

Abstract
Background
Proteins are composed of one or more amino acid chains and exhibit several structure levels. IDPs (intrinsically disordered proteins) represent a class of proteins that do not fold into any particular conformation and exist as dynamic ensembles in their native state. Due to their intrinsic adaptability, IDPs participate in many regulatory biological processes, including parasite immune escape. Using the information from trypanosomatids proteomes, we developed a pipeline for the identification, characterization and analysis of IDPs. The pipeline employs six disorder prediction methodologies and integrates structural and functional annotation information, subcellular location prediction and physicochemical properties. At the core of the IDP pipeline, there is a relational database that describes the protein disorder knowledge in a logically consistent manner.
Results
The results obtained from the IDP pipeline showed that Leishmania and Trypanosoma species have approximately 70% and 55% IDPs, respectively. Our results indicate that IDPs in trypanosomatids contain disorder-promoting amino acids and order-promoting amino acids. The functional annotation analysis demonstrated enrichment of selected Gene Ontology terms. A relevant association was observed between the disordered residue numbers within predicted IDPs and their subcellular location, lack of transmembrane domains and lack of predicted function. We validated our computational findings with 2D electrophoresis designed for IDP identification and found that 100% of the identified protein spots were predicted in silico.
Conclusions
Because there is no pipeline or database addressing IDPs in trypanosomatids, the pipeline described here represents the first attempt to establish possible correlations between protein function and structural disorder in these eukaryotes. Interestingly, all significant associations detected in the contingency analysis were observed when the protein disorder content reached approximately 40%. The exploratory data analysis allowed us to develop hypotheses regarding the IDPs’ association with key biological features of these parasites, including transcription and transcriptional regulation, RNA processing and splicing, and cytoskeleton.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-15-1100) contains supplementary material, which is available to authorized users.
Keywords: Intrinsically disordered proteins (IDPs), Trypanosomatids, IDP prediction
Background
Intrinsically disordered proteins
The traditional view of protein structure/function connection holds as one of its principles the concept that the biological function of a protein is critically dependent on a well-defined 3D conformational structure.
Despite the existence of studies that date back over 20 years [1, 2] demonstrating the existence of proteins or protein domains without a defined structure, the generality of this phenomenon was reported in 1999 [3] and has only recently been extensively studied (see Additional file 1).
In the early 1990s, the existence of functionally active proteins lacking a stable conformation under physiological conditions was evidenced by several studies. These proteins, currently known as IDPs (intrinsically disordered proteins), were identified in both prokaryotes and eukaryotes, including mammals. Some IDPs are either fully disordered or completely structured, but most have intrinsically disordered regions (IDRs) that comprise unstructured segments whose amino acid composition prevents autonomous folding [4].
Only 32% of the crystallized structures available in the PDB (Protein Data Bank) [5] are completely devoid of disorder, indicating that a stable 3D fold represents the exception rather than the rule [6]. Some authors have even suggested that intrinsic protein disorder may require a reassessment of the protein-structure-function paradigm [7].
The number of IDPs and IDRs in various proteomes is very large. For example, nearly 75% of the signaling proteins in mammals are predicted to contain long disordered regions of more than 30 residues, approximately half of their total proteins are predicted to contain such long disordered regions, and approximately 25% of their proteins are predicted to be fully disordered. The results observed by Dunker and colleagues [8] show that for Eukaryota genomes such as Drosophila melanogaster and Homo sapiens, the percentage of proteins with contiguous disordered segments with lengths greater than 30 amino acids is 36.6% and 35.2%, respectively. In contrast, it has been reported [9] that general patterns distinguishing disordered regions and amino acid compositions present in non-parasitic protozoa such as T. thermophila are lacking. For instance, parasitic protozoan datasets are significantly depleted in tryptophan (W) and enriched in lysine (K) [9]. IDPs and IDRs show an amazing structural variability as well as a very wide variety of functions, although the unfoldome and unfoldomics concepts were only recently introduced [10].
A peculiar feature of IDPs is that as a consequence of their “lack” of a defined structure, they are associated with high accessibility of their polypeptide chains. Thus, this class of proteins is prone to extensive post-translational modifications, such as phosphorylation, acetylation, and ubiquitination, that can modulate their biological activities [11–13].
The growing list of features attributed to disordered regions and proteins suggests that they act as molecular rheostats to support a continuum of conformational states and transitions. These features enable IDRs and IDPs to mediate highly specific interactions with multiple binding partners [4]. IDPs require interaction/binding with other biomolecules. This process involves a disorder-to-order transition that favors the participation of IDPs in different cellular pathways, including regulatory cascades [14–16]. In this context, IDPs and IDRs play varied roles in regulating the function of their binding partners and in promoting the assembly of supramolecular complexes [10].
Interestingly, the proteins involved in host/parasite interaction processes in Plasmodium falciparum have been described as IDPs. The structural plasticity of IDPs allows promiscuous interactions and may contribute in different ways to parasite invasion and survival within the host, either by inhibiting the generation of effective antibody mediated-responses or by facilitating interaction with host molecules necessary for a successful infection [17].
Model organism
The Family Trypanosomatidae includes organisms of the genera Leishmania, Phytomonas, Crithidia, Blastocrithidia, Herpetomonas and Trypanosoma. Two members of these genera are medically important (Leishmania and Trypanosoma).
The African trypanosomes (Trypanosoma brucei, T. congolense and T. vivax) are endemic in rural areas in sub-Saharan Africa and cause sleeping sickness in humans. This disease can be fatal if untreated [18, 19]. Trypanosoma cruzi is endemic in South and Central America and is the etiological agent of Chagas disease [18, 20].
Parasites of the genus Leishmania cause leishmaniasis, a multifaceted disease presenting a wide spectrum of clinical manifestations. In humans, disease presentation varies from self-healing cutaneous lesions to visceral disorders that may lead to death if untreated [18].
The range of drugs available for treatment is limited, and drug resistance has been observed [21].
Due to the important medical and public health aspects of trypanosomatids, this work investigated the genomic information on L. braziliensis, L. major, L. infantum, L. mexicana, L. tarentolae, T. cruzi and T. brucei in a wide in silico comparative analysis for the identification and characterization of IDPs.
IDP prediction
Due to the functional plasticity of IDPs [3, 22–27] and the problems associated with their recognition, several different computational methodologies and analysis tools have been developed to enable the systematic identification of the structural disorder of proteins from different predicted proteomes [28].
Despite the existence of numerous algorithms, the computational identification of IDPs still represents a major challenge, mainly because of the absence of a proper consensus definition on how to identify an IDP [3, 29–33].
As general features, proteins classified as IDPs possess low-complexity regions, low hydrophobic amino acid content, and therefore highly polar characteristics and charged amino acids [34, 35]. These attributes are consistent with the inability of these proteins to adopt a globular conformation and have motivated the evolution of several algorithms designed to predict disordered regions in proteins [34, 36–40]. In addition to these features, other peculiarities have been employed in the identification of protein structural disorders, such as the preference for specific amino acids and the high variability of these amino acids in sequences [32, 37, 41]. Another major factor is related to the length of disordered regions required for identification as an IDP, as prediction accuracy is higher for long disordered regions (larger than 40 base pairs) [42].
Given the wide range of features, multifactorial computational approaches are still scarce and unable to predict all the characteristics described above in silico. Notably, specific methodologies have been described [43] that perform better with certain protein characteristics. Pryor et al. [43] evaluated the performance of 13 disorder predictor programs to predict disordered regions located in integral membrane proteins. As a result, it has become clear that combinatorial approaches should provide more complete answers to the problem of predicting IDPs [38].
NMR (nuclear magnetic resonance) studies have shown that the degree of disorder in proteins can vary greatly, ranging from the total absence of secondary and tertiary structure [44, 45] to the presence of partial secondary structures [3, 27, 46], which further complicates predictions. Recently, single-cell experiments with IDPs have provided new insights into the biophysics and complexity of these proteins. These studies promise to better illuminate the critical roles of the biophysics underlying protein disorder [47].
Single-molecule experiments address three broad classes of structural and functional complexity: a) the conformational features and dynamics of monomeric IDPs; b) IDP interactions with binding partners and concomitant folding; and c) the more complex behavior of IDPs, with a specific focus on binding-modulated function by interaction with multiple partners.
Due to the complexity in predicting these regions, we used the ROC (receiver operating characteristic) curve [48] methodology to identify the best combination of structural disorder predictors, which was then used in a computational pipeline applying ab initio techniques for genome-scale prediction in trypanosomatids.
Methods
The IDP pipeline was developed to run in a Linux environment implemented in the Perl language using the MySQL Database Management System. Version 2.3 of the Leishmania braziliensis, Leishmania major, Leishmania infantum, Trypanosoma brucei and Trypanosoma cruzi proteomes and version 4.0 of the Leishmania mexicana and Leishmania tarentolae proteomes were downloaded from TriTrypDB (http://tritrypdb.org/tritrypdb/).
IDP pipeline
Of note, protein disorder can be defined in many ways depending on the research focus and experimental method used. In this work, we considered an IDP as a given predicted protein that has a region of at least 40 consecutive disordered residues. This definition is not ideal, and this issue represents a field of intense debate that has already been reported in the paper “Assessment of protein disorder region prediction in CASP 10” [48].
The automated pipeline was developed employing different methods of prediction, enabling the identification and characterization of IDPs in different organisms (Figure 1). The predicted proteomes of the studied organisms were the only input to the IDP pipeline; as a result, it generated a MySQL database with IDP characterization information, two files with the analysis of the results (descriptive and contingency analysis), and a set of directories containing the outputs from each algorithm.
Figure 1.
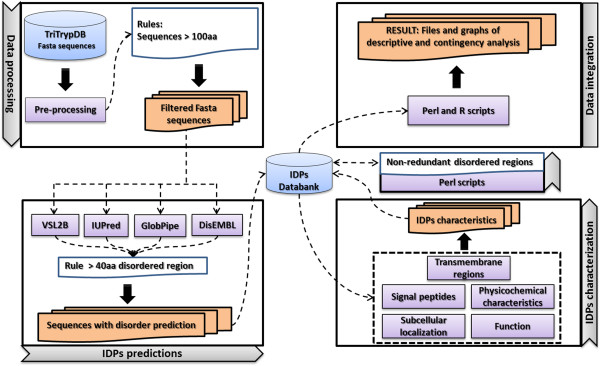
IDP pipeline. The IDP pipeline starts with the pre-processing of FASTA sequences with verification of the sequence length and possible annotation errors. The IDP prediction was made using four different programs. Sequences with regions of at least 40 amino acids were predicted as disordered. The identified IDPs were characterized according to different features. Next, the non-redundant disordered regions were considered for each possible combination of disorder prediction programs. Finally, the descriptive and contingency analyses were conducted. The data generated by the IDP pipeline were stored in a MySQL database for analysis during the process and for future requests.
Pre-processing sequences
Sequences that have possible annotation errors such as an absence of initial methionine, internal stop codons and illegal characters (*, X, B, Z and U), most likely inserted during the process of automatic annotation, were removed.
Because the prediction accuracy is higher for long disordered regions (larger than 40 base pairs), a sequence length cutoff of 100 bp (base pairs) was applied.
IDP prediction
Disorder predictors that were publically available for download and local execution were selected. Four predictors were selected, spanning six different methodologies of disorder prediction: DisEMBL (implements three methods) [37], GlobPipe [49], IUPred [50] and VSL2B [51].
IDP feature prediction
The Phobius algorithm [52] was used to predict transmembrane regions and signal peptides.
Physicochemical predictions were made by the program pepstats from the EMBOSS package (The European Molecular Biology Open Software Suite).
WoLF PSORT was used for the prediction of subcellular locations [53].
The functional annotation of predicted IDPs was based on the Gene Ontology vocabulary of functional classification. The program chosen to perform functional annotation was Blast2GO Pipeline Version B2G4Pipe [54].
The classification of proteins as “hypothetical” or “with predicted function” was based on sequence similarity searches using BLAST (Basic Local Alignment Search Tool) [55] against the non-redundant database (nr) of proteins from NCBI (National Center for Biotechnology Information). The e-value cutoff applied was 1.0 × 10-6.
Functional enrichment analysis
The Perl library GO::TermFinder was used to identify the enrichment of functional terms in IDPs [56]. This algorithm compares genes that code for IDPs to a list of previously functionally annotated genes. A statistical test was used to check for association strength. The hypergeometric distribution and the Bonferroni correction for multiple hypotheses were employed to correct p-values. We defined 0.05 as the minimum p-value for the functional annotation of IDP genes.
Prediction of consensus regions
The protein disorder consensus regions were obtained considering a) the overlap degree and b) the consecutiveness of the regions. In the first case, the regions were grouped, generating a single consensus region. In the second case, an overlap of the subsequent regions was necessary within a margin of 10% of their length around the region boundaries (see Additional file 2).
Localization of protein disorder in a given protein
Each disordered region was classified by its localization along the whole protein sequence length using the following terms: a) N terminal, b) C terminal, c) intermediate, and d) spanning from the C to N end. The N and C terminal regions were defined as 15% of sequence ends. The number and percentage of disordered residues were also calculated.
Descriptive analysis
The descriptive analysis summarizes some key information that resulted from the IDP pipeline. This analysis comprises thirteen descriptive results, including the number of proteins larger than the minimum length chosen by the user, the number of proteins that start with methionine and contain no annotation errors, and the number of IDPs predicted by the best combination of predictors. For optimal viewing of this information, graphs were generated for each of the items described above.
Calculating amino acid frequencies
To analyze the frequency of amino acids present in disordered regions compared with globular regions, the method described by Romero and colleagues was employed [34]. For amino acid frequency estimation in globular regions, the dataset PDB_S25 version May 2010 [57], available at http://bioinfo.tg.fh-giessen.de/pdbselect/, was used. The PDB_S25 dataset consists of non-redundant protein chains, where any two chains share more than 25% sequence identity.
Contingency analysis
The contingency analysis evaluated the association between two variables and predicted whether the frequency of related variables was above or below an expected value obtained through the likelihood ratio chi-square test [58]. We defined 0.05 as the maximum p-value to consider an association to be significant. The analysis was performed in the R environment (http://www.r-project.org/) using the VCD (Visualizing Categorical Data) package. For each found association, an association plot was created [59].
Best combination of structural disorder predictors
To assess the performance of structural disorder prediction algorithms in trypanosomatids, individual and combined algorithm predictions were evaluated. Thus, when we refer to a predictor, we are quoting either an individual predictor or a given combination of predictors.
Considering multiple predictions, the “consensus disorder prediction” was obtained by taking into account the longest overlapped prediction region. A minimum of one amino acid overlap was required to join two different predictions.
ROC analysis [48] was performed using single regions or “consensus disorder predictions” to determine the best combination of predictors.
The positive control dataset was obtained from the Disprot database (version 4.9) (http://www.disprot.org), which stores experimental information about protein disorders. Considering that in this database, each disordered region can be identified by different experimental techniques and that experimentally validated regions can be superposed, consensus information was created to remove positive and negative prediction redundancy (see Additional file 2).
A true positive (TP) was defined every time a prediction was fully inserted (10% tolerance) within the coordinates defined by the Disprot database. A true negative (TN) was defined whenever a prediction was not generated for an ordered (structured) region. A false positive (FP) occurred every time a prediction was generated for an ordered region. Finally, a false negative (FN) was generated whenever a prediction was not generated for a disordered region (see Additional file 3). These values made it possible to calculate two essential metrics of classification performance: True Positive Rate (TPR) = TP/(TP+FN) and False Positive Rate (FPR) = FP/(FP + TN).
Ethics statement
Experiments were performed in compliance with the Ethical Principles in Animal Research adopted by Brazilian College of Animal Experimentation (COBEA) and was approved by the College of Medicine of Ribeirão Preto of the University of São Paulo – Ethical Commission of Ethics in Animal Research (CETEA) under protocol number 118/2008.
Experimental analysis
To validate the computational predictions, we employed two different experimental approaches described in the literature and developed specifically for IDP identification [60, 61].
The first one, Csizmók et al. (an unconventional two-dimensional electrophoresis (2-DE)), was applied to protein extracts of L. major. Proteins were resolved on a 7.5% polyacrylamide native gel for the first dimension (heating for 10 min at 100°C) and on a 5-20% large-format (18 × 16 cm) polyacrylamide gel containing 8 M urea for the second dimension. The two-dimensional gel was stained with colloidal Coomassie (Coomassie blue G-250) for 30 hours. The IDPs were identified near the diagonal line.
The second one, Galea et al. (an IDP enrichment methodology to protein extracts), was applied in L. major and L. braziliensis protein extracts. The purified proteins enriched by heating at 100°C for one hour were subjected to conventional 2-DE. Proteins were focalized in pH 3-5.6 (L. major) and pH 4-7 (L. braziliensis) strips (13 cm, nonlinear gradient), and SDS-PAGE was performed using 12.5% polyacrylamide gels stained with colloidal Coomassie for 30 hours.
Results and discussion
Because these pathogenic organisms are ancestral eukaryotes, the relevance of their disordered proteins in terms of their participation in different biological processes is applicable to higher organisms. Thus, our main objective was to analyze the IDP content in trypanosomatids. Our study approach adopted the development of an automatic pipeline that can be applied not only for trypanosomatids but for any organism. The developed IDP pipeline was applied to seven trypanosomatids proteomes (L. major, L. braziliensis, L. infantum, L. mexicana, L. tarentolae, T. cruzi and T. brucei).
The best combinatorial approach for IDP prediction in trypanosomatid genomes
As discussed above, a number of approaches can be used to predict protein disorder, each presenting its own strengths and weaknesses. To determine which algorithm combination presented the best performance, we built gold standard positive and negative datasets with sequences from the Disprot database (http://www.disprot.org). The performance evaluation was estimated using the ROC graphs approach to select the most efficient algorithm combination. Our results (Figure 2) indicated that the best algorithm combination for structural disorder prediction included REM465, GlobPipe, IUPred and VSL2B (TPR=0.670 and FPR=0.402). This particular combination presented the lowest number of false positives and a high number of false negatives among the five best predictor combinations. Therefore, this combination does not make misclassifications but tends to classify regions with a high certainty of being disorderly. In this respect, it is especially important to combine predictors because the combination can override the specific weaknesses of each predictor alone. As a direct consequence, the presence of FP classifications might result in erroneous profiling of proteins in the association analysis between disordered regions and other features. We observed that among the five best-performing combinations, those with the highest count of FP predictions included the Hotloops predictor. The individual performance of this predictor showed that Hotloops predictions had a higher level of FPs compared with the other predictors. Therefore, we chose an algorithm combination that did not include Hotloops.
Figure 2.
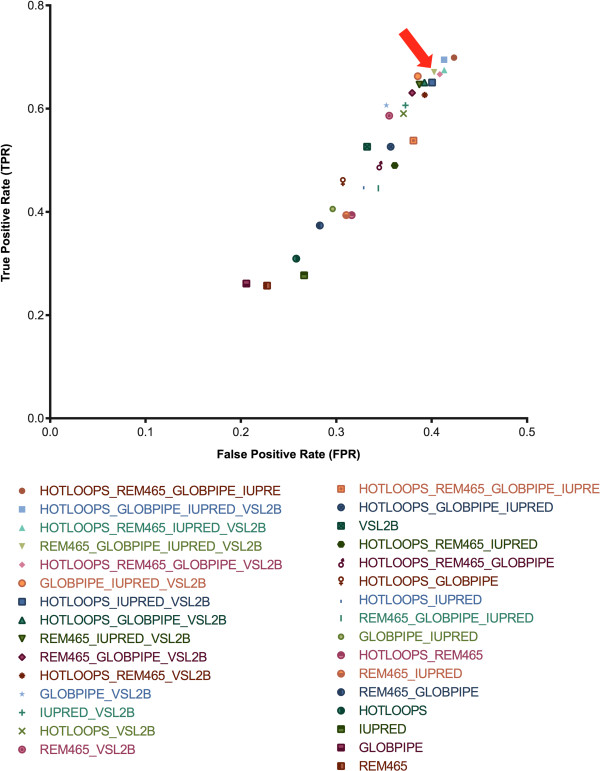
ROC graph. The Disprot database was used to establish the positive and negative datasets to find the most efficient algorithm combination. The TPR and FPR values of all possible combinations among the selected disordered predictors are presented. The chosen combination is indicated with a red arrow. The chosen algorithm combination for structural disorder prediction included REM465, GlobPipe, IUPred and VSL2B (True Positive Rate = 0.670 and False Positive Rate = 0.402). This particular combination presented the lowest count of false positives and a high count of false negatives among the five best predictor combinations.
Thus, despite not having the largest TPR among the five best performing combinations, we believe that this combination is ideal for our analysis.
IDPs in trypanosomatids
Disordered proteins commonly exist without a stable three-dimensional structure and are characterized by highly flexible conformations. Analysis of protein disorder in several genomic sequences has shown that the occurrence of disordered regions of substantial size (more than 40 residues) is surprisingly common in functional proteins [23, 62]. The existence of disordered functional proteins such as hormones [63] and the experimental observation of such proteins in normal cells [64] demonstrates the crucial role of this protein class.
Disordered proteins play an important role in many biological processes, such as the regulation of transcription, translation and signal transduction [3, 25, 65, 66], and their existence is a strong argument for a reassessment of the structure-function paradigm [7].
Our results indicated that approximately 70% and 55% of Leishmania and Trypanosoma protein content, respectively, could be classified as disordered (protein disordered regions greater than or equal to 40 consecutive residues) after the pre-processing filtering step (see Additional files 4 and 5). Such high protein disorder content is not unexpected because similar results were found in other protozoan parasites such as Toxoplasma gondii (almost 70%) and Plasmodium falciparum (almost 50%) [17]. It is important to highlight that similar to trypanosomatids, three of the four apicomplexan species in which IDPs were found to be most abundant (P. falciparum, P. vivax, and T. gondii) represent major and widely distributed human pathogens [17]. Taken together, these results suggest a potential correlation between protein disorder and the genomic plasticity required to interact with different hosts, as previously described by Feng et al. [17].
Another interesting finding is the correlation between functional annotation and protein disorder. In this context, it is important to note that approximately 60% of trypanosomatid genomes consist of hypothetical proteins. Our results indicate that 60-70% of proteins predicted as IDPs have no predicted function (see Additional file 6) inferred by functional sequence similarity annotation methodologies. Considering that the predominantly used methodologies for automatic genome annotation (sequence similarity) have the low-complexity filter enabled as a default setting and matrices that are used as training dataset proteins with different composition profiles (most are globular proteins), our results suggest that this approach is inefficient for the functional annotation of disordered proteins.
A recent publication by Oates and colleagues (D2P2: database of disordered protein predictions) [67] included predictions for several trypanosomatid genomes and implemented a distinctive approach to estimating prediction agreement among the evaluated algorithms.
In contrast to the D2P2 database that evaluated consensus predictions using agreement among the different predictions, our methodology employed gold standard positive and negative datasets obtained from broad mining in public domain databases, including the PDB and Disprot, for the organisms studied. This approach added an essential confidence layer pertaining to the accuracy of predictions for trypanosomatids that is impossible to achieve when considering the 1765 complete proteomes included in D2P2.
IDPs attributes in trypanosomatids
Considering that only slight differences were observed in sequence attributes among the analyzed predicted proteomes, we summarize the most essential ones here.
In Leishmania species, almost 34% of residues were disordered, whereas in Trypanosoma species, 22% of residues were disordered (Figure 3). In comparison, Feng and collaborators [17] found that 10.3% of residues were disordered in P. falciparum. The observed differences may be related to differences in GC content (approximately 60% for trypanosomatids and approximately 20% for P. falciparum) and/or to the use of a prediction approach that involved just one predictor (Hot-loops of DisEMBL) in the work with the Apicomplexa genome and four predictors (REM465, GlobPipe, IUPred and VSL2B) for trypanosomatid genomes. For Leishmania and Trypanosoma species, the great majority (70%) of IDPs contained less than 50% disordered residues (see Additional file 7). Almost 30% of disordered regions ranged from 40 to 60 amino acids in length, with almost 50% of all disordered regions reaching 80 amino acids in length (see Additional file 8).
Figure 3.
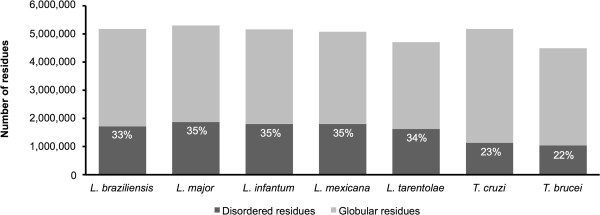
Number of disordered residues. The considered number of disordered residues was predicted by the chosen combination of disorder prediction programs (REM465, GlobPipe, IUPred and VSL2B).
Signal peptide analysis (Figure 4) showed that 45% of the IDPs presented signal peptides for the nucleus, followed by 15% for the mitochondria, 14% for the cytoplasm, 13% for the plasma membrane and 6% for the extracellular space. A previous study with the yeast proteome reported that proteins containing disorder are often located in the cell nucleus [68], suggesting that this occurs because the nucleus represents physical protection for unfolded structures against the cytoplasmic degradation apparatus.
Figure 4.
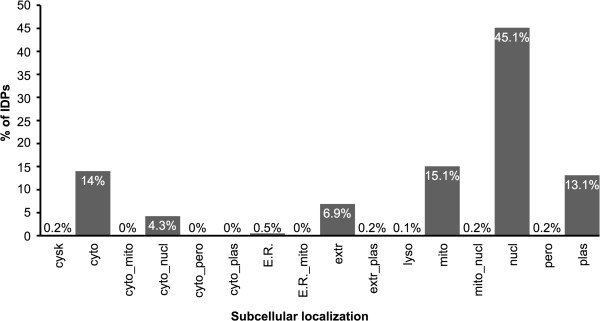
Subcellular localization of IDPs in L. braziliensis. The IDP subcellular localization prediction was performed using the algorithm WolfPSort with the terms cysk (cytoskeleton), cyto (cytosol), cyto_mito (cytosol or mitochondria), cyto_nucl (cytosolic or nuclear), cyto_pero (cytosol or peroxisome) cyto_plas (cytosol or plasma membrane), ER (endoplasmic reticulum), ER_mito (endoplasmic reticulum or mitochondria), extr (extracellular), extr_plas (extracellular or plasma membrane), lyso (lysosome), myth (mitochondria), mito_nucl (mitochondria or nuclear), nuclear (nuclear), pero (peroxisome) and plasma (plasma membrane).
Transmembrane domain predictions for Leishmania and Trypanosoma IDPs showed that approximately 50% and 65% have up to three transmembrane domains, respectively.
As can be observed in Figure 5, there is a tendency for the disordered regions to be located in the intermediate portion of the sequences (35%), followed by the C-terminal end (31%). L. mexicana and L. tarentolae represent outliers of this general profile, and we speculate that the obtained results are associated with the preliminary annotation status of the version of the genome used in this analysis. Additionally, T. cruzi had a much higher number of disordered regions (612 regions) spanning from the C to N-terminal region compared with the other six organisms. We speculate that this bias is associated with the fact that over 50% of the T. cruzi genome consists of repeated sequences [69].
Figure 5.
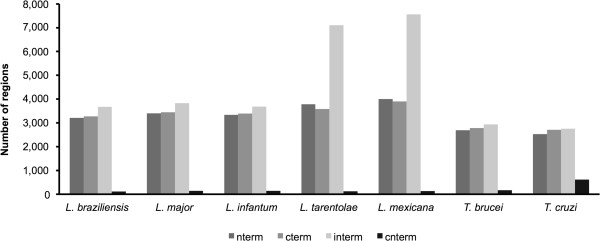
Location of disordered regions in proteins. The disordered regions were classified by the following terms: nterm (N terminal), cterm (C terminal), interm (intermediate) and cnterm (spanning from the C to N end). The N and C terminal regions were defined as 15% of sequence ends. The X-axis represents the name of the organism. The Y-axis indicates the number of disordered regions.
Amino acid frequencies
A comparative analysis of amino acid frequencies in globular protein regions was performed based on PDB_S25 data (http://bioinfo.tg.fh-giessen.de/pdbselect/). Our results suggest a general rule for trypanosomatids IDPs: a) P, Q, E, R and S amino acids are enriched, and b) W, Y, F, V, I, L and C amino acids are depleted (see Additional file 9).
These disordered regions are depleted in hydrophobic amino acids (I, L and V) and aromatic amino acids (W, Y and F), which typically constitute the hydrophobic core of globular proteins [10]. The decreased content of cysteine (C) in disordered regions supports the lack of formation of hydrophobic cores of globular proteins because this amino acid frequently occurs in sites of activating or stabilizing disulfide bridges important for maintaining protein structure in proteins and are thus not necessary in IDPs [25]. Consequently, L, I, F, V, W, Y and C can be considered order promoters.
Amino acids that function as disorder promoters in trypanosomatids are P, Q, E, R and S. For example, proline (P) enrichment is related to the lack of structure because this amino acid is known to oppose the formation of rigid secondary structures. P is actively involved in protein-protein interaction regions [70] and shows a strong preference for open conformation regions, which suggests a functional dimension for the prevalence of this amino acid in IDPs that strongly depends on the target recognition [25].
Disordered proteins in trypanosomatid genomes have low concentrations of aromatic (F, Y and W) and hydrophobic (I, L, F, W and V) amino acids and high concentrations of polar (S, T, Q, R, D and E) and charged (R, D and E) amino acids. Minor variations in the overall behavior may also depend on the experimental method used to identify the region (NMR, X-ray crystallography and circular dichroism) [71], the disordered region size [72], the structural disorder predictor [10] and the location of the disordered region along the sequences (N-terminal, C-terminal and intermediate) [73].
IDP functions in trypanosomatids
Figure 6A shows that the terms ‘binding’ (corrected p-value: 0) and ‘catalytic activity’ (corrected p-value: 2.14e-221) are the most common among the 20 most highly enriched terms in the GO molecular function category. GO defines the term ‘binding’ as the selective, non-covalent, often stoichiometric interaction of a molecule with one or more specific sites on another molecule. GO defines ‘catalytic activity’ as the catalysis of a biochemical reaction at physiological temperatures in a biologically catalyzed reaction. The reactants are known as substrates and the catalysts as enzymes.
Figure 6.
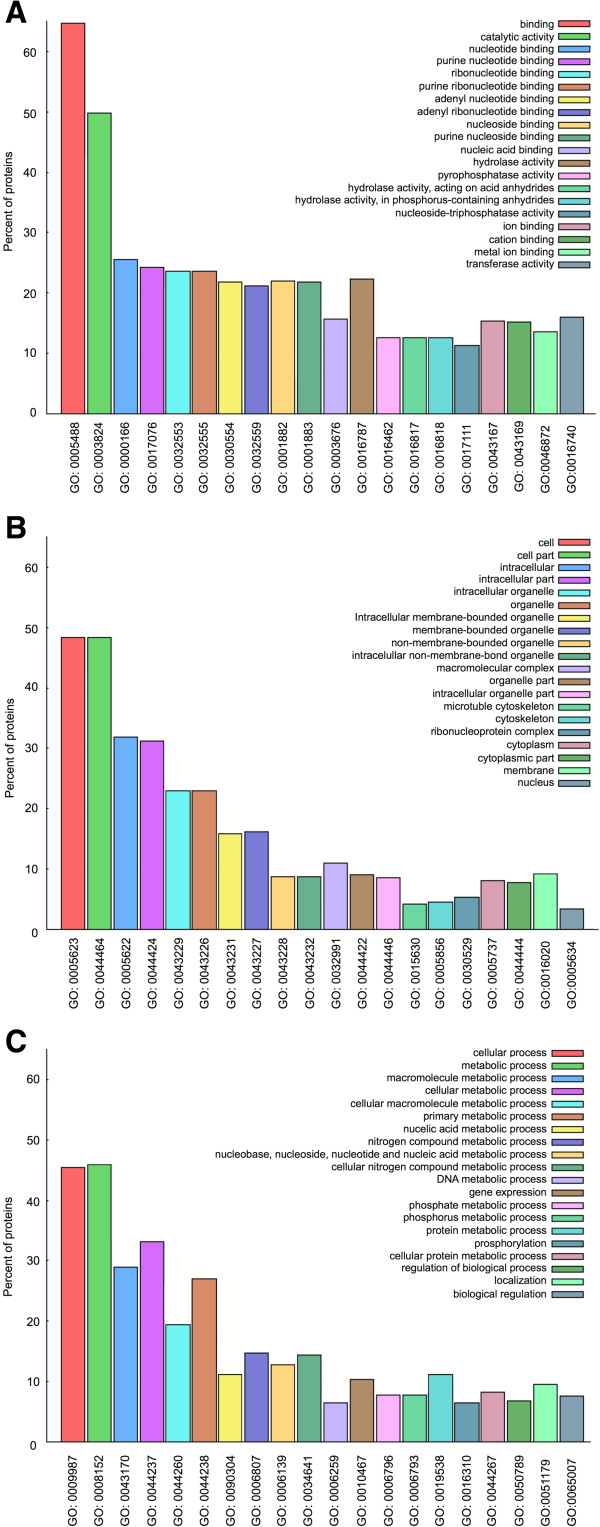
GO enrichment analysis. The Perl library GO::TermFinder was used for the enrichment analysis. A p-value ≤ 0.05 was considered significant. The X-axis indicates the GO number; a description of each is included in the legend. The Y-axis indicates the percentage of proteins. The first 20 enriched GO terms of L. braziliensis IDPs are included for A) molecular function, B) cellular component and C) biological process.
For cellular component category analysis (Figure 6B), we observed that the terms ‘part of the cell’ (corrected p-value: 3.72e-240) and ‘intracellular’ (corrected p-value: 1.38e-132) were the most common. According to the GO definition, ‘part of the cell’ is any constituent part of a cell, defined as the basic structure and functional unit of all organisms. ‘Intracellular’ is the live content of a cell, which is contained by (but not including) the plasma membrane, usually excluding large vacuoles and masses of material ingested or secreted.
For the biological process category (Figure 6C), the terms ‘metabolic process’ (corrected p-value: 6.33e-202) and ‘cellular process’ (corrected p-value: 7.91e-229) were highlighted. According to the GO definition, ‘metabolic processes’ are chemical reactions and pathways including anabolism and catabolism by which living organisms transform chemicals. The definition of ‘cellular process’ is any process performed at the cellular level but not necessarily restricted to a single cell; for example, cellular communication is a cellular process.
Experimental data have revealed that for many proteins, the performed function depends on the degree of unstructuration instead of the degree of protein structuration [74, 75]. Taking this into account, among the many GO terms that we have shown to be significantly enriched (p-value less than 0.05) in IDPs and given their relevance for trypanosomatids, we chose to highlight the following:
Transcription and transcriptional regulation: DNA-protein and protein-protein interactions are the core processes during the transcription process. Several examples of IDPs involved in transcriptional regulation have been reported [15, 22]. For example, the activation domain C-terminus of the proto-oncoprotein (bZIP) is unstructured and flexible and effectively suppresses transcription in vitro [76]. In trypanosomatids, post-transcriptional control is the predominant means by which gene expression is regulated. In fact, it is thought that genes are transcribed and processed continuously and then regulated, either by selective transport to the cytoplasm, mRNA stability or the selection of mRNA sequences for translation. mRNA stability has been widely studied in trypanosomatids, with most data focusing on demonstrating the role of the 3’UTR (3’ untranslated region) and interacting proteins. Much of the field of regulation of gene expression in trypanosomatids needs to be elucidated and investigated. In this context, we suggest that IDPs may play an important role.
RNA processing and splicing: In trypanosomatids, trans-splicing is responsible for processing the polycistronic pre-mRNA, resulting in individual messages. Thus, each mRNA contains the spliced leader sequence (SL) at the 5’ end and the poly-A tail at the 3’ end. Pre-mRNA processing in trypanosomatids is catalyzed by the spliceosome, a high-molecular-weight machinery comprising ribonucleoproteins (RNPs) and other proteins. Proteins involved in spliceosome “assembly” (by facilitating the recruitment of its components) are rich in serine and arginine residues, two amino acids enriched in trypanosomatids IDPs.
Cytoskeleton: A set of protein structures, filaments and microtubules determine the structure and shape of the cell and contribute to the cytoskeleton. IDPs play crucial roles in the assembly and function of the cytoskeleton. The degree of disorder of cytoskeleton proteins is comparable to those observed in proteins involved in cell signaling and regulation [65].
Flagellum: Leishmania and Trypanosoma are flagellate protozoa whose life cycle involves the sequential infection of vector and mammalian hosts. During these processes, complex morphological and biochemical changes occur successively. These processes are particularly important for Leishmania, which possesses both an extracellular flagellated form and an intracellular form without flagella in the mammalian host.
In bacteria, the main flagellum component is a filament called flagellin, ranging from 12 to 25 nm in diameter. Structural disorder plays a crucial role in the assembly of the bacterial flagella [24]. Based on these observations, we suggest that protein terminal disorder is associated with the flagellum in trypanosomatids, and this disorder may play important roles in a manner similar to that occurring in flagellin.
Contingency analysis
With the predictions made from different disorder algorithms and several other analyses, each predicted IDP loaded in the database showed 56 associated variables. To verify the existence of significant associations between protein disorder and other identified biological features, we performed a multivariate investigation using the contingency analysis strategy approach. This approach was applied to thirteen chosen features (protein length, protein annotation, protein localization, protein molecular weight, protein charge, isoelectric point, number of C-terminal disordered regions, number of N-terminal disordered regions, number of intermediate disordered regions, number of CN-terminal disordered regions, percent of disordered residues, number of transmembrane domains and number of disordered regions) to generate a contingency table. The characteristics that were significantly associated with the percentage of disordered residues were a) predicted/hypothetical, b) isoelectric point, c) location and d) transmembrane domains.
A very interesting inverse behavior was observed in all significant associations (p-value less than 0.05) when the protein disorder content reached approximately 40%.
One of these significant associations correlates with two classification terms: a) the percentage of disordered residues and b) functional protein annotation (predicted or hypothetical) (Figure 7A).
Figure 7.
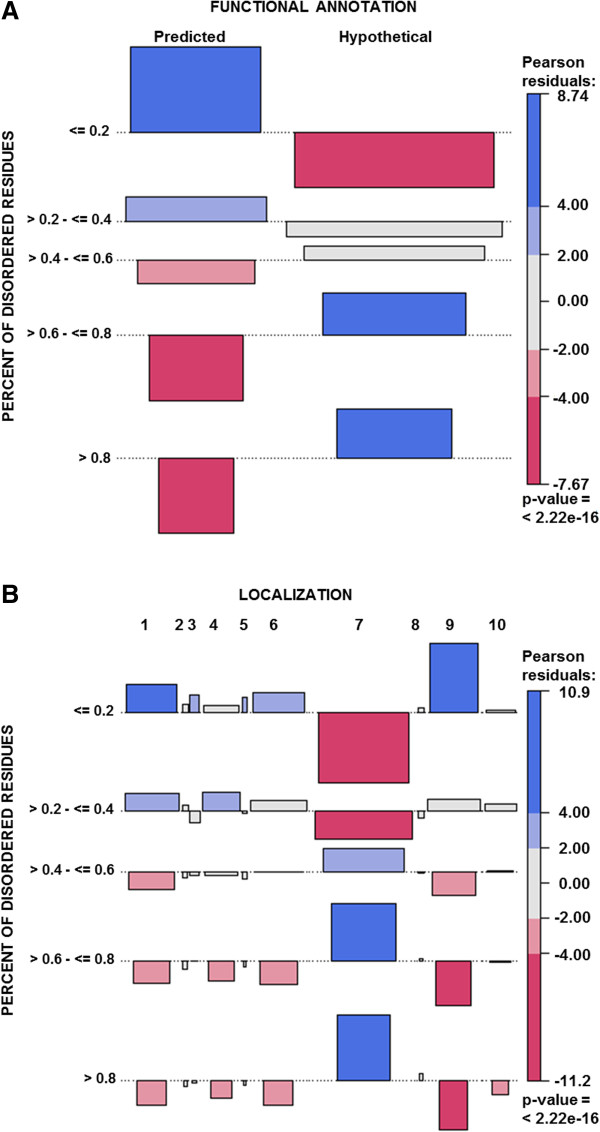
Associations with L. braziliensis IDPs. The VCD package was used, considering a p-value ≤0.05 as a significant association. The colors represent whether the frequency is higher (blue) or lower (pink) than expected. The numbers represent the categories of attributes. The association between the percentage of disordered residues and A) the functional annotation of the protein (predicted or hypothetical) and B) the predicted location as 1- cytosol; 2- cytoskeleton; 3- endoplasmic reticulum; 4- extracellular; 5- lysosome; 6- mitochondria; 7- nuclear; 8- peroxisome; 9- plasma membrane and 10- without prediction.
As the disorder content increased up to 40%, the predicted function annotation became more frequent than expected. For values greater than 40%, we observed a higher hypothetical protein annotation frequency than expected. Approximately 60% of trypanosomatid genomes consisted of hypothetical proteins with no significant similarity with public domain databases. Considering the current scenario of genome automatic annotation, we suggest a direct association of our findings, specifically the increasing of structural disorder percentage, with the high content of unpredicted protein functions in trypanosomatid genomes.
Another significant association correlated the classification term isoelectric point and the percentage of disordered residues. In more than 40% of disordered content, a higher than expected frequency of very basic proteins (pH>9) was observed (see Additional file 10).
The classification term subcellular location also revealed a significant association with the percentage of disordered residues (Figure 7B). When the disordered residue content reached 40%, a higher frequency than expected occurred in the classes plasma membrane, cytosol and mitochondria. In contrast, when the percent of disordered residues was above 40%, a higher increase in the nuclear location class was observed.
The last significant association correlated the classification term transmembrane domain and the percentage of disordered residues. IDPs with more than 40% disordered content tended to contain transmembrane domains at a lower frequency than expected (see Additional file 11).
Experimental analysis
To confirm the computational predictions, two special gel electrophoresis methods were employed to separate and/or enrich protein preparations with IDPs (one was developed by Csizmók et al. and the other by Galea et al.). The first methodology involved a special two-dimensional electrophoresis and was performed only for the L. major proteome (see Additional file 12A) because it required a large amount of parasite culture to obtain a sufficient protein extract. The second approach, based on previous IDPs enrichment, was applied to the L. major and L. braziliensis proteomes (see Additional file 12B and C).
After IDP fractionation, 17 protein spots were selected from the gels and identified by mass spectrometry (MALDI-TOF/TOF analyzer). The results (see Additional file 13) indicated that 100% (15/15) of the identified proteins were recognized as disordered proteins in silico by the best combination of structural disorder predictors (REM465, GlobPipe, IUPRED and VSL2B).
This finding indicated that the pipeline met its goal of integrative IDP prediction in addition to data integration with a safe, automatic and organized computational characterization.
Conclusions
In this work, a central pipeline for the identification, characterization and analysis of IDPs in trypanosomatids was developed. Predictions were experimentally validated, and the pipeline met its goal of data integration by performing a safe, automatic, organized and integrative IDP prediction and computational characterization. The results presented highlight the following: a) the high content of IDPs, approximately 70% for Leishmania and 50% for Trypanosoma species; b) that different features such as functional annotation (predicted/hypothetical), isoelectric point, location (nucleus) and transmembrane domains are significantly associated with the percentage of disordered residues, as confirmed by the contingency analysis; and c) that the terms ‘binding’ and ‘catalytic activity’ >are the most common ones for molecular function category analysis. Thus, the IDP scenario in trypanosomatids may prove to be relevant to the understanding of host-parasite interaction and the peculiarities of pathogens’ biology. In addition, the information regarding the IDP content in trypanosomatids may drive new studies of gene function and the evolution of these ancestral and pathogenic eukaryotes. The aim of this work is not to develop a web server but to perform a deep analysis of IDP content in trypanosomatid genomes. The database and developed pipeline are available to the research community upon request.
Availability of supporting data
The dataset supporting the results of this article is included within the article (and its additional files).
Electronic supplementary material
Additional file 1: Number of publications related to IDPs. The following terms were searched: intrinsically disordered proteins, intrinsically unfolded proteins, intrinsically unstructured proteins and natively unfolded proteins. (PDF 11 KB)
Additional file 2: Consensus disorder prediction. Consensus of disordered prediction. (PDF 28 KB)
Additional file 3: Considered hits and errors. Considered hits and errors. (PDF 31 KB)
Additional file 4: Pre-processing results. Number and percentage of sequences that went into each step of the IDP pipeline pre-processing. (PDF 10 KB)
Additional file 5: IDPs information. Identified IDPs for each organism. (PDF 3 MB)
Additional file 6: IDPs functional annotation. Classification of the identified IDPs (hypothetical or with predicted function). (PDF 10 KB)
Additional file 7: Percent of disordered residues in L. braziliensis. Percentage of disordered residues in L. braziliensis. (PDF 11 KB)
Additional file 8: Percent of disordered regions in L. braziliensis. Percentage of disordered regions in L. braziliensis. (PDF 11 KB)
Additional file 9: Frequency of IDP amino acids relative to globular amino acids in L. braziliensis. Frequency of IDP amino acids relative to globular amino acids in L. braziliensis. (PDF 12 KB)
Additional file 10: Association between the percentage of disordered residues and isoelectric points in L. braziliensis. The colors represent whether the frequency is higher (blue) or lower (pink) than expected. The numbers represent the categories of attributes. (PDF 30 KB)
Additional file 11: Association between the percentage of disordered residues and transmembrane domains in L. braziliensis. The colors represent whether the frequency is higher (blue) or lower (pink) than expected. The numbers represent the categories of attributes. (PDF 31 KB)
Additional file 12: IDPs identification gels. A) L. major 2D electrophoresis gel; the IDPs are located near the diagonal line; B) L. major 2D electrophoresis with IDP enrichment; and C) L. braziliensis 2D electrophoresis with IDP enrichment. (PDF 268 KB)
Additional file 13: Experimental validation. The IDP spot number is provided in Figure S8A, S8B and S8C. (PDF 14 KB)
Acknowledgments
This work was supported by Fundação de Amparo à Pesquisa do Estado de Minas Gerais (http://www.fapemig.br) [APQ-01661-13 and APQ-01085-12 to JCR] and Conselho Nacional de Pesquisa e Desenvolvimento (http://www.cnpq.br) [301652/2012-0 and 486618/2013 to JCR]. AKC’s laboratory received financial support from FAPESP (http://www.fapesp.br) [2006/50323-7].
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
PCR conducted the research, designed the study, and wrote the manuscript. RT participated in the design of the study. JST carried out the 2D gels. VSA helped to draft the manuscript. AKC provided helpful discussions of the results and the draft of the manuscript. JCR supervised the research and wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Patrícia de Cássia Ruy, Email: patiruy@gmail.com.
Raul Torrieri, Email: raul.ibm@gmail.com.
Juliano Simões Toledo, Email: jstoledo@gmail.com.
Viviane de Souza Alves, Email: gouveia_va@yahoo.com.
Angela Kaysel Cruz, Email: akcruz@fmrp.usp.br.
Jeronimo Conceição Ruiz, Email: jeronimo@cpqrr.fiocruz.br.
References
- 1.Lynch WP, Riseman VM, Bretscher A. Smooth muscle caldesmon is an extended flexible monomeric protein in solution that can readily undergo reversible intra- and intermolecular sulfhydryl cross-linking. A Mech caldesmon’s F-actin bundling activity. J Biol Chem. 1987;262(15):7429–7437. [PubMed] [Google Scholar]
- 2.Barlow PN, Vidal JC, Lister MD, Hancock AJ, Sigler PB. Synthesis and some properties of constrained short-chain phosphatidylcholine analogues: (+)- and (-)-(1,3/2)-1-O-(phosphocholine)2,3-O- dihexanoylcyclopentane-1,2,3-triol. Chem Phys Lipids. 1988;46(3):157–164. doi: 10.1016/0009-3084(88)90017-5. [DOI] [PubMed] [Google Scholar]
- 3.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J Mol Biol. 1999;293(2):321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 4.Buljan M, Chalancon G, Dunker AK, Bateman A, Balaji S, Fuxreiter M, Babu MM. Alternative splicing of intrinsically disordered regions and rewiring of protein interactions. Curr Opin Struct Biol. 2013;23(3):443–450. doi: 10.1016/j.sbi.2013.03.006. [DOI] [PubMed] [Google Scholar]
- 5.Berman HM, Bhat TN, Bourne PE, Feng Z, Gilliland G, Weissig H, Westbrook J. The Protein Data Bank and the challenge of structural genomics. Nat Struct Biol. 2000;7(Suppl):957–959. doi: 10.1038/80734. [DOI] [PubMed] [Google Scholar]
- 6.Obradovic Z, Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK. Predicting intrinsic disorder from amino acid sequence. Proteins. 2003;53(Suppl 6):566–572. doi: 10.1002/prot.10532. [DOI] [PubMed] [Google Scholar]
- 7.Dunker AK, Obradovic Z. The protein trinity–linking function and disorder. Nat Biotechnol. 2001;19(9):805–806. doi: 10.1038/nbt0901-805. [DOI] [PubMed] [Google Scholar]
- 8.Dunker AK, Obradovic Z, Romero P, Garner EC, Brown CJ. Intrinsic protein disorder in complete genomes. Genome Inform Ser Workshop Genome Inform. 2000;11:161–171. [PubMed] [Google Scholar]
- 9.Mohan A, Sullivan WJ, Radivojac P, Dunker AK, Uversky VN. Intrinsic disorder in pathogenic and non-pathogenic microbes: discovering and analyzing the unfoldomes of early-branching eukaryotes. Mol Biosyst. 2008;4(4):328–340. doi: 10.1039/b719168e. [DOI] [PubMed] [Google Scholar]
- 10.Uversky VN. The mysterious unfoldome: structureless, underappreciated, yet vital part of any given proteome. J Biomed Biotechnol. 2010;2010:568068. doi: 10.1155/2010/568068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishida N, Hara T, Kamura T, Yoshida M, Nakayama K, Nakayama KI. Phosphorylation of p27Kip1 on serine 10 is required for its binding to CRM1 and nuclear export. J Biol Chem. 2002;277(17):14355–14358. doi: 10.1074/jbc.C100762200. [DOI] [PubMed] [Google Scholar]
- 12.Blain SW, Massagué J. Breast cancer banishes p27 from nucleus. Nat Med. 2002;8(10):1076–1078. doi: 10.1038/nm1002-1076. [DOI] [PubMed] [Google Scholar]
- 13.Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr Biol. 1999;9(12):661–664. doi: 10.1016/S0960-9822(99)80290-5. [DOI] [PubMed] [Google Scholar]
- 14.Kriwacki RW, Hengst L, Tennant L, Reed SI, Wright PE. Structural studies of p21Waf1/Cip1/Sdi1 in the free and Cdk2-bound state: conformational disorder mediates binding diversity. Proc Natl Acad Sci U S A. 1996;93(21):11504–11509. doi: 10.1073/pnas.93.21.11504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dyson HJ, Wright PE. Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol. 2002;12(1):54–60. doi: 10.1016/S0959-440X(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 16.Lacy ER, Filippov I, Lewis WS, Otieno S, Xiao L, Weiss S, Hengst L, Kriwacki RW. p27 binds cyclin-CDK complexes through a sequential mechanism involving binding-induced protein folding. Nat Struct Mol Biol. 2004;11(4):358–364. doi: 10.1038/nsmb746. [DOI] [PubMed] [Google Scholar]
- 17.Feng Z, Zhang X, Han P, Arora N, Anders R, Norton R. Abundance of intrinsically unstructured proteins in P. falciparum and other apicomplexan parasite proteomes. Mol Biochem Parasitol. 2006;150(2):256–267. doi: 10.1016/j.molbiopara.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 18.Aslett M, Aurrecoechea C, Berriman M, Brestelli J, Brunk BP, Carrington M, Depledge DP, Fischer S, Gajria B, Gao X, et al. TriTrypDB: a functional genomic resource for the Trypanosomatidae. Nucleic Acids Res. 2010;38(Database issue):D457–462. doi: 10.1093/nar/gkp851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cox FE. History of sleeping sickness (African trypanosomiasis) Infect Dis Clin North Am. 2004;18(2):231–245. doi: 10.1016/j.idc.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 20.Hotez PJ, Bottazzi ME, Franco-Paredes C, Ault SK, Periago MR. The neglected tropical diseases of Latin America and the Caribbean: a review of disease burden and distribution and a roadmap for control and elimination. PLoS Negl Trop Dis. 2008;2(9):e300. doi: 10.1371/journal.pntd.0000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.El-On J. Current status and perspectives of the immunotherapy of leishmaniasis. Isr Med Assoc J. 2009;11(10):623–628. [PubMed] [Google Scholar]
- 22.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6(3):197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 23.Dunker A, Brown C, Lawson J, Iakoucheva L, Obradović Z. Intrinsic disorder and protein function. Biochemistry. 2002;41(21):6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 24.Namba K. Roles of partly unfolded conformations in macromolecular self-assembly. Genes Cells. 2001;6(1):1–12. doi: 10.1046/j.1365-2443.2001.00384.x. [DOI] [PubMed] [Google Scholar]
- 25.Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci. 2002;27(10):527–533. doi: 10.1016/S0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- 26.Tompa P, Csermely P. The role of structural disorder in the function of RNA and protein chaperones. FASEB J. 2004;18(11):1169–1175. doi: 10.1096/fj.04-1584rev. [DOI] [PubMed] [Google Scholar]
- 27.Uversky VN, Permyakov SE, Zagranichny VE, Rodionov IL, Fink AL, Cherskaya AM, Wasserman LA, Permyakov EA. Effect of zinc and temperature on the conformation of the gamma subunit of retinal phosphodiesterase: a natively unfolded protein. J Proteome Res. 2002;1(2):149–159. doi: 10.1021/pr0155127. [DOI] [PubMed] [Google Scholar]
- 28.Bracken C, Iakoucheva LM, Romero PR, Dunker AK. Combining prediction, computation and experiment for the characterization of protein disorder. Curr Opin Struct Biol. 2004;14(5):570–576. doi: 10.1016/j.sbi.2004.08.003. [DOI] [PubMed] [Google Scholar]
- 29.Linderstrom-Lang K, Schellman J. Protein Structure and Enzyme Activity. In: Boyer PD, Lardy H, Myrback K, editors. The Enzymes. New York: Academic Press; 1959. pp. 443–510. [Google Scholar]
- 30.Pullen RA, Jenkins JA, Tickle IJ, Wood SP, Blundell TL. The relation of polypeptide hormone structure and flexibility to receptor binding: the relevance of X-ray studies on insulins, glucagon and human placental lactogen. Mol Cell Biochem. 1975;8(1):5–20. doi: 10.1007/BF01731645. [DOI] [PubMed] [Google Scholar]
- 31.Cary PD, Moss T, Bradbury EM. High-resolution proton-magnetic-resonance studies of chromatin core particles. Eur J Biochem. 1978;89(2):475–482. doi: 10.1111/j.1432-1033.1978.tb12551.x. [DOI] [PubMed] [Google Scholar]
- 32.Dunker A, Lawson J, Brown C, Williams R, Romero P, Oh J, Oldfield C, Campen A, Ratliff C, Hipps K, Ausio J, Nissen M, Reeves R, Kang C, Kissinger C, Bailey R, Griswold M, Chiu W, Garner E, Obradovic Z. Intrinsically disordered protein. J Mol Graph Model. 2001;19(1):26–59. doi: 10.1016/S1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 33.Daughdrill GW, Pielak GJ, Uversky VN, Cortese MS, Dunker K. Protein Folding Handbook. vol. Volume 1. Basel, Switzerland: Wiley-VCH; 2005. Natively Disordered Proteins. [Google Scholar]
- 34.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, Dunker AK. Sequence complexity of disordered protein. Proteins. 2001;42(1):38–48. doi: 10.1002/1097-0134(20010101)42:1<38::AID-PROT50>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 35.Vucetic S, Brown C, Dunker A, Obradovic Z. Flavors of protein disorder. Proteins. 2003;52(4):573–584. doi: 10.1002/prot.10437. [DOI] [PubMed] [Google Scholar]
- 36.Uversky VN, Gillespie JR, Fink AL. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41(3):415–427. doi: 10.1002/1097-0134(20001115)41:3<415::AID-PROT130>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 37.Linding R, Jensen L, Diella F, Bork P, Gibson T, Russell R. Protein disorder prediction: implications for structural proteomics. Structure. 2003;11(11):1453–1459. doi: 10.1016/j.str.2003.10.002. [DOI] [PubMed] [Google Scholar]
- 38.Ferron F, Longhi S, Canard B, Karlin D. A practical overview of protein disorder prediction methods. Proteins. 2006;65(1):1–14. doi: 10.1002/prot.21075. [DOI] [PubMed] [Google Scholar]
- 39.Dosztányi Z, Mészáros B, Simon I. Bioinformatical approaches to characterize intrinsically disordered/unstructured proteins. Brief Bioinform. 2010;11(2):225–243. doi: 10.1093/bib/bbp061. [DOI] [PubMed] [Google Scholar]
- 40.Oldfield CJ, Cheng Y, Cortese MS, Brown CJ, Uversky VN, Dunker AK. Comparing and combining predictors of mostly disordered proteins. Biochemistry. 2005;44(6):1989–2000. doi: 10.1021/bi047993o. [DOI] [PubMed] [Google Scholar]
- 41.Brown C, Takayama S, Campen A, Vise P, Marshall T, Oldfield C, Williams C, Dunker A. Evolutionary rate heterogeneity in proteins with long disordered regions. J Mol Evol. 2002;55(1):104–110. doi: 10.1007/s00239-001-2309-6. [DOI] [PubMed] [Google Scholar]
- 42.Han P, Zhang X, Feng ZP. Predicting disordered regions in proteins using the profiles of amino acid indices. BMC Bioinformatics. 2009;10(Suppl 1):S42. doi: 10.1186/1471-2105-10-S1-S42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pryor EE, Wiener MC. A critical evaluation of in silico methods for detection of membrane protein intrinsic disorder. Biophys J. 2014;106(8):1638–1649. doi: 10.1016/j.bpj.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abercrombie BD, Kneale GG, Crane-Robinson C, Bradbury EM, Goodwin GH, Walker JM, Johns EW. Studies on the conformational properties of the high-mobility-group chromosomal protein HMG 17 and its interaction with DNA. Eur J Biochem. 1978;84(1):173–177. doi: 10.1111/j.1432-1033.1978.tb12154.x. [DOI] [PubMed] [Google Scholar]
- 45.Penkett CJ, Redfield C, Dodd I, Hubbard J, McBay DL, Mossakowska DE, Smith RA, Dobson CM, Smith LJ. NMR analysis of main-chain conformational preferences in an unfolded fibronectin-binding protein. J Mol Biol. 1997;274(2):152–159. doi: 10.1006/jmbi.1997.1369. [DOI] [PubMed] [Google Scholar]
- 46.Bai Y, Chung J, Dyson HJ, Wright PE. Structural and dynamic characterization of an unfolded state of poplar apo-plastocyanin formed under nondenaturing conditions. Protein Sci. 2001;10(5):1056–1066. doi: 10.1110/ps.00601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee T, Moran-Gutierrez CR, Deniz AA. Semin Cell Dev Biol. 2014. Probing protein disorder and complexity at single-molecule resolution. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Green DM, Swets JM. Signal detection theory and psychophysics. New York: John Wiley and Sons Inc; 1966. [Google Scholar]
- 49.Linding R, Russell R, Neduva V, Gibson T. GlobPlot: exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003;31(13):3701–3708. doi: 10.1093/nar/gkg519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dosztányi Z, Csizmók V, Tompa P, Simon I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J Mol Biol. 2005;347(4):827–839. doi: 10.1016/j.jmb.2005.01.071. [DOI] [PubMed] [Google Scholar]
- 51.Peng K, Radivojac P, Vucetic S, Dunker A, Obradovic Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinformatics. 2006;7:208. doi: 10.1186/1471-2105-7-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Käll L, Krogh A, Sonnhammer EL. A combined transmembrane topology and signal peptide prediction method. J Mol Biol. 2004;338(5):1027–1036. doi: 10.1016/j.jmb.2004.03.016. [DOI] [PubMed] [Google Scholar]
- 53.Horton P, Park KJ, Obayashi T, Fujita N, Harada H, Adams-Collier CJ, Nakai K. WoLF PSORT: protein localization predictor. Nucleic Acids Res. 2007;35(Web Server issue):W585–587. doi: 10.1093/nar/gkm259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- 55.Altschul S, Gish W, Miller W, Myers E, Lipman D. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 56.Boyle EI, Weng S, Gollub J, Jin H, Botstein D, Cherry JM, Sherlock G. GO::TermFinder–open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics. 2004;20(18):3710–3715. doi: 10.1093/bioinformatics/bth456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hobohm U, Sander C. Enlarged representative set of protein structures. Protein Sci. 1994;3(3):522–524. doi: 10.1002/pro.5560030317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Agresti A. Categorical Data Analysis. 2. Hoboken, NJ, USA: John Wiley & Sons, Inc.; 2002. [Google Scholar]
- 59.Friendly M. SAS User Group International Conference Proceedings, 17, 190–200: 1992. Canada: York University, Downsview, ONT; 1992. Graphical methods for categorical data. [Google Scholar]
- 60.Csizmók V, Szollosi E, Friedrich P, Tompa P. A novel two-dimensional electrophoresis technique for the identification of intrinsically unstructured proteins. Mol Cell Proteomics. 2006;5(2):265–273. doi: 10.1074/mcp.M500181-MCP200. [DOI] [PubMed] [Google Scholar]
- 61.Galea C, Pagala V, Obenauer J, Park C, Slaughter C, Kriwacki R. Proteomic studies of the intrinsically unstructured mammalian proteome. J Proteome Res. 2006;5(10):2839–2848. doi: 10.1021/pr060328c. [DOI] [PubMed] [Google Scholar]
- 62.Uversky VN. Natively unfolded proteins: a point where biology waits for physics. Protein Sci. 2002;11(4):739–756. doi: 10.1110/ps.4210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Boesch C, Bundi A, Oppliger M, Wüthrich K. 1H nuclear-magnetic-resonance studies of the molecular conformation of monomeric glucagon in aqueous solution. Eur J Biochem. 1978;91(1):209–214. doi: 10.1111/j.1432-1033.1978.tb20953.x. [DOI] [PubMed] [Google Scholar]
- 64.Daniels AJ, Williams RJ, Wright PE. The character of the stored molecules in chromaffin granules of the adrenal medulla: a nuclear magnetic resonance study. Neuroscience. 1978;3(6):573–585. doi: 10.1016/0306-4522(78)90022-2. [DOI] [PubMed] [Google Scholar]
- 65.Iakoucheva L, Brown C, Lawson J, Obradović Z, Dunker A. Intrinsic disorder in cell-signaling and cancer-associated proteins. J Mol Biol. 2002;323(3):573–584. doi: 10.1016/S0022-2836(02)00969-5. [DOI] [PubMed] [Google Scholar]
- 66.Iakoucheva LM, Radivojac P, Brown CJ, O’Connor TR, Sikes JG, Obradovic Z, Dunker AK. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004;32(3):1037–1049. doi: 10.1093/nar/gkh253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oates ME, Romero P, Ishida T, Ghalwash M, Mizianty MJ, Xue B, Dosztányi Z, Uversky VN, Obradovic Z, Kurgan L, Dunker A, Gough J. D2P2: database of disordered protein predictions. Nucleic Acids Res. 2013;41(Database issue):D508–516. doi: 10.1093/nar/gks1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ward JJ, Sodhi JS, McGuffin LJ, Buxton BF, Jones DT. Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J Mol Biol. 2004;337(3):635–645. doi: 10.1016/j.jmb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 69.El-Sayed NM, Myler PJ, Bartholomeu DC, Nilsson D, Aggarwal G, Tran AN, Ghedin E, Worthey EA, Delcher AL, Blandin G, Westenberger SJ, Caler E, Cerqueira GC, Branche C, Haas B, Anupama A, Arner E, Aslund L, Attipoe P, Bontempi E, Bringaud F, Burton P, Cadag E, Campbell DA, Carrington M, Crabtree J, Darban H, da Silveira JF, de Jong P, Edwards K. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science. 2005;309(5733):409–415. doi: 10.1126/science.1112631. [DOI] [PubMed] [Google Scholar]
- 70.Williamson MP. The structure and function of proline-rich regions in proteins. Biochem J. 1994;297(Pt 2):249–260. doi: 10.1042/bj2970249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Williams RM, Obradovi Z, Mathura V, Braun W, Garner EC, Young J, Takayama S, Brown CJ, Dunker AK. Pac Symp Biocomput. 2001. The protein non-folding problem: amino acid determinants of intrinsic order and disorder; pp. 89–100. [DOI] [PubMed] [Google Scholar]
- 72.Radivojac P, Obradovic Z, Smith DK, Zhu G, Vucetic S, Brown CJ, Lawson JD, Dunker AK. Protein flexibility and intrinsic disorder. Protein Sci. 2004;13(1):71–80. doi: 10.1110/ps.03128904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li X, Romero P, Rani M, Dunker AK, Obradovic Z. Predicting protein disorder for N-, C-, and internal regions. Genome Inform Ser Workshop Genome Inform. 1999;10:30–40. [PubMed] [Google Scholar]
- 74.Sugase K, Dyson HJ, Wright PE. Mechanism of coupled folding and binding of an intrinsically disordered protein. Nature. 2007;447(7147):1021–1025. doi: 10.1038/nature05858. [DOI] [PubMed] [Google Scholar]
- 75.Galea CA, Nourse A, Wang Y, Sivakolundu SG, Heller WT, Kriwacki RW. Role of intrinsic flexibility in signal transduction mediated by the cell cycle regulator, p27 Kip1. J Mol Biol. 2008;376(3):827–838. doi: 10.1016/j.jmb.2007.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Campbell KM, Terrell AR, Laybourn PJ, Lumb KJ. Intrinsic structural disorder of the C-terminal activation domain from the bZIP transcription factor Fos. Biochemistry. 2000;39(10):2708–2713. doi: 10.1021/bi9923555. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Number of publications related to IDPs. The following terms were searched: intrinsically disordered proteins, intrinsically unfolded proteins, intrinsically unstructured proteins and natively unfolded proteins. (PDF 11 KB)
Additional file 2: Consensus disorder prediction. Consensus of disordered prediction. (PDF 28 KB)
Additional file 3: Considered hits and errors. Considered hits and errors. (PDF 31 KB)
Additional file 4: Pre-processing results. Number and percentage of sequences that went into each step of the IDP pipeline pre-processing. (PDF 10 KB)
Additional file 5: IDPs information. Identified IDPs for each organism. (PDF 3 MB)
Additional file 6: IDPs functional annotation. Classification of the identified IDPs (hypothetical or with predicted function). (PDF 10 KB)
Additional file 7: Percent of disordered residues in L. braziliensis. Percentage of disordered residues in L. braziliensis. (PDF 11 KB)
Additional file 8: Percent of disordered regions in L. braziliensis. Percentage of disordered regions in L. braziliensis. (PDF 11 KB)
Additional file 9: Frequency of IDP amino acids relative to globular amino acids in L. braziliensis. Frequency of IDP amino acids relative to globular amino acids in L. braziliensis. (PDF 12 KB)
Additional file 10: Association between the percentage of disordered residues and isoelectric points in L. braziliensis. The colors represent whether the frequency is higher (blue) or lower (pink) than expected. The numbers represent the categories of attributes. (PDF 30 KB)
Additional file 11: Association between the percentage of disordered residues and transmembrane domains in L. braziliensis. The colors represent whether the frequency is higher (blue) or lower (pink) than expected. The numbers represent the categories of attributes. (PDF 31 KB)
Additional file 12: IDPs identification gels. A) L. major 2D electrophoresis gel; the IDPs are located near the diagonal line; B) L. major 2D electrophoresis with IDP enrichment; and C) L. braziliensis 2D electrophoresis with IDP enrichment. (PDF 268 KB)
Additional file 13: Experimental validation. The IDP spot number is provided in Figure S8A, S8B and S8C. (PDF 14 KB)