

Abstract
Amyloidosis is an uncommon disease that is characterized by abnormal extracellular deposition of misfolded protein fibrils leading to organ dysfunction. The deposited proteins display common chemical and histologic properties but can vary dramatically in their origin. Kidney disease is a common manifestation in patients with systemic amyloidosis with a number of amyloidogenic proteins discovered in kidney biopsy specimens. The emergence of mass spectrometry-based proteomics has added to the diagnostic accuracy and overall understanding of amyloidosis. This in-depth review discusses the general histopathologic features of renal amyloidosis and includes an in-depth discussion of specific forms of amyloid affecting the kidney.
Keywords: AA, AL, ALECT2, amyloidosis, hereditary amyloidosis
Introduction
Amyloidosis represents a family of disorders defined by the extracellular deposition of protein fibrils with a characteristic β-pleated sheet conformation. Kidney disease is a common manifestation and a major contributor to morbidity in these patients. Rudolph Virchow first used the term amyloid in 1854 [1], and in 1971, monoclonal light chain became the first chemically characterized amyloid protein [2]. Mass spectrometry-based proteomics has greatly increased the ability to diagnose and type amyloid, and to date, up to 30 amyloidogenic proteins have been identified [1,3,4]. Although many proteins have been implicated in amyloidosis, amyloid fibrils all share common chemical and ultrastructural properties including organization into insoluble beta-pleated sheets identified by X-ray crystallography [5, 6]. This review will focus on the renal manifestations of amyloidosis including pathophysiologic mechanisms, histologic features, protein identification and morphologic and clinical characteristics of common and rare forms of amyloidosis.
Histologic diagnosis and identification of amyloid
The histologic features of amyloid are the same regardless of its composition. On H&E-stained tissue sections, amyloid is identified as extracellular amorphous material that is lightly eosinophilic. These deposits often stain weakly for periodic acid Schiff (PAS), demonstrate a blue-to-gray hue on the trichrome stain and are typically negative on the Jones methenamine silver (JMS) stain (Figure 1). These tinctorial properties contrast with the histologic appearance of collagen, a major component of basement membranes, mesangial matrix and areas of sclerosis, which demonstrates strong positivity for PAS and JMS. All types of amyloid show affinity for Congo red dye, demonstrating an orange-red appearance by light microscopy and characteristic apple-green birefringence upon polarization (Figure 2). For optimal results, the tissue sections for the Congo red stain need to be cut thicker (>5 μm) than the 2- to 4-μm sections used for routine light microscopy stains. Thioflavin T also binds beta-sheet-rich structures but is less specific than Congo red [3]. By electron microscopy, amyloid deposits are composed of haphazardly oriented non-branching fibrils ranging from 7 to 10 nm in diameter.
Fig. 1.

Amyloid typically shows a lightly eosinophilic appearance upon staining with hematoxylin and eosin (A), weak staining with PAS (B), negative silver staining (C) and demonstrates a blue-gray hue on the trichrome stain (D).
Fig. 2.
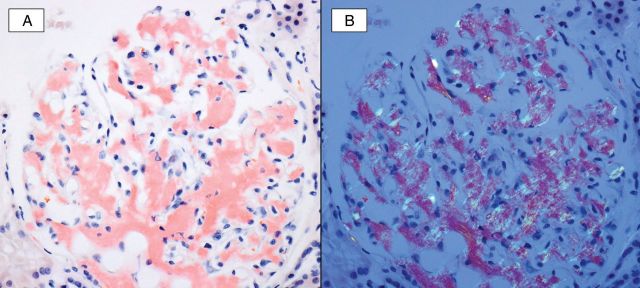
A Congo red study (A) highlights global mesangial and segmental capillary wall staining of amorphous deposits. Upon polarization (B) the Congophilic material shows characteristic apple-green birefringence, confirming the presence of amyloid.
In the kidney, amyloid deposits may be found in any of the parenchymal compartments, including glomeruli, tubules, interstitium and/or vessels. Glomeruli are most commonly involved, with 97% of cases in a study showing glomerular deposition [7]. In most instances, amyloid accumulation involves the mesangium before the capillary walls. In early cases, the process can be subtle and involves only a few mesangial regions and therefore can easily be missed by routine histologic evaluation. More extensive involvement results in marked expansion of the mesangium, which can take on a nodular appearance and mimic mesangial sclerotic processes such as diabetic glomerulosclerosis. However, the often negative staining with PAS and JMS is more typical of amyloid. Occasionally, subepithelial or intramembranous deposits of amyloid, composed of fibrils arranged perpendicular to the glomerular basement membrane, may show widely spaced subepithelial spikes (so-called amyloid spicules) (Figure 3). These spicules are silver positive and tend to be more focal and longer than the ‘spikes’ seen in membranous nephropathy. Progression of amyloid deposition eventually causes complete effacement of the glomerular architecture. Glomerular involvement is frequently accompanied by deposits in the tubulointerstitium and/or renal vessels, particularly interlobular arteries and arterioles. The localization of the amyloid affects the clinical presentation. In cases with primarily glomerular involvement, proteinuria often dominates whereas kidney insufficiency may be the primary manifestation when tubulointerstitial or vascular deposits are more abundant. In rare cases, renal medullary deposits are substantial and result in urine concentrating defects with polyuria and nocturia. As is often the case, amyloid deposits may be seen in multiple compartments with a mixed clinical picture.
Fig. 3.

Focal subepithelial spicules are seen at high power (A, JMS). Ultrastructural examination (B) reveals abundant amyloid fibrils, which are densely packed within the spicules (small image).
Although all forms of amyloidosis share common histologic and chemical properties, the pathogenesis and treatment strategy can vary significantly depending on the precursor protein. Typing can often be performed by routine immunofluorescence or immunohistochemical stains in the more common forms of amyloidosis. Immunofluorescence staining for immunoglobulins (Igs) and light chains is routinely performed on kidney biopsy specimens and in most cases can identify monotypic light chains (Figure 4). Occasional cases of light chain amyloidosis (AL) may show inconclusive or negative staining [7–9]. Routine immunofluorescence antibodies for kappa and lambda light chains can target domains that may be altered or deleted in the amyloidogenic forms. Studies have shown that up to 36% of AL in the kidney show absent light chain staining [10].
Fig. 4.

Immunofluorescence microscopy in a case of light chain amyloidosis shows strong staining of amorphous glomerular deposits for lambda light chain (A) without staining for kappa light chain (B).
The recent emergence of laser microdissection and mass spectrometry-based proteomic analysis (LMD/MS) has proven to be an accurate and useful tool in the typing of amyloid [4]. Said et al. reported the largest series of renal amyloidosis to date, and the origin of amyloid was discovered in >97% of cases with the aid of LMD/MS. Proteomics-based identification of amyloid is recommended in cases where routine immunofluorescence and/or immunohistochemical testing cannot definitively type the amyloid deposits (Table 1) [4, 7]. LMD/MS requires the aid of an experienced laboratory, and detailed methods for LMD/MS have been previously published [4, 11]. In brief, LMD/MS is performed by first identifying Congo red-positive areas in the biopsy, which may include glomeruli, vessel walls and/or interstitial deposits. These foci are microdissected and removed from the tissue core followed by digestion into tryptic peptides. The resulting peptides are analyzed using liquid chromatography electrospray tandem mass spectrometry, and a list of proteins is generated from the peptide sequences. The total number of mass spectra collected and matched to a specific protein is indicated by the ‘spectra’ value (Figure 5). Higher spectra values indicate greater protein abundance and greater confidence in the accuracy of protein identification [4, 11].
Table 1.
Recommended indications for use of laser microdissection/mass spectrometry-based proteomics for amyloid typing in kidney biopsies
| Absence of tissue available for immunofluorescence microscopy |
| Negative immunofluorescence staining for kappa and lambda light chains AND negative immunohistochemical staining for serum amyloid A protein |
| Equal immunofluorescence staining for both kappa and lambda light chains. |
| Strong immunofluorescence staining for immunoglobulin heavy chains, with or without light chain staining. |
| Positive immunofluorescence staining for kappa and/or lambda light chains AND positive immunohistochemical staining for serum amyloid A protein. |
| Equivocal Congo red staining. |
Table reproduced (with permission) from Said et al. [7].
Fig. 5.
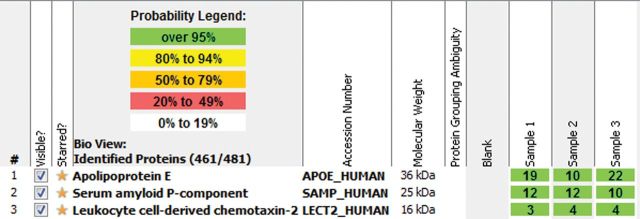
Representative mass spectroscopy data on a case of LECT2 amyloidosis. Analysis reveals high spectra for LECT2, apolipoprotein E and serum amyloid P-component within amyloid deposits. Courtesy of Dr. Sanjeev Sethi (Mayo Clinic, Rochester MN, USA).
The renal amyloidoses
Many amyloidogenic proteins have the potential to cause kidney disease (Table 2). The mechanisms of amyloidogenesis in these cases are variable and include abnormal protein production, overproduction or decreased excretion of wild-type proteins and hereditary mutation. The most common type of renal amyloidosis is associated with plasma cell dyscrasias and is composed of components of Igs. These are most often derived from monoclonal light chain (AL) but rarely are composed of Ig heavy chain (AH) or both Ig heavy and light chain (AHL) [13, 14]. Amyloidosis arising from serum amyloid A (SAA) constitutes the second most prevalent type of amyloid (after AL) and typically arises in the setting of chronic inflammatory conditions. Amyloidogenic leukocyte chemotactic factor 2 (ALECT2) has been reported in two large series as the third most common form of renal amyloid; however, this finding is likely dependent on geography given its higher prevalence in patients of Mexican descent [7, 15]. Other forms of renal amyloid include fibrinogen Aα chain (AFib), lysozyme (ALys), apolipoproteins AI (AApo AI), AII (AApo AII), AIV (AApo AIV), transthyretin (ATTR) and gelsolin (AGel).
Table 2.
Reported precursor proteins in renal amyloidosis
| Amyloid type | Renal distribution | Extra-renal involvement |
|---|---|---|
| Ig-related (AL, AH, and AHL) | All compartments: glomeruli most common | All organs with exception of central nervous system |
| AA | All compartments: glomeruli most common | All organs with exception of central nervous system |
| ALect2 | All compartments: interstitium most common | Lung, liver, adrenal gland, spleen and colon |
| AFib | Glomerular deposition: extra-glomerular deposits are less common | Adrenal gland, spleen and peripheral nervous system |
| ALys | All compartments: glomeruli and arterioles most common | Liver, GI tract, spleen, lymph node, skin and salivary gland |
| AApo AI | All compartments: medullary interstitium most common | Liver, heart, skin, larynx, palate, peripheral nervous system and testes |
| AApo AII | All compartments: glomeruli and small vessels most common | Adrenal glands and small vessels of other organs |
| AApo AIV | Medullary interstitium | Hearta |
| ATTR | All compartments: glomeruli and arterioles most common | Peripheral nervous system, autonomic nervous system, heart, GI tract and eye |
| AGel | Predominantly glomerular | Cornea, peripheral nervous system and skin |
Ig, immunoglobulin; AL, light chain amyloid; AH, heavy chain amyloid; AHL, heavy and light chain amyloid; AA, amyloid A; ALECT2, leukocyte chemotactic factor-2 amyloid; AFib, fibrinogen Aα chain amyloid; ALys, lysozyme amyloid; AApo AI, apolipoprotein AI amyloid; AApo AII, apolipoprotein AII amyloid; AApo AIV, apolipoprotein AIV amyloid; ATTR, transthyretin amyloid; AGel, gelsolin amyloid.
aFrom Bergström et al. [12].
AL, AH and AHL
AL is the most common form of amyloid in developed countries. In two recent studies from the USA and Italy, AL accounted for 81 and 68% of renal amyloidosis cases, respectively [7, 16]. AH and AHL are uncommon forms of amyloidosis and are included in the category of Ig-related amyloidosis. Ig-related amyloidosis is associated with plasma cell dyscrasias and can rarely be seen with other lymphoproliferative disorders. In a study of 190 patients with multiple myeloma undergoing kidney biopsy, 40 patients (21%) demonstrated amyloidosis, of which 88% were AL [17]. Lambda light chain is more commonly the amyloidogenic protein than kappa light chain by a ratio of 3:1 [18, 19].
AL amyloid fibrils are composed of either fully intact light chains (including both constant and variable domains) or the variable domain only. Mutations in the N-terminus (contained within the variable domain) and of the V λ VI subgroup may result in increased amyloidogenic properties [13, 20]. Post-translational protein modification and proteolysis may also play a role in amyloid deposition [21]. The pathogenesis of AH and AHL is less understood, but similar mechanisms have been hypothesized [14].
Kidney disease is the most common presenting manifestation of systemic Ig-related amyloidosis occurring in up to 50% of patients. The average age at diagnosis is 65 years, and many studies have demonstrated a slight male predominance with a male-to-female ratio of ∼2:1 [7, 14, 16, 22]. When the kidney is involved, renal insufficiency is common, accounting for 47% of cases in a study [7]. Proteinuria has been reported in upwards of 73% of patients with Ig-related renal amyloid with full nephrotic syndrome in 25–68% [13, 16]. Patients with AL-λ tend to present with lower serum creatinine, and higher degrees of proteinuria than those with AL-κ. In comparison with AL amyloid, AH and AHL are more frequently associated with hematuria, but no significant difference in serum creatinine, estimated glomerular filtration rate (eGFR), 24-h urine protein or percentage of patients with nephrotic syndrome has been established. Outside of the kidney, AH and AHL are associated with less-frequent cardiac involvement and patients are more likely to have overt multiple myeloma than those with AL [14].
Histologically, AL/AH/AHL can involve all renal parenchymal compartments, with the glomerulus being the most commonly affected. Nasr et al. found no significant differences in the parenchymal distribution between AL and AH/AHL. Some histologic differences were identified including four cases (25%) of AH/AHL with PAS-positive amyloid deposits [14]. Uncommonly, AL deposits may be limited to the vasculature, and this finding was associated with higher serum creatinine at presentation and lower proteinuria than diffuse AL [23]. An unusual case of intratubular AL-λ amyloidosis presenting with acute kidney injury has also been reported without glomerular, interstitial or vascular involvement [24]. Occasionally, amyloidosis can be seen in combination with other monoclonal Ig-associated diseases such as light chain cast nephropathy and light chain deposition disease [25].
The treatment of Ig-related amyloidosis is typically directed at the underlying plasma cell dyscrasia. Untreated patients with AL have a median survival of 12 months after diagnosis and <5 months in those with cardiac involvement. With advances in anti-plasma cell therapy and stem cell transplantation, the prognosis has improved. Skinner et al. demonstrated a prolonged median survival of 4.6 years in 312 patients treated with high-dose melphalan followed by autologous stem cell transplant (ASCT) [26]. In a study by Leung et al., renal response in patients with AL following high-dose melphalan and ASCT was seen in 43% of patients and was associated with the degree of baseline proteinuria [27]. Kidney transplantation has been performed sparingly in patients with AL. Pinney et al. reported a series including 25 patients with AL who underwent kidney transplant. The median graft survival was 5.8 years with 5- and 10-year graft survival rates of 74 and 25%, respectively. Twenty-eight percent of grafts demonstrated recurrent renal amyloid; however, no graft loss was attributed to amyloid nephropathy. At the time of transplantation, those without at least a partial clonal response to treatment had a significantly worse graft survival [28].
AA
Amyloid A amyloidosis is the second most common form of systemic amyloidosis. In two studies from the USA, AA constituted 7 and 12.5% of renal biopsies with amyloidosis [4, 7]. AA is more prevalent in Europe where it has been reported in up to 30–40% of patients with renal amyloid deposition [16, 29, 30]. The amyloid fibrils of AA are derived from SAA protein, which is an apolipoprotein component of high-density lipoprotein (HDL) and an acute-phase reactant [31]. SAA is produced by hepatocytes and is under transcriptional regulation of pro-inflammatory cytokines [32]. In a minority of patients with a sustained inflammatory stimulus and SAA overproduction, protein misfolding with subsequent amyloid deposition may occur. The underlying inflammatory stimuli associated with AA include chronic inflammatory arthritis, infection, periodic fever syndromes (such as familial Mediterranean fever), inflammatory bowel disease, neoplasia and Castleman's disease [29, 33–38]. Lachmann et al. reported a median gap of 17 years between the onset of inflammatory disease and the diagnosis of AA amyloidosis [33].
The SAA gene family is located on the short arm of chromosome 11, with three expressed gene products. The SAA1 and SAA2 genes are predominantly expressed as part of the acute-phase response and are the primary isotypes deposited in AA. SAA4 expression is constitutively active and not an acute-phase reactant [32]. A case of AA secondary to SAA4 mutation in the absence of chronic inflammation has been reported [39].
The average age of patients with AA is ∼10 years, younger than those with AL, and AA represents the most common form of amyloidosis in children [40]. Kidney disease is the most common clinical manifestation of AA with up to 97% of patients in a study presenting with >500 mg of proteinuria per day or serum creatinine of >132.6 μmol/L (1.5 mg/dL) [33]. Full nephrotic syndrome has been reported in 54–63% of patients and elevated serum creatinine in 75–81% [7, 29].
Histologic localization of AA deposits within the renal parenchyma is similar to AL. Glomerular deposition is the most common and extra-glomerular deposits, including tubular basement membrane, interstitial and vascular deposits are seen in 12, 73 and 88% of cases, respectively, in a study [7]. Verine et al. reported an amyloid-associated inflammatory response in 30 of 68 cases. Nineteen showed a granulomatous reaction around vascular and interstitial deposits and 17 demonstrated glomerular endocapillary leukocytes. Twelve of these demonstrated a crescentic and/or necrotizing glomerular injury [29]. The possibility of a superimposed glomerulonephritis was raised, and anti-neutrophil cytoplasmic antibody serology was negative in 8 of 12 patients tested. Histologic identification of AA can be performed using immunohistochemistry (Figure 6). Occasionally, distinction between AA and Ig-related amyloidosis can be difficult due to variable nonspecific immunofluorescence staining for Ig components and complement within amyloid deposits [7, 8]. In such cases, LMD/MS can be helpful in identifying the precursor protein.
Fig. 6.
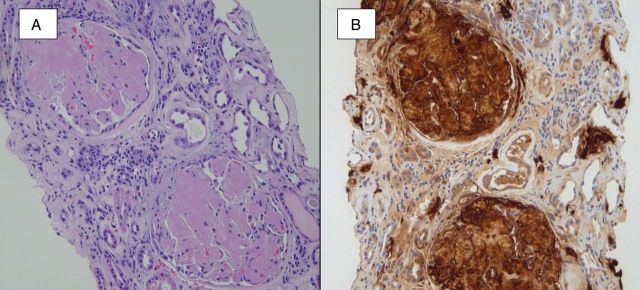
A hematoxylin- and eosin-stained section (A) shows the accumulation of amorphous and lightly eosinophilic material within glomeruli, arterioles and interstitial areas. A Congo red stain (not shown) was positive for amyloid, and immunofluorescence staining for light chains (not shown) was negative. An immunohistochemical stain for SAA protein (B) confirmed the presence of AA amyloidosis.
The treatment of AA amyloidosis is primarily directed at the underlying chronic inflammatory condition with the goal of decreasing production of SAA. Lachmann et al. showed that AA amyloid deposits had the potential to regress when circulating SAA could be kept at low levels [33]. Improvement of renal function was typically a slow process, often taking months to even years; however, recurrence of the inflammatory stimulus resulted in rapid progression of kidney disease [31, 33]. In patients with AA amyloidosis secondary to rheumatic disease, immunosuppressive therapy has been a mainstay of treatment including targeted therapy against tumor necrosis factor (TNF) and more recently interleukin-6 (IL-6). Okuda et al. compared treatment with tocilizumab (an IL-6 inhibitor) and anti-TNF therapy in patients with AA amyloidosis secondary to rheumatic disease. Those treated with tocilizumab showed greater control of SAA, improvement in GFR, and higher rates of clinical remission than those treated with anti-TNF therapy [41]. In patients with other underlying inflammatory conditions, disease-specific therapies have shown benefit. These include colchicine in patients with familial Mediterranean fever, anti-microbial therapy in those with chronic infection and surgical resection of tumors [34, 35]. Another promising treatment approach targets amyloid deposition itself. Eprodisate, which has structural similarities to heparin sulfate, acts to disrupt the interaction between amyloidogenic fibrils and glycosaminoglycans. It has been shown in a multicenter study to slow the progression of kidney disease in AA amyloidosis [42]. In those who progress to end stage renal disease (ESRD), kidney transplant is an accepted form of renal replacement therapy. In a study including 43 patients with AA amyloidosis receiving kidney transplant, Pinney et al. reported 5- and 10-year graft survival rates of 86 and 59%, respectively. Nine patients developed recurrent amyloid in their graft and two experienced graft failure as a result. As would be expected, serum SAA concentrations were significantly higher in those with recurrent amyloid than those without [28].
ALECT2
Leukocyte chemotactic factor 2 (LECT2) is a recent addition to the growing list of amyloidogenic proteins [43]. LECT2 is produced by hepatocytes and primarily acts as a neutrophil chemotactic factor and may also play a role in tissue repair and growth following damage [44, 45]. In two recent studies from the USA, ALECT2 was the third most common form of renal amyloidosis accounting for 3% of renal amyloidosis cases [7, 15]. The mechanism of amyloidogenesis is not fully understood, but a possible genetic role has been suspected. Murphy et al. performed gene sequencing on four patients with ALECT2 and found that all four were homozygous for the G allele resulting from an amino acid substitution (Ile40Val) [46]. The majority of reported cases have been identified in patients of Mexican descent, also arguing for a genetic role [47]. Clinical disease related to ALECT2 has largely been limited to the kidney with one report demonstrating incidentally discovered ALECT2 deposits within hepatic, splenic, colonic and adrenal tissue [48]. ALECT2 deposition within the lung and kidney presenting as pulmonary-renal syndrome has also been reported [49].
Renal ALECT2 more often presents with renal insufficiency than with heavy proteinuria. Said et al. reported 13 cases of ALECT2 and found that 17% presented with nephrotic syndrome whereas 92% presented with renal insufficiency [7]. This clinical presentation correlates with the histologic distribution of amyloid deposits. Deposition of ALECT2 is seen in all renal compartments; however, interstitial deposits often dominate (Figure 7) [7, 15, 46, 50]. Identification of ALECT2 is best performed by LMD/MS; however, in our experience, immunohistochemical characterization is a useful tool when performed by an experienced laboratory [49].
Fig. 7.
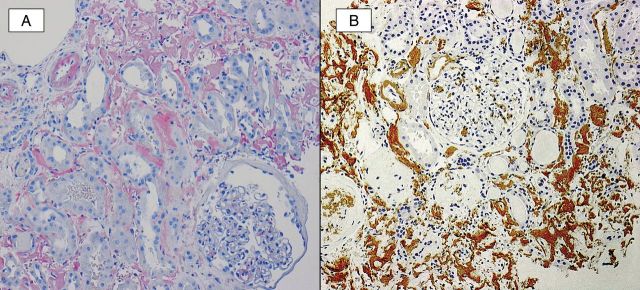
A Congo red study (A) demonstrates strong staining of amyloid deposits within the interstitium, vasculature and mesangial areas. An immunohistochemical stain (B) for LECT2 is positive in these deposits.
Hereditary amyloidosis
The hereditary amyloidoses are a rare but clinically important and varied group of disorders, which are primarily inherited in an autosomal-dominant fashion. Implicated precursor proteins include fibrinogen Aα chain (AFib), lysozyme (ALys), apolipoproteins AI (AApo AI) and AII (AApo AII), transthyretin (ATTR) and gelsolin (AGel). In a study of 350 patients originally diagnosed with AL, genetic analysis demonstrated hereditary amyloidosis in 34 cases (9.7%) [9]. Identification of precursor proteins in hereditary forms of amyloidosis requires a high index of suspicion given that specific immunohistochemical stains are often not available. Like other types of amyloid, LMD/MS has proven useful in identifying the precursor protein, particularly in conjunction with genetic testing for specific mutations.
Fibrinogen Aα chain amyloidosis was first described in 1993 and primarily manifests as progressive kidney disease [51]. At least nine mutations have been identified with the Glu526Val variant accounting for 90% of patients in a study [52]. Amyloid fibrils are composed of fragments from the carboxyl terminal of the fibrinogen Aα chain with all reported mutations involving this region [52, 53]. Gillmore et al. has reported the largest series of patients to date with a median age at presentation of 58 years. Proteinuria was a universal manifestation, and kidney insufficiency was present in 54% [52]. Kidney biopsy often shows marked glomerular deposition with frequent obliteration of capillary lumina and little to no extra-glomerular deposition [7, 52, 54, 55]. Immunofluorescence microscopy can be useful in diagnosing AFib with nodular mesangial staining for fibrinogen suggesting the diagnosis [54]. Extra-renal deposition is uncommon and primarily occurs late in the disease course. The majority of patients follow a progressive course to ESRD with a median time from presentation to ESRD of 4.6 years [52]. The primary treatment options in patients with end stage kidney disease from AFib include isolated kidney transplantation and combined liver and kidney transplantation [56, 57]. In 10 patients receiving an isolated kidney transplant, Pinney et al. reported 5- and 10-year graft survival rates of 85 and 30%, respectively. Recurrent amyloid was identified in seven patients. Nine patients underwent combined liver and kidney transplant with 5- and 10-year graft survival of 63 and 31%, respectively. No patients in this group developed recurrent amyloidosis [28].
Lysozyme is a bacteriolytic enzyme that is synthesized by hepatocytes and leukocytes including neutrophils and macrophages. Variant lysozyme leading to systemic amyloidosis was first reported in 1993 and has since been documented in families of English, French and Italian descent [58–62]. At least four pathogenic mutations have been described, all located within exon 2 of the lysozyme gene. Amyloid deposition can be widespread and affected organs include the kidney, liver, gastrointestinal tract, spleen, lymph nodes, salivary glands and skin. Extra-renal manifestations include GI bleeding, megaloblastic anemia due to malabsorption and hepatic rupture [59]. Renal involvement has been described in many but not all reports of ALys amyloidosis. Renal manifestations range from mild proteinuria to progressive kidney insufficiency requiring renal replacement therapy. Renal biopsies have shown amyloid deposition within all compartments; however, the glomerulus and arterioles appear to be the most frequently and most severely affected. Interstitial deposits within the cortex and medulla have also been reported [59, 61, 62]. Progression of ALys to ESRD appears to be a slow process. Pinney et al. reported three patients with ALys who received renal transplants with a median time of progression to ESRD of 10.4 years. All three patients had good graft function at the time of publication without evidence of recurrent amyloid [28]. Two patients in a family reported by Yazaki et al. received kidney transplants with long-term follow-up revealing good function [62].
Apolipoprotein AI and AII variants are well-described forms of hereditary systemic amyloidosis with renal involvement [53]. Both apolipoprotein AI and AII are synthesized in the liver and small intestine and are structural components of HDL [63]. In the case of AApo AI, at least 19 mutations have been described within the APOAI gene resulting in amyloidosis [64]. Mutations near the N-terminus are associated with renal, hepatic and occasionally cardiac deposition whereas mutations near the C-terminus are associated with cardiac, cutaneous and laryngeal deposits [53, 63, 64]. In males, testicular AApo AI deposition can occur and hypogonadism may be the first sign of disease [65]. Kidney disease in AApo AI often manifests as slowly progressive renal failure with minimal proteinuria; however, nephrotic syndrome has been reported. Renal amyloid deposits have mainly been reported within the medullary interstitium with sparing of the renal cortex [66]. Two cases of AApo AI with more extensive glomerular and vascular involvement have also been described [30, 67]. Amyloidogenic apolipoprotein AII was first reported in 2001, and all of the reported mutations have occurred at the stop codon resulting in a 21 amino acid extension of the wild-type protein [68, 69]. Clinically, kidney disease dominates and patients typically present with progressive renal failure and proteinuria. Histologically, AApo AII amyloid deposits are most frequently identified within the glomerulus and small vessels. Extra-renal amyloid deposits have been identified in autopsy cases, primarily within vessel walls [53, 68]. A case of apolipoprotein AIV amyloidosis (AApo AIV) has recently been described with predominantly medullary amyloid deposition [70]. Amyloidosis due to variant apolipoprotein AI, AII or AIV is very rare and accounted for <1% of renal amyloid biopsies in two studies [7, 30]. No specific treatment is available for AApo AI, AII or AIV amyloidosis, and kidney transplantation has proven effective in cases progressing to organ failure. Fourteen patients receiving either isolated kidney or combined liver–kidney or heart–kidney transplants for AApo AI were included in a study by Pinney et al. with a median graft survival of 13.1 years. The 5- and 10-year survival was 100 and 77%, respectively, with only one patient losing their graft to recurrent amyloidosis [28]. Few reports of kidney transplantation in AApo AII amyloidosis are available, but long-term graft survival of up to 10 years has been described [71].
Transthyretin amyloidosis is the most common type of familial amyloidosis and is primarily associated with peripheral neuropathy and dysautonomia. Other sites of involvement include the heart, skin, gastrointestinal tract and kidneys. Transthyretin is predominantly synthesized in the liver and at least 100 different mutations leading to amyloidosis have been described in the TTR gene with up to 15 nephropathic variants [72]. The TTR Val30Met variant is the most common and has a worldwide distribution. Wild-type transthyretin also has the potential to form amyloid fibrils and is implicated in systemic senile amyloidosis. The age of onset can be quite variable, and a family history may be difficult to uncover due to late-onset disease [9, 72]. Kidney involvement is often heralded by microalbuminuria, and in a study, 50% of patients with microalbuminuria progressed to overt nephropathy and 32% of those showed progression to ESRD [73]. On kidney biopsy, ATTR deposits have been identified in all renal compartments. Within the glomerulus, the mesangial areas are most commonly involved followed by the arterioles at the vascular pole [73]. Tubulointerstitial deposits are a common finding and are often most prominent in the renal medulla [74, 75]. The long-term outcome of those with progressive nephropathy is poor with an average survival of 21 months after the initiation of dialysis in a study [76]. Definitive treatment involves liver transplantation to halt production of mutated transthyretin. Results following liver transplant are not always favorable and likely related to the underlying degree of amyloid deposition and organ dysfunction at the time of transplant. Combined liver–kidney transplantation has been performed on a number of patients with hereditary ATTR, and in a study, six patients treated with a combined transplant showed no recurrence of proteinuria after 84 months [77].
Gelsolin amyloidosis (AGel) is a rare form of hereditary systemic amyloidosis that primarily manifests as an autosomal-dominant polyneuropathy syndrome. The cardinal clinical manifestations of AGel amyloidosis include lattice corneal dystrophy, progressive neuropathies and cutis laxa [78, 79]. Kidney disease is uncommon but has been reported. In those with homozygous mutations in the gelsolin gene, renal manifestations, including proteinuria and progressive renal failure, may be more severe and occur earlier than those with heterozygous mutations [80]. At least three mutations resulting in a variant gelsolin protein have been reported, and two of these (Asp187Asn and Gly194Arg) have been reported with kidney involvement [80–83]. Kidney biopsy findings in patients with AGel amyloidosis include predominantly glomerular amyloid deposits [81, 82]. Sethi et al. reported a case of AGel amyloidosis with unusual ultrastructural features including parallel and swirling organization of amyloid fibrils [82]. Like other forms of hereditary amyloidosis, a specific therapy for gelsolin amyloidosis does not exist. Kidney transplant has been reported with stable graft function 3–6 years post-transplant [78, 80].
Summary
In summary, renal amyloidosis is an uncommon disorder, accounting for ∼2% of native kidney biopsies [7, 30]. All forms of amyloid share common histologic and ultrastructural findings but are composed of a variety of different precursor proteins. Determination of the composition of amyloid deposits is of paramount importance as it drives treatment and predicts outcome and the likelihood of extra-renal disease. Advances in diagnostic techniques, including the emergence of laser microdissection and mass spectroscopy-based proteomic analysis, have resulted in greater accuracy in amyloid identification.
Conflict of interest statement
None declared.
References
- 1.Hazenberg BP. Amyloidosis: a clinical overview. Rheum Dis Clin N Am. 2013;39:323–345. doi: 10.1016/j.rdc.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 2.Glenner GG, Terry W, Harada M, et al. Amyloid fibril proteins: proof of homology with immunoglobulin light chains by sequence analyses. Science. 1971;172:1150–1151. doi: 10.1126/science.172.3988.1150. [DOI] [PubMed] [Google Scholar]
- 3.Leung N, Nasr SH, Sethi S. How I treat amyloidosis: the importance of accurate diagnosis and amyloid typing. Blood. 2012;120:3206–3213. doi: 10.1182/blood-2012-03-413682. [DOI] [PubMed] [Google Scholar]
- 4.Sethi S, Vrana JA, Theis JD, et al. Laser microdissection and mass spectrometry-based proteomics aids the diagnosis and typing of renal amyloidosis. Kidney Int. 2012;82:226–234. doi: 10.1038/ki.2012.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dember LM. Amyloidosis-associated kidney disease. J Am Soc Nephrol. 2006;17:3458–3471. doi: 10.1681/ASN.2006050460. [DOI] [PubMed] [Google Scholar]
- 6.Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med. 2003;349:583–596. doi: 10.1056/NEJMra023144. [DOI] [PubMed] [Google Scholar]
- 7.Said SM, Sethi S, Valeri AM, et al. Renal amyloidosis: origin and clinicopathologic correlations of 474 recent cases. Clin J Am Soc Nephrol. 2013;8:1515–1523. doi: 10.2215/CJN.10491012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Satoskar AA, Burdge K, Cowden DJ, et al. Typing of amyloidosis in renal biopsies: diagnostic pitfalls. Arch Pathol Lab Med. 2007;131:917–922. doi: 10.5858/2007-131-917-TOAIRB. [DOI] [PubMed] [Google Scholar]
- 9.Lachmann HJ, Booth DR, Booth SE, et al. Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med. 2002;346:1786–1791. doi: 10.1056/NEJMoa013354. [DOI] [PubMed] [Google Scholar]
- 10.Novak L, Cook WJ, Herrera GA, et al. AL-amyloidosis is underdiagnosed in renal biopsies. Nephrol Dial Transplant. 2004;19:3050–3053. doi: 10.1093/ndt/gfh503. [DOI] [PubMed] [Google Scholar]
- 11.Vrana JA, Gamez JD, Madden BJ, et al. Classification of amyloidosis by laser microdissection and mass spectrometry-based proteomic analysis in clinical biopsy specimens. Blood. 2009;114:4957–4959. doi: 10.1182/blood-2009-07-230722. [DOI] [PubMed] [Google Scholar]
- 12.Bergström J, et al. Two different types of amyloid deposits—apolipoprotein A-IV and transthyretin—in a patient with systemic amyloidosis. Lab Invest. 2004;84(8):981–988. doi: 10.1038/labinvest.3700124. [DOI] [PubMed] [Google Scholar]
- 13.Markowitz GS. Dysproteinemia and the kidney. Adv Anat Pathol. 2004;11:49–63. doi: 10.1097/00125480-200401000-00005. [DOI] [PubMed] [Google Scholar]
- 14.Nasr SH, Said SM, Valeri AM, et al. The diagnosis and characteristics of renal heavy-chain and heavy/light-chain amyloidosis and their comparison with renal light-chain amyloidosis. Kidney Int. 2013;83:463–470. doi: 10.1038/ki.2012.414. [DOI] [PubMed] [Google Scholar]
- 15.Larsen CP, Walker PD, Weiss DT, et al. Prevalence and morphology of leukocyte chemotactic factor 2-associated amyloid in renal biopsies. Kidney Int. 2010;77:816–819. doi: 10.1038/ki.2010.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bergesio F, Ciciani AM, Manganaro M, et al. Renal involvement in systemic amyloidosis: an Italian collaborative study on survival and renal outcome. Nephrol Dial Transplant. 2008;23:941–951. doi: 10.1093/ndt/gfm684. [DOI] [PubMed] [Google Scholar]
- 17.Nasr SH, Valeri AM, Sethi S, et al. Clinicopathologic correlations in multiple myeloma: a case series of 190 patients with kidney biopsies. Am J Kidney Dis. 2012;59:786–794. doi: 10.1053/j.ajkd.2011.12.028. [DOI] [PubMed] [Google Scholar]
- 18.Gertz MA, Lacy MQ, Dispenzieri A. Immunoglobulin light chain amyloidosis and the kidney. Kidney Int. 2002;61:1–9. doi: 10.1046/j.1523-1755.2002.00085.x. [DOI] [PubMed] [Google Scholar]
- 19.Gertz MA, Leung N, Lacy MQ, et al. Clinical outcome of immunoglobulin light chain amyloidosis affecting the kidney. Nephrol Dial Transplant. 2009;24:3132–3137. doi: 10.1093/ndt/gfp201. [DOI] [PubMed] [Google Scholar]
- 20.Abraham RS, Geyer SM, Price-Troska TL, et al. Immunoglobulin light chain variable (V) region genes influence clinical presentation and outcome in light chain-associated amyloidosis (AL) Blood. 2003;101:3801–3808. doi: 10.1182/blood-2002-09-2707. [DOI] [PubMed] [Google Scholar]
- 21.Enqvist S, Sletten K, Westermark P. Fibril protein fragmentation pattern in systemic AL-amyloidosis. J Pathol. 2009;219:473–480. doi: 10.1002/path.2607. [DOI] [PubMed] [Google Scholar]
- 22.Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol. 1995;32:45–59. [PubMed] [Google Scholar]
- 23.Eirin A, Irazabal MV, Gertz MA, et al. Clinical features of patients with immunoglobulin light chain amyloidosis (AL) with vascular-limited deposition in the kidney. Nephrol Dial Transplant. 2012;27:1097–1101. doi: 10.1093/ndt/gfr381. [DOI] [PubMed] [Google Scholar]
- 24.El-Zoghby Z, Lager D, Gregoire J, et al. Intra-tubular amyloidosis. Kidney Int. 2007;72:1282–1288. doi: 10.1038/sj.ki.5002411. [DOI] [PubMed] [Google Scholar]
- 25.Lorenz EC, Sethi S, Poshusta TL, et al. Renal failure due to combined cast nephropathy, amyloidosis and light-chain deposition disease. Nephrol Dial Transplant. 2010;25:1340–1343. doi: 10.1093/ndt/gfp735. [DOI] [PubMed] [Google Scholar]
- 26.Skinner M, Sanchorawala V, Seldin DC, et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Int Med. 2004;140:85–93. doi: 10.7326/0003-4819-140-2-200401200-00008. [DOI] [PubMed] [Google Scholar]
- 27.Leung N, Dispenzieri A, Lacy MQ, et al. Severity of baseline proteinuria predicts renal response in immunoglobulin light chain-associated amyloidosis after autologous stem cell transplantation. Clin J Am Soc Nephrol. 2007;2:440–444. doi: 10.2215/CJN.02450706. [DOI] [PubMed] [Google Scholar]
- 28.Pinney JH, Lachmann HJ, Sattianayagam PT, et al. Renal transplantation in systemic amyloidosis-importance of amyloid fibril type and precursor protein abundance. Am J Transplant. 2013;13:433–441. doi: 10.1111/j.1600-6143.2012.04326.x. [DOI] [PubMed] [Google Scholar]
- 29.Verine J, Mourad N, Desseaux K, et al. Clinical and histological characteristics of renal AA amyloidosis: a retrospective study of 68 cases with a special interest to amyloid-associated inflammatory response. Hum Pathol. 2007;38:1798–1809. doi: 10.1016/j.humpath.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 30.von Hutten H, Mihatsch M, Lobeck H, et al. Prevalence and origin of amyloid in kidney biopsies. Am J Surg Pathol. 2009;33:1198–1205. doi: 10.1097/PAS.0b013e3181abdfa7. [DOI] [PubMed] [Google Scholar]
- 31.Simons JP, Al-Shawi R, Ellmerich S, et al. Pathogenetic mechanisms of amyloid A amyloidosis. P Natl Acad Sci USA. 2013;110:16115–16120. doi: 10.1073/pnas.1306621110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Urieli-Shoval S, Linke RP, Matzner Y. Expression and function of serum amyloid A, a major acute-phase protein, in normal and disease states. Curr Opin Hematol. 2000;7:64–69. doi: 10.1097/00062752-200001000-00012. [DOI] [PubMed] [Google Scholar]
- 33.Lachmann HJ, Goodman HJ, Gilbertson JA, et al. Natural history and outcome in systemic AA amyloidosis. N Engl J Med. 2007;356:2361–2371. doi: 10.1056/NEJMoa070265. [DOI] [PubMed] [Google Scholar]
- 34.Lachmann HJ, Gilbertson JA, Gillmore JD, et al. Unicentric Castleman's disease complicated by systemic AA amyloidosis: a curable disease. QJM. 2002;95:211–218. doi: 10.1093/qjmed/95.4.211. [DOI] [PubMed] [Google Scholar]
- 35.Lane T, Loeffler JM, Rowczenio DM, et al. AA amyloidosis complicating the hereditary periodic fever syndromes. Arthritis Rheum. 2013;65:1116–1121. doi: 10.1002/art.37827. [DOI] [PubMed] [Google Scholar]
- 36.Saha A, Theis JD, Vrana JA, et al. AA amyloidosis associated with hepatitis B. Nephrol Dial Transplant. 2011;26:2407–2412. doi: 10.1093/ndt/gfr224. [DOI] [PubMed] [Google Scholar]
- 37.Tank SJ, Chima RS, Shah V, et al. Renal amyloidosis following tuberculosis. Indian J Pediatr. 2000;67:679–681. doi: 10.1007/BF02762183. [DOI] [PubMed] [Google Scholar]
- 38.Sethi S, El Ters M, Vootukuru S, et al. Recurrent AA amyloidosis in a kidney transplant. Am J Kidney Dis. 2011;57:941–944. doi: 10.1053/j.ajkd.2011.02.383. [DOI] [PubMed] [Google Scholar]
- 39.Murphy CL, Wang S, Kestler DP, et al. AA amyloidosis associated with a mutated serum amyloid A4 protein. Amyloid. 2009;16:84–88. doi: 10.1080/13506120902879905. [DOI] [PubMed] [Google Scholar]
- 40.Bilginer Y, Akpolat T, Ozen S. Renal amyloidosis in children. Pediatr Nephrol. 2011;26:1215–1227. doi: 10.1007/s00467-011-1797-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Okuda Y, Ohnishi M, Matoba K, et al. Comparison of the clinical utility of tocilizumab and anti-TNF therapy in AA amyloidosis complicating rheumatic diseases. Mod Rheumatol. 2014;24:137–143. doi: 10.3109/14397595.2013.854048. [DOI] [PubMed] [Google Scholar]
- 42.Dember LM, Hawkins PN, Hazenberg BP, et al. Eprodisate for the treatment of renal disease in AA amyloidosis. N Engl J Med. 2007;356:2349–2360. doi: 10.1056/NEJMoa065644. [DOI] [PubMed] [Google Scholar]
- 43.Benson MD, James S, Scott K, et al. Leukocyte chemotactic factor 2: a novel renal amyloid protein. Kidney Int. 2008;74:218–222. doi: 10.1038/ki.2008.152. [DOI] [PubMed] [Google Scholar]
- 44.Yamagoe S, Kameoka Y, Hashimoto K, et al. Molecular cloning, structural characterization, and chromosomal mapping of the human LECT2 gene. Genomics. 1998;48:324–329. doi: 10.1006/geno.1997.5198. [DOI] [PubMed] [Google Scholar]
- 45.Nagai H, Hamada T, Uchida T, et al. Systemic expression of a newly recognized protein, LECT2, in the human body. Pathol Int. 1998;48:882–886. doi: 10.1111/j.1440-1827.1998.tb03855.x. [DOI] [PubMed] [Google Scholar]
- 46.Murphy CL, Wang S, Kestler D, et al. Leukocyte chemotactic factor 2 (LECT2)-associated renal amyloidosis: a case series. Am J Kidney Dis. 2010;56:1100–1107. doi: 10.1053/j.ajkd.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Said SM, Sethi S, Cornell LD, et al. Renal ALECT2 Amyloidosis: a clinicopathologic study of 17 patients. Mod Pathol. 2013;26(Supp 2):392A. [Google Scholar]
- 48.Dogan A, Thies J, Vrana J, et al. Clinical and pathological phenotype of leukocyte cell-derived chemotaxin-2 (LECT2) amyloidosis (ALECT2) [abstract OP-058] Amyloid. 2010;17(suppl 1):69–70. [Google Scholar]
- 49.Khalighi M, Yue A, Hwang M, et al. Leukocyte chemotactic factor 2 (LECT2) amyloidosis presenting as pulmonary-renal syndrome: a case report and review of the literature. Clin Kidney J. 2013;6:618–621. doi: 10.1093/ckj/sft126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holanda DG, Acharya VK, Dogan A, et al. Atypical presentation of atypical amyloid. Nephrol Dial Transplant. 2011;26:373–376. doi: 10.1093/ndt/gfq638. [DOI] [PubMed] [Google Scholar]
- 51.Benson MD, Liepnieks J, Uemichi T, et al. Hereditary renal amyloidosis associated with a mutant fibrinogen alpha-chain. Nat Gen. 1993;3:252–255. doi: 10.1038/ng0393-252. [DOI] [PubMed] [Google Scholar]
- 52.Gillmore JD, Lachmann HJ, Rowczenio D, et al. Diagnosis, pathogenesis, treatment, and prognosis of hereditary fibrinogen A alpha-chain amyloidosis. J Am Soc Nephrol. 2009;20:444–451. doi: 10.1681/ASN.2008060614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benson MD. Ostertag revisited: the inherited systemic amyloidoses without neuropathy. Amyloid. 2005;12:75–87. doi: 10.1080/13506120500106925. [DOI] [PubMed] [Google Scholar]
- 54.Sethi S, Fervenza FC, Miller D, et al. Recurrence of amyloidosis in a kidney transplant. Am J Kidney Dis. 2010;56:394–398. doi: 10.1053/j.ajkd.2009.10.061. [DOI] [PubMed] [Google Scholar]
- 55.Miller DV, Dogan A, Sethi S. New-onset proteinuria with massive amorphous glomerular deposits. Am J Kidney Dis. 2010;55:749–754. doi: 10.1053/j.ajkd.2009.05.015. [DOI] [PubMed] [Google Scholar]
- 56.Mousson C, Heyd B, Justrabo E, et al. Successful hepatorenal transplantation in hereditary amyloidosis caused by a frame-shift mutation in fibrinogen alpha-chain gene. Am J Transplant. 2006;6:632–635. doi: 10.1111/j.1600-6143.2005.01199.x. [DOI] [PubMed] [Google Scholar]
- 57.Zeldenrust S, Gertz M, Uemichi T, et al. Orthotopic liver transplantation for hereditary fibrinogen amyloidosis. Transplantation. 2003;75:560–561. doi: 10.1097/01.TP.0000046526.10003.EC. [DOI] [PubMed] [Google Scholar]
- 58.Pepys MB, Hawkins PN, Booth DR, et al. Human lysozyme gene mutations cause hereditary systemic amyloidosis. Nature. 1993;362:553–557. doi: 10.1038/362553a0. [DOI] [PubMed] [Google Scholar]
- 59.Granel B, Valleix S, Serratrice J, et al. Lysozyme amyloidosis: report of 4 cases and a review of the literature. Medicine. 2006;85:66–73. doi: 10.1097/01.md.0000200467.51816.6d. [DOI] [PubMed] [Google Scholar]
- 60.Gillmore JD, Booth DR, Madhoo S, et al. Hereditary renal amyloidosis associated with variant lysozyme in a large English family. Nephrol Dial Transplant. 1999;14:2639–2644. doi: 10.1093/ndt/14.11.2639. [DOI] [PubMed] [Google Scholar]
- 61.Valleix S, Drunat S, Philit JB, et al. Hereditary renal amyloidosis caused by a new variant lysozyme W64R in a French family. Kidney Int. 2002;61:907–912. doi: 10.1046/j.1523-1755.2002.00205.x. [DOI] [PubMed] [Google Scholar]
- 62.Yazaki M, Farrell SA, Benson MD. A novel lysozyme mutation Phe57Ile associated with hereditary renal amyloidosis. Kidney Int. 2003;63:1652–1657. doi: 10.1046/j.1523-1755.2003.00904.x. [DOI] [PubMed] [Google Scholar]
- 63.Obici L, Franceschini G, Calabresi L, et al. Structure, function and amyloidogenic propensity of apolipoprotein A-I. Amyloid. 2006;13:191–205. doi: 10.1080/13506120600960288. [DOI] [PubMed] [Google Scholar]
- 64.Rowczenio D, Dogan A, Theis JD, et al. Amyloidogenicity and clinical phenotype associated with five novel mutations in apolipoprotein A-I. Am J Pathol. 2011;179:1978–1987. doi: 10.1016/j.ajpath.2011.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scalvini T, Martini PR, Obici L, et al. Infertility and hypergonadotropic hypogonadism as first evidence of hereditary apolipoprotein A-I amyloidosis. J Urology. 2007;178:344–348. doi: 10.1016/j.juro.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 66.Gregorini G, Izzi C, Obici L, et al. Renal apolipoprotein A-I amyloidosis: a rare and usually ignored cause of hereditary tubulointerstitial nephritis. J Am Soc Nephrol. 2005;16:3680–3686. doi: 10.1681/ASN.2005040382. [DOI] [PubMed] [Google Scholar]
- 67.Murphy CL, Wang S, Weaver K, et al. Renal apolipoprotein A-I amyloidosis associated with a novel mutant Leu64Pro. Am J Kidney Dis. 2004;44:1103–1109. doi: 10.1053/j.ajkd.2004.08.033. [DOI] [PubMed] [Google Scholar]
- 68.Yazaki M, Liepnieks JJ, Barats MS, et al. Hereditary systemic amyloidosis associated with a new apolipoprotein AII stop codon mutation Stop78Arg. Kidney Int. 2003;64:11–16. doi: 10.1046/j.1523-1755.2003.00047.x. [DOI] [PubMed] [Google Scholar]
- 69.Benson MD, Liepnieks JJ, Yazaki M, et al. A new human hereditary amyloidosis: the result of a stop-codon mutation in the apolipoprotein AII gene. Genomics. 2001;72:272–277. doi: 10.1006/geno.2000.6499. [DOI] [PubMed] [Google Scholar]
- 70.Sethi S, Theis JD, Shiller SM, et al. Medullary amyloidosis associated with apolipoprotein A-IV deposition. Kidney Int. 2012;81:201–206. doi: 10.1038/ki.2011.316. [DOI] [PubMed] [Google Scholar]
- 71.Magy N, Liepnieks JJ, Yazaki M, et al. Renal transplantation for apolipoprotein AII amyloidosis. Amyloid. 2003;10:224–228. doi: 10.3109/13506120309041739. [DOI] [PubMed] [Google Scholar]
- 72.Lobato L, Rocha A. Transthyretin amyloidosis and the kidney. Clin J Am Soc Nephrol. 2012;7:1337–1346. doi: 10.2215/CJN.08720811. [DOI] [PubMed] [Google Scholar]
- 73.Lobato L, Beirao I, Silva M, et al. Familial ATTR amyloidosis: microalbuminuria as a predictor of symptomatic disease and clinical nephropathy. Nephrol Dial Transplant. 2003;18:532–538. doi: 10.1093/ndt/18.3.532. [DOI] [PubMed] [Google Scholar]
- 74.Lobato L, Beirao I, Guimaraes SM, et al. Familial amyloid polyneuropathy type I (Portuguese): distribution and characterization of renal amyloid deposits. Am J Kidney Dis. 1998;31:940–946. doi: 10.1053/ajkd.1998.v31.pm9631837. [DOI] [PubMed] [Google Scholar]
- 75.Oguchi K, Takei Y, Ikeda S. Value of renal biopsy in the prognosis of liver transplantation in familial amyloid polyneuropathy ATTR Val30Met patients. Amyloid. 2006;13:99–107. doi: 10.1080/13506120600722662. [DOI] [PubMed] [Google Scholar]
- 76.Lobato L, Beirao I, Silva M, et al. End-stage renal disease and dialysis in hereditary amyloidosis TTR V30M: presentation, survival and prognostic factors. Amyloid. 2004;11:27–37. doi: 10.1080/13506120410001673884. [DOI] [PubMed] [Google Scholar]
- 77.Lobato L, Beirao I, Seca R, et al. Combined liver-kidney transplantation in familial amyloidotic polyneuropathy TTR V30M: nephrological assessment. Amyloid. 2011;18(Suppl 1):190–192. doi: 10.3109/13506129.2011.574354071. [DOI] [PubMed] [Google Scholar]
- 78.Shoja MM, Ardalan MR, Tubbs RS, et al. Outcome of renal transplant in hereditary gelsolin amyloidosis. Am J Med Sci. 2009;337:370–372. doi: 10.1097/MAJ.0b013e3181a4199c. [DOI] [PubMed] [Google Scholar]
- 79.Meretoja J. Familial systemic paramyloidosis with lattice dystrophy of the cornea, progressive cranial neuropathy, skin changes and various internal symptoms. A previously unrecognized heritable syndrome. Ann Clin Res. 1969;1:314–324. [PubMed] [Google Scholar]
- 80.Maury CP, Kere J, Tolvanen R, et al. Homozygosity for the Asn187 gelsolin mutation in Finnish-type familial amyloidosis is associated with severe renal disease. Genomics. 1992;13:902–903. doi: 10.1016/0888-7543(92)90183-s. [DOI] [PubMed] [Google Scholar]
- 81.Ardalan MR, Shoja MM, Kiuru-Enari S. Amyloidosis-related nephrotic syndrome due to a G654A gelsolin mutation: the first report from the Middle East. Nephrol Dial Transplant. 2007;22:272–275. doi: 10.1093/ndt/gfl548. [DOI] [PubMed] [Google Scholar]
- 82.Sethi S, Theis JD, Quint P, et al. Renal amyloidosis associated with a novel sequence variant of gelsolin. Am J Kidney Dis. 2013;61:161–166. doi: 10.1053/j.ajkd.2012.07.016. [DOI] [PubMed] [Google Scholar]
- 83.de la Chapelle A, Tolvanen R, Boysen G, et al. Gelsolin-derived familial amyloidosis caused by asparagine or tyrosine substitution for aspartic acid at residue 187. Nat Gen. 1992;2:157–160. doi: 10.1038/ng1092-157. [DOI] [PubMed] [Google Scholar]