

Novel function and mechanism of the neurotransmitter dopamine on the inhibition of tumor-induced monocytic MDSCs.
Keywords: iNOS, nitric oxide, MDSC, neurotransmitter, cancer, immune suppression
Abstract
MDSCs accumulate in tumor-bearing animals and cancer patients and are a major factor responsible for cancer-induced immunosuppression that limits effective cancer immunotherapy. Strategies aimed at effectively inhibiting the function of MDSCs are expected to enhance host anti-tumor immunity and improve cancer immunotherapy significantly. The neurotransmitter DA has been found to have anti-cancer activity, but the underlying mechanism is poorly understood. In this study, we sought to investigate the therapeutic mechanism and efficacy of DA on the inhibition of cancer development via the regulation of MDSC functions. The regulation of the suppressive function of Gr-1+CD115+ MDSCs by DA was determined by use of murine syngeneic LLC and B16 graft models treated with DA in vivo, as well as Gr-1+CD115+ MDSCs isolated from these model treated with DA ex vivo. Here, we show that Gr-1+CD115+ monocytic MDSCs express D1-like DA receptors. DA dramatically attenuated the inhibitory function of tumor-induced monocytic MDSCs on T cell proliferation and IFN-γ production via D1-like DA receptors and retarded tumor growth. DA and other D1 receptor agonists inhibited IFN-γ-induced NO production by MDSCs from tumor-bearing mice and cancer patients. Decreased NO production was, in part, mediated via the suppression of p-ERK and p-JNK. In conclusion, the neurotransmitter DA potently inhibits the suppressive function of MDSC and enhances anti-tumor immunity. Our finding provides a mechanistic basis for the use of DA or D1-like receptor agonists to overcome tumor-induced immunosuppression in cancer immunotherapy.
Introduction
Effective anti-tumor immunity is often hampered by tumor-induced immune suppression [1]. Growing evidence suggests that MDSCs, a heterogeneous group of host immune cells comprised of immature macrophages, DCs, and granulocytes with immunosuppressive functions [2–4], are a major factor that limits the effectiveness of cancer immunotherapy and represent an appealing target for therapeutic intervention [5, 6]. In healthy individuals, MDSCs primarily reside in the bone marrow and rapidly differentiate into mature myeloid cells after emigration to peripheral lymphoid organs [6]. Tumor-derived factors can induce the expansion and accumulation of MDSCs in the bone marrow, spleen, and blood and at the sites of tumor and inhibit MDSC differentiation and maturation, thus endowing them with potent, immunosuppressive functions [7]. A significant increase in MDSCs is observed in tumor-bearing mice, as well as patients with a variety of cancers [8–11]. More importantly, increased levels of circulating MDSCs have been closely correlated with clinically advanced stages of cancer and extensive metastatic tumor burdens [11].
MDSCs facilitate tumor growth and metastasis, mainly by suppressing effector T-cell activities via IFN-γ-dependent production of NO [12], the depletion of arginine [13], and the expression of ROS [14]. Murine MDSCs exhibit the CD11b+Gr-1+ phenotype, and their human counterpart is identified as being Lin‒HLA-DR‒CD33+ or CD33+CD14‒CD11b+ [15, 16]. Recently, two distinct subpopulations of MDSCs, Ly-6G+Ly-6Clow granulocytic MDSCs and Ly-6G‒/lowLy-6Chigh monocytic MDSCs, have been described [9, 17, 18]. It was demonstrated that CD115+Gr-1+CD11b+ monocytic MDSCs possess a stronger suppressive function on T cells than granulocytic MDSCs [19, 20] and promoted the activation and expansion of tumor-specific Tregs. Given the immunosuppressive functions of MDSCs, strategies aimed at the elimination or inhibition of MDSCs may significantly improve anti-tumor responses and the efficacy of cancer immunotherapy [21].
DA is a member of the catecholamine family of neurotransmitters and functions in the CNS to regulate motility and mental functions, such as cognition, motor control, mood, and pain perception. Recent experimental and epidemiologic studies have suggested that DA may play an important role in regulating host anti-tumor immune responses. Administration of DA to tumor-bearing mice dramatically increases the efficacy of anti-cancer drugs on breast and colon cancers [22]. Patients with Parkinson’s disease treated with DA receptor agonists have significantly reduced risk of smoking-related cancers and moderately reduced risk of several nonsmoking-related cancers [23, 24]. However, the mechanism underlying the inhibitory effect of DA on cancer development is poorly understood.
The diverse physiologic effects of DA are mediated by at least 5 distinct DA receptors, D1–D5. These receptors belong to the G protein-coupled receptor family and fall into two groups, namely D1-like receptors, including D1 and D5, and D2-like receptors, including D2–D4. An emerging number of studies have demonstrated the immunomodulatory functions of DA. For example, antagonizing D1-like receptors could attenuate Th17-mediated immune diseases, such as experimental autoimmune encephalomyelitis and neutrophilic airway inflammation [25, 26]. In addition, systemic administration of DA or its D1-type agonist could induce neuroprotection after CNS injury by alleviating Treg-mediated immune suppression [27]. In this regard, it was shown that DA receptors are expressed on a variety of immune cells, including T- and B-lymphocytes, neutrophils, monocytes, and NK cells [28].
In light of those findings, we hypothesized that the immunomodulatory function of DA may underlie its anti-tumor activity. In the current study, we focused our investigation on the role and mechanism of DA in the inhibition of tumor-induced MDSCs.
MATERIALS AND METHODS
Mice
Four- to 6-week-old C57BL/6 mice were purchased from the Experimental Animal Center of the Chinese Academy of Sciences (Shanghai, China) and housed under specific pathogen-free conditions. All experiments were performed according to the Guide for the Care and Use of Medical Laboratory Animals, issued by the Ministry of Health of China, and approved by the Shanghai Medical Laboratory Animal Care and Use Committee.
Cell lines
The LLC cell line and the B16 melanoma cell line, originally derived from the C57BL/6 mice, were obtained from the Type Culture Collection of the Chinese Academy of Sciences. Cells were cultured in DMEM (Invitrogen, Carlsbad, CA, USA), supplemented with 10% FBS (Hyclone, Logan, UT, USA) and 1% antibiotics (Invitrogen) at 37°C with 5% CO2.
Lung cancer patient blood samples
Blood was obtained from lung cancer patients (stages III–IV) after informed consent, according to a protocol approved by the Institutional Review Board of Xinhua Hospital, School of Medicine, Shanghai Jiao Tong University (Shanghai, China). PBMCs of 6 patients were separated by Ficoll-Paque Plus (GE Healthcare, Pittsburgh, PA, USA) density gradient centrifugation.
In vivo tumor models and DA treatment
LLC cells (1 × 106) or 5 × 105 B16 cells in 100 μl PBS were s.c. inoculated into the right flank of C57BL/6 mice. Animals were divided randomly into 2 groups on Day 8 after inoculation. The vehicle group was treated with PBS, whereas the DA group was given 50 mg/kg/day DA (Sigma-Aldrich, St. Louis, MO, USA) i.p. for 7 consecutive days [22]. Tumor volume was measured twice/week. All experiments were performed in triplicate.
MDSC depletion
Anti-Gr-1 (clone RB6-8C5; eBioscience, San Diego, CA, USA) and anti-Ly-6G (clone 1A8; BioLegend, San Diego, CA, USA) were used for the depletion of MDSCs, based on previous studies [29, 30]. In brief, on Day 8 after tumor inoculation, mice with palpable tumors were injected i.p. with 200 µg anti-Gr-1 or anti-Ly-6G mAb every 48 h until Day 14. On Day 9, one-half of the mice were given 50 mg/kg/day DA i.p. for 7 consecutive days. Tumor volume was measured every other day.
MDSC purification
Spleens and bone marrow of mice with high tumor burden were harvested. Following the removal of RBCs, bone marrow cells and splenocytes were fractionated by centrifugation on a Percoll density gradient (GE Healthcare) [19]. The cell layer between 50 and 60% Percoll was collected and washed twice with PBS. CD115+ cells were isolated by use of a CD115 microbead kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Cells with a purity of >90% were used for experiments. Lin‒CD33+HLA-DR‒ MDSCs were sorted on an FACSAria II sorter (BD Biosciences, San Jose, CA, USA).
In vitro restimulation of T cells
Splenocytes from LLC-bearing mice, treated or untreated with DA, were cocultured with mitomycin C-inactivated LLC cells at a ratio of 10:1 for 24 h. Brefeldin A was added to the culture for an additional 5 h. The cells were collected and stained for surface CD4 or CD8 and intracellular IFN-γ.
Flow cytometry
Cells were incubated with an Fc-blocking antibody (clone 2.4G2; BD Biosciences) for 20 min at 4°C and then stained with primary antibodies for 30 min at 4°C, washed, and incubated with secondary antibodies, if necessary, for 20 min at 4°C. Anti-CD115-PE, anti-Ly-6C-FITC, anti-CD3-APC, anti-CD4-PE, anti-CD8-PE, anti-CD80-APC, anti-CD86-APC, anti-MHC-APC, anti-CD11c-APC, anti-IFN-γ-APC, and isotype-matched mAb were purchased from eBioscience; anti-D1 receptor, anti-D5 receptor, mouse anti-rabbit IgG-PerCP, and donkey anti-goat IgG-PerCP were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). For apoptosis determination, freshly isolated MDSCs were cultured at a concentration of 1 × 106 cells/well and were left untreated or treated with 50 μM DA for 4 h or 10 μM DA for 24 h at 37°C. Apoptosis was determined by use of an Annexin V apoptosis kit (BD Biosciences). Data were acquired on a FACSCalibur flow cytometer (BD Biosciences) and analyzed by use of FlowJo (Tree Star, Ashland, OR, USA).
RT-PCR
Total RNA from MDSCs was purified by use of a PureYield RNA Midiprep Kit (Promega, Madison, WI, USA), and 500 ng total RNA was reverse transcribed into cDNA by use of Transcriptor First Strand cDNA Synthesis Kit (Roche, Branchburg, NJ, USA). The expression of various DA receptors was determined by regular PCR, with initial denaturation at 94°C for 5 min, followed by 38 cycles of denaturation at 94°C for 45 s, annealing at 57°C for 45 s, extension at 72°C for 1 min, and final elongation at 72°C for 10 min by use of primers: Drd1, forward 5′-TGTCCCTGCTTATCCTGTCC-3′, reverse 5′-CTGCCTTCGGAGTCATCTTC-3′; Drd2, forward 5′-GAGAAGGCTTTGCAGACCAC-3′, reverse 5′-AGGACAGGACCCAGACAATG-3′; Drd3, forward 5′-CCCTCAGCAGTCTTCCTGTC-3′, reverse 5′-AGTCCTCTCCACTTGGCTCA-3′; Drd4, forward 5′-CTGCAGACACCCACCAACTA-3′, reverse 5′-AAGGAGCAGACGGACGAGTA-3′; Drd5, forward 5′-CCACTGCTTCCATCCTGAAT-3′, reverse 5′-TGCGCGTGTAGGTCACTATC-3′.
CFSE labeling
Splenocytes from C57BL/6 mice were suspended in prewarmed PBS with 0.1% BSA and incubated with 5 μM CFSE (Invitrogen) at 37°C for 10 min, followed by quenching with 5 vol ice-cold culture medium and washing three times with fresh medium.
Suppression assay
CFSE-labeled C57BL/6 tumor-free mouse splenocytes (5 × 105) were cocultured with various numbers of CD115+ cells, isolated from control or DA-treated tumor-bearing mice in the presence of 1 μg/ml anti-CD3 (clone 500A2; eBioscience) and 2 μg/ml anti-CD28 (clone 37.51; eBioscience) for 4 days. Alternatively, purified CD115+ cells, untreated or pretreated for 4 h with 50 µM DA or the DA receptor agonist SKF38393 or Quinpirole (Sigma-Aldrich), were used for the coculture. At the end of 4 days, CFSE signal on CD3+ T cells was analyzed by flow cytometry, and NO levels in the supernatant were measured by use of Greiss reagents (Sigma-Aldrich).
NO measurement
Intracellular NO production was detected by use of the NO-specific fluorogenic probe DAF-FM (Invitrogen). Lin‒CD33+HLA-DR‒ cells isolated from lung cancer patients were stimulated by vehicle or 50 ng/ml IFN-γ (eBioscience) for 24 h in the presence or absence of 10 μM D1 agonist SKF38393 (Sigma-Aldrich). The cells were then loaded with 5 μM DAF-FM. Following washing with cold PBS for 3 times, intracellular NO levels were analyzed by flow cytometry based on the DAF-FM signal. To confirm the role of ERK and JNK in IFN-γ-induced NO production by MDSCs, freshly isolated MDSCs from tumor-bearing mice were treated with IFN-γ alone or incubated with signaling inhibitors U0126 (10 µM) or SP600125 (25 μM) for 30 min before IFN-γ stimulation. NO levels in the supernatant were measured by use of Griess reagents (Sigma-Aldrich), as described previously [31]. In brief, culture supernatants were mixed with an equal volume of Griess reagent (0.1% N-1-naphthylethylenediamine dihydrochloride and 1% sulphanilamide in 5% phosphoric acid) and incubated at room temperature for 10 min. Absorbance was measured at 550 nm in a microplate reader. Nitrite concentrations were calculated based on a standard curve derived from the reaction of NaNO2 in the assay.
Immunoblotting
MDSCs, freshly isolated from tumor-bearing mice, were treated with or without 50 μM DA. Thirty minutes later, 50 ng/ml IFN-γ was added to the culture. The treatment was allowed to proceed for another 1 or 3 h. Cells were lysed in a lysis buffer containing protease and phosphatase inhibitors (Thermo Fisher Scientific, Waltham, MA, USA). Cell lysates containing 25 μg protein were resolved by SDS-PAGE, transferred to PVDF membranes, and probed with antibodies against p-ERK, p-p38, or p-JNK (Cell Signaling Technology, Beverly, MA, USA). Membranes were then washed for 30 min with a stripping buffer (Thermo Fisher Scientific) and reprobed with antibodies against total ERK, p38, JNK, and β-actin (Cell Signaling Technology). Antibody-reactive bands were detected by ECL (Thermo Fisher Scientific) and quantified by laser densitometry (Bio-Rad Laboratories, Hercules, CA, USA). For detection of iNOS level, cells were treated with IFN-γ for 24 h in the presence or absence of 5 or 10 μM DA.
Statistics
Values were expressed as mean ± sem. Statistical significance was assessed by 2-tailed unpaired Student’s t-test and one-way ANOVA for 2-group and multiple-group comparisons, respectively. Differences were deemed statistically significant for P < 0.05.
RESULTS
DA inhibits tumor growth and the function of Gr-1+CD115+ MDSCs in vivo
We used mouse syngeneic LLC and B16 graft models to study the mechanism of the anti-tumor effect of DA. DA administration inhibited tumor growth in vivo in both models, as evidenced by the significantly smaller (P < 0.01) tumor size in DA-treated mice compared with PBS-treated control mice (Fig. 1A).
Figure 1. DA inhibits tumor growth and the suppressive function of Gr-1+CD115+ MDSCs in vivo.
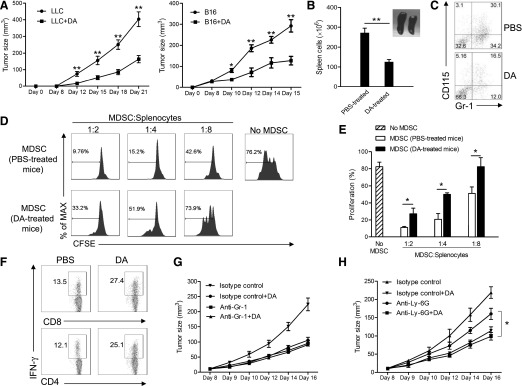
(A) Tumor volume in mice treated with PBS (n = 8) or DA (n = 8). *P < 0.05, and **P < 0.01. (B) Statistical comparison of splenocyte numbers in PBS-treated and DA-treated tumor-bearing mice. A representative image of the spleens of PBS- and DA-treated tumor-bearing mice, 21 days after tumor cell inoculation, is shown. **P < 0.01. (C) Flow cytometry analysis of the percentage of Gr-1+CD115+ MDSCs in the spleens of PBS- and DA-treated tumor-bearing mice. (D) Flow cytometry of CFSE dilution in CD3+ T cells in naive splenocytes stimulated with anti-CD3 and anti-CD28 and cocultured with CD115+ splenic MDSCs from PBS- or DA-treated tumor-bearing mice for 4 days. Numbers indicate the percentage of CFSElow T cells in total CD3+ T cells. (E) Statistical analysis of the percentages of CFSElow T cells in total CD3+ T cells after 4-day coculture with CD115+ MDSCs from PBS- or DA-treated tumor-bearing mice. (F) Flow cytometry of IFN-γ expression by splenic CD4+ and CD8+ T cells in PBS- or DA-treated tumor-bearing mice. (G) Tumor volume in mice treated with anti-Gr-1 mAb or isotype control antibody, with (n = 6) or without (n = 6) DA. (H) Tumor volume in mice treated with anti-Ly-6G mAb or isotype control antibody, with (n = 6) or without (n = 6) DA. (E and H) *P < 0.05. Representative data from 3 independent experiments, yielding similar results, are shown. Error bars indicate sem.
The delayed tumor growth in DA-treated mice could be contributed by the inhibition of tumor-induced immunosuppression by DA, leading to enhanced host anti-tumor immunity. Indeed, tumor-induced splenomegaly was reduced in DA-treated mice (Fig. 1B), suggesting reduced accumulation of MDSCs in the spleen (Fig. 1C). As MDSCs are a major factor responsible for tumor-induced immunosuppression in vivo [6], we compared the suppressive function of tumor-induced MDSCs from PBS- and DA-treated tumor-bearing mice at the end of the experiment. Gr-1+CD115+ MDSCs from PBS-treated mice strongly suppressed the proliferation of cocultured splenic T cells isolated from tumor-free mice, whereas the suppressive function of MDSCs from DA-treated mice was reduced significantly (P < 0.05; Fig. 1D and E and Supplemental Fig. 1). Consistently, the spleens of DA-treated tumor-bearing mice harbored significantly higher fractions of IFN-γ-producing CD4+ and CD8+ T cells than the spleens of PBS-treated control mice (Fig. 1F).
To ascertain that the anti-tumor effect of DA was mediated via its inhibition of MDSCs, we depleted total MDSCs by use of an anti-Gr-1 antibody or the granulocytic subset of MDSCs by use of an anti-Ly-6G antibody. Depletion of Gr-1+ but not Ly-6G+ granulocytic MDSCs completely abrogated the further inhibitory effect of DA on tumor growth (Fig. 1G and H). In addition, much stronger suppressive activity was observed in Gr-1+CD115+ cells than Gr-1+CD115‒ cells from tumor-bearing mice ex vivo (Supplemental Fig. 2A), and the suppressive function of Gr-1+CD115‒ cells was slightly reduced by DA treatment in tumor-bearing mice, which was much less pronounced than that of Gr-1+CD115+ cells (Supplemental Fig. 2B). Treatment of purified CD115‒ MDSCs in vitro with DA produced a similar effect (data not shown). These data suggest that DA retards tumor growth by inhibiting the functions of Gr-1+CD115+ MDSCs and thereby, promoting effector CD4+ and CD8+ T-cell function in tumor-bearing mice.
DA directly inhibits Gr-1+CD115+ MDSC function in vitro
To determine if DA directly inhibits the function of MDSCs, we pretreated Gr-1+CD115+ MDSCs from tumor-bearing mice with DA and cocultured them with splenic T cells. The proliferation of cocultured CD3+ T cells was inhibited significantly by untreated MDSCs, whereas DA inhibited the suppressive function of MDSCs on T-cell proliferation in a dose-dependent manner (Fig. 2A and B). The diminished, suppressive activity of DA-treated MDSCs was not a result of increased apoptosis (Fig. 2C), and there was no increased apoptosis of MDSCs after prolonged DA treatment for up to 24 h (data not shown). Therefore, DA directly inhibits the function of Gr-1+CD115+ MDSCs through a mechanism other than triggering MDSC apoptosis.
Figure 2. DA attenuates MDSC-mediated suppression of T-cell proliferation in vitro.

(A) Flow cytometry of CFSE dilution in CD3+ T cells in naive splenocytes stimulated with anti-CD3 and anti-CD28 and cultured for 4 days alone or with CD115+ splenic MDSCs from mice with large tumor burdens and untreated or pretreated for 4 h with 25 µM or 50 µM DA. Numbers indicate the percentage of CFSElow T cells in total CD3+ T cells. (B) Statistical analysis of the percentages of CFSElow T cells in CD3+ T cells cultured alone or with untreated or DA-treated MDSCs. *P < 0.05, and ***P < 0.001. (C) Flow cytometry of propidium iodide (PI) and Annexin V staining of CD115+ MDSCs, untreated or treated with 50 µM DA for 4 h. The data shown are representative of 3 independent experiments giving similar results. Error bars indicate sem.
DA inhibits Gr-1+CD115+ MDSC function via D1-like receptors
The physiologic functions of DA are mediated by DA receptors that fall into two subfamilies, namely D1-like receptors (including D1 and D5) and D2-like receptors (including D2–D4) [32]. Activation of D1 increases intracellular cAMP levels, whereas activation of D2 has been shown generally to decrease intracellular cAMP levels, thus triggering opposite effects [32]. Gr-1+CD115+ MDSCs express the messages of D1-like receptors D1 and D5, whereas the expression of D2-like receptors was undetectable (Fig. 3A). Treatment of Gr-1+CD115+ MDSCs with the D1-like receptor agonist SKF38393 inhibited their suppressive function on the proliferation of cocultured T cells, mimicking the effect of DA, whereas treatment with the D2-like receptor agonist Quinpirole had no effect (Fig. 3B). These results indicate that DA inhibits the suppressive function of Gr-1+CD115+ MDSCs through D1-like receptors.
Figure 3. DA inhibits Gr-1+CD115+ MDSC function through D1-like DA receptors.

(A) RT-PCR analysis of the expression of DA receptor genes in Gr-1+CD115+ MDSCs isolated from tumor-bearing mice. The intensity of the band of DA receptor 1 (Drd1) was defined as 1. N.D., Not detected. (B) Flow cytometry of CFSE dilution in CD3+ T cells in naive splenocytes stimulated with anti-CD3 and anti-CD28 and cultured for 4 days alone or with CD115+ splenic MDSCs purified from mice with large tumor burdens and untreated or pretreated for 4 h with 50 µM SKF38393 or Quinpirole. Numbers indicate the percentage of CFSElow T cells in total CD3+ T cells. Representative data from 3 independent experiments, yielding similar results, are shown. Error bars indicate sem.
Decreased NO production and enhanced differentiation underlie the inhibition of Gr-1+CD115+ MDSCs by DA in vivo
Monocytic MDSCs express high levels of NO [17]. The immunosuppressive activity of monocytic MDSCs on T cells could be blocked substantially by NG-L-monomethyl arginine, an inhibitor of iNOS [17], suggesting that NO production is a major mechanism by which monocytic MDSCs inhibit T-cell function. We thus investigated if the inhibition of Gr-1+CD115+ MDSC function by DA also involves decreased NO production. Gr-1+CD115+ MDSCs from DA-treated tumor-bearing mice produced significantly less NO than those from untreated tumor-bearing mice after coculturing with syngeneic splenocytes (Fig. 4A). DA and the D1 receptor agonist SKF38393, but not the D2 receptor agonist Quinpirole, inhibited NO production by CD115+ MDSCs from tumor-bearing mice (Fig. 4B). As it was shown that monocytic MDSCs exert their suppressive functions through NO production in an IFN-γ-dependent manner [16], we assessed further if DA or its D1 receptor agonists could inhibit IFN-γ-induced NO production by MDSCs. Stimulation with IFN-γ triggered strong NO secretion by CD115+ MDSCs, which was inhibited by DA or SKF38393 but not Quinpirole (Fig. 4C).
Figure 4. DA inhibits NO production and promotes the maturation of Gr-1+CD115+ MDSCs.
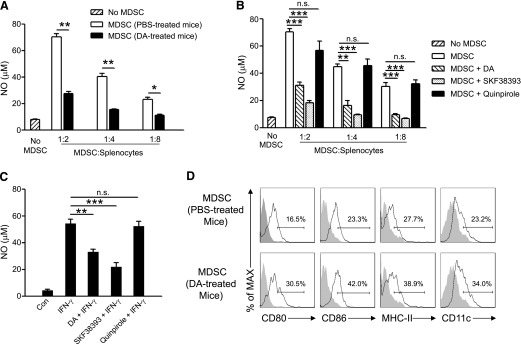
(A) NO production by purified CD115+ MDSCs from PBS- or DA-treated tumor-bearing mice cocultured with syngeneic splenocytes for 4 days in the presence of anti-CD3 and anti-CD28. (B) NO production by CD115+ MDSCs isolated from tumor-bearing mice and treated with 50 µM DA, SKF38393, or Quinpirole for 4 h and then cocultured with syngeneic splenocytes for 4 days. (C) NO production by CD115+ MDSCs isolated from tumor-bearing mice and stimulated with 50 ng/ml IFN-γ for 48 h in the presence or absence of 10 µM DA, SKF38393, or Quinpirole. MDSCs without any treatment served as control (Con). *P < 0.05, **P < 0.01, and ***P < 0.001; n.s., no statistically significant difference (P > 0.05). (D) Flow cytometry of CD80, CD86, MHC-II, and CD11c expression by Gr-1+CD115+ MDSCs in PBS- or DA-treated tumor-bearing mice. The data shown are representative of 4 independent experiments giving similar results. Error bars indicate sem.
The differentiation of MDSCs into mature myeloid cells results in a reduction in their suppressive function [33, 34]. The administration of DA to tumor-bearing mice promoted the maturation of Gr-1+CD115+ MDSCs, as evidenced by the increased expression of CD80, CD86, and MHC-II and promoted their differentiation into mature DCs (Fig. 4D and Supplemental Fig. 3). Treatment of purified CD115+ MDSCs ex vivo with DA for 24 h produced a similar effect (data not shown). Taken together, DA abrogates the immunosuppressive function of monocytic MDSCs, at least by inhibiting their NO production and promoting their maturation in vivo.
DA inhibits ERK and JNK signaling in monocytic MDSCs
We sought to understand better the mechanism underlying the regulation of NO production by DA in monocytic MDSCs. iNOS is a key enzyme that mediates the production of NO in MDSCs [13]. DA treatment inhibited IFN-γ-induced iNOS expression in MDSCs in a dose-dependent manner (Fig. 5A). The signaling through MAPKs was shown to induce the expression of iNOS [35]. We found that DA did not have a notable inhibitory effect on p-p38 but substantially down-regulated p-ERK1/2 and p-JNK (Fig. 5B). Indeed, monocytic MDSCs treated with the ERK1/2 inhibitor U0126 or the JNK inhibitor SP600125 were unable to produce NO in response to IFN-γ (Fig. 5C), which was similar to the effect produced by DA or its D1 receptor agonist SKF38393 (Fig. 4C). These results suggest that the inhibitory effect of DA on NO production by monocytic MDSCs may involve the down-regulation of ERK and JNK signaling.
Figure 5. DA inhibits ERK and JNK signaling in monocytic MDSCs.

(A) Immunoblot (upper) of iNOS expression by purified CD115+ MDSCs from tumor-bearing mice, stimulated with or without 50 ng/ml IFN-γ for 24 h in the presence or absence of 5 or 10 µM DA. (Lower) The intensity of the bands was quantitated. *P < 0.05, and **P < 0.01; (B) Immunoblot of p-p38, total p38, p-ERK, total ERK, p-JNK, and total JNK by purified CD115+ MDSCs stimulated with 50 ng/ml IFN-γ for 1 or 3 h in the presence or absence of 50 µM DA. β-Actin was immunoblotted as a loading control in A and B. (C) NO production by purified CD115+ MDSCs pretreated with 10 µM U0126 or 25 µM SP600125 for 30 min, followed by 24 h stimulation with 50 ng/ml IFN-γ. Data representing 4 independent experiments, yielding similar results, are shown. ***P < 0.001. Error bars indicate sem.
MDSCs of cancer patients express D1-like receptors, and their agonist inhibits NO production
We verified further the expression of D1-like receptors on MDSCs from the peripheral blood of lung cancer patients. We sorted Lin‒CD33+HLA-DR‒ cells, which are commonly defined as MDSCs in humans and generally agreed to be suppressive [6, 36]. Lin‒CD33+HLA-DR‒ MDSCs from all the patients analyzed expressed D1 and D5 receptors (Fig. 6A). The D1-like receptor agonist SKF38393 significantly inhibited IFN-γ-induced NO production by human MDSCs (Fig. 6B and C). These results suggest that DA inhibits the function of human MDSCs via mechanisms similar to those in murine MDSCs.
Figure 6. MDSCs in cancer patients express D1-like DA receptors, and their agonist inhibits NO production.
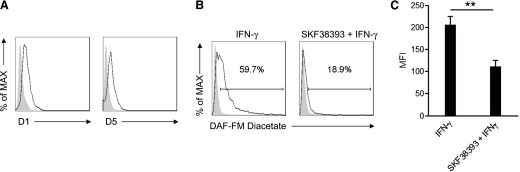
(A) D1 and D5 expression by Lin‒ CD33+HLA-DR‒ MDSCs, sorted from peripheral blood of lung cancer patients. Shaded histograms indicate the staining by use of isotype control antibodies. (B) Flow cytometric measurement of intracellular NO levels by Lin‒CD33+HLA-DR‒ MDSCs from lung cancer patients pretreated with vehicle or 10 µM SKF38393 and then stimulated with 50 ng/ml IFN-γ for 24 h. Shaded histograms indicate the fluorescence of cells without IFN-γ stimulation. (C) The mean fluorescence intensity (MFI) of intracellular NO level in human MDSCs stimulated with IFN-γ alone or with IFN-γ and SKF38393. The data shown summarize 3 independent experiments. **P < 0.01. Error bars indicate sem.
DISCUSSION
It has become increasingly clear that successful cancer immunotherapy will be achieved only with a strategy that also aims to overcome tumor-induced immunosuppression. The critical role of tumor-induced MDSCs in interfering with effective host anti-tumor immunity has been established by a large number of studies [16]. Several therapeutic strategies that target tumor-induced MDSCs are currently being explored experimentally, including the depletion of MDSCs with anti-Gr-1 mAb [37], promotion of MDSC differentiation with all-trans retinoic acid [38], and the inhibition of the suppressive function or tumor-promoting activity of MDSCs by compounds, such as the phosphodiesterase inhibitor sildenafil or cyclooxygenase-2 inhibitors [39, 40]. However, the feasibility of translating these strategies into the clinic and their possible side-effects on other critical immunocompetent cells require further evaluation, necessitating additional therapeutic strategies to be explored. In this study, we showed that administration of DA at low, nontoxic doses [41] could inhibit tumor growth and effectively reduce the suppressive function of tumor-induced Gr-1+CD115+ monocytic MDSCs on host T-cell proliferation and effector function. Ex vivo and in vitro experiments showed that DA directly attenuated the immunosuppressive functions of monocytic MDSCs via signaling through D1-like DA receptors (Fig. 7).
Figure 7. Model of DA-mediated regulation of MDSC function and enhancement of anti-tumor immunity.

Gr-1+CD115+ monocytic MDSCs express the D1-like receptors D1 and D5. The activation of D1 and D5 on MDSCs by DA and other D1-like receptor agonists inhibits ERK and JNK activation in response to IFN-γ stimulation, thereby inhibiting the production of NO, a pivotal mediator of the suppressive activity of MDSCs on T-cell proliferation and IFN-γ production. The attenuation of NO production by MDSCs leads to enhanced effector T-cell function and anti-tumor immunity. IFNGR, IFN-γ receptor.
In addition, the numbers of MDSCs in the spleen of tumor-bearing mice reduced after DA treatment, which may contribute to the retarded tumor growth; however, the decrease of MDSC numbers could also result from a reduced tumor burden, as many studies have shown that MDSC numbers in tumor-bearing hosts correlate with tumor size. We performed MDSC depletion experiments and showed that the anti-tumor effect of DA depends largely on monocytic MDSCs. Together with in vitro data showing that pretreatment of monocytic MDSCs from tumor-bearing mice with DA significantly inhibited their suppressive function without inducing cell death, these data support the conclusion that DA retarded tumor growth, mainly through inhibiting the suppressive function of monocytic MDSCs. The possibility that DA may directly reduce the MDSC numbers in tumor bearers needs to be explored further.
An intriguing parallel to the expression of D1-like receptors in MDSCs was described for Tregs, which also expressed significantly more D1-like receptors than effector T cells [27]. DA, by signaling through D1-like receptors, caused the down-regulation of Treg function in a mouse model of CNS injury [27]. As Tregs and MDSCs represent two major immunosuppressive cell types that are expanded in patients with advanced malignancies and critically contribute to cancer immune evasion [42], such a result, together with ours reported here, offers mechanistic explanations to the enhancement of host anti-tumor response by DA via the inhibition of immunosuppressive Tregs and MDSCs. With the consideration that MDSCs can further promote the induction, activation, and expansion of tumor-specific Tregs [19], and in vitro and in vivo inhibition of MDSC could abrogate Treg proliferation and tumor-induced immune tolerance [43], we reason that DA may promote the anti-tumor response also by down-regulating Treg function indirectly through the inhibition of MDSCs. Whether DA-mediated enhancement of the host anti-tumor response involves additional cell types is currently being investigated.
Functionally, MDSCs suppress effector T-cell responses through NO and ROS production and arginase-1-mediated L-arginine depletion [16]. Recent studies have shown that monocytic MDSCs express high levels of NO and little ROS, and their immunosuppressive activity can be blocked substantially by iNOS inhibitors or significantly impaired in iNOS-deficient hosts [17], supporting NO as a pivotal effector of the suppressive activity of monocytic MDSCs. MDSC-derived NO suppresses effector T-cell function via mechanisms that include the inhibition of JAK3 and STAT5 signaling [44], MHC-II expression [45], and the induction of apoptosis [46]. Animal experiments suggested further that NO may reduce leukocyte adhesion to tumor-associated endothelium and thereby, impair T-cell homing to metastatic tumor nodules [47]. Our results demonstrate that DA, at nontoxic concentrations, efficiently targets this critical, immunosuppressive effector of murine monocytic MDSCs. The analogous human MDSCs are also suppressive to T-cell proliferation and express iNOS, but the absence of a marker homologous to mouse Gr-1 has made them less clearly defined [6, 36]. In humans, MDSCs are most commonly defined as CD33+HLA-DR‒ and lineage (CD3, CD19, CD56)-negative cells that are generally agreed to be suppressive [6, 36]. Some studies also describe these cells as expressing CD11b or within a CD15+ population [6, 48]. More importantly, our data showed that Lin‒CD33+HLA-DR‒ cells from the peripheral blood of cancer patients also respond to DA in a similar fashion, providing a strong basis for pharmacokinetic studies and clinical trials to test the efficacy of DA or D1-like receptor agonists to overcome tumor-induced immunosuppression in cancer treatment.
The inhibition of NO production by monocytic MDSCs by DA involves the down-regulation of ERK and JNK signaling. The ERK pathway is often aberrantly up-regulated in human cancers and likely contributes to tumor survival and the acquisition and maintenance of tumor stem cell-like properties [49]. Inhibition of ERK signaling through multiple approaches, such as the use of the small molecule inhibitor U0126, the ectopic expression of dominant-negative MEK1, and the knockdown of ERK1 or ERK2, could achieve robust reduction in the propagation of prostate cancer stem-like cells [50]. Moreover, VEGF, a molecule expressed by many human tumors to promote tumor angiogenesis, can also be induced via the MAPK pathway [22]. DA could inhibit VEGF-induced tumor angiogenesis by down-regulating MAPK signaling [22]. The combined inhibitory effects of DA on both tumor angiogenesis and MDSC function may therefore render DA a superior drug candidate for cancer therapy. Indeed, our data showed that the inhibition of NO production by DA was relatively more moderate compared with the inhibition by ERK and JNK inhibitors, indicating that DA may represent a more physiologic immunomodulator than ERK and JNK inhibitors for cancer treatment.
Our finding highlights a possible endogenous cross-talk mechanism between the CNS and the immune system and might explain the intriguing observation of the relatively lower cancer incidence in schizophrenia patients [51], who have increased plasma DA levels. As DA cannot cross the blood-brain barrier, how brain-derived DA may gain access to and regulate MDSC function in cancer patients remains unclear. It is possible that the CNS DA level may influence the synthesis and release of other neurochemicals with immunomodulatory functions [52]. Alternatively, dopaminergic innervation through sympathetic nerves of primary and secondary lymphoid organs, such as the thymus, spleen, lymph nodes, and bone marrow, may subject MDSCs under the regulation of nervous system-derived DA. Endogenous DA can also be derived from many peripheral organs [53]. Many types of immune cells, such as T cells, DCs, macrophages, and neutrophils [54], have the capacity to synthesize DA, enabling them to regulate the function of other DA-responsive immune cells in an autocrine or paracrine manner. Of note, Tregs were recently found to contain substantial amounts of DA that can act on D1-like receptors in an autocrine manner to suppress IL-10 and TGF-β synthesis [55]. Whether MDSCs may produce and release DA to mediate immune regulation in a fashion similar to Tregs remains to be investigated.
Taken together, our study identifies a previously unknown function of the neurotransmitter DA as a potent inhibitor of tumor-induced monocytic MDSCs by signaling through D1-like receptors. With the consideration that DA is a well-characterized drug with manageable toxicity and has been used for the treatment of nonmalignant diseases, our study offers a strong and mechanistic basis for pharmacokinetic studies and clinical trials to evaluate the efficacy of DA or D1-like receptor agonists to overcome tumor-induced immunosuppression in cancer treatment.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from National Natural Science Foundation of China (81201602), Shanghai Pujiang Program (12PJ1407600), Scientific Foundation of Shanghai Health Bureau (20114138), and Scientific Research Foundation for Returned Scholars (Ministry of Education of China; to J.W.) and Shanghai Key Laboratory of Pediatric Gastroenterology and Nutrition (11DZ2260500; to W.C.).
Glossary
- APC
allophycocyanin
- DA
dopamine
- DAF-FM
4-amino-5-methylamino-2′,7′-difluorofluorescein
- DC
dendritic cell
- LLC
Lewis lung carcinoma
- MDSC
myeloid-derived suppressor cell
- MHC-II
MHC class II
- p
phosphorylated
- ROS
reactive oxygen species
- Treg
regulatory T cell
- VEGF
vascular endothelial growth factor
Footnotes
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
AUTHORSHIP
J.W. designed and directed research, performed research, analyzed data, and wrote the manuscript. R.Z. performed research and discussed results. N.T., Z.G., and J.Z. performed research. Y.C. analyzed data and discussed results. K.C. discussed results and revised the manuscript. W.C. designed and directed research.
DISCLOSURES
No potential conflicts of interest were disclosed.
REFERENCES
- 1.Gattinoni L., Powell D. J. Jr, Rosenberg S. A., Restifo N. P. (2006) Adoptive immunotherapy for cancer: building on success. Nat. Rev. Immunol. 6, 383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kusmartsev S., Gabrilovich D. I. (2006) Role of immature myeloid cells in mechanisms of immune evasion in cancer. Cancer Immunol. Immunother. 55, 237–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rabinovich G. A., Gabrilovich D., Sotomayor E. M. (2007) Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 25, 267–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Talmadge J. E. (2007) Pathways mediating the expansion and immunosuppressive activity of myeloid-derived suppressor cells and their relevance to cancer therapy. Clin. Cancer Res. 13, 5243–5248. [DOI] [PubMed] [Google Scholar]
- 5.Shojaei F., Wu X., Malik A. K., Zhong C., Baldwin M. E., Schanz S., Fuh G., Gerber H. P., Ferrara N. (2007) Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat. Biotechnol. 25, 911–920. [DOI] [PubMed] [Google Scholar]
- 6.Gabrilovich D. I., Nagaraj S. (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 9, 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kusmartsev S., Gabrilovich D. I. (2003) Inhibition of myeloid cell differentiation in cancer: the role of reactive oxygen species. J. Leukoc. Biol. 74, 186–196. [DOI] [PubMed] [Google Scholar]
- 8.Melani C., Chiodoni C., Forni G., Colombo M. P. (2003) Myeloid cell expansion elicited by the progression of spontaneous mammary carcinomas in c-erbB-2 transgenic BALB/c mice suppresses immune reactivity. Blood 102, 2138–2145. [DOI] [PubMed] [Google Scholar]
- 9.Youn J. I., Nagaraj S., Collazo M., Gabrilovich D. I. (2008) Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 181, 5791–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Almand B., Clark J. I., Nikitina E., van Beynen J., English N. R., Knight S. C., Carbone D. P., Gabrilovich D. I. (2001) Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J. Immunol. 166, 678–689. [DOI] [PubMed] [Google Scholar]
- 11.Diaz-Montero C. M., Salem M. L., Nishimura M. I., Garrett-Mayer E., Cole D. J., Montero A. J. (2009) Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 58, 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kusmartsev S. A., Li Y., Chen S. H. (2000) Gr-1+ myeloid cells derived from tumor-bearing mice inhibit primary T cell activation induced through CD3/CD28 costimulation. J. Immunol. 165, 779–785. [DOI] [PubMed] [Google Scholar]
- 13.Bronte V., Serafini P., Mazzoni A., Segal D. M., Zanovello P. (2003) L-Arginine metabolism in myeloid cells controls T-lymphocyte functions. Trends Immunol. 24, 302–306. [DOI] [PubMed] [Google Scholar]
- 14.Kusmartsev S., Nefedova Y., Yoder D., Gabrilovich D. I. (2004) Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J. Immunol. 172, 989–999. [DOI] [PubMed] [Google Scholar]
- 15.Murdoch C., Muthana M., Coffelt S. B., Lewis C. E. (2008) The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 8, 618–631. [DOI] [PubMed] [Google Scholar]
- 16.Ostrand-Rosenberg S., Sinha P. (2009) Myeloid-derived suppressor cells: linking inflammation and cancer. J. Immunol. 182, 4499–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Movahedi K., Guilliams M., Van den Bossche J., Van den Bergh R., Gysemans C., Beschin A., De Baetselier P., Van Ginderachter J. A. (2008) Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 111, 4233–4244. [DOI] [PubMed] [Google Scholar]
- 18.Lesokhin A. M., Hohl T. M., Kitano S., Cortez C., Hirschhorn-Cymerman D., Avogadri F., Rizzuto G. A., Lazarus J. J., Pamer E. G., Houghton A. N., Merghoub T., Wolchok J. D. (2012) Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 72, 876–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang B., Pan P. Y., Li Q., Sato A. I., Levy D. E., Bromberg J., Divino C. M., Chen S. H. (2006) Gr-1+CD115+ immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res. 66, 1123–1131. [DOI] [PubMed] [Google Scholar]
- 20.Garcia M. R., Ledgerwood L., Yang Y., Xu J., Lal G., Burrell B., Ma G., Hashimoto D., Li Y., Boros P., Grisotto M., van Rooijen N., Matesanz R., Tacke F., Ginhoux F., Ding Y., Chen S. H., Randolph G., Merad M., Bromberg J. S., Ochando J. C. (2010) Monocytic suppressive cells mediate cardiovascular transplantation tolerance in mice. J. Clin. Invest. 120, 2486–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kao J., Ko E. C., Eisenstein S., Sikora A. G., Fu S., Chen S. H. (2011) Targeting immune suppressing myeloid-derived suppressor cells in oncology. Crit. Rev. Oncol. Hematol. 77, 12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sarkar C., Chakroborty D., Chowdhury U. R., Dasgupta P. S., Basu S. (2008) Dopamine increases the efficacy of anticancer drugs in breast and colon cancer preclinical models. Clin. Cancer Res. 14, 2502–2510. [DOI] [PubMed] [Google Scholar]
- 23.Olsen J. H., Friis S., Frederiksen K., McLaughlin J. K., Mellemkjaer L., Møller H. (2005) Atypical cancer pattern in patients with Parkinson’s disease. Br. J. Cancer 92, 201–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rugbjerg K., Friis S., Lassen C. F., Ritz B., Olsen J. H. (2012) Malignant melanoma, breast cancer and other cancers in patients with Parkinson’s disease. Int. J. Cancer 131, 1904–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakano K., Higashi T., Hashimoto K., Takagi R., Tanaka Y., Matsushita S. (2008) Antagonizing dopamine D1-like receptor inhibits Th17 cell differentiation: preventive and therapeutic effects on experimental autoimmune encephalomyelitis. Biochem. Biophys. Res. Commun. 373, 286–291. [DOI] [PubMed] [Google Scholar]
- 26.Nakagome K., Imamura M., Okada H., Kawahata K., Inoue T., Hashimoto K., Harada H., Higashi T., Takagi R., Nakano K., Hagiwara K., Kanazawa M., Dohi M., Nagata M., Matsushita S. (2011) Dopamine D1-like receptor antagonist attenuates Th17-mediated immune response and ovalbumin antigen-induced neutrophilic airway inflammation. J. Immunol. 186, 5975–5982. [DOI] [PubMed] [Google Scholar]
- 27.Kipnis J., Cardon M., Avidan H., Lewitus G. M., Mordechay S., Rolls A., Shani Y., Schwartz M. (2004) Dopamine, through the extracellular signal-regulated kinase pathway, downregulates CD4+CD25+ regulatory T-cell activity: implications for neurodegeneration. J. Neurosci. 24, 6133–6143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McKenna F., McLaughlin P. J., Lewis B. J., Sibbring G. C., Cummerson J. A., Bowen-Jones D., Moots R. J. (2002) Dopamine receptor expression on human T- and B-lymphocytes, monocytes, neutrophils, eosinophils and NK cells: a flow cytometric study. J. Neuroimmunol. 132, 34–40. [DOI] [PubMed] [Google Scholar]
- 29.Seung L. P., Rowley D. A., Dubey P., Schreiber H. (1995) Synergy between T-cell immunity and inhibition of paracrine stimulation causes tumor rejection. Proc. Natl. Acad. Sci. USA 92, 6254–6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xia S., Sha H., Yang L., Ji Y., Ostrand-Rosenberg S., Qi L. (2011) Gr-1+ CD11b+ myeloid-derived suppressor cells suppress inflammation and promote insulin sensitivity in obesity. J. Biol. Chem. 286, 23591–23599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang W. J., Wei H., Hagen T., Frei B. (2007) Alpha-lipoic acid attenuates LPS-induced inflammatory responses by activating the phosphoinositide 3-kinase/Akt signaling pathway. Proc. Natl. Acad. Sci. USA 104, 4077–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Missale C., Nash S. R., Robinson S. W., Jaber M., Caron M. G. (1998) Dopamine receptors: from structure to function. Physiol. Rev. 78, 189–225. [DOI] [PubMed] [Google Scholar]
- 33.Zoglmeier C., Bauer H., Nörenberg D., Wedekind G., Bittner P., Sandholzer N., Rapp M., Anz D., Endres S., Bourquin C. (2011) CpG blocks immunosuppression by myeloid-derived suppressor cells in tumor-bearing mice. Clin. Cancer Res. 17, 1765–1775. [DOI] [PubMed] [Google Scholar]
- 34.Kusmartsev S., Gabrilovich D. I. (2002) Immature myeloid cells and cancer-associated immune suppression. Cancer Immunol. Immunother. 51, 293–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chan E. D., Riches D. W. (2001) IFN-γ + LPS induction of iNOS is modulated by ERK, JNK/SAPK, and p38(MAPK) in a mouse macrophage cell line. Am. J. Physiol. Cell Physiol. 280, C441–C450. [DOI] [PubMed] [Google Scholar]
- 36.Lechner M. G., Liebertz D. J., Epstein A. L. (2010) Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J. Immunol. 185, 2273–2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ko H. J., Kim Y. J., Kim Y. S., Chang W. S., Ko S. Y., Chang S. Y., Sakaguchi S., Kang C. Y. (2007) A combination of chemoimmunotherapies can efficiently break self-tolerance and induce antitumor immunity in a tolerogenic murine tumor model. Cancer Res. 67, 7477–7486. [DOI] [PubMed] [Google Scholar]
- 38.Nefedova Y., Fishman M., Sherman S., Wang X., Beg A. A., Gabrilovich D. I. (2007) Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res. 67, 11021–11028. [DOI] [PubMed] [Google Scholar]
- 39.Serafini P., Meckel K., Kelso M., Noonan K., Califano J., Koch W., Dolcetti L., Bronte V., Borrello I. (2006) Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J. Exp. Med. 203, 2691–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rodriguez P. C., Hernandez C. P., Quiceno D., Dubinett S. M., Zabaleta J., Ochoa J. B., Gilbert J., Ochoa A. C. (2005) Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J. Exp. Med. 202, 931–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zaroslinski J. F., Possley L. H., Schwartz R. A., Morris R. N., Carone F. A., Browne R. K. (1977) The pharmacology and subacute toxicology of dopamine. Proc. R. Soc. Med. 70 (Suppl 2), 2–6. [PMC free article] [PubMed] [Google Scholar]
- 42.Pan P. Y., Ma G., Weber K. J., Ozao-Choy J., Wang G., Yin B., Divino C. M., Chen S. H. (2010) Immune stimulatory receptor CD40 is required for T-cell suppression and T regulatory cell activation mediated by myeloid-derived suppressor cells in cancer. Cancer Res. 70, 99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Serafini P., Mgebroff S., Noonan K., Borrello I. (2008) Myeloid-derived suppressor cells promote cross-tolerance in B-cell lymphoma by expanding regulatory T cells. Cancer Res. 68, 5439–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bingisser R. M., Tilbrook P. A., Holt P. G., Kees U. R. (1998) Macrophage-derived nitric oxide regulates T cell activation via reversible disruption of the Jak3/STAT5 signaling pathway. J. Immunol. 160, 5729–5734. [PubMed] [Google Scholar]
- 45.Harari O., Liao J. K. (2004) Inhibition of MHC II gene transcription by nitric oxide and antioxidants. Curr. Pharm. Des. 10, 893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rivoltini L., Carrabba M., Huber V., Castelli C., Novellino L., Dalerba P., Mortarini R., Arancia G., Anichini A., Fais S., Parmiani G. (2002) Immunity to cancer: attack and escape in T lymphocyte-tumor cell interaction. Immunol. Rev. 188, 97–113. [DOI] [PubMed] [Google Scholar]
- 47.Gehad A. E., Lichtman M. K., Schmults C. D., Teague J. E., Calarese A. W., Jiang Y., Watanabe R., Clark R. A. (2012) Nitric oxide-producing myeloid-derived suppressor cells inhibit vascular E-selectin expression in human squamous cell carcinomas. J. Invest. Dermatol. 132, 2642–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmielau J., Finn O. J. (2001) Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of T-cell function in advanced cancer patients. Cancer Res. 61, 4756–4760. [PubMed] [Google Scholar]
- 49.Tabu K., Kimura T., Sasai K., Wang L., Bizen N., Nishihara H., Taga T., Tanaka S. (2010) Analysis of an alternative human CD133 promoter reveals the implication of Ras/ERK pathway in tumor stem-like hallmarks. Mol. Cancer 9, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rybak A. P., Ingram A. J., Tang D. (2013) Propagation of human prostate cancer stem-like cells occurs through EGFR-mediated ERK activation. PLoS ONE 8, e61716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cohen M., Dembling B., Schorling J. (2002) The association between schizophrenia and cancer: a population-based mortality study. Schizophr. Res. 57, 139–146. [DOI] [PubMed] [Google Scholar]
- 52.Asanuma M., Ogawa N., Sora Y. H., Pongdhana K., Haba K., Mori A. (1990) Alterations of somatostatin and its modulation by levodopa in MPTP-treated mouse brain. J. Neurol. Sci. 100, 155–160. [DOI] [PubMed] [Google Scholar]
- 53.Goldstein D. S., Mezey E., Yamamoto T., Aneman A., Friberg P., Eisenhofer G. (1995) Is there a third peripheral catecholaminergic system? Endogenous dopamine as an autocrine/paracrine substance derived from plasma DOPA and inactivated by conjugation. Hypertens. Res. 18 (Suppl 1), S93–S99. [DOI] [PubMed] [Google Scholar]
- 54.Sarkar C., Basu B., Chakroborty D., Dasgupta P. S., Basu S. (2010) The immunoregulatory role of dopamine: an update. Brain Behav. Immun. 24, 525–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cosentino M., Fietta A. M., Ferrari M., Rasini E., Bombelli R., Carcano E., Saporiti F., Meloni F., Marino F., Lecchini S. (2007) Human CD4+CD25+ regulatory T cells selectively express tyrosine hydroxylase and contain endogenous catecholamines subserving an autocrine/paracrine inhibitory functional loop. Blood 109, 632–642. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.