that vendors often do not provide the sequence of the PCR primers.
CURRENT LIMITATIONS OF THE PROTOCOL INCLUDE
In its present form, the throughput of the protocol is limited by the laborious collection of individual neurons.
The success of the experiment depends on the quality of the primers (Box 1). Once a primer pair has been confirmed to be adequate in terms of specificity, it provides a tool that can be used repeatedly with confidence.
Due to the minute quantities of starting material, certain transcripts may fall below the detection limit.
Limited to effectively processing 200–300 probes per cell – not applicable for whole-genome expression analysis.
The quantitative nature of the results is not yet clear – can likely be used for qualitative analysis, but further work is necessary to evaluate whether it is truly quantitative (due to biological variability – cell size; processing of single cells for preparation of RNA etc’).
mRNA transcripts do not necessarily correlate with protein expression; there may be a discrepancy between the two.
PROCEDURE
CRITICAL STEP Use RNase free reagents and RNA protective technique throughout the protocol (clean surfaces of working area, wear gloves, avoid mouth pipetting, RNase-free filter tips and tubes etc’)
Cell aspiration TIMING 5 minutes per cell
-
1|
Prepare PCR strip tubes with 5 µl 2XCellsDirect reaction mix per tube. Keep on ice.
-
2|
Mount pipette (with a tip opening between 3 to 5 µm) on micromanipulator connected to tubing to enable transfer of pressure through the pipette.
-
3|
Visually identify the isolated cell of choice under the microscope, use micromanipulator to approach it with positive pressure (applied through a 3 ml syringe) to clear its surroundings and aspirate it into patch pipette, ensuring minimal contamination by surrounding cells or cell debris.
!CAUTION Use minimal pressure to aspirate the cell, so as to raise a minimal amount of fluid into the pipette.
? TROUBLESHOOTING
-
4|
Retract pipette and remove holder with syringe from setup (maintaining connection to syringe). Lower pipette tip into solution in bottom of PCR tube. Apply positive pressure to eject the cell into the solution, while retracting the tip of the pipette out of the solution. Freeze immediately on dry ice.
! CAUTION Use minimal pressure to eject the cell into the solution, so as not to blow the solution out of the tube. Maintaining positive pressure while retracting the pipette is important, so as to minimize capillary action that could drive solution back into the tip of the pipette.
CRITICAL STEP Each micropipette can be used only once for transferring each individual cell into reaction buffer.
CRITICAL STEP It is essential to include a negative control sample for the qPCR analysis, in which no individual cell is picked; instead, transfer external solution equivalent to the carryover volume collected during single-cell transfer into reaction buffer. The negative control will enable detection of contamination in subsequent steps of the procedure.
? TROUBLESHOOTING
# Pause point – cells can be stored for several months at −80C until processing.
Specific Target Amplification TIMING 6 h
-
5|
Prepare primer pairs at 20 µM each primer. Mix primers used for specific-target amplification (up to 100 primer pairs) bringing the final concentration of each primer to 200nM (if using less than 100 primer pairs, dilute accordingly with RNase-free water). Large stock solutions of this primer mix may be made and stored @ −20°C if the same primers are to be used continuously.
Note: if more than 100 genes are to be assayed, higher stock concentrations of primer pairs should be made (above 20 µM).
-
6|Prepare reaction mix.
Component Volume (µl) (per sample) RT Platinum Taq 0.2 Primer mix (200 nM) 2.5 RNase free H2O 1.3 -
7|
Thaw cells from dry ice directly in 50°C bath for 15 seconds, then place on ice.
-
8|Add 4 µl / well of reaction mix and put in thermocycler, pre-programmed to run:
Time Temperature Purpose 15 minutes 50°C Reverse Transcription 2 minutes 95°C Denaturation Cycle 18–20 times: 15 seconds 95°C Denaturation 4 minutes 60°C Annealing and elongation Indefinitely 4°C End -
9|Add 4 µl of EXOSAP-IT and return to PCR machine pre-programmed to perform:
Time Temperature Purpose 15 minutes 37°C Digest primers and dNTPs 15 minutes 80°C Denature enzyme # Pause point – cDNA can be stored indefinitely at −20°C until further processing.
qPCR analysis of samples TIMING 5 h
-
10|Prepare qPCR premix (according to vendor’s protocol):
Component Volume (µl) (per sample) TaqMan Gene Expression Master Mix 2.5 DNA Binding Dye Sample Loading Reagent 0.25 EvaGreen DNA binding dye 0.25 -
11|In prelabeled PCR-strip tubes prepare samples:
Component Volume (µl) (per sample) Reaction Mix 3 EXOSAP-IT treated samples 2 Briefly vortex mix and spin down. Store on ice until loading.
-
12|In separate set of prelabeled PCR strip tubes prepare assays:
Component Volume (µl) (per sample) Assay Loading Reagent 2.5 qPCR primer pairs (20 µM) 2.5 Briefly vortex mix and spin down. Store on ice until loading.
-
13|
Prime Fluidigm Biomark qPCR chip on IFC controller according to vendor’s instructions. (~15 min)
! CAUTION Chip must be loaded within an hour of priming so as to reduce loss of pressure within the chip.
? TROUBLESHOOTING
-
14|
Pipette 5 µL of each assay and 5 µL of each sample into their respective inlets on the chip.
▲ CRITICAL STEP Use caution when pipetting the samples as bubbles can be introduced. A fresh sterile tip can be used to remove or pop bubbles by partial aspiration of the bubble, while pulling it out of the well. Care should be taken to avoid cross-contamination or loss of reaction volume.
? TROUBLESHOOTING
-
15|
Load mix onto Fluidigm Biomark qPCR chip in IFC controller according to manufacturor’s protocol.
? TROUBLESHOOTING
-
16|
Remove dust particles from surface of chip by applying scotch tape to the surface, smoothening on to surface of chip, and removing carefully, ensuring that the surface of the chip is completely clean.
! CAUTION Do not blow dust particles away as this could introduce contaminations. Only remove with scotch tape.
-
17|Load chip onto Biomark and initiate qPCR reaction; Include melting curve analysis.
Time Temperature Purpose 2 minutes 50°C Thermal mixing 30 minutes 70°C Thermal mixing 10 minutes 25°C Thermal mixing 2 minutes 50°C Uracil-N-glycosylase activation 10 minutes 95°C Hotstart for Taq polymerase Cycle 35 times 15 seconds 95°C Denaturation 1 minute 60°C Annealing and elongation Ramp 60°C to 95°C Melting curve ? TROUBLESHOOTING
-
18|
Initialize chip scanning. Allow initial ROX scanning to proceed. Open ROX scan files in appropriate software. Scans should demonstrate equivalent ROX loading in all wells. Dust particles will show up as streaks on top of the ROX signal and should be removed by repeating step 17. If no problems appear, proceed with qPCR reaction.
Data analysis and visualization TIMING 4h
-
19|
Analyze data utilizing vendor software. Crucial aspects to pay attention to: compare melting curves from target amplification to melting curves obtained from reference reactions used for dilution curve analysis (see primer choice box).
? TROUBLESHOOTING
-
20|
Data visualization. Vendor software enables heatmap visualization of results, as well as export to spreadsheets for further analysis. Our preference is to visualize the raw data, utilizing the Heatmapviewer component of the Genepattern package available from the Broad Institute (http://genepattern.broadinstitute.org).
ANTICIPATED RESULTS
The Biomark system is equipped with user-friendly qPCR analysis software, in which results may be presented either as a heatmap or spreadsheet, and all the key parameters of the reaction such as the threshold cycles and melting tempature are listed. Due to the high-throughput nature of the assay, we routinely include a number of internal controls, such as Actin, GAPDH and MAP2, as well as neuron-specific reference genes, such as TUBB3, NCAM, and Synapsin 1, with the expectation that essentially the whole population of neuronal cells assayed will express these reference genes. The signal obtained from the internal controls enables quality assurance for multiple steps in the process, from the aspiration of single cells, through reverse-transcription, specific target amplification and qPCR. Furthermore, a non-template control should be routinely included in order to identify potential sources of contamination. Identification of cellular transformation to a specific neuronal subtype is made possible by use of neuron subtype-specific probes (Figure 2). If the protocol is followed carefully, a success rate of close to 100% (as evaluated based on amplification of reference genes in the single cells) is expected. While we have routinely performed 2–3 replicates per reaction (either loading the samples or the probes in replicates) and have essentially always observed consistent results, we recommend performing the assay in duplicate, unless there are experimental limitations.
FIGURE 2.
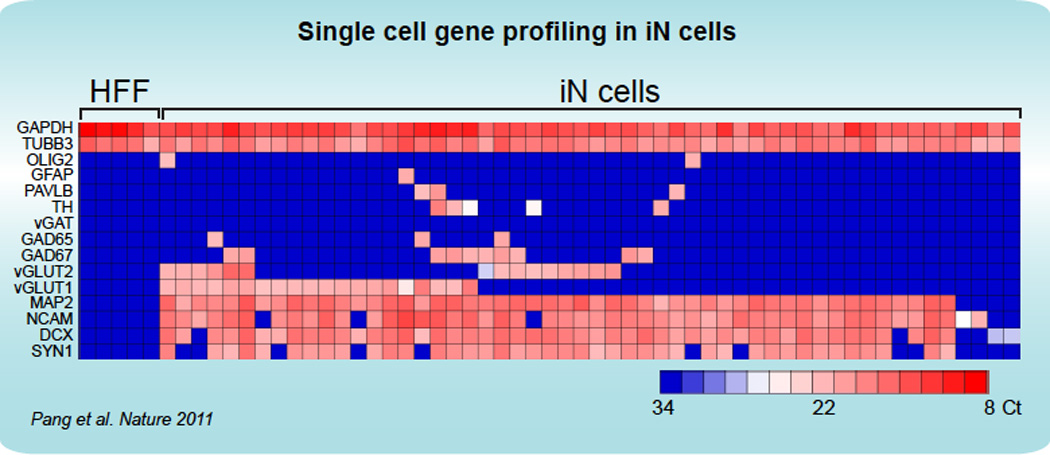
Single-cell gene expression profiling using Fluidigm dynamic arrays. Rows represent the evaluated genes and columns represent individual cells. Heat map (blue to red) represents the threshold Ct values as indicated. HFF = Human Fetal Fibroblast; iN = inducible Neuron. (from Pang et al., Nature 2011)
TIMING
Day 1
Steps 1–4, single cell aspiration. 5 minutes a cell; 6 h for ~50 cells.
Day 2
Steps 5–9, Specific Target Amplification, 6 h
Steps 10–18, qPCR analysis of samples, 5 h
Day 3
Steps 19–20, Data analysis and visualization, 4h
TROUBLESHOOTING
| Step | Problem | Possible reasons | Solution |
|---|---|---|---|
| 3 14 |
No amplification detected for a single sample. |
|
Do it again! |
| 3 | Results appear contaminated (e.g. neurons appear to be contaminated by cells from another source) | Additional material was aspirated, contaminating the reaction. | Caution in collecting sample. |
| 4 | Amplification in no-template control | Solutions may be contaminated. | Study melting curves carefully to determine extent of contamination. Possibly need to rerun reaction with new reagents. |
| 14 | Bubbles forming in sample/assay inlets | Chip does not maintain pressure. |
|
| 14 | Many bubbles in sample / assay wells | Bubbles are introduced during pipetting. | |
| 13–16 | No reaction detected in any sample |
|
|
| 13–16 | ROX passive reference is uneven | Problem in loading the chip | Make sure to load chip within 1 hr of priming and ensure no bubbles are formed while loading chip. |
| 19 | Multiple peaks for single primer pair in melting curves |
|
|
| 19 | Multiple peaks in multiple primer pair melting curves | EXOSAP-IT did not fully digest primers. | Ensure proper mixing prior to incubation for EXOSAP-IT reaction. If problem persists, repeat reaction with new EXOSAP-IT reagent. |
| 19 | No amplification detected for a single primer pair | Primers are not optimal. | Redesign primer pair. |
MATERIALS
REAGENTS
BioMark 48.48 or 96.96 Dynamic-Array (Fluidigm, Cat. No. BMK-M-48.48; BMK-M-96.96)
Gene expression 48.48 or 96.96 Dynamic Array Sample & Loading Reagent Kit (Fluidigm, Cat. No.85000800; Cat. No. 85000802)
CellsDirect reaction mix (Applied Biosystems, Cat. No. 11753-100)
TaqMan Gene Expression Master Mix (Applied Biosystems, Cat. No. 4369016)
Preamp Master Mix (Applied Biosystems, Cat. No. 4384267)
DNA Binding Dye Sample Loading Reagent (Fluidigm, Cat. No. 100-0388)
EvaGreen DNA binding dye (Biotium, Cat. No. 31000)
Assay Loading Reagent (Fluidigm, Cat. No. 85000736)
Low EDTA (0.1 mM EDTA) TE Buffer (Teknova, Cat. No. T0227
ExoSAP-IT (Affymetrix USB, Cat. No.78201)
Sodium chloride (NaCl; Sigma-Aldrich, Cat. No. S3014)
Potassium chloride (KCl; Merck, Cat. No. 104938)
Calcium chloride, dihydrate (CaCl2, 2H2O; Merck, Cat. No. 208291)
Magnesium chloride, hexahydrate (MgCl2, 6H2O; AppliChem, Cat. No. A3618)
HEPES (Sigma-Aldrich, Cat. No.H3375)
D-(+)-Glucose (Sigma-Aldrich, Cat. No. G8270)
Potassium D-gluconate (Sigma-Aldrich, Cat. No. G4500)
Ethylene glycol-bis (2-aminoethylether)-N,N,N´,N´-tetraacetic acid (EGTA; Sigma-Aldrich, Cat. No.. E3889).
Scotch tape
REAGENT SETUP
Extracellular solution
The bath solution contained (in mM): NaCl 140, KCl 5, CaCl2 2, MgCl2 2, HEPES 10, and glucose 10, pH 7.4. The final osmolarity of the solution should be 310 ± 10 mOsm.
CRITICAL Store the freshly prepared solution at 4 °C for up to 1 week.
Primer and sample organization Due to the relatively high-throughput nature of the protocol (analyzing up to 96 samples across 96 probes), the organization of the components is extremely important. Our preference is to use tubes of different colors for the samples and the probes, organize them consistently (e.g. strips of 8, organized on 96 well plates, and labeled with a simple code (e.g. numbers for probes and letters for samples).
Glass pipette preparation
Glass pipette was prepared using PC10 two stage puller. By adjusting the heating temperature of the two steps, the tip of the glass pipette will come to ~2–3 µm diameter (~0.5 MΩ).
EQUIPMENT
!CAUTION All equipment should be treated as potentially contaminated with RNAse.
Inverted tissue culture microscope with phase contrast and epifluorescence equipped with ×10 and ×40 objectives (Axiovert 40 CFL, Zeiss) (Olympus BX51W upright microscope (Olympus); ×4 Objective (Olympus XL Fluoro ×4/340 objective, 0.28 NA, Olympus)
TMC 65–560 anti-vibration table (Technical Manufacturing Corporation)
Glass micropipettes (Inner diameter 1.0 ± 0.05 mm; outer diameter 1.5 ± 0.05 mm; Garner Glass Company, cat. no. KG33)
PC-10 Glass Microelectrode Puller (Narishige)
PCR tube strips (USA scientific)
Tubing
10 ml syringe (BD Bioscience)
Micromanipulator (MPC-385, Sutter)
Fluidigm Biomark system
Fluidigm IFC controller.
FIGURE 1.
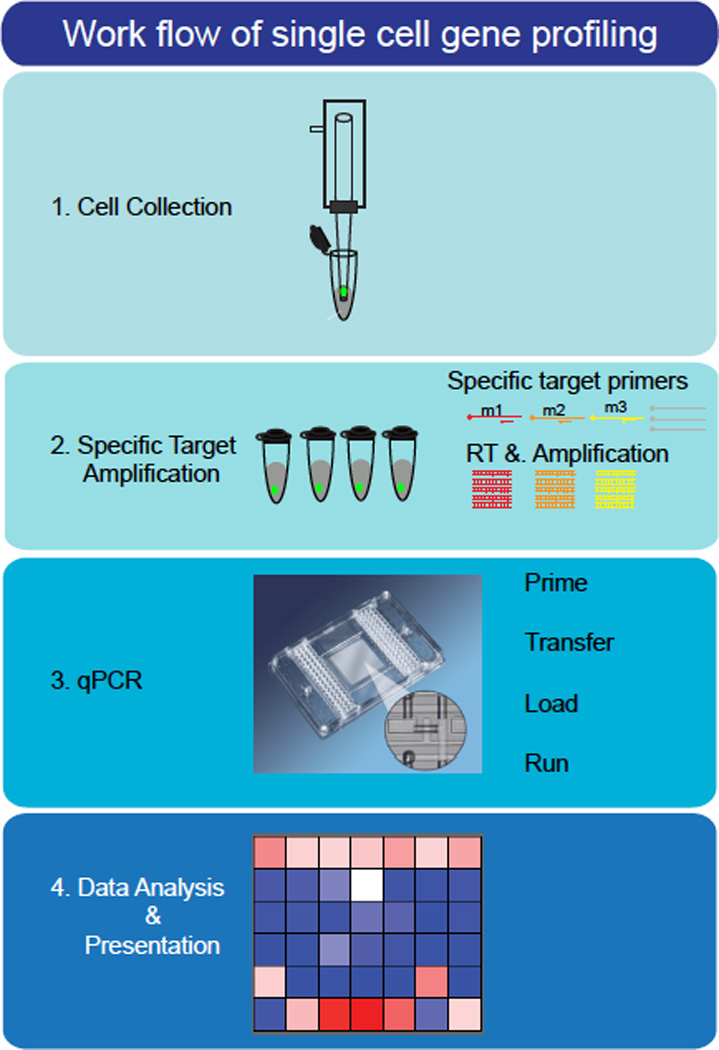
A schematic workflow for single cell gene profiling. The procedure for single cell gene profiling consists of four major steps: 1. Collecting cells (for detailed explanation see Box 2); 2. Specific target amplifications (STA) using a mix of gene-specific primer pairs. M1 to M3 represent genes of interest, gray mRNAs represent other genes. Target RNAs are subject to reverse transcription and amplification; 3. Quantitative PCR, performed on the Fluidigm Biomark system. Samples are loaded on one side of the array, while probes are loaded on the opposing side, with mixing occurring only in the appropriate wells, allowing high-throughput parallel analysis of multiple samples across multiple probes; 4. Data analysis and presentation. At this point, quality assurance is performed to ensure the specificity and reliability of the results, followed by data analysis and presentation (potentially in the form of a heatmap).
BOX FIGURE 1.
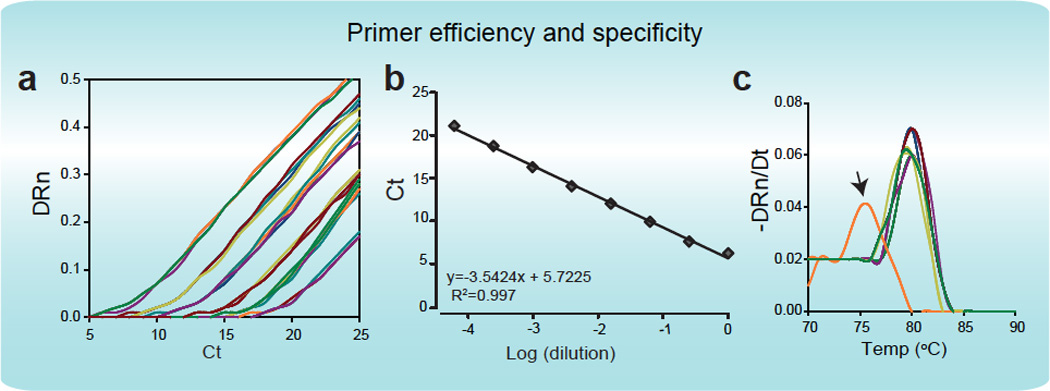
Assessment of primer efficiency and specificity. a, An example of amplification curves using Fluidigm IFC arrays. 6 4-fold sequential dilutions of total human brain RNA are shown, probed for expression of vGLUT2. b, The efficiency of the primer pair for vGLUT2 was assessed by plotting the cycle threshold value (Ct) at each concentration against the logarithm of the fold dilution of the sample. The slope of a linear-regression trendline is indicative of primer efficiency, as defined in the text. c, An example of melting curve analysis, demonstrating the specific amplification of the target gene in the majority of the single cell samples tested, as well as a non-specific amplification observed in a single sample (indicated by an arrow).
BOX FIGURE 2.
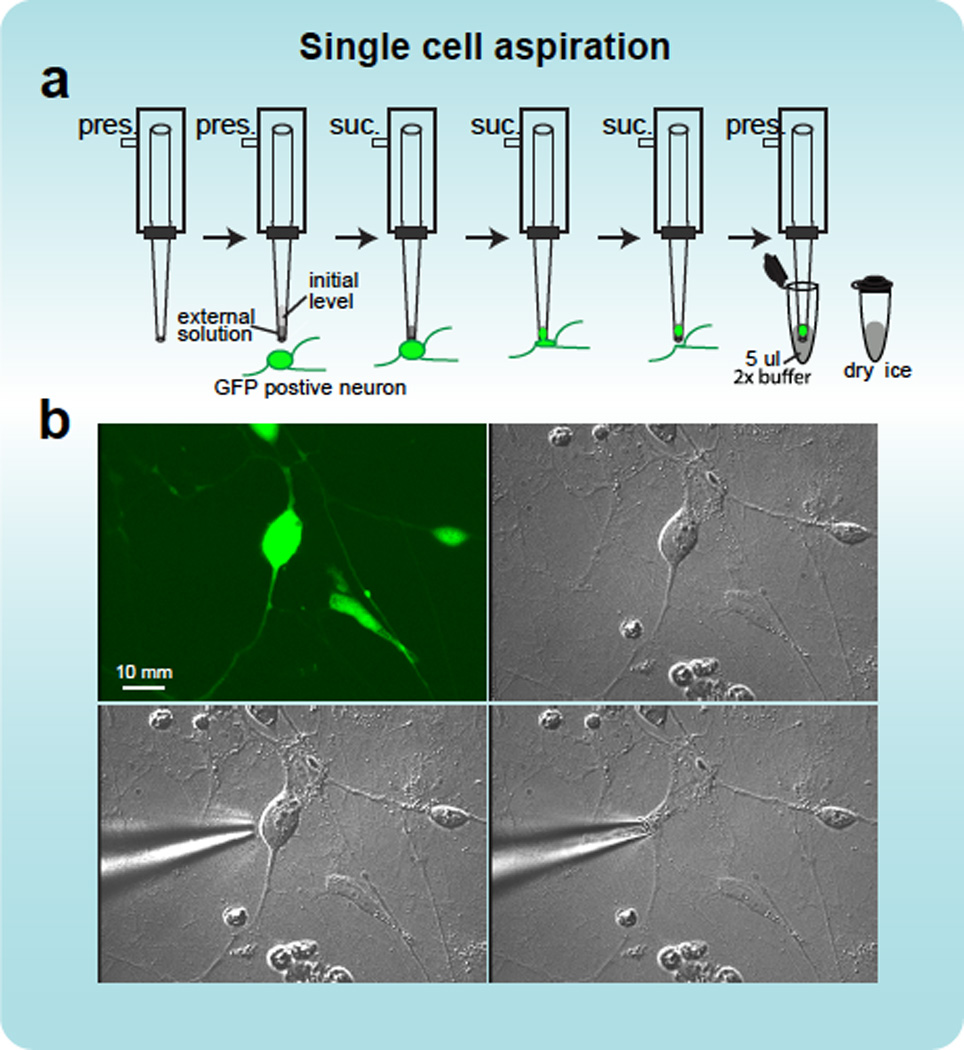
Aspiration of single cells. a, A schematic illustration of the method for collection single cells using a patch pipette. Initially, a patch pipette with a tip opening of 2 to 3 µm (~0.5 Mohm resistance) is fixed to the pipette holder, which is attached through plastic tubing to a 3 ml syringe. Slight positive pressure (pres.) is applied through the syringe while inserting the pipette into the media on the dish containing the target neuronal cells. Target cells are identified by their GFP expression and neuronal morphology, as assessed by differential-interference contrast (DIC) imaging. The positive pressure driven through the pipette while it is lowered into the solution reduces the influx of liquid into the pipette due to capillary action. Under visual direction through the microscope, a micromanipulator is used to position the pipette close to the cell body. Once the tip touches the cell, the cell will normally adhere to the tip, at this point the pressure is relieved, and slight suction (suc.) applied, enabling gradual aspiration of the cell into the pipette tip. The pipette is then retracted, and while positioning it in a pre-prepared PCR tube with 5 µl reaction buffer, the syringe is used to apply positive pressure to expel the cell into the solution in the tube. b, Fluorescent and DIC image of the collection of an iN cell as described.
Box 2: Micromanipulator assisted manual collection of single neuron.
Neurons are highly polarized cells, characterized by lengthy axonal projections and dendritic arbors. These morphological features cause the collecting of single neurons to be more challenging than that of other cell types such as lymphocytes. Our chosen methodology utilizes the equipment available in a standard electrophysiology laboratory, for manual selection and aspiration of target neuronal cells in culture. Thus, standard patch-clamp setups equipped with a fluorescent microscope and manipulator, are used for targeted collection of the soma of a single neurons, minimizing the solution volume and the potential for contamination by other cell types. Furthermore, this setup potentially enables the analysis of the morphological and electrophysiological features of the cells parallel to their transcriptional profiling. In this box, we provide a schematic illustration of the methodology for collection of single neuronal cells.
ACKNOWLEDGEMENTS
A.C. acknowledges the generous support of the AXA Research Fund. Z.P.P. is supported by Brain and Behavior Research Foundation (NARSAD Young Investigator Award). We thank Dr. Nan Yang for help in experimental work. We thank Dr. Shawn Chavez for initial instruction in the use of the Fluidigm Biomark system, as well as Dr. Renee Reijo Pera for access to the Fluidigm Biomark system in her lab. We would like to thank members of the Malenka and Sudhof labs for their comments on the manuscript.
Footnotes
Author contributions
A.C. and Z.P.P. conceived the protocol, collected the data, presented the figures, and wrote the manuscript. T.C.S., M.W. and R.C. M supervised the project and contributed to the writing.
REFERENCES
- 1.Dolmetsch R, Geschwind DH. The Human Brain in a Dish: The Promise of iPSC-Derived Neurons. Cell. 2011;145(6):831. doi: 10.1016/j.cell.2011.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li HH, et al. Amplification and analysis of DNA sequences in single human sperm and diploid cells. Nature. 1988;335(6189):414. doi: 10.1038/335414a0. [DOI] [PubMed] [Google Scholar]
- 3.Cauli B, et al. Molecular and physiological diversity of cortical nonpyramidal cells. J Neurosci. 1997;17(10):3894. doi: 10.1523/JNEUROSCI.17-10-03894.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cauli Bruno, Lambolez Bertrand. Unravelling Single Cell Genomics. The Royal Society of Chemistry; 2010. p. 81. [Google Scholar]
- 5.Koirala S, Corfas G. Identification of novel glial genes by single-cell transcriptional profiling of Bergmann glial cells from mouse cerebellum. PLoS One. 2011;5(2):e9198. doi: 10.1371/journal.pone.0009198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lambolez B, et al. AMPA receptor subunits expressed by single Purkinje cells. Neuron. 1992;9(2):247. doi: 10.1016/0896-6273(92)90164-9. [DOI] [PubMed] [Google Scholar]
- 7.Surmeier DJ, et al. Dopamine receptor subtypes colocalize in rat striatonigral neurons. Proc Natl Acad Sci U S A. 1992;89(21):10178. doi: 10.1073/pnas.89.21.10178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tietjen I, Rihel J, Dulac CG. Single-cell transcriptional profiles and spatial patterning of the mammalian olfactory epithelium. Int J Dev Biol. 2005;49(2–3):201. doi: 10.1387/ijdb.041939it. [DOI] [PubMed] [Google Scholar]
- 9.Tietjen I, et al. Single-cell transcriptional analysis of neuronal progenitors. Neuron. 2003;38(2):161. doi: 10.1016/s0896-6273(03)00229-0. [DOI] [PubMed] [Google Scholar]
- 10.Mackler SA, Brooks BP, Eberwine JH. Stimulus-induced coordinate changes in mRNA abundance in single postsynaptic hippocampal CA1 neurons. Neuron. 1992;9(3):539. doi: 10.1016/0896-6273(92)90191-f. [DOI] [PubMed] [Google Scholar]
- 11.Mackler SA, Eberwine JH. Diversity of glutamate receptor subunit mRNA expression within live hippocampal CA1 neurons. Mol Pharmacol. 1993;44(2):308. [PubMed] [Google Scholar]
- 12.Ginsberg SD, et al. Single-cell gene expression analysis: implications for neurodegenerative and neuropsychiatric disorders. Neurochem Res. 2004;29(6):1053. doi: 10.1023/b:nere.0000023593.77052.f7. [DOI] [PubMed] [Google Scholar]
- 13.Sucher NJ, Deitcher DL. PCR and patch-clamp analysis of single neurons. Neuron. 1995;14(6):1095. doi: 10.1016/0896-6273(95)90257-0. [DOI] [PubMed] [Google Scholar]
- 14.Lao KQ, et al. mRNA-sequencing whole transcriptome analysis of a single cell on the SOLiD system. J Biomol Tech. 2009;20(5):266. [PMC free article] [PubMed] [Google Scholar]
- 15.Tang F, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods. 2009;6(5):377. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- 16.White AK, et al. High-throughput microfluidic single-cell RT-qPCR. Proc Natl Acad Sci U S A. 2011 doi: 10.1073/pnas.1019446108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Spurgeon SL, Jones RC, Ramakrishnan R. High throughput gene expression measurement with real time PCR in a microfluidic dynamic array. PLoS One. 2008;3(2):e1662. doi: 10.1371/journal.pone.0001662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pang ZP, et al. Induction of human neuronal cells by defined transcription factors. Nature. 2011;476(7359):220. doi: 10.1038/nature10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoo AS, et al. MicroRNA-mediated conversion of human fibroblasts to neurons. Nature. 2011;476(7359):228. doi: 10.1038/nature10323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Narsinh KH, et al. Single cell transcriptional profiling reveals heterogeneity of human induced pluripotent stem cells. J Clin Invest. 2011;121(3):1217. doi: 10.1172/JCI44635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liss B, Roeper J. Correlating function and gene expression of individual basal ganglia neurons. Trends Neurosci. 2004;27(8):475. doi: 10.1016/j.tins.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 22.Weng JY, Lin YC, Lien CC. Cell type-specific expression of acid-sensing ion channels in hippocampal interneurons. J Neurosci. 2010;30(19):6548. doi: 10.1523/JNEUROSCI.0582-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McClung CA, Nestler EJ. Neuroplasticity mediated by altered gene expression. Neuropsychopharmacology. 2008;33(1):3. doi: 10.1038/sj.npp.1301544. [DOI] [PubMed] [Google Scholar]
- 24.Lammel S, Ion DI, Roeper J, Malenka RC. Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron. 2011;70(5):855. doi: 10.1016/j.neuron.2011.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lobo MK, et al. Cell type-specific loss of BDNF signaling mimics optogenetic control of cocaine reward. Science. 2011;330(6002):385. doi: 10.1126/science.1188472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Allen SE, Darnell RB, Lipscombe D. The neuronal splicing factor Nova controls alternative splicing in N-type and P-type CaV2 calcium channels. Channels (Austin) 2010;4(6):483. doi: 10.4161/chan.4.6.12868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bharadwaj R, Kolodkin AL. Descrambling Dscam diversity. Cell. 2006;125(3):421. doi: 10.1016/j.cell.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 28.Hodne K, Haug TM, Weltzien FA. Single-cell qPCR on dispersed primary pituitary cells - an optimized protocol. BMC Mol Biol. 2010;11:82. doi: 10.1186/1471-2199-11-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris J, Singh JM, Eberwine JH. Transcriptome analysis of single cells. J Vis Exp. 2011;(50) doi: 10.3791/2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Esumi S, Kaneko R, Kawamura Y, Yagi T. Split single-cell RT-PCR analysis of Purkinje cells. Nat Protoc. 2006;1(4):2143. doi: 10.1038/nprot.2006.343. [DOI] [PubMed] [Google Scholar]
- 31.Li Y, et al. An improved one-tube RT-PCR protocol for analyzing single-cell gene expression in individual mammalian cells. Anal Bioanal Chem. 2010;397(5):1853. doi: 10.1007/s00216-010-3754-0. [DOI] [PubMed] [Google Scholar]
- 32.Bendall SC, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332(6030):687. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bustin SA, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55(4):611. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 34.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 35.Thornton B, Basu C. Real-time PCR (qPCR) primer design using free online software. Biochem Mol Biol Educ. 2011;39(2):145. doi: 10.1002/bmb.20461. [DOI] [PubMed] [Google Scholar]