

Abstract
In patients with multiple myeloma, the heparan sulfate proteoglycan syndecan-1 (CD138) is shed from the surface of tumor cells and accumulates in the serum and within the extracellular matrix of the bone marrow where it promotes tumor growth and metastasis. In the present study we discovered that commonly used anti-myeloma drugs stimulate syndecan-1 shedding both in vitro and in animals bearing myeloma tumors. Enhanced shedding is accompanied by increased syndecan-1 synthesis prior to drug induced tumor cell death. Addition of a caspase inhibitor blocks the drug-induced shedding of syndecan-1 in vitro indicating that shedding is linked to the onset of apoptosis. ADAMs inhibitors or siRNA targeting ADAMs blocked drug-induced shedding suggesting that up regulation or activation of ADAMs is responsible for cleaving syndecan-1 from the tumor cell surface. These results reveal that myeloma chemotherapy stimulates synthesis and shedding of syndecan-1, a potentially negative side effect that may lead to accumulation of high levels of syndecan-1 to establish a microenvironment that nurtures relapse and promotes tumor progression. Interestingly, we also found that chemotherapeutic drugs stimulated syndecan-1 shedding from pancreatic cancer cells as well, indicating that drug-induced shedding of syndecan-1 may occur in many cancer types. Overall, our results indicate that use of metalloproteinase inhibitors (to inhibit syndecan-1 shedding) in combination with chemotherapy may represent a novel therapeutic strategy to prevent re-establishment of a microenvironment conducive for tumor relapse.
Keywords: Chemotherapy, multiple myeloma, shedding, shed syndecan-1, batimastat, hepatocyte growth factor
1. Introduction
Syndecan-1 is a transmembrane heparan sulfate proteoglycan expressed by numerous cell types (Bernfield et al., 1992). On the cell surface, syndecan-1 interacts with growth factor receptors and integrins to control cellular behaviors such as adhesion, invasion and signaling (Beauvais and Rapraeger, 2004). Syndecan-1 is also found in the nucleus of some cells where it can regulate gene transcription (Purushothaman et al., 2011). Syndecan-1 on the cell surface can be proteolytically cleaved, releasing the extracellular (ectodomain) core protein that bears both heparan sulfate and chondroitin sulfate chains (Bass et al., 2009). Cells shed syndecan-1 constitutively at low levels but shedding is accelerated in response to growth factors, chemokines, microbial toxins, insulin and cellular stress (Hayashida et al., 2010). These different stimuli trigger several signaling pathways that elevate protease activity to drive syndecan-1 shedding. Known as sheddases, these proteases include matrix metalloproteinases (MMPs), membrane-type MMPs, ADAMs (A disintegrin and metalloproteinase) and ADAMTS (ADAMs with thrombospondin motifs) (Manon-Jensen et al., 2010). In addition, phosphorylation of the syndecan-1 cytoplasmic domain by protein tyrosine kinases (Reiland et al., 1996), dissociation of Rab5 from the syndecan-1 cytoplasmic domain (Hayashida et al., 2008) or reduction of glycosaminoglycan content, also trigger syndecan-1 shedding (Ramani et al., 2012).
Shed syndecan-1 is present at high levels in some disease states including inflammation, microbial infections, and cancer (Choi et al., 2010; Manon-Jensen et al., 2010). In murine models of inflammation, high levels of shed syndecan-1 direct neutrophil migration, chemokine signaling and the subsequent disease outcome (Kliment et al., 2009; Li et al., 2002). In microbial infections, proteins secreted by pathogens elevate shed syndecan-1 (Park et al., 2001) which in turn inhibits host-derived antimicrobial peptides and promote infection (Park et al., 2000). In multiple myeloma, high levels of tumor-derived shed syndecan-1 drive growth, metastasis and tumor angiogenesis (Purushothaman et al., 2010; Ramani et al., 2013; Yang et al., 2005). Moreover, following therapy, high levels of shed syndecan-1 become trapped with the bone marrow extracellular matrix of myeloma patients (Bayer-Garner et al., 2001). Similar to myeloma, high levels of syndecan-1 expression and shedding have also been reported for pancreatic cancer (Ding et al., 2005). In breast cancer, shed syndecan-1 is shown to support tumor proliferation (Su et al., 2007) (Nikolova et al., 2009). Shed syndecan-1 wields its powerful effect on tumor behavior primarily through conditioning the tumor microenvironment. By enhancing growth factor signaling within host cells (e.g., endothelial cells and osteoblasts), shed syndecan-1 primes the stroma to foster rampant tumor growth (Purushothaman et al., 2010; Ramani et al., 2011). Shed syndecan-1 is a reliable prognostic marker in some cancers. For example, a high level of shed syndecan-1 in the serum of lung cancer and myeloma patients correlates with a poor outcome (Joensuu et al., 2002; Seidel et al., 2000). In addition, tumor and stromal syndecan-1 expression is also a strong predictor of poor response to chemotherapy (Gotte et al., 2006; Tokes et al., 2009).
Our present understanding of mechanisms driving syndecan-1 shedding in cancers is limited. It has been demonstrated that FGF-2 triggers MMP-7 mediated shedding of syndecan-1 (Ding et al., 2005) and that heparanase enhances shedding of syndecan-1 in part to due up regulation of MMP-9 (Purushothaman et al., 2008). There is mounting evidence that chemotherapeutic drugs routinely used to treat cancer patients can result in release of factors that enhance tumor growth. In the present study we demonstrate that chemotherapy dramatically upregulates syndecan-1 shedding in vitro and in vivo. Therapy-generated shed syndecan-1 is biologically active and modulates growth factor signaling in functional assays. The elevated shedding in response to chemotherapy is observed for both myeloma and pancreatic cancer. Chemotherapy-driven syndecan-1 shedding requires caspase activity and additionally triggers an increase in syndecan-1 synthesis. Blocking the activity of different metalloproteinases, particularly ADAMs, during chemotherapy diminishes the generation of shed syndecan-1. Use of batimastat (BB-94), a broad-based metalloproteinase inhibitor during chemotherapy, abolished therapy-induced shedding. Further, both the synthesis and shedding of syndecan-1 are elevated in chemoresistant myeloma cells. These results suggest a mechanism whereby chemotherapy induced shedding of syndecan-1 might lead to establishment of a tumor microenvironment that supports tumor survival and relapse accompanied by robust tumor growth.
2. Results
2.1. Chemotherapy elevates the level of shed syndecan-1 in vitro and in vivo
In myeloma, high levels of shed syndecan-1 promote tumor growth, angiogenesis, and bone disease (Ramani et al., 2013). Previous work has shown that following chemotherapy, myeloma tumor cells exhibit a reduction in the level of cell surface syndecan-1 and in myeloma patients there is an increase in shed syndecan-1 bound within the bone marrow extracellular matrix following therapy (Bayer-Garner et al., 2001; Jourdan et al., 1998; Tagoug et al., 2013). These findings suggest that chemotherapy stimulates shedding of syndecan-1 which then binds to the bone marrow. To test this possibility, we treated different myeloma cell lines with proteoasome inhibitor bortezomib (Bort), a chemotherapeutic agent widely used in myeloma patients. Following treatment with Bort, a high level of syndecan-1 accumulated in the medium conditioned by myeloma cell lines (Fig. 1A). Irrespective of the basal level of shed syndecan-1 which varied across the different myeloma cell lines, [lowest – (OCI-My5 - 26.76 ± 3.78 ng/ml); highest – (MM1.S -123.00 ± 17.94 ng/ml)], treatment with Bort significantly elevated syndecan-1 shedding. To confirm that the syndecan-1 released in response to chemotherapy is indeed the shed form of the proteoglycan and not a full-length molecule released by dying tumor cells, we probed the conditioned medium after chemotherapy using a polyclonal antibody to syndecan-1 and also with an antibody that recognizes only the cytoplasmic domain of syndecan-1. Blots revealed that the syndecan-1 in conditioned medium lacks the cytoplasmic domain (Fig. 1B), proving that it is the shed form of the proteoglycan. The chemotherapy-induced increase in shed syndecan-1 was not exclusive to Bort and was observed with other chemotherapeutic drugs as well (Fig. 1C). Treatment with doxorubicin (10 μM), dexamethasone (100 μM), cisplatin (10 μM), and carfilzomib (50 nM) significantly elevated shed syndecan-1 (Fig. 1C). We next assessed whether chemotherapy had a similar effect on shed syndecan-1 in vivo. Treating mice bearing disseminated myeloma tumors with Bort dramatically increased the circulating level of shed syndecan-1, mimicking our in vitro results (Fig. 1D). To extend this finding to other cell types, Panc-1, a pancreatic cancer cell line and HS-5, a non-transformed bone marrow stromal cell line were treated with Bort and doxorubicin (Dox). While both these drugs significantly elevated the level of shed syndecan-1 in Panc-1 cells, only a modest effect was observed with HS-5 (Fig. 1E).
FIGURE 1. Chemotherapy elevates the level of shed syndecan-1 in vitro and in vivo.
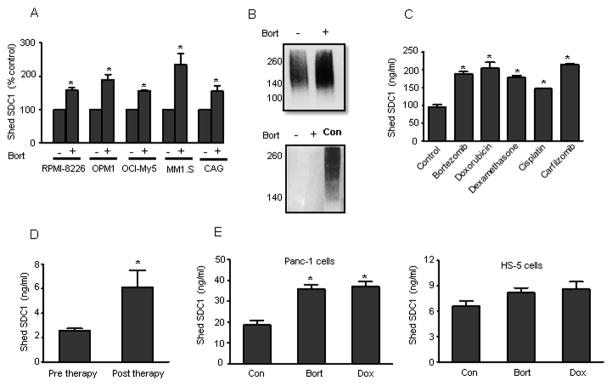
(A) 5 X 105 cells/ml of human myeloma cell lines (RPMI-8226, OPM1, OCI-My5, MM1.S, and CAG) were seeded in serum-free medium for 4 h and then bortezomib (100 nM) was added and incubated further for 16 h. Untreated cells served as the control. The level of shed syndecan-1 in the conditioned medium from each cell line was determined by ELISA. Data are mean ± S.E. of three independent experiments. *, p < 0.01 versus their respective untreated controls. (B) MM1.S cells were plated at a density of 5 X 105 cells/ml in serum free medium for 4 h and then bortezomib (100 nM) was added and incubated further for 16 h. Untreated cells served as the control. Conditioned medium from the two groups were collected separately and concentrated. Equal protein from concentrated conditioned media was analyzed by immunoblotting for syndecan-1 using either polyclonal antibody to syndecan-1 (upper panel) or an antibody that recognizes the cytoplasmic domain of syndecan-1 (lower panel). MM1.S total cell extracts were used as a positive control (control). (C) 5 X 105 cells/ml of MM1.S cells were seeded in serum-free medium for 4 h and then followed by treatment with chemotherapeutic agents for 16 h. Shed syndecan-1 levels in the media were determined. *, p < 0.05 versus untreated control. (D) Disseminated tumors in SCID mice (n=6) were established by intravenous injection of CAG, human myeloma cells. Sera were collected from mice before chemotherapy treatment (pre therapy) and 72 h later the mice were treated with bortezomib (1mg/kg) for 16 h and sera were collected again (post therapy). The level of shed syndecan-1 in sera was determined. Data are mean ± S.E. *, p < 0.001 versus pre therapy. (E) 5 X 105 cells of different origins: Panc-1 (pancreatic cancer), HS-5 (human bone marrow stromal cells), were seeded in serum-free medium for 4 h and then in the presence of bortezomib (1 uM) or doxorubicin (8 uM) for 16 h. Untreated cells of each cell line were included as control. At the end of incubation, the level of shed syndecan-1 in the conditioned medium was determined. Data are mean ± S.E. of three independent experiments. *, p < 0.005 versus untreated controls.
2.2. Response to chemotherapy enhances both syndecan-1 shedding and synthesis and is dependent on activation of caspase
We next examined more closely how chemotherapy-induced cytotoxicity in tumor cells elevates shed syndecan-1. We treated MM1.S cells with increasing doses of Bort and Dox. Interestingly, only concentrations that affected cell viability (Bort - 5 nM, Dox - 4 uM) after 16 h of treatment, significantly elevated the level of shed syndecan-1 (Fig. 2A), suggesting that the process of tumor cell killing by chemotherapy leads to a burst of syndecan-1 shedding. Previous work has demonstrated that triggering syndecan-1 shedding in myeloma causes an increase in synthesis of syndecan-1 (Ramani et al., 2012; Yang et al., 2007). To determine if the enhanced shedding was accompanied by an increase in syndecan-1 synthesis we examined the level of syndecan-1 transcript 8 h after initial exposure to drug. At this time point tumor cell viability has not been reduced by drug treatment (data not shown). Results indicate a significant increase in syndecan-1 synthesis following treatment with either Bort or Dox (Fig. 2B). Together these data indicate that an early response to chemotherapy is enhanced syndecan-1 synthesis.
FIGURE 2. Tumor cell response to chemotherapy leads to increased shedding and synthesis of syndecan-1 that is dependent on caspase activation.
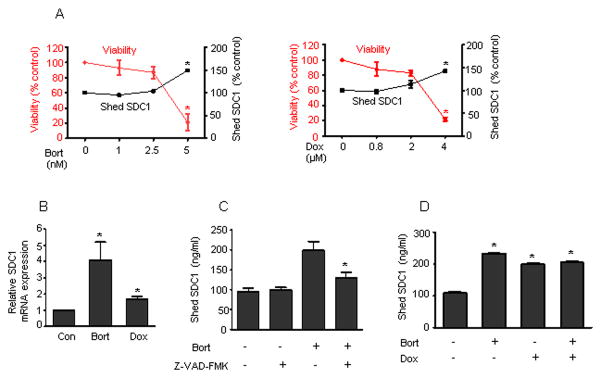
(A) MM1.S cells were plated at a density of 5 X 105 cells/ml in serum free medium for 16 h with different concentrations of bortezomib (1, 2.5, 5 nM) or doxorubicin (0.8, 2, 4 uM). Cell viability was assessed by MTT assay and the level of shed syndecan-1 was determined. Untreated MM1.S cells served as the control. The experiments were performed in quadruplicate and the data presented are mean ± S.E. of three independent experiments. *, p < 0.005 versus untreated controls. (B) MM1.S cells were seeded in serum free medium for 8 h in the presence of bortezomib (5 nM) or doxorubicin (4 uM). SDC1 mRNA transcript from each sample was determined by real time PCR and normalized against a standard housekeeping gene (18S rRNA). Syndecan-1 transcript levels in untreated cells served as the control. Data are mean ± SE of three independent experiments. *p < 0.01 versus untreated control. (C) 5 X 105 MM1.S cells were seeded in serum free medium for 6 h in the presence of DMSO or caspase inhibitor Z-VAD-FMK (1 uM). At the end of incubation, bortezomib (100 nM) was added and incubated for 16 h. The level of shed syndecan-1 in the conditioned medium was then determined. Data are mean ± S.E. of three independent experiments. *, p < 0.05 versus bortezomib treated cells. (D) MM1.S cells were seeded in serum free medium for 4 h and then bortezomib (100 nM) and/or doxorubicin (8 uM) were added and further incubated for 16h. The level of shed syndecan-1 in the conditioned medium was determined. Untreated cells served as the control. Data are mean ± S.E. of three independent experiments. *, p < 0.01 versus untreated control.
Caspase activation leading to cell death is a major mechanism of action, common to numerous chemotherapeutic drugs. To test a role for caspase, we treated MM1.S cells with Bort either in the presence or absence of Z-VAD-FMK, a pan caspase inhibitor. Blocking caspase activity significantly reduced chemotherapy-induced shedding of syndecan-1 (Fig. 2C). This indicates that syndecan-1 shedding is not a direct, immediate response to drug, but rather occurs once the cell enters the process leading to cell death. We next probed whether different chemotherapeutic drugs were acting through identical or distinct mechanisms to elevate shedding of syndecan-1. Bort and Dox are drugs belonging to entirely different families of therapeutic agents and mediate tumor cell killing via distinct mechanisms (Carvalho et al., 2009; Paramore and Frantz, 2003). However, adding the two drugs together did not have an additive effect on shedding of syndecan-1 (Fig. 2D), suggesting that these chemotherapeutic drugs share a common pathway to trigger shedding.
2.3. Chemotherapy-induced syndecan-1 shedding is mediated predominantly by ADAMs
Studies have shown that chemotherapeutic drugs elevate the expression and activity of proteases to drive the shedding of cell surface molecules (Goffin et al., 2010; Kyula et al., 2010; Vahdat et al., 2010). To identify the sheddases involved in generating shed syndecan-1 during therapy, we treated myeloma cell lines MM1.R and MM1.S with bortezomib either in the presence or absence of DMSO, ADAM10 inhibitor – GI254023X (GI), ADAM inhibitor – TAPI-0, and a broad metalloproteinase inhibitor – batimastat (BB-94). All three inhibitors significantly blocked the basal shedding of syndecan-1 in both the myeloma cell lines tested (Fig. 3A). Blocking ADAM10 and especially ADAM family of proteases significantly decreased the level of shed syndecan-1 generated by Bort (Fig. 3A). In both the cell lines, treatment with BB-94, a broad based metalloproteinase inhibitor, completely blocked shed syndecan-1 generation triggered by Bort (Fig. 3A). Treatment with chemotherapy has been shown to decrease the level of cell surface syndecan-1 (Jourdan et al., 1998; Tagoug et al., 2013). To confirm that it is the process of shedding that causes the decrease in cell surface syndecan-1 post-chemotherapy, we treated myeloma cells with 5 nM Bort (a concentration that significantly elevates shed syndecan-1) (Fig. 2A) in the presence or absence of BB-94. Treatment with Bort decreased myeloma cell surface syndecan-1, whereas in the presence of Bort and BB-94 syndecan-1 was retained at the cell surface (Fig. 3B). To further confirm the role of ADAM family of proteases, MM1.S cells were co-transfected with siRNA targeting both ADAM10 and ADAM17. PCR analysis demonstrated a 61.2 ± 4.5 % decrease in ADAM10 expression and 49.8 ± 4.4% decrease in ADAM17 transcript levels (n=3 experiments) compared to cells transfected with scrambled non-targeting siRNA. Targeting both these proteases simultaneously significantly decreased Bort-induced shedding of syndecan-1 (Fig. 3C). In contrast to myeloma, blocking ADAM10 alone failed to block Bort-induced syndecan-1 shedding in pancreatic cancer cells (Fig. 3D), though pancreatic cancer cells are shown to express ADAM10 (Gaida et al., 2010). Similar to myeloma, both TAPI-0 and BB-94 significantly blocked the effect of Bort on syndecan-1 shedding (Fig. 3D) in pancreatic cancer cells. This data demonstrates that the repertoire of proteases involved in chemotherapy-induced syndecan-1 shedding varies among different cancers.
FIGURE 3. Chemotherapy-driven shedding of syndecan-1 is mediated predominantly by ADAMs.

(A) 5 X 105 MM1.S or MM1.R cells were seeded in serum free medium for 6 h in the presence of DMSO, GI (10 μM), TAPI (10 μM), or BB-94 (5 μM) separately and then treated with bortezomib (100 nM) for further 16 h. At the end of incubation, the level of shed syndecan-1 in the conditioned medium was determined. Data are mean ± S.E. of three independent experiments. *, p < 0.005 versus untreated control, #, p < 0.005 versus bortezomib + DMSO treated group. (B) Cell surface syndecan-1 on MM1.S after different treatments was analyzed by flow cytometry using anti-syndecan-1 antibody: bortezomib (dotted line), bortezomib + batimastat (solid line), untreated (dashed line). Controls included cells stained with IgG control antibody (shaded). (C) MM1.S cells were transfected with either control siRNA or co-transfected with ADAM17 and ADAM10 siRNA and incubated overnight. The transfected cells were then treated with bortezomib (100 nM) for 14 h. At the end of incubation, the level of shed syndecan-1 in the conditioned medium was determined. Data are mean ± S.E. of three independent experiments. *, p < 0.05 versus untreated cells transfected with control siRNA, #, p < 0.005 versus cells transfected with control siRNA and treated with bortezomib. (D) 5 X 105 Panc-1 cells were seeded in serum free medium for 6 hours in the presence of DMSO, GI (10 μM), TAPI (10 μM), or BB94 (5 μM) and then treated with bortezomib (100 nM) for further 16 h. At the end of incubation, the level of shed syndecan-1 in the conditioned medium was determined. Data are mean ± S.E. of three independent experiments. *, p < 0.005 versus untreated cells, #, p < 0.005 versus bortezomib + DMSO treated group.
2.4. Chemotherapy-generated shed syndecan-1 enhances growth factor activity
To determine if the shed syndecan-1 that is induced by chemotherapy retains its biological activity, we used an established bioassay that measures the ability of syndecan-1 to enhance hepatocyte growth factor (HGF)-induced secretion of IL-11 in osteoblast-like Saos-2 cells (Hjertner et al., 1999). Conditioned media from untreated or Bort treated MM1.S cells do not have any appreciable levels of HGF (unpublished observations) and therefore did not stimulate IL-11 secretion from Saos-2 cells in absence of exogenous HGF (Fig. 4A, lanes 3, 4). HGF added to conditioned medium from Bort-treated cells (Bort CM) stimulated significantly higher levels of IL-11 secretion than did HGF when added to conditioned medium from untreated cells (Con CM) (Fig. 4A, lanes 5, 6). To determine whether the shed syndecan-1 in the Bort CM is responsible for enhancing HGF activity, i) we immunodepleted syndecan-1 from Bort CM, which dramatically reduced IL-11 production (Fig. 4A, lanes 7, 8), ii) as an additional confirmation, Bort CM was treated with heparitinase (HepIII), a bacterial enzyme that extensively degrades the heparan sulfate chains on syndecan-1 and thereby blocks its ability to modulate HGF activity (Ramani et al., 2011). HGF added to Hep III treated Bort CM produced significantly less IL-11 than HGF mixed with untreated Bort CM (Fig. 4A, lanes 6, 9). These data collectively demonstrate that the shed syndecan-1 in the conditioned medium from chemotherapy treated cells is biologically active and can regulate HGF function. This data further demonstrates the ability of chemotherapy to modulate growth factor activity via shed syndecan-1. As blocking metalloproteinases diminishes the increase in shed syndecan-1 accompanying chemotherapy (Fig. 3A), addition of TAPI or BB-94 alongside bortezomib decreases the ability of chemotherapy to modulate HGF activity and in turn IL-11 production (Fig. 4B).
FIGURE 4. Chemotherapy-induced shed syndecan-1 is biologically active.
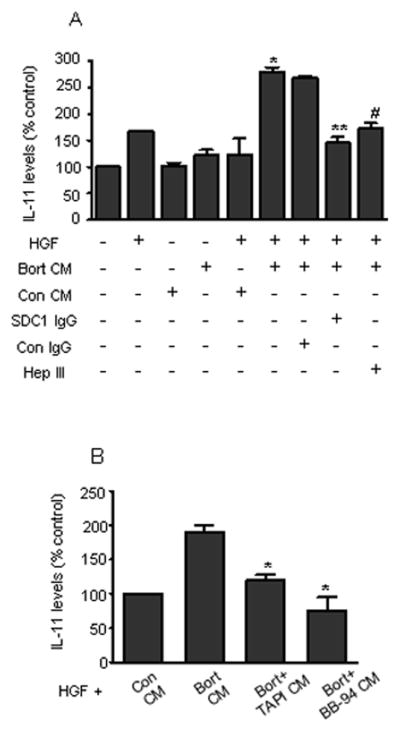
(A) Serum-free media conditioned for 16 h by MM1.S cells either untreated (Con CM – having low shed syndecan-1) or treated with 100 nM bortezomib (Bort CM – having high level of shed syndecan-1) were used in this experiment. Further, conditioned medium from bortezomib treated cells was immunodepleted of syndecan-1 using specific antibody (SDC1 IgG) or treated with isotype control antibody (Con IgG). A separate aliquot of Bort CM was treated with heparitinase (Hep III), a bacterial enzyme that extensively degrades heparan sulfate chains. The treated and untreated conditioned media were then added to Saos-2 cells in culture either alone or along with 1 ng HGF and further incubated for 24 h. At the end of incubation, medium from Saos-2 was removed and the level of IL-11 secreted by the Saos-2 cells was determined. Data are mean ± S.E. of three independent experiments. * p < 0.01 versus Con CM+HGF, ** p< 0.01 versus Bort CM treated with Con IgG and HGF, # p<0.05 versus Bort CM+HGF. (B) MM1.S cells were plated at a density of 5 X 105 cells/ml in serum free medium for 16 h. Different groups included cells that were either left untreated or treated with bortezomib (100 nM) alone or with bortezomib and either TAPI-0 (10 μM) or BB-94 (5 μM). Conditioned medium from different groups was collected and added to Saos-2 cells in culture along with 1 ng HGF for 24 h. At the end of incubation, the levels of IL-11 secreted by the Saos-2 cells were determined. Data are mean ± S.E. of three independent experiments. * p < 0.01 versus Bort CM+HGF.
2.5. Chemoresistant myeloma cells express high levels of shed syndecan-1
Because chemotherapy elevated syndecan-1 shedding, we tested whether cells that have acquired resistance to therapy have an elevated level of syndecan-1 shedding. Results revealed that the level of shed syndecan-1 in the conditioned medium from chemoresistant myeloma cell lines RPMI-DOX4 (doxorubicin resistant) and RPMI-LR5 (melphalan resistant) is significantly higher than their chemosensitive parental cell line, RPMI-WT (Fig. 5A). In spite of a difference in shed syndecan-1, the level of cell surface syndecan-1 was almost identical between the three cell lines (Fig. 5B). Testing for total metalloproteinase activity revealed ~2 fold high protease activity in RPMI-LR5 compared to RPMI-WT (Fig. 5C). The level of syndecan-1 transcript in RPMI-LR5 cells was also significantly higher than RPMI-WT cells (Fig. 5D). Elevated syndecan-1 synthesis in RPMI-LR5 cells might explain how these cells maintain a constant level of cell surface syndecan-1 comparable to RPMI-WT in spite of high shedding. Treating RPMI-LR5 cells with TAPI or BB-94 significantly blocked syndecan-1 shedding (Fig. 5E). Though RPMI-LR5 and RPMI-DOX4 are resistant to melphalan and doxorubicin respectively, they are sensitive to bortezomib. Treating the two chemoresistant cell lines with bortezomib elevated shed syndecan-1 significantly (Fig. 5F), demonstrating that chemoresistant cells respond to other chemotherapeutic agents by accelerating syndecan-1 shedding.
FIGURE 5. Chemoresistant myeloma cell lines synthesize and shed high levels of syndecan-1.
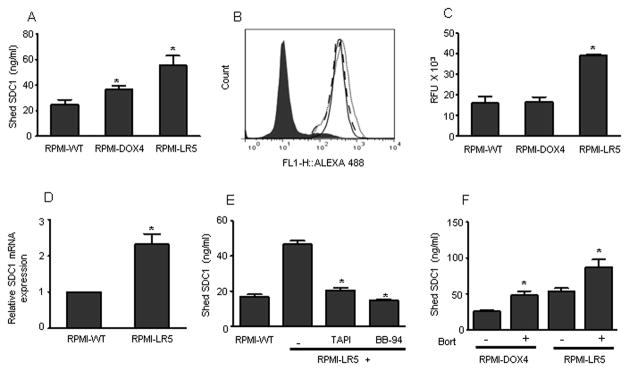
(A) 5 X 105 RPMI-WT parental cells and the chemoresistant cell lines RPMI-DOX4 and RPMI-LR5 were seeded in serum free medium for 16 h. At the end of incubation, the level of shed syndecan-1 in the conditioned medium was determined. Data are mean ± S.E. of three independent experiments. *, p < 0.05 versus RPMI-WT. (B) Cell surface expression of syndecan-1 on each cell line was determined by staining with BA38 antibody and analyzed by flow cytometry: IgG control antibody (shaded), RPMI-WT (dotted line), RPMI-DOX4 cells (solid line), RPMI-LR5 cells (dashed line). (C) 5 X 105 RPMI-WT cells and the chemoresistant cell lines RPMI-DOX4 and RPMI-LR5 were seeded in HBSS buffer and the total metalloproteinase activity was measured after adding flurogenic substrate, Mca-KPLGL-Dpa-AR-NH2. Data are mean ± S.E. of three independent experiments. *, p < 0.005 versus RPMI-WT. (D) RPMI-WT and RPMI-LR5 cells were seeded in serum free medium for 8 h. At the end of incubation, the level of syndecan-1 mRNA transcript from each sample was determined by real time PCR and normalized against a standard housekeeping gene (18S rRNA). Data are mean ± S.E. of three independent experiments. *p<0.005 versus RPMI-WT. (E) 5 X 105 RPMI-WT or RPMI-LR5 cells were seeded in serum free medium and incubated for 16 h. Treatment groups included RPMI-LR5 cells incubated with TAPI (10 μM) or BB-94 (5 μM) separately. At the end of incubation, the level of shed syndecan-1 in the conditioned medium was determined. Data are mean ± S.E. of three independent experiments. *, p < 0.005 versus untreated RPMI-LR5 cells. (F) 5 X 105 cells/ml of RPMI-DOX4 or RPMI-LR5 were seeded in serum-free medium for 4 h and then in the presence of bortezomib (100 nM) for 16 h. Untreated cells served as control. The level of shed syndecan-1 in the conditioned medium from each cell line was determined. Data are mean ± S.E. of three independent experiments. *, p < 0.01 versus their untreated control.
3. Discussion
The present study reveals that exposure of myeloma tumor cells to chemotherapeutic drugs widely used in myeloma patients potently stimulates shedding of syndecan-1. We found that i) chemotherapeutic drugs elevate syndecan-1 shedding and synthesis by myeloma tumor cells, ii) blocking metalloproteinases inhibits chemotherapy-induced syndecan-1 shedding, iii) chemotherapy-induced shed syndecan-1 is biologically active, and iv) myeloma tumor cells resistant to chemotherapy synthesize and shed high levels of syndecan-1. These findings are important because shed syndecan-1 is known to actively promote myeloma tumor growth, angiogenesis, and metastasis (Ramani et al., 2013) and thus chemotherapy induced shedding of syndecan-1 likely supports and drives the onset of highly aggressive disease.
Although it was previously demonstrated that chemotherapeutic drugs can cause the loss of syndecan-1 from the cell surface (Jourdan et al., 1998; Tagoug et al., 2013), it was not known if this loss was due to a reduction in syndecan-1 synthesis, an increase in syndecan-1 degradation or due to stimulation of syndecan-1 shedding. Our results reveal that the decrease in cell surface syndecan-1 post-chemotherapy is due, at least in part, to enhanced shedding, driven by metalloproteinases. The decrease in cell surface syndecan-1 is not due to a decrease in synthesis of the proteoglycan, as our data demonstrates that in fact syndecan-1 synthesis increases post chemotherapy (Fig. 2B). This observation is in line with our prior work demonstrating that enhancing shedding elevates syndecan-1 synthesis (Ramani et al., 2012; Yang et al., 2007). However, even among drugs that stimulate similar levels of shedding (e.g., bortezomib and doxorubicin); the corresponding increase in syndecan-1 core protein synthesis was strikingly different (Fig 2B and 2D). This suggests that some drugs may uncouple the mechanism linking syndecan-1 shedding and synthesis.
Syndecan-1 shed in response to chemotherapy remains biologically active and is capable of enhancing HGF activity as determined by functional assays (Fig. 4A). We found that shed syndecan-1 enhanced HGF/c-met signaling to stimulate IL-11, a key regulator of skeletal biology. This indicates that a consequence of chemotherapy-driven shedding could be an amplification of the HGF/c-met/IL-11 axis by shed syndecan-1, worsening the bone disease in myeloma. The finding that shed syndecan-1 can enhance HGF activity is also important because HGF, which can enhance myeloma tumor cell proliferation and inhibit apoptosis, is the most highly expressed chemokine present in many myeloma patients (Borset et al., 1996; Derksen et al., 2003; Zhan et al., 2002). In addition to its role in potentiating HGF activity, shed syndecan-1 also enhances VEGF signaling and thereby accelerates angiogenesis (Lamorte et al., 2012; Purushothaman et al., 2010).
Tumor cells that survive chemotherapy cause relapse and the eventual death of the patient (Lonial et al., 2011). These surviving tumor cells often emerge as highly aggressive cells that fuel rapid disease progression. Interestingly, we find that myeloma cell lines selected for their ability to resist chemotherapy shed higher levels of syndecan-1 than their non-chemoresistant counterparts (Fig. 5A). Thus, in addition to chemotherapy causing an initial burst of syndecan-1 shedding, the cells that survive chemotherapy and continue to grow exhibit elevated levels of shedding. This enhanced shedding likely contributes to the rapid disease progression seen following reemergence of a tumor after chemotherapy. We have previously demonstrated that high levels of syndecan-1 are present in fibrotic bone marrow of myeloma patients post-treatment (Bayer-Garner et al., 2001). This likely is the result of shed syndecan-1 binding to bone marrow extracellular matrix molecules such as collagens and fibronectin. We speculate that this trapped syndecan-1, via its heparan sulfate chains, binds to and retains growth factors thereby providing an enriched microenvironment to nurture tumor relapse. Thus, to minimize the amount of syndecan-1 present within the bone marrow microenvironment, our findings suggest that metalloproteinase inhibitors (to inhibit syndecan-1 shedding) could be used in combination with standard myeloma chemotherapy. This combination could significantly dampen tumor relapse and prevent aggressive progression of this cancer.
4. Experimental procedures
4.1. Cell Lines and Reagents
Myeloma cell lines RPMI-8226, MM1.S, MM1.R, OCI-My5, CAG, and OPM1 were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS). The cell lines utilized and the investigator who kindly provided the lines were: MM1.S and MM1.R cells (Drs. Nancy Krett and Steven Rosen, Northwestern University), OCI-My5, OPM1 (Dr. Fenghuang Zhan, University of Iowa), chemoresistant cell lines RPMI-LR5 (Dr. William Dalton, Moffitt Cancer Center), RPMI-DOX4 (Dr. Erming Tiam, University of Arkansas for Medical Sciences), CAG (Dr. Joshua Epstein, University of Arkansas for Medical Sciences), Panc-1 (Dr. Lacey McNally, University of Alabama at Birmingham), and Saos-2 (Dr. Majd Zayzafoon, University of Alabama at Birmingham). RPMI-8226 and HS-5 were obtained from the American Type Culture Collection (Manassas, VA). Myeloma cell lines were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum. Panc-1, Saos-2 and HS-5 were cultured in DMEM supplemented with 10% FBS. Bortezomib, cisplatin, and carfilzomib were from SelleckChem. (Houston, TX). ADAM10 inhibitor, GI254023X was a kind gift from Dr. Andreas Ludwig, Aachen, Germany. Doxorubicin and dexamethasone were from Sigma-Aldrich (St Louis, MO). Caspase inhibitor (Z-VAD-FMK), and ADAM inhibitor (TAPI-0) were from Calbiochem (San Diego, CA), MMP substrate (Mca-KPLGL-Dpa-AR-NH2) was from R&D systems (Minneapolis, MN).
4.2. ADAM17 and ADAM10 siRNA transfection
Prior to transfection of siRNA, MM1.S cells were plated in fresh complete medium and grown for 24 h. The cells were then transfected with Trilencer-27-siRNA (Origene, Rockville, MD) targeting ADAM10 (ID 11487), ADAM17 (ID 6868) or control siRNA (Trilencer-27 universal scrambled negative control siRNA) using Amaxa NucleofectorTM (Amaxa, Cologne, Germany), program S-023 and buffer V. The transfected cells were then incubated in complete medium overnight prior to use in experiments. Decrease in ADAM10 and ADAM10 transcript levels after siRNA transfection was confirmed by real-time PCR as described (Pruessmeyer et al., 2010).
4.3. Flow Cytometry
For flow cytometry, cells were collected by centrifugation and washed twice in phosphate-buffered saline (PBS). Cell pellets were resuspended in anti-human syndecan-1 antibody, clone BA-38 (Cell Sciences, Canton, MA) diluted in PBS and incubated at 4°C for 1 h. Cells were then washed three times in PBS and incubated with secondary antibody conjugated to Alexa Fluor 448 or Alexa Fluor 647 (Invitrogen, Carlsbad, CA) at 4°C for 1 h. Following incubation, the cells were washed three times in PBS and fixed with 4% paraformaldehyde prior to analysis. Cells stained with appropriate isotype-matched IgG antibody served as control.
4.4. Animal studies
CB.17 scid/scid mice obtained from Charles River Breeding Laboratories (Wilmington, MA) were housed and monitored in the animal facility at UAB. Mice were maintained in laminar flow rooms with constant temperature and humidity and monitored regularly. All the animals were handled as per protocols and procedures approved by Animal Care and Use Committee of UAB. Briefly, human myeloma cells, CAG (3 million/mice) were injected intravenously. Three weeks after injection, sera were collected from mice and stored at −80°C. 72 h later Bort (2 mg/kg) was delivered intravenously. 16 h after Bort treatment, sera were collected again. Using specific ELISA, levels of human immunoglobulin κ light chain (Bethyl Laboratories, Montgomery, TX) and syndecan-1 (Cell Sciences, Norwood, MA) in sera were measured following the manufacturer’s protocol.
4.5. MTT assay
MM1.S cells were seeded at a concentration of 2.5 X 104 cells/well in a 96 well plate either treated with different concentrations of Bort or Dox for 16 h. Untreated cells served as control. Cell viability was measured using CytoTox 96® Non-Radioactive Cytotoxicity Assay (Promega Corporation, Madison, WI) as per manufacturer’s instructions.
4.6. IL-11 bioassay
Saos-2 cells were seeded in 24-well plates at 5 X 104 cells per well in complete growth medium. After overnight incubation, the monolayer was washed once with sterile PBS prior to treatment. In most of the experiments, treated or untreated conditioned media were mixed with an equal volume of RPMI 1640 media and incubated with the pre-washed Saos-2 monolayer for at least 24 h. At the end of the incubation, conditioned media from Saos-2 monolayers were collected and centrifuged to remove cell debris, and the levels of IL-11 were determined using human IL-11 DuoSet ELISA (R&D Systems, Minneapolis, MN) as per the manufacturer’s instructions. To determine the role of shed syndecan-1 in conditioned medium from Bort treated cells, shed syndecan-1 in the conditioned medium was immunodepleted using 2 μg/ml polyclonal anti-human syndecan-1 antibody (R&D Systems) or goat IgG as a control, bound to protein G-Sepharose beads. Heparan sulfate chains from the shed syndecan-1 were removed by addition of 5 milliunits/ml heparinase III (Seikagaku, Kogyo, Japan) at 37°C for 2 h. Conditioned media after different treatments were mixed with 1 ng of recombinant human HGF (R&D Systems) and incubated for 15 minutes prior to addition to Saos-2 cells in culture, and incubated for 24 h.
4.7. Western blotting
Cells were incubated in serum free medium either untreated or in the presence of Bort (100 nM) for 16 h. Conditioned medium was collected; cells and cellular debris were separated by centrifuging the conditioned medium at 1000 rpm for 5 min. Conditioned medium was concentrated using Centriplus columns with a 30-kDa cutoff value (Millipore Corp., Bedford, MA). Protein concentration of concentrated medium was quantified by a BCA protein assay reagent kit (Pierce, Rockford, IL). Equal amounts of protein were loaded onto 4–20% gradient SDS-polyacrylamide gels (Bio-Rad, Richmond, CA), transferred to a positively charged nylon membrane (Nytran SPC, Schleicher & Schuell, Keene, NH), and probed with either polyclonal goat anti-human syndecan-1 (R&D Systems) or cytoplasmic domain specific anti-SDC1 antibody (Biovision, Mountain View, CA) followed by horseradish peroxidase-conjugated secondary anti-mouse antibody (GE Healthcare, Piscataway, NJ). Immunoreactive bands were detected using enhanced chemiluminescence (GE Healthcare).
4.8. Preparation of conditioned medium
In all experiments, cells were seeded and incubated in complete medium 24 h prior to being tested. On the day of experiments, cells were washed in serum-free medium (SFM) twice and incubated in SFM prior to further experimentation. For adherent cells (HS-5, Panc-1), equal numbers were seeded in complete growth medium in a 12-well plate. After overnight incubation, the monolayers were washed with serum-free medium at least twice prior to addition of chemotherapeutic drugs.
4.9. RNA Extraction and Real-time PCR for human syndecan-1
5 X 105 MM1.S cells were seeded in serum free medium and incubated for 8 h either in the presence of Bort or Dox. Untreated cells served as control. At the end of the incubation, cells were washed once with PBS and RNA was extracted using RNeasy columns (Qiagen, Valencia, CA). One microgram of total RNA was reverse transcribed to cDNA using Maxima® First Strand cDNA synthesis kit for RT-qPCR (Thermo scientific, Rockford, IL) as per the manufacturer’s protocol. 25 ng of diluted cDNA was mixed with 2X IQ™ SYBR® green supermix (Bio-Rad) along with gene specific primers (200 nM final concentration). The primers used were 5′-TCTGACAACTTCTCCGGCTC-3′ (forward) and 5′-CCACTTCTGGCAGGACTAC A-3′ (reverse) for syndecan-1 and 5′-CGGCGACGACCCATTCGAAC-3′ (forward) and 5′-GAAT CGAACCCTGATTCCCCGT C-3′ (reverse) for 18S rRNA. The cycle parameters included initial denaturation at 95 °C for 3 min followed by 45 cycles of 95 °C for 10 s and 55 °C for 60 s, followed by one cycle of 95 °C for 60 s and 55 °C for 60 s. To ensure specific amplification, a melt curve was generated at the end of PCR for each sample. The PCR cycle at which the fluorescence exceeded a set threshold (CT), for each sample was determined by the iCycler software. Data were analyzed according to the comparative CT method, as described previously (Schmittgen and Livak, 2008), using internal control (18S rRNA) transcript levels to normalize differences in sample loading and preparation.
4.10. Syndecan-1 ELISA
The level of shed syndecan-1 in the conditioned medium after different treatments was assessed by an ELISA using an Eli-pair kit specific for human syndecan-1 core protein (Cell Sciences). The standard curve was linear between 8 and 256 ng/ml, and all samples were diluted to concentrations within that range. All of the samples were run in duplicate.
Acknowledgments
This work was supported by NIH CA135075 and CA138340 to RDS. We thank Dr. Jianbo He for help with in vivo experiments. We also thank the Rheumatic Diseases Core Center and the Comprehensive Flow Cytometry Core (funded by -P30 AR48311).
Abbreviations
- SDC1
Syndecan-1
- Bort
Bortezomib
- Dox
Doxorubicin
- HGF
Hepatocyte growth factor
- IL-11
Interleukin-11
References
- Bass MD, Morgan MR, Humphries MJ. Syndecans shed their reputation as inert molecules. Sci Signal. 2009;2:pe18. doi: 10.1126/scisignal.264pe18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer-Garner IB, Sanderson RD, Dhodapkar MV, Owens RB, Wilson CS. Syndecan-1 (CD138) immunoreactivity in bone marrow biopsies of multiple myeloma: shed syndecan-1 accumulates in fibrotic regions. Mod Pathol. 2001;14:1052–1058. doi: 10.1038/modpathol.3880435. [DOI] [PubMed] [Google Scholar]
- Beauvais DM, Rapraeger AC. Syndecans in tumor cell adhesion and signaling. Reprod Biol Endocrinol. 2004;2:3. doi: 10.1186/1477-7827-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernfield M, Kokenyesi R, Kato M, Hinkes MT, Spring J, Gallo RL, Lose EJ. Biology of the syndecans: a family of transmembrane heparan sulfate proteoglycans. Annu Rev Cell Biol. 1992;8:365–393. doi: 10.1146/annurev.cb.08.110192.002053. [DOI] [PubMed] [Google Scholar]
- Borset M, Hjorth-Hansen H, Seidel C, Sundan A, Waage A. Hepatocyte growth factor and its receptor c-met in multiple myeloma. Blood. 1996;88:3998–4004. [PubMed] [Google Scholar]
- Carvalho C, Santos RX, Cardoso S, Correia S, Oliveira PJ, Santos MS, Moreira PI. Doxorubicin: the good, the bad and the ugly effect. Curr Med Chem. 2009;16:3267–3285. doi: 10.2174/092986709788803312. [DOI] [PubMed] [Google Scholar]
- Choi S, Lee H, Choi JR, Oh ES. Shedding; towards a new paradigm of syndecan function in cancer. BMB Rep. 2010;43:305–310. doi: 10.5483/bmbrep.2010.43.5.305. [DOI] [PubMed] [Google Scholar]
- Derksen PW, de Gorter DJ, Meijer HP, Bende RJ, van Dijk M, Lokhorst HM, Bloem AC, Spaargaren M, Pals ST. The hepatocyte growth factor/Met pathway controls proliferation and apoptosis in multiple myeloma. Leukemia. 2003;17:764–774. doi: 10.1038/sj.leu.2402875. [DOI] [PubMed] [Google Scholar]
- Ding K, Lopez-Burks M, Sanchez-Duran JA, Korc M, Lander AD. Growth factor-induced shedding of syndecan-1 confers glypican-1 dependence on mitogenic responses of cancer cells. J Cell Biol. 2005;171:729–738. doi: 10.1083/jcb.200508010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaida MM, Haag N, Gunther F, Tschaharganeh DF, Schirmacher P, Friess H, Giese NA, Schmidt J, Wente MN. Expression of A disintegrin and metalloprotease 10 in pancreatic carcinoma. Int J Mol Med. 2010;26:281–288. doi: 10.3892/ijmm_00000463. [DOI] [PubMed] [Google Scholar]
- Goffin L, Seguin-Estevez Q, Alvarez M, Reith W, Chizzolini C. Transcriptional regulation of matrix metalloproteinase-1 and collagen 1A2 explains the anti-fibrotic effect exerted by proteasome inhibition in human dermal fibroblasts. Arthritis Res Ther. 2010;12:R73. doi: 10.1186/ar2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotte M, Kersting C, Ruggiero M, Tio J, Tulusan AH, Kiesel L, Wulfing P. Predictive value of syndecan-1 expression for the response to neoadjuvant chemotherapy of primary breast cancer. Anticancer Res. 2006;26:621–627. [PubMed] [Google Scholar]
- Hayashida K, Bartlett AH, Chen Y, Park PW. Molecular and cellular mechanisms of ectodomain shedding. Anat Rec. 2010;293:925–937. doi: 10.1002/ar.20757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashida K, Stahl PD, Park PW. Syndecan-1 ectodomain shedding is regulated by the small GTPase Rab5. J Biol Chem. 2008;283:35435–35444. doi: 10.1074/jbc.M804172200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjertner O, Torgersen ML, Seidel C, Hjorth-Hansen H, Waage A, Borset M, Sundan A. Hepatocyte growth factor (HGF) induces interleukin-11 secretion from osteoblasts: a possible role for HGF in myeloma-associated osteolytic bone disease. Blood. 1999;94:3883–3888. [PubMed] [Google Scholar]
- Joensuu H, Anttonen A, Eriksson M, Makitaro R, Alfthan H, Kinnula V, Leppa S. Soluble syndecan-1 and serum basic fibroblast growth factor are new prognostic factors in lung cancer. Cancer Res. 2002;62:5210–5217. [PubMed] [Google Scholar]
- Jourdan M, Ferlin M, Legouffe E, Horvathova M, Liautard J, Rossi JF, Wijdenes J, Brochier J, Klein B. The myeloma cell antigen syndecan-1 is lost by apoptotic myeloma cells. Br J Haematol. 1998;100:637–646. doi: 10.1046/j.1365-2141.1998.00623.x. [DOI] [PubMed] [Google Scholar]
- Kliment CR, Englert JM, Gochuico BR, Yu G, Kaminski N, Rosas I, Oury TD. Oxidative stress alters syndecan-1 distribution in lungs with pulmonary fibrosis. J Biol Chem. 2009;284:3537–3545. doi: 10.1074/jbc.M807001200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyula JN, Van Schaeybroeck S, Doherty J, Fenning CS, Longley DB, Johnston PG. Chemotherapy-induced activation of ADAM-17: a novel mechanism of drug resistance in colorectal cancer. Clin Cancer Res. 2010;16:3378–3389. doi: 10.1158/1078-0432.CCR-10-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamorte S, Ferrero S, Aschero S, Monitillo L, Bussolati B, Omede P, Ladetto M, Camussi G. Syndecan-1 promotes the angiogenic phenotype of multiple myeloma endothelial cells. Leukemia. 2012;26:1081–1090. doi: 10.1038/leu.2011.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Park PW, Wilson CL, Parks WC. Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell. 2002;111:635–646. doi: 10.1016/s0092-8674(02)01079-6. [DOI] [PubMed] [Google Scholar]
- Lonial S, Mitsiades CS, Richardson PG. Treatment options for relapsed and refractory multiple myeloma. Clin Cancer Res. 2011;17:1264–1277. doi: 10.1158/1078-0432.CCR-10-1805. [DOI] [PubMed] [Google Scholar]
- Manon-Jensen T, Itoh Y, Couchman JR. Proteoglycans in health and disease: the multiple roles of syndecan shedding. Febs J. 2010;277:3876–3889. doi: 10.1111/j.1742-4658.2010.07798.x. [DOI] [PubMed] [Google Scholar]
- Nikolova V, Koo CY, Ibrahim SA, Wang Z, Spillmann D, Dreier R, Kelsch R, Fischgrabe J, Smollich M, Rossi LH, Sibrowski W, Wulfing P, Kiesel L, Yip GW, Gotte M. Differential roles for membrane-bound and soluble syndecan-1 (CD138) in breast cancer progression. Carcinogenesis. 2009;30:397–407. doi: 10.1093/carcin/bgp001. [DOI] [PubMed] [Google Scholar]
- Paramore A, Frantz S. Bortezomib. Nat Rev Drug Discov. 2003;2:611–612. doi: 10.1038/nrd1159. [DOI] [PubMed] [Google Scholar]
- Park PW, Pier GB, Hinkes MT, Bernfield M. Exploitation of syndecan-1 shedding by Pseudomonas aeruginosa enhances virulence. Nature. 2001;411:98–102. doi: 10.1038/35075100. [DOI] [PubMed] [Google Scholar]
- Park PW, Pier GB, Preston MJ, Goldberger O, Fitzgerald ML, Bernfield M. Syndecan-1 shedding is enhanced by LasA, a secreted virulence factor of Pseudomonas aeruginosa. J Biol Chem. 2000;275:3057–3064. doi: 10.1074/jbc.275.5.3057. [DOI] [PubMed] [Google Scholar]
- Pruessmeyer J, Martin C, Hess FM, Schwarz N, Schmidt S, Kogel T, Hoettecke N, Schmidt B, Sechi A, Uhlig S, Ludwig A. A disintegrin and metalloproteinase 17 (ADAM17) mediates inflammation-induced shedding of syndecan-1 and -4 by lung epithelial cells. J Biol Chem. 2010;285:555–564. doi: 10.1074/jbc.M109.059394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushothaman A, Chen L, Yang Y, Sanderson RD. Heparanase stimulation of protease expression implicates it as a master regulator of the aggressive tumor phenotype in myeloma. J Biol Chem. 2008;283:32628–32636. doi: 10.1074/jbc.M806266200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushothaman A, Hurst DR, Pisano C, Mizumoto S, Sugahara K, Sanderson RD. Heparanase-mediated loss of nuclear syndecan-1 enhances histone acetyltransferase (HAT) activity to promote expression of genes that drive an aggressive tumor phenotype. J Biol Chem. 2011;286:30377–30383. doi: 10.1074/jbc.M111.254789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purushothaman A, Uyama T, Kobayashi F, Yamada S, Sugahara K, Rapraeger AC, Sanderson RD. Heparanase-enhanced shedding of syndecan-1 by myeloma cells promotes endothelial invasion and angiogenesis. Blood. 2010;115:2449–2457. doi: 10.1182/blood-2009-07-234757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani VC, Pruett PS, Thompson CA, DeLucas LD, Sanderson RD. Heparan sulfate chains of syndecan-1 regulate ectodomain shedding. J Biol Chem. 2012;287:9952–9961. doi: 10.1074/jbc.M111.330803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani VC, Purushothaman A, Stewart MD, Thompson CA, Vlodavsky I, Au JL, Sanderson RD. The heparanase/syndecan-1 axis in cancer: mechanisms and therapies. Febs J. 2013;280:2294–2306. doi: 10.1111/febs.12168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramani VC, Yang Y, Ren Y, Nan L, Sanderson RD. Heparanase plays a dual role in driving hepatocyte growth factor (HGF) signaling by enhancing HGF expression and activity. J Biol Chem. 2011;286:6490–6499. doi: 10.1074/jbc.M110.183277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiland J, Ott VL, Lebakken CS, Yeaman C, McCarthy J, Rapraeger AC. Pervanadate activation of intracellular kinases leads to tyrosine phosphorylation and shedding of syndecan-1. Biochem J. 1996;319:39–47. doi: 10.1042/bj3190039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Seidel C, Sundan A, Hjorth M, Turesson I, Dahl IM, Abildgaard N, Waage A, Borset M. Serum syndecan-1: a new independent prognostic marker in multiple myeloma. Blood. 2000;95:388–392. [PubMed] [Google Scholar]
- Su G, Blaine SA, Qiao D, Friedl A. Shedding of syndecan-1 by stromal fibroblasts stimulates human breast cancer cell proliferation via FGF2 activation. J Biol Chem. 2007;282:14906–14915. doi: 10.1074/jbc.M611739200. [DOI] [PubMed] [Google Scholar]
- Tagoug I, Plesa A, Dumontet C. Bortezomib influences the expression of malignant plasma cells membrane antigens. Eur J Pharmacol. 2013;706:11–16. doi: 10.1016/j.ejphar.2013.02.002. [DOI] [PubMed] [Google Scholar]
- Tokes AM, Szasz AM, Farkas A, Toth AI, Dank M, Harsanyi L, Molnar BA, Molnar IA, Laszlo Z, Rusz Z, Kulka J. Stromal matrix protein expression following preoperative systemic therapy in breast cancer. Clin Cancer Res. 2009;15:731–739. doi: 10.1158/1078-0432.CCR-08-1523. [DOI] [PubMed] [Google Scholar]
- Vahdat AM, Reiners KS, Simhadri VL, Eichenauer DA, Boll B, Chalaris A, Simhadri VR, Wiegmann K, Krell HW, Rose-John S, Engert A, von Strandmann EP, Hansen HP. TNF-alpha-converting enzyme (TACE/ADAM17)-dependent loss of CD30 induced by proteasome inhibition through reactive oxygen species. Leukemia. 2010;24:51–57. doi: 10.1038/leu.2009.230. [DOI] [PubMed] [Google Scholar]
- Yang Y, Macleod V, Bendre M, Huang Y, Theus AM, Miao HQ, Kussie P, Yaccoby S, Epstein J, Suva LJ, Kelly T, Sanderson RD. Heparanase promotes the spontaneous metastasis of myeloma cells to bone. Blood. 2005;105:1303–1309. doi: 10.1182/blood-2004-06-2141. [DOI] [PubMed] [Google Scholar]
- Yang Y, Macleod V, Miao HQ, Theus A, Zhan F, Shaughnessy JD, Jr, Sawyer J, Li JP, Zcharia E, Vlodavsky I, Sanderson RD. Heparanase enhances syndecan-1 shedding: a novel mechanism for stimulation of tumor growth and metastasis. J Biol Chem. 2007;282:13326–13333. doi: 10.1074/jbc.M611259200. [DOI] [PubMed] [Google Scholar]
- Zhan F, Hardin J, Kordsmeier B, Bumm K, Zheng M, Tian E, Sanderson R, Yang Y, Wilson C, Zangari M, Anaissie E, Morris C, Muwalla F, van Rhee F, Fassas A, Crowley J, Tricot G, Barlogie B, Shaughnessy J., Jr Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood. 2002;99:1745–1757. doi: 10.1182/blood.v99.5.1745. [DOI] [PubMed] [Google Scholar]