

Many viruses make noncoding (nc)RNAs and the challenge is to define their functions. In this review, Tycowski et al. summarize what has been learned so far about the origin and function of virus-encoded ncRNAs.
Keywords: virus, ribonucleoprotein, miRNA, lncRNA, sisRNA, gene regulation
Abstract
Eukaryotic cells produce several classes of long and small noncoding RNA (ncRNA). Many DNA and RNA viruses synthesize their own ncRNAs. Like their host counterparts, viral ncRNAs associate with proteins that are essential for their stability, function, or both. Diverse biological roles—including the regulation of viral replication, viral persistence, host immune evasion, and cellular transformation—have been ascribed to viral ncRNAs. In this review, we focus on the multitude of functions played by ncRNAs produced by animal viruses. We also discuss their biogenesis and mechanisms of action.
Like their host cells, many—but not all—viruses make noncoding RNAs (ncRNAs). As in the case of cellular ncRNAs, the challenge is to define functions for viral ncRNAs. What we have learned so far offers several guidelines for undertaking this mission.
Because viral genomes are limited in size, any space expended on a ncRNA is rationed. Thus, viral ncRNAs should play important roles in enhancing the viral life cycle or counteracting the defenses that the host organism raises against viral infection.
The viruses that have acquired the most ncRNAs are the herpesviruses, perhaps because they have relatively large genomes and an unusual lifestyle. These vertebrate viruses are characterized by two states: lytic (actively replicating) and latent (where the viral dsDNA genome is maintained in host cells for extended periods while producing only a few viral gene products). Some herpesviruses have oncogenic potential.
Over evolutionary time, viruses actively exchange genetic material with their host cells. However, just because a ncRNA expressed by a virus exhibits hallmarks of a known class of host cell ncRNAs does not mean that it necessarily functions comparably. Viruses are extremely clever at altering the bits of host cell genome that they acquire to meet their own needs.
As for all investigations of viral infection, studying viral ncRNAs has richly enhanced our understanding of their host cells. In particular, surprising insights into the evolutionary relationships between viruses and their hosts have emerged. With increasing frequency, studies of viral ncRNAs have led to novel insights into host cell functioning.
In some cases, synthesizing a ncRNA rather than a protein may be the preferred mode of accomplishing a function for a virus. RNAs are less immunogenic and therefore can more easily slip under the radar of the host cell immune system.
All viral ncRNAs—like host ncRNAs—associate with proteins that are integral to their function. These are usually encoded by the host genome. Normally, viral ncRNAs are discretely localized within the infected cell—in the nucleus, in subnuclear bodies, in the cytoplasm, or in cytoplasmic organelles. Both associated proteins and subcellular location provide additional hints and tools for tracking down viral ncRNA function.
The rate of assigning functions to viral ncRNAs has accelerated markedly since we last reviewed this topic (Steitz et al. 2010). Much can be ascribed to powerful technological advances that uncover the in vivo associations of viral ncRNAs in molecular detail. Here we focus on progress made in the past 5 years elucidating the roles of abundant ncRNAs produced by animal viruses. We do not consider RNAs of low abundance detected in recent high-throughput sequencing (HITS) efforts, which have not been characterized functionally. We emphasize the diverse roles of viral ncRNAs and emerging insights into how they may have evolved from their cellular counterparts.
Viral ncRNAs of RNA polymerase III (Pol III) origin: few, but many
Viral ncRNAs transcribed by Pol III are not widespread but are usually expressed at high copy numbers in infected cells. In adenovirus, a dsDNA virus that causes a variety of human infections, two virus-associated (VA) RNAs (∼107 molecules per cell) (Table 1) were the first viral ncRNAs identified (Reich et al. 1966; Steitz et al. 2010). These ∼160-nucleotide-(nt)-long RNAs (Fig. 1A) were shown to be transcribed by RNA Pol III based on their sensitivity to high but not low levels of α-amanitin (Soderlund et al. 1976; Weinmann et al. 1976). VA RNAs are molecularly best characterized for their role in counteracting the host antiviral defense through inhibition of protein kinase R (PKR). PKR is activated by binding to dsRNA, originating from viral transcription, whereupon it phosphorylates the translation initiation factor eIF2 to inhibit protein synthesis in virus-infected cells (de Haro et al. 1996). The cytoplasmic VA RNAI competes with dsRNA for binding to PKR but, instead of stimulating, inhibits activation of PKR, thereby enabling viral protein synthesis (Mathews and Shenk 1991; Wilson et al. 2014). VA RNAs can also act as substrates for the RNase III enzyme Dicer, with the products being incorporated into functional Argonaute (AGO)-containing RNA-induced silencing complexes (RISCs) (Aparicio et al. 2006, 2010). Because of the high copy number of VA RNAs in infected cells, cellular microRNA (miRNA) biogenesis may be severely compromised (Lu and Cullen 2004; Andersson et al. 2005). However, whether VA RNA-derived miRNAs benefit the adenovirus life cycle remains unclear (Kamel et al. 2013).
Table 1.
Viral ncRNAs excluding miRNAs
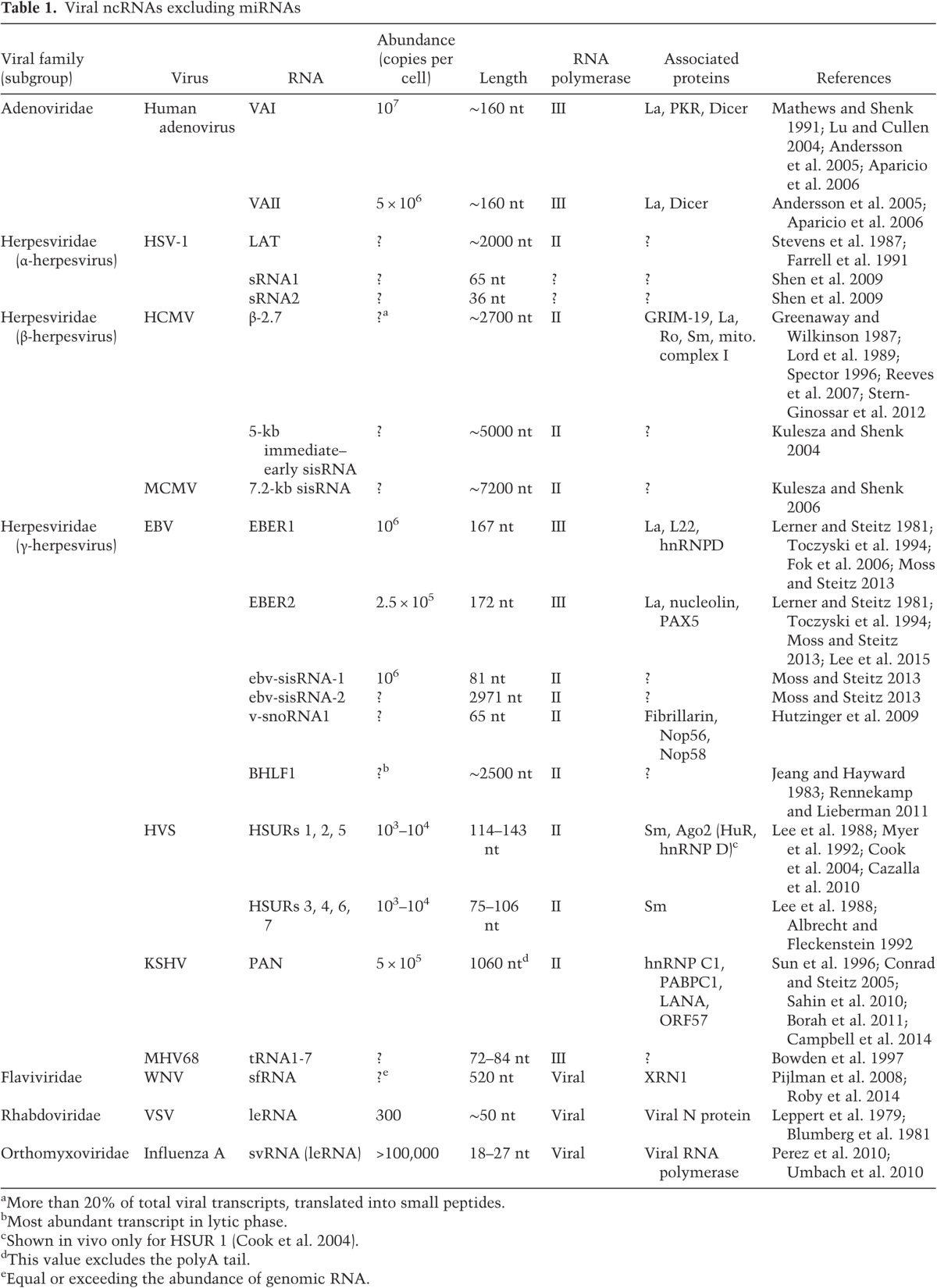
Figure 1.
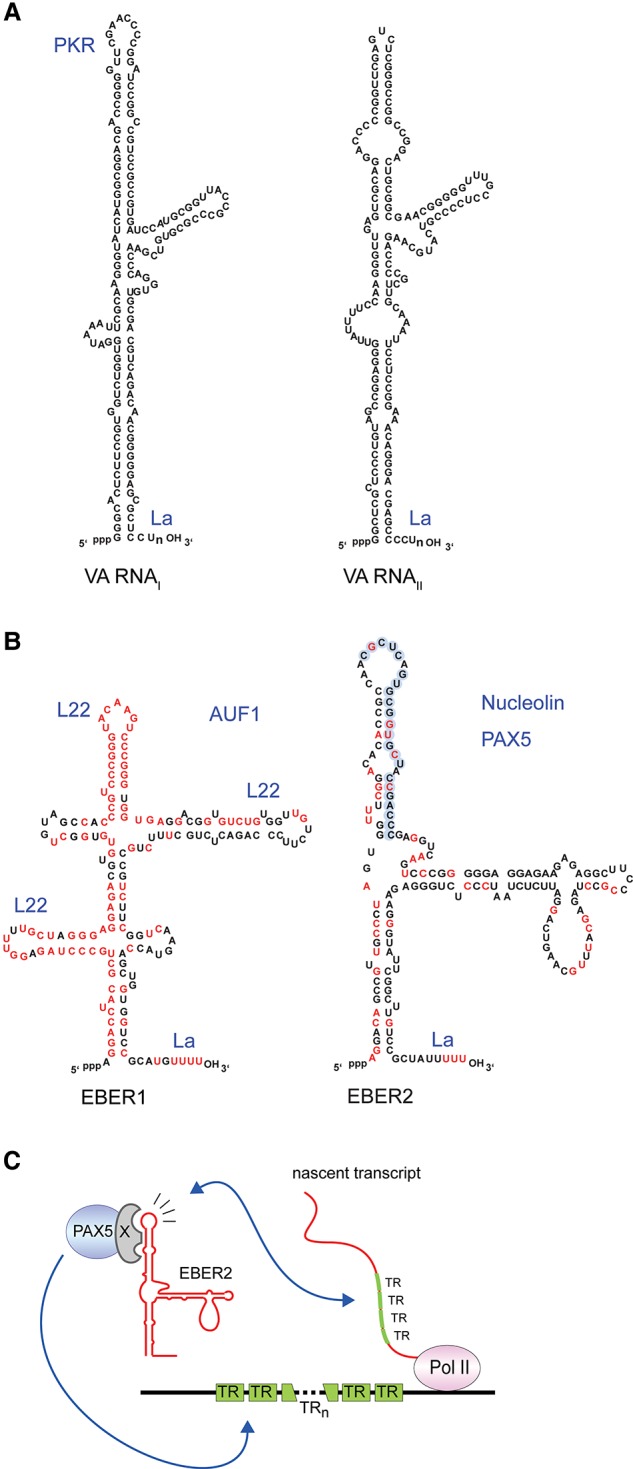
(A) VA RNAI and RNAII. The PKR-binding site on VA RNAI is indicated. (B) Epstein-Barr virus (EBV)-encoded RNA 1 (EBER1) and EBER2. Both RNAs are highly conserved among related viruses (conserved nucleotides are shown in red). Known interaction sites of binding proteins are indicated. L22 binds three stem–loops of EBER1, while La binds the polyU tract at the 3′ end of both EBERs. The exact binding sites of AU-rich element-binding factor 1 (AUF1) and nucleolin have not been determined. Paired box protein 5 (PAX5) likely binds EBER2 indirectly. The EBER2 nucleotides highlighted in blue engage in RNA–RNA interactions with the terminal repeat (TR) transcript. (C) Recruitment mechanism of the EBER2–PAX5 complex to the TR regions of the EBV genome (Lee et al. 2015). EBER2 and PAX5 interact indirectly through an unknown bridging factor (denoted as X). Base-pairing of EBER2 with nascent TR transcripts acts to recruit and/or stabilize PAX5 binding to TR DNA. The variable number of TRs (≤20) in the EBV genome is indicated by TRn.
Epstein-Barr virus (EBV), an oncogenic γ-herpesvirus that causes infectious mononucleosis as well as B-cell lymphomas, synthesizes two abundant ncRNAs called EBV-encoded RNA 1 (EBER1) and EBER2 (Fig. 1B; Rosa et al. 1981). EBERs were discovered because they bind the autoantigen La (Lerner et al. 1981), an abundant cellular RNA chaperone that recognizes the U-rich 3′ end of all RNA Pol III transcripts (Wolin and Cedervall 2002), including VA RNAI and VA RNAII (Francoeur and Mathews 1982). The similarities between EBERs and VA RNAs—namely, their size, being highly structured RNA Pol III products, their interaction with La, and their high abundance (EBERs accumulate to ∼106 copies per infected cell)—prompted attempts to demonstrate a homologous function. EBERs were reported to substitute for VA RNA in vivo (Bhat and Thimmappaya 1983) as well as bind and inhibit PKR in vitro (Clarke et al. 1990). However, EBERs reside in the nucleoplasm and do not undergo nucleo–cytoplasmic shuttling (Howe and Steitz 1986; Fok et al. 2006), raising the question of whether in vitro binding of the nuclear EBERs to cytoplasmic PKR is physiologically relevant.
Further studies of EBER deletion strains generated contradictory observations regarding B-cell growth and transformation (Swaminathan et al. 1991; Yajima et al. 2005; Gregorovic et al. 2011). EBERs have been reported to selectively induce the expression of the cytokines interleukins 9 and 10, anti-apoptotic protein Bcl-2, and insulin-like growth factor 1 in a cell type-dependent manner (Komano et al. 1999; Kitagawa et al. 2000; Yang et al. 2004; Iwakiri et al. 2005). The molecular mechanisms underlying these gene expression changes have not been addressed, but further advances in understanding EBERs’ molecular modes of action have been made recently.
The interacting proteins of EBER1 include La, the ribosomal protein L22, and AU-rich element-binding factor 1 (AUF1/heterogeneous nuclear ribonucleoprotein D [hnRNPD]) (Fig. 1B; Toczyski and Steitz 1991; Lee et al. 2012). Except for La, which aids in proper folding and stability of RNA Pol III transcripts, the significance of EBER1's interaction with the other two proteins is unknown. EBER1 binding to L22 in EBV-infected cells as well as in cells expressing only EBER1 results in striking relocalization of L22 from the nucleolus to the nucleoplasm (Toczyski et al. 1994; Gregorovic et al. 2011), a phenotype that suggests important consequences beyond the observation that the EBER1–L22 interaction promotes colony formation in soft agar (Houmani et al. 2009). Given the high copy number of EBER1 in EBV-infected cells, it was proposed that sequestering AUF1 leads to interference with AUF1 functions, such as regulating the stability of AU-rich element-containing mRNAs or suppressing senescence and maintaining telomeres (Lee et al. 2012; Pont et al. 2012). EBER1 in complex with La has been reported to be secreted and extracellularly activate Toll-like receptor 3 signaling (Iwakiri et al. 2009), yet how the secretion of the naked (i.e., unvesiculated) EBER1–La complex is achieved mechanistically remains unclear. In a related study, EBERs were found associated with exosomes (Ahmed et al. 2014), which are secretory vesicles that bypass the ER–Golgi network. Further studies are required to address whether secretion of EBERs is physiologically relevant or merely a consequence of DNA damage response-induced exosome production elicited by EBV infection (Yu et al. 2006; Nikitin et al. 2010).
EBER2 is less well studied and is slightly lower in abundance than EBER1 in infected B cells (∼2.5 × 105 molecules) (Moss and Steitz 2013). Given its nuclear location, we recently probed EBER2's association with chromatin (Lee et al. 2015) using capture hybridization analysis of RNA targets (CHART), a technique similar to chromatin immunoprecipitation that allows RNA localization to specific sites on DNA (Simon et al. 2011). EBER2 was found to bind to the so-called terminal repeats (TRs) of the EBV genome, overlapping previously identified binding sites for the cellular transcription factor paired box protein 5 (PAX5) (Arvey et al. 2012). PAX5 is a well-characterized master regulator of B-lymphocyte development and function (Medvedovic et al. 2011). EBER2 and PAX5 were further shown to physically interact and act in concert to regulate the expression of a subset of EBV latent genes (Lee et al. 2015). As a consequence, efficient viral lytic replication is compromised. This observation may be related to the interaction of EBER2 with nucleolin (V Fok and J Steitz, unpubl.), as during the replication of human cytomegalovirus (HCMV), a β-herpesvirus, the viral homolog of the host DNA polymerase processivity factor (UL44) associates with nucleolin (Bender et al. 2014). Because EBV lytic replication has been implicated in promoting tumor formation (Katsumura et al. 2011), uncovering a connection to DNA replication may relate to the tumorigenic potential of EBER2 (Komano et al. 1999).
More detailed analyses (Lee et al. 2015) revealed that the recruitment of EBER2 and PAX5 to their binding sites on EBV DNA depends on RNA–RNA interactions between a region within EBER2 and nascent transcripts derived from the TR regions (Fig. 1C). Thus, EBER2 acts as a recruiting or stabilizing element for PAX5 binding to its target sites on the EBV genome. Here, using an EBV RNA rather than a protein to facilitate PAX5 targeting makes sense for a limited viral genome: Whereas protein domains, usually of ≥60 amino acids, typically recognize dsDNA consensus sequences of <10 nt, a complementary ncRNA requires only the same number of nucleotides to base-pair with another RNA. Since viruses adopt mechanisms from their hosts over evolutionary time, these recent revelations suggest the possibility that cellular ncRNAs may use equivalent strategies based on RNA–RNA interactions for guiding transcription factors to their chromatin target sites.
Apart from VA RNAs and EBERs, other known viral RNA Pol III transcripts include miRNA precursors, such as the murine γ-herpesviral tRNA–pre-miRNA chimeras (Bowden et al. 1997; Bogerd et al. 2010; Diebel et al. 2010) and retroviral pre-miRNAs (Kincaid et al. 2012), and are discussed below.
Herpesvirus saimiri (HVS) U-rich RNAs (HSURs): ncRNA warfare
HSURs (Fig. 2) are produced by HVS, an oncogenic T-lymphotropic γ-herpesvirus that causes fatal T-cell lymphomas and leukemias in new world primates and transforms human primary T cells in vitro (Albrecht and Fleckenstein 1992; Ensser and Fleckenstein 2005). They are the most abundant viral transcripts in latently infected marmoset T cells (Lee et al. 1988; Wassarman et al. 1989). The HSURs resemble the cellular Sm-class small nuclear RNAs (snRNAs) that function in splicing; they bind the same Sm proteins as the splicing snRNAs and are assembled into RNP complexes through the same biogenesis pathway (Golembe et al. 2005). Expression levels of the HSURs are regulated by cellular ARE-binding proteins, such as hnRNPD and HuR (Myer et al. 1992; Fan et al. 1997; Cook et al. 2004). Although HVS strains with a deletion of the HSUR genes are capable of transforming T lymphocytes, cells infected with a mutant virus that lacks HSURs 1 and 2 grow significantly slower than cells infected with the wild-type virus (Murthy et al. 1989). Microarray analysis revealed that host genes involved in T-cell activation are up-regulated by HSURs 1 and 2 (Cook et al. 2005).
Figure 2.

Nucleotide sequences and predicted secondary structures of HSURs 1–7 and hvs-pre-miR-HSURs 2, 4, and 5. Predicted base-pairing interactions between HSURs 1, 2, or 5 and host miR-142-3p and miR-16 as well as the experimentally determined interaction between HSUR 1 and miR-27 are shown with the miRNA seed sequences colored red. Arrowheads indicate the 3′ ends of the mature HSURs in the HSUR–pre-miRNA chimeras. Mature HVS miRNAs are shaded gray. The Sm-binding site and 3′ box sequences are boxed.
More recently, HSURs 1 and 2 were discovered to interact with several host miRNAs, including miR-27, miR-16, and miR-142-3p (Fig. 2; Cazalla et al. 2010). One of these, miR-27, is targeted for rapid degradation via base-pairing with HSUR 1 (Cazalla et al. 2010). Possibly during evolution, HVS acquired one of the host splicing snRNA genes and then repurposed this splicing snRNP for RNA degradation. The same miRNA, miR-27, is also targeted for degradation by murine CMV (MCMV) using a similar antisense RNA-based mechanism (Buck et al. 2010; Libri et al. 2012; Marcinowski et al. 2012).
A recent AGO high-throughput sequencing of RNA isolated by cross-linking immunoprecipitation (HITS-CLIP) study of HVS-transformed T cells revealed that miR-27 directly down-regulates T-cell activation proteins and many components of the T-cell receptor (TCR) signaling pathway (Guo et al. 2014). Because miR-27 is a repressor of T-cell activation, degradation of miR-27 mediated by HSUR 1 promotes activation and presumably proliferation of HVS-infected host T cells. Interestingly, distantly related T-cell oncogenic Macaviruses, such as Alcelaphine Herpesvirus 1 (AlHV-1) and Ovine Herpesvirus 2 (OvHV-2), do not degrade miR-27 but instead encode viral homologs of key miR-27 target genes (Guo et al. 2014). The fact that two groups of oncogenic viruses use parallel strategies to up-regulate the same set of host proteins highlights the importance of these proteins to the viral life cycle and host T-cell activation. Similar parallel strategies of miRNA degradation (mediated by antisense RNAs) versus co-option of miRNA target genes are also evident in other viruses that infect different species, such as in the MCMV versus HCMV (β-herpesvirus) (Guo et al. 2014). Perhaps these parallels can be exploited to discover new viral ncRNAs. The functions of other HSURs are unknown.
Viral miRNAs: small RNAs, big roles
Like their host cells, many DNA and RNA viruses express miRNAs. Herpesviruses encode the largest number of miRNAs, with Rhesus lymphocryptovirus (rLCV; closely related to EBV) holding the record at 68 miRNAs (with the potential for 70 from 35 pre-miRNAs) (Riley et al. 2010). Viral miRNAs are incorporated into RISCs by binding the host AGO family proteins and then down-regulate protein production from viral and host genes. Use of viral miRNAs instead of viral proteins to manipulate gene expression has several advantages: although miRNAs are short (∼21 nt), they are capable of regulating a large number of mRNA targets via base-pairing with their seed sequences (nucleotides 2–8); moreover, viral miRNAs are less likely than proteins to be recognized by the host immune system.
Starting with the discovery of the first viral miRNA (Pfeffer et al. 2004), multiple studies suggest at least three important roles for viral miRNAs that are described below: regulation of (1) viral persistence, (2) proliferation and long-term survival of host cells, and (3) host immune evasion.
-
Most herpesviral miRNAs are expressed during the latent stage of the viral life cycle (Grey 2014) and are known to modulate several key viral latent-to-lytic regulators. For example, HCMV-encoded miR-UL112-1 down-regulates the major immediate–early gene IE72, leading to decreased expression of viral genes involved in replication, thereby maintaining latency (Grey et al. 2007; Murphy et al. 2008). A similar strategy is used by the γ-herpesvirus Kaposi's sarcoma (KS)-associated herpesvirus (KSHV), the causative agent of several human cancers and lymphoproliferative disorders, such as KS, multicentric Castleman's disease (MCD), and primary effusion lymphoma (PEL) (Ganem 2006). Viral miR-K12-9* targets the important immediate–early transcriptional activator RTA (ORF50) to dampen its expression, preventing promiscuous lytic reactivation from latency (Bellare and Ganem 2009); more recent studies suggest that this miRNA-mediated regulation may be indirect (Lei et al. 2010; Lu et al. 2010). Likewise, the ovine herpesvirus miRNA OvHV-2-miR-5 targets ORF50 to maintain latency (Riaz et al. 2014). Also, in EBV-infected cells, viral miR-BART20-5p promotes latency by down-regulating two EBV immediate–early genes: BZLF1 and BRLF1 (Jung et al. 2014). In addition, one of the viral miRNAs (miR-H2-3p) processed from the latency-associated transcript (LAT) expressed by Herpes simplex virus 1 (HSV-1), an α-herpesvirus, is perfectly complementary to the viral immediate–early transactivator ICP0 mRNA and acts as an siRNA to down-regulate ICP0 mRNA and protein levels (Umbach et al. 2008).
Although in the above examples viral miRNAs target viral mRNAs, viral miRNAs also contribute to viral persistence by modulating host mRNA expression. KSHV miR-K12-1 inhibits viral lytic replication by down-regulating the IκB protein and thereby activating NF-κB signaling (Lei et al. 2010). Another KSHV miRNA, miR-K12-4-5p, was found to target retinoblastoma (Rb)-like protein 2 (Rbl2), a repressor of DNA methyltransferases 3a and 3b, and thus globally regulate the epigenetic state of infected cells (Lu et al. 2010). EBV BART-18-5p suppresses mitogen-activated protein kinase (MAPK) signaling by targeting MAPK kinase kinase 2 (MAP3K2), important for the initiation of lytic viral replication (Qiu and Thorley-Lawson 2014).
-
For many oncogenic viruses, miRNAs contribute significantly to the virally induced gene expression program important for host cell growth and survival. For example, several EBV and KSHV miRNAs repress levels of proapoptotic proteins such as PUMA, TOMM22, BCLAF1, and caspase 3 (Choy et al. 2008; Ziegelbauer et al. 2009; Buck et al. 2010; Suffert et al. 2011). KSHV miR-K12-1 also down-regulates p21, an inhibitor of cell cycle progression, to promote virally induced oncogenesis (Gottwein and Cullen 2010). Genome-wide AGO HITS-CLIP (Chi et al. 2009) and photoactivatable-ribonucleoside-enhanced CLIP (PAR-CLIP) (Hafner et al. 2010) studies revealed that targets of EBV and KSHV miRNAs are enriched in apoptosis and cell cycle pathways (Gottwein et al. 2011; Haecker et al. 2012; Riley et al. 2012; Skalsky et al. 2012). Studies of EBV genomic deletions revealed that several miRNAs expressed from the BHRF1 locus (miR-BHRF1s) play critical roles in B-cell transformation by inhibiting apoptosis and promoting cell cycle progression (Seto et al. 2010; Feederle et al. 2011). Consistently, expression of EBV miRNAs from the BamHI A rightward transcript (BART) locus is sufficient to prevent apoptosis in Burkitt's lymphoma (BL) cells or transformed primary B lymphocytes that have lost the EBV genome (Vereide et al. 2014). Similarly, genetic deletion and complementation studies of KSHV miRNAs have shown that they promote cell cycle progression and inhibit apoptosis, necessary for cellular transformation (Moody et al. 2013).
Interestingly, many γ-herpesviruses, including KSHV and Marek's disease virus type 1 (MDV-1), as well as retroviruses like simian foamy virus (SFV) express homologs of cellular miR-155 (for review, see Guo and Steitz 2014b). These viral miR-155 mimics target the same set of host mRNAs as cellular miR-155 to promote virally induced cell transformation. The critical role of viral miR-155 has been demonstrated in genetic studies using MDV-1 as a model. Deletion or seed mutation of the MDV-1 miR-155 homolog miR-M4 abolishes the oncogenicity of MDV-1; the mutant phenotype could be rescued by expression of host miR-155 (Zhao et al. 2011).
Viral miRNAs attenuate host antiviral immune mechanisms, enhancing viral survival. For example, KSHV miR-K12-10a suppresses the host proinflammatory response by down-regulating levels of the tumor necrosis factor (TNF)-like weak inducer of apoptosis (TWEAK) receptor (TWEAKR) protein (Abend et al. 2010). miR-K12-9 and miR-K12-5 directly target IRAK1 and MYD88, respectively, components of the interleukin-1 receptor and Toll-like receptor signaling cascade, leading to reduced inflammatory cytokine expression (Abend et al. 2012). Likewise, HCMV clinical strain-specific miR-UL148D modulates the host immune response by directly regulating a host chemokine called regulated on activation, normal T-cell expressed, and secreted (RANTES) (Kim et al. 2012). Another two KSHV miRNAs, miR-K12-11 and miR-K12-5, repress production of activation-induced cytidine deaminase (AID), which marks infected host cells for elimination by natural killer (NK) cells (Bekerman et al. 2013). HCMV miR-UL112-1 down-regulates production of the major histocompatibility complex class I-related chain B (MICB) by synergistically binding nearby cellular miR-376a to MICB mRNA; this decreases killing of infected cells by NK cells through the NKG2D pathway (Stern-Ginossar et al. 2007; Nachmani et al. 2010). Interestingly, different miRNAs expressed by KSHV and EBV also directly bind MICB mRNA and repress protein production, suggesting a common strategy used by diverse herpesviruses to evade NK cell-mediated host immune defense (Nachmani et al. 2009). In addition, HCMV miR-US4-1 specifically down-regulates the aminopeptidase ERAP1, involved in the MHC I antigen presentation pathway, thereby evading the host cytotoxic T-lymphocyte-mediated antiviral response (Kim et al. 2011). Finally, HCMV miR-UL112-1, miR-US5-1, and miR-US5-2 target multiple genes in the host secretory pathway to modulate the secretion of several host proinflammatory cytokines (Hook et al. 2014).
Small DNA oncoviruses, such as polyomaviruses, also produce miRNAs. For example, Simian virus 40 (SV40) makes two miRNAs that are perfectly complementary to early viral mRNAs, targeting them for cleavage; deletion of these viral miRNAs renders SV40-infected cells more susceptible to cytotoxic T-cell-mediated killing in cell culture (Sullivan et al. 2005). However, more recent studies in an animal model of SV40 infection suggest that the SV40 miRNAs control viral replication rather than host immune evasion and cellular transformation (Zhang et al. 2014).
In many of the above studies, it is not clear whether the reported effects of the viral miRNAs are direct or indirect. Identifying direct mRNA targets for any miRNA is difficult because target recognition requires base-pairing only with the miRNA seed region, which is very short (≤8 nt). The problem is even more challenging for viral miRNAs because, unlike host, viral miRNA-binding sites are poorly conserved. Therefore, certain cross-linking approaches such as AGO HITS-CLIP and PAR-CLIP have proven most reliable for target identification. Other methods, such as biotin-tagged miRNA approaches, work to various extents (Guo and Steitz 2014a; Tan et al. 2014; Imig et al. 2015).
Less trodden paths for viral miRNA biogenesis
Most cellular and viral miRNAs are produced from long RNA Pol II transcripts through stepwise cleavages by two RNase III enzymes—Drosha and Dicer—in the nucleus and cytoplasm, respectively (Fig. 3A; Ha and Kim 2014). As for many other complex biological processes, exceptions to the canonical pathway have been discovered for the biogenesis of several cellular miRNAs (for review, see Xie and Steitz 2014). Examples include Drosha-independent but splicing-dependent pre-miRNAs called mirtrons (Ruby et al. 2007; Okamura et al. 2008), m7G-capped pre-miRNAs and endogenous shRNAs that are transcribed directly as pre-miRNAs (Babiarz et al. 2008; Xie et al. 2013), and AGO2 slicing activity-dependent miR-451, which bypasses Dicer cleavage (Cheloufi et al. 2010; Cifuentes et al. 2010). Not surprisingly, viruses use various alternative mechanisms to produce their own miRNAs. However, none of the above-mentioned cellular pathways have been assigned to viral miRNAs. Thus, viruses seemingly invent their own alternative miRNA biogenesis pathways in creative ways, sometimes by using cellular machineries that normally act on other types of RNAs.
Figure 3.

Viral miRNA biogenesis pathways. (A) The canonical pathway. pri-miRNAs are typically transcribed by RNA Pol II, 5′-capped, and 3′-polyadenylated. The Microprocessor complex (Drosha and DGCR8) cleaves pri-miRNAs to release pre-miRNA hairpins, which are exported by Exportin-5 and processed by Dicer into mature miRNA duplexes in the cytoplasm. One miRNA strand is preferentially selected by AGO to form RISCs. (B) Drosha-independent miRNA biogenesis in animal viruses. MHV68 pri-miRNAs are tRNA–pre-miRNA chimeras that are processed by RNaseZ at the 5′ end of the first pre-miRNA hairpin. The enzyme that separates the two pre-miRNAs is unknown. HVS pri-miRNAs are snRNA–pre-miRNA chimeras that are processed by Integrator to release the pre-miRNA. The 3′ end formation mechanism for HVS pre-miRNAs remains elusive. Bovine leukemia virus (BLV) and some SFV miRNAs are derived from pre-miRNAs that are directly transcribed by RNA Pol III as endogenous shRNAs. All viral pre-miRNAs pictured are exported by XPO5 and processed by Dicer.
Because the vast majority of viral miRNAs are encoded by DNA viruses, it is not surprising that noncanonical viral miRNA biogenesis pathways were first discovered in γ-herpesviruses, including murine γ-herpesvirus 68 (MHV68; also known as Murid herpesvirus 4) and HVS (Figs. 2, 3B; Bogerd et al. 2010; Cazalla et al. 2011). These two viruses use similar strategies: pre-miRNA hairpins are cotranscribed downstream from other ncRNAs and are initially processed by the enzyme that is responsible for the 3′ end formation of the upstream ncRNA, thereby skipping the Drosha cleavage step. In the case of MHV68 miRNA biogenesis, pre-miRNAs are located downstream from viral tRNAs (Fig. 3; Bogerd et al. 2010; Diebel et al. 2010; Reese et al. 2010). There are usually two pre-miRNA hairpins cotranscribed with the tRNA, with the first pre-miRNA generating more abundant miRNAs. This arrangement is special in at least two ways. First, unlike most miRNAs, MHV68-encoded miRNAs are transcribed by RNA Pol III instead of Pol II; the polyU tract appearing at the 3′ end of the second pre-miRNA is a hallmark of transcript termination by Pol III. Second, the tRNA–miRNA chimeric transcript serves as the pri-miRNA and is processed by host RNaseZ to separate the pre-miRNAs from the tRNA (Bogerd et al. 2010). The enzyme that separates the two pre-miRNAs is currently unknown.
HVS pre-miRNAs are cotranscribed downstream from HSURs instead of viral tRNAs (Fig. 2). Therefore, HVS miRNAs are transcribed by RNA Pol II and depend on snRNA biogenesis elements, including a special snRNA promoter and the snRNA-specific 3′ end processing signal: the 3′ box. The Integrator complex, which is responsible for processing the upstream ncRNA, cleaves to release the downstream viral pre-miRNAs (Cazalla et al. 2011). The detailed 5′ and 3′ end formation mechanisms for HVS pre-miRNAs are not fully characterized: Integrator presumably cleaves upstream of the 3′ box, leaving a 5′ end-tailed pre-miRNA that may be trimmed by Integrator (Int 11 is homologous to CPSF-73, which has both endonuclease and exonuclease activities) (Baillat et al. 2005; Dominski et al. 2005) or another exoribonuclease. The enzyme that cleaves the 3′ end of the pre-miRNA remains elusive. Integrator-processed pre-miRNAs are then exported by Exportin-5 and further processed by Dicer, as are canonical pre-miRNAs. The ncRNA–pre-miRNA chimeric strategy developed by MHV68 and HVS might be an economic way to pack miRNAs as “by-products” of other RNA pathways into compact viral genomes. Alternatively, the upstream ncRNA and the downstream miRNAs may contribute to a common biological function benefiting the virus.
Until recently, whether RNA viruses express miRNAs had been controversial. It is now clear that at least four retroviruses generate viral miRNAs (Kincaid et al. 2012, 2014; Whisnant et al. 2014; Yao et al. 2014). Surprisingly, only avian leukosis virus (ALV) uses the canonical biogenesis pathway to express its one miRNA (Yao et al. 2014). The other three viruses use RNA Pol III to transcribe pri-miRNAs from the proviral genome. These are then processed by the noncanonical pathways described below, which avoid potentially detrimental cleavage of the viral genome or mRNA by Drosha (Kincaid et al. 2012, 2014; Whisnant et al. 2014).
The miRNA biogenesis pathway used by bovine leukemia virus (BLV) is analogous to a well-known strategy used by researchers to generate shRNAs for RNAi (Fig. 3B; Medina and Joshi 1999). BLV expresses pre-miRNAs as subgenomic RNA Pol III transcripts driven by type II Pol III promoters (Burke et al. 2014). Here, the 5′ and 3′ ends of the pre-miRNA hairpins are defined by Pol III transcription initiation and termination, as are man-made U6 promoter-driven shRNAs. These pre-miRNAs are structurally similar to canonical pre-miRNAs that are cleaved by Drosha and are exported by Exportin-5. A triphosphatase activity is then implicated in converting the 5′ triphosphate of the 5p-miRNA into a 5′ monophosphate concurrent with or after Dicer action (Burke et al. 2014).
In SFV, miRNAs transcribed by RNA Pol III are encoded in long TR (LTR) regions (Kincaid et al. 2014). Some SFV miRNAs are processed from pri-miRNA transcripts containing hairpins that were initially predicted to be too short for Drosha recognition. However, knocking down Drosha significantly decreases viral pre-miRNAs expressed from a transfected plasmid in 293T cells, while pre-miRNAs accumulate when Drosha and DGCR8 are overexpressed (Kincaid et al. 2014). Therefore, SFV pri-miRNAs appear to be bona fide Drosha substrates, perhaps suggesting that Drosha acts differently on viral Pol III pri-miRNAs. Computational analysis has predicted that SFV also expresses BLV-like pre-miRNAs.
All three bovine foamy virus (BFV) miRNAs are embedded in RNA Pol III transcripts structurally similar to SFV pri-miRNAs (Whisnant et al. 2014). Whether Drosha is involved in their processing requires further investigation.
Given these many deviant pathways and the realization that viruses often acquire mechanisms from the host, it is surprising that there are almost no common noncanonical miRNA biogenesis pathways shared by viruses and their hosts. The only exception so far was discovered in an attempt to identify host miRNAs that follow a biogenesis pathway similar to that of MHV68 miRNAs: Small RNAs derived from regions downstream from tRNA genes were detected by Northern blotting in mouse 3T12 cells (Reese et al. 2010). On the other hand, several artificial miRNA biogenesis pathways have been devised using RNA viruses, providing proof-of-principle evidence for additional pathways. For example, cytoplasmic RNA viruses, such as Sindbis virus and tick-borne encephalitis virus, can be engineered to produce functional miRNAs (Rouha et al. 2010; Shapiro et al. 2010). Such artificial pri-miRNAs are successfully processed despite the fact that these viruses replicate exclusively in the cytoplasm, suggesting that more unexpected miRNA biogenesis mechanisms may be discovered in the future.
Abundant long ncRNAs (lncRNAs) that sPAN the herpesviruses
In lytic KSHV-infected cells, the most abundant transcript is a lncRNA called polyadenylated nuclear (PAN) RNA (Sun et al. 1996; Zhong and Ganem 1997). PAN RNA is an early gene product comprising nearly 80% of total polyadenylated cellular transcripts (∼500,000 copies per cell). This high copy number is attributable to two features of the ∼1.1-kb PAN transcript: MTA/ORF57 binding near the 5′ end and a triple-helical stabilization element (ENE) upstream of the 3′ polyA tail (Fig. 4; Conrad and Steitz 2005; Mitton-Fry et al. 2010; Sahin et al. 2010). Bioinformatic studies have revealed ENEs in genomic regions syntenic to the PAN locus in three other γ-herpesviruses (rhesus rhadinovirus [RRV], equine herpesvirus 2 [EHV2], and retroperitoneal fibromatosis-associated herpesvirus Macaca nemestrina [RFHVMn]), suggesting the presence of PAN RNA homologs (Tycowski et al. 2012; KT Tycowski, unpubl.). Indeed, a PAN-like RNA was identified in RRV-infected cells. Bioinformatic searches also revealed ENE structures in insect polydnaviruses and dicistroviruses and a protist mimivirus (Tycowski et al. 2012). Importantly, the discovery of viral ENEs led to the identification of analogous triplex-forming stabilizing elements in two vertebrate lncRNAs: MALAT1 and MENβ (Brown et al. 2012, 2014; Wilusz et al. 2012).
Figure 4.
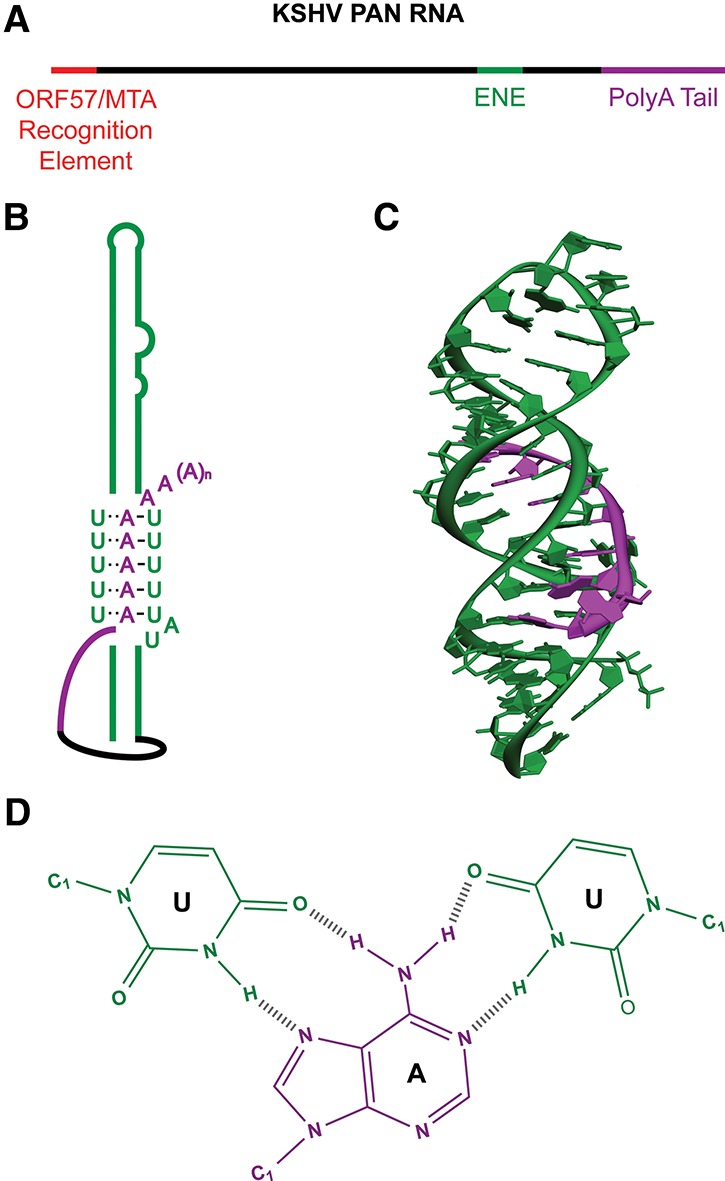
(A) KSHV PAN RNA contains two key stabilization elements: the ORF57/MTA recognition element for ORF57/MTA binding at the 5′ end and a stabilization element known as the ENE near the 3′ end. (B) The ENE clamps the polyA tail of PAN RNA. (C) The X-ray structure revealed that the oligoA (purple) binds both sides of the U-rich internal loop within a stem–loop structure of the PAN RNA (green) to form an RNA triple helix (Protein Data Bank [PDB] 3P22) (Mitton-Fry et al. 2010). (D) Watson-Crick and Hoogsten H bonds stabilize the triple-helical structure and prevent access of deadenylases to the polyA tail of PAN RNA.
PAN RNA is essential for virion production. In KSHV-infected B lymphocytes, oligonucleotide targeted knockdown of PAN RNA reduced the expression of late lytic genes but not viral DNA replication (Borah et al. 2011). These findings were confirmed in human embryonic kidney epithelial cells, where a KSHV bacmid with a deletion that removes 60% of PAN RNA at the 3′ end did not produce viral particles but also showed decreased protein expression for immediate–early RTA/ORF50 and the early K-bZIP genes (Rossetto and Pari 2012). Another study replicated the oligonucleotide targeted knockdown of PAN RNA and reported decreases in viral mRNA expression at the global level (Campbell et al. 2014). The conclusions of these three studies thus differed on the extent to which PAN RNA regulates the viral genome. Clearly, it would be ideal to conduct analyses of a complete PAN knockout, which is problematic because the 5′ end of KSHV PAN RNA overlaps the viral K-7 survivin mRNA.
A potential mechanism for PAN RNA function emerged from a chromatin isolation by RNA purification (ChIRP) (Chu et al. 2011) study of KSHV-infected human B cells (Rossetto and Pari 2012). On the promoter of the transitional viral transactivator RTA/ORF50, PAN RNA interacts with demethylases JMJD3 and UTX. The ChIRP technique was extended in a global high-throughput manner and revealed ubiquitous PAN RNA binding along the entire viral genome (Rossetto et al. 2013). Confirmation using a related technique, such as CHART, might clarify the specificity of PAN RNA's binding to viral chromatin. Compelling in vivo data suggest that PAN RNA also binds to the KSHV latency-associated nuclear antigen (LANA) and that this association inhibits LANA's interaction with histone H3 in vitro but has no effect on LANA protein levels (Campbell et al. 2014). Further studies are needed to better understand the significance of PAN RNA's interaction with LANA during reactivation from latency.
Other herpesviruses also express abundant lncRNAs that are important to the viral life cycle. In human cells infected by the β-herpesvirus HCMV, a highly abundant ∼2.7-kb RNA (β2.7) comprises >20% of the total viral transcriptome (Greenaway and Wilkinson 1987; Spector 1996). It prevents mitochondria-induced apoptosis, enabling steady ATP production for viral processes and persistent infection (Reeves et al. 2007; Stern-Ginossar et al. 2012). Notably, ribosome profiling and mass spectrometry reveal that β2.7 is also translated into small peptides and therefore has a coding function (Stern-Ginossar et al. 2012). The most abundant lytic transcript of another human γ-herpesvirus, EBV, is a nuclear ∼2.5-kb RNA from the early BHLF1 gene (Jeang and Hayward 1983). One study suggests its involvement in viral DNA replication (Rennekamp and Lieberman 2011).
Intronic ncRNAs: molecular treasures from ‘junk’ sequences
The discovery of many stable intronic sequence RNAs (sisRNAs) has accelerated in recent years (Hesselberth 2013), fostering the notion that sequences “discarded” during splicing can be repurposed to serve physiological roles. This strategy is especially apt for viruses, whose genomes are severely size-restricted and would benefit maximally by evolving functions for splicing by-products.
ncRNAs comprised of stable introns were first discovered in the α-herpesvirus HSV-1 (Stevens et al. 1987). During latency, HSV-1 produces only one gene product in high abundance: the LAT ncRNA (Stevens et al. 1987). The ∼8.3-kb LAT is capped, polyadenylated, and spliced, forming an ∼6.3-kb exonic product (Kang et al. 2006) and an ∼2-kb intron (Farrell et al. 1991). The exonic sequence is further processed to generate four viral miRNAs (Umbach et al. 2008) as well as two sRNAs (62 nt and 36 nt) that map to the first 1.5 kb of the LAT (Peng et al. 2008). The LAT-derived miRNAs and sRNAs apparently function in the maintenance of latent infection; they inhibit lytic reactivation (Umbach et al. 2008) and apoptosis (Peng et al. 2008), respectively.
The excised intronic LAT persists and accumulates to high levels in infected cells (Farrell et al. 1991; Kang et al. 2006), making it a sisRNA (Gardner et al. 2012). Interestingly, the functional form of the LAT sisRNA appears to be the branched lariat (Wu et al. 1996; Rodahl and Haarr 1997), which is formed at a nonconsensus branch site sequence (Mukerjee et al. 2004) using guanosine (rather than adenosine) as the branch point (Zabolotny et al. 1997). Selection of this unusual branch site is guided by a thermodynamically stable (16-base-pair) RNA hairpin directly upstream of the consensus splice acceptor polypyrimidine tract (Krummenacher et al. 1997). This unusual organization of the LAT sisRNA branch point is believed to be responsible for its molecular stability, as disruption of the stem–loop results in alternative branch point usage and degradation of the LAT intron (Krummenacher et al. 1997). Expression of the HSV-1 LAT sisRNA maintains infection by inhibiting apoptosis of neuronal cells (Perng et al. 2000; Inman et al. 2001) and silencing viral lytic gene expression through alteration of the heterochromatin structure at viral promoters (Cliffe et al. 2009). The precise role of the LAT sisRNA in this process is not clearly defined; it may recruit polycomb-repressive complexes (PRCs) (Kwiatkowski et al. 2009), similar to other lncRNAs (Tsai et al. 2010). Beyond HSV-1, LAT homologs have been found in other α-herpesviruses—(e.g., HSV-2; Mitchell et al. 1990), bovine herpesvirus 1 (Rock et al. 1987), and simian varicella virus (Ou et al. 2007))—but not in β-herpesviruses or γ-herpesviruses.
On the other hand, other sisRNAs have been identified in HCMV and MCMV (β-herpesviruses) and, more recently, EBV (a γ-herpesvirus). Similar to the HSV-1 LAT sisRNA, stability may be conferred on the CMV sisRNAs by preservation of the lariat structure after splicing (Schwarz and Kulesza 2014). HCMV infection proceeds with the expression of various immediate–early viral gene products that are essential for replication (White and Spector 2007). A 5-kb immediate–early RNA from HCMV was discovered to be intronic in origin (Kulesza and Shenk 2004) and accumulate to high abundance in the nucleus. A homologous, but larger, 7.2-kb sisRNA was later discovered in MCMV and is apparently essential for the progression from acute to persistent infection; mutant viruses that do not produce this RNA fail to enter the persistent phase, and thus the CMV sisRNA appears to be an important virulence factor (Kulesza and Shenk 2006).
EBV sisRNAs that arise from a region of the EBV genome known as the W repeats were recently identified (Moss and Steitz 2013). The W repeats are transcribed during a highly oncogenic form of latency (latency program III) and in a rare form of latency (Wp restricted) observed in ∼15% of endemic BL (Kelly et al. 2009). Five to eight W repeats are present in naturally occurring EBV sequences; the number is optimized for viral fitness (Tierney et al. 2011). They appear at the 5′ end of the ∼100-kb primary transcript for the EBV nuclear antigen (EBNA) mRNAs, which are generated through alternative splicing. The W repeat region is organized such that two short coding exons are separated by short (81-nt) and long (2791-nt) introns (Fig. 5). Analysis of small RNA sequencing (RNA-seq) and ribosome-depleted RNA-seq data identified the short (Moss and Steitz 2013) and long (Moss et al. 2014) introns as sisRNAs ebv-sisRNA-1 and ebv-sisRNA-2, respectively.
Figure 5.
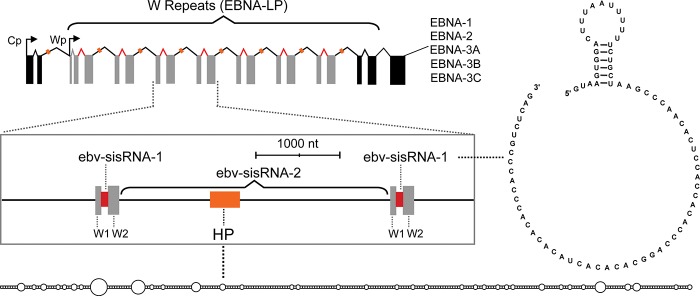
Cartoon of the primary transcript encoding the EBNAs. The W repeat region (encoding EBNA-LP) is indicated with gray boxes representing the W1 and W2 coding exons. The short intron generating ebv-sisRNA-1 is in red, and the long intron is in black (with the large hairpin [HP] in orange). The boxed region zooms in on ∼1.5 W repeats and is annotated as above. Dotted black lines point to cartoon structures of the large 586-nt hairpin and the sequence/secondary structure of ebv-sisRNA-1.
ebv-sisRNA-1 is the third most abundant viral ncRNA after the EBERs (∼20% of the level of EBER1) (Moss and Steitz 2013) and is localized to the nucleus of latently infected B cells. Unlike the LAT intron, ebv-sisRNA-1 is small, comprising all 81 nt of the excised short intron. Also unlike the LAT intron, the dominant form of ebv-sisRNA-1 is a debranched linear molecule; thus, the high stability of ebv-sisRNA-1 cannot arise from an unusual branch structure. The ebv-sisRNA-1 sequence is conserved in lymphocryptoviruses (one of the two major clades in the γ-herpesviruses), as is a short (23-nt) hairpin structure, where compensatory mutations maintain base-pairing (suggesting functional importance). The short hairpin presents a conserved U-rich motif in the loop region, while the downstream sequence is unstructured and contains a completely conserved CA-rich region. Both motifs are likely protein interaction sites.
The extent of ebv-sisRNA-2 has yet to be fully defined; however, abundant sequencing reads cover the long W repeat intron—a reproducible observation in multiple EBV-infected cell lines (Cao et al. 2015). Interestingly, read coverage persists in polyA-selected libraries, suggesting that this sequence may also be present in larger stable transcripts spanning the W repeat region (Cao et al. 2015). A notable feature of ebv-sisRNA-2 is the presence of a predicted conserved and thermodynamically stable RNA secondary structure (Moss and Steitz 2013). Most striking is a massive (586-nt) hairpin that is extensively conserved in structure, but not sequence, between divergent lymphocryptoviruses. Similar massive hairpins have been observed in the untranslated regions (UTRs) of human and nematode mRNAs, where they are substrates for ADAR-editing enzymes (Eggington et al. 2011). It is unusual to find such a large hairpin in a viral RNA.
Another EBV intron-derived ncRNA is a viral snoRNA (v-snoRNA1) (Hutzinger et al. 2009). This RNA has sequence and structural similarity to canonical C/D box snoRNAs and is conserved in all EBV strains as well as in the closely related rLCV. v-snoRNA1 is processed from a 4.5-kb intron within the BARTs and is located 100 nt downstream from the EBV miRNA BART2 (Edwards et al. 2008); it can be further processed into a 24-nt sRNA (v-snoRNA124pp). Binding of v-snoRNA124pp to a complementary sequence in the 3′ UTR of the viral DNA polymerase (BALF5) mRNA leads to cleavage of the mRNA, suggesting that it acts as an siRNA to regulate viral replication.
Subgenomic ncRNAs from ssRNA viruses
A different kind of ncRNA, subgenomic flaviviral RNA (sfRNA), is produced by positive-strand RNA viruses belonging to the genus Flavivirus, which includes such arthropod-borne human pathogens as dengue virus (DENV), West Nile virus (WNV), and yellow fever virus (YFV). An ∼300- to 500-nt-long sfRNA is generated by incomplete degradation of the 3′ UTR of genomic RNA (gRNA) by the cellular 5′ → 3′ exonuclease XRN1 (Fig. 6; for review, see Bidet and Garcia-Blanco 2014; Roby et al. 2014).
Figure 6.
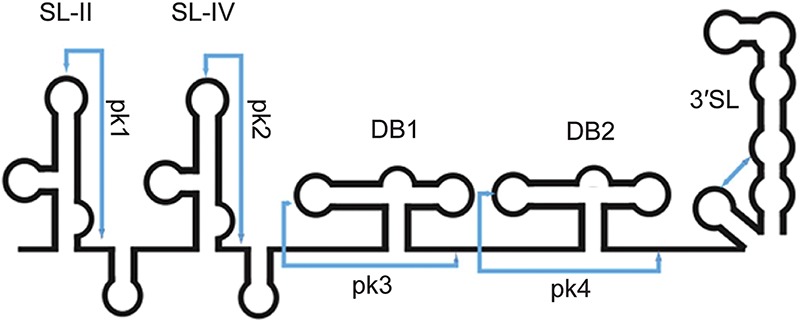
Schematic representation of the secondary structure of sfRNA from WNV. The elements shown were all experimentally verified in DENV and some in WNV (Chapman et al. 2014b). (SL) Stem–loop; (DB) dumbbell. Pseudoknot (pk) interactions are indicated by blue lines. Adapted from Macrae (2014), with permission from Elsevier. (Not to scale.)
The 3′ UTRs of Flaviviruses possess a collection of highly conserved structures, some of which are able to stall XRN1 progression to generate stable sfRNAs. The 5′-proximal stem–loop structure called SL-II constitutes the major block to XRN1 progression and thus demarcates the 5′ end of sfRNA (Pijlman et al. 2008; Chapman et al. 2014b). Extensive secondary structure mapping and mutational analysis of DENV sfRNA revealed a three-way junction in SL-II that is critical for halting XRN1 (Chapman et al. 2014b). Subsequently, the crystal structure of an XRN1-resistant segment from Murray Valley encephalitis virus revealed a precise RNA structure-based mechanism for XRN1 stalling: The RNA fold organized around the three-way junction adopts a ring-like conformation, with the 5′ end of the XRN1-resistant structure passing through the ring (Chapman et al. 2014a). All studied Flaviviruses, except YFV, possess a second conserved XRN1-resistant structure starting with SL-IV (Fig. 6; Pijlman et al. 2008); however, its role in the viral life cycle is not clear because, in wild-type viruses, XRN1 is nearly completely stalled by SL-II, and shorter sfRNAs accumulate to very low levels. The dengue isolate DV2-43, where two shorter 5′-trimmed species are more abundant than the full-size sfRNA (Liu et al. 2010), is the sole exception. sfRNA is required for Flavivirus cytopathicity and pathogenicity (Pijlman et al. 2008). It has been demonstrated to (1) interfere with cellular RNA decay pathways by inhibiting XRN1 (Moon et al. 2012), (2) dampen the antiviral activity of type I interferon (Schuessler et al. 2012), and (3) inhibit the RNAi pathway in both the vertebrate and arthropod (most frequently insect) hosts, most likely by serving as a decoy substrate for Dicer (Schnettler et al. 2012). Inhibition of the host interferon response appears to be, at least in some Flaviviruses, achieved by binding and inactivating cellular regulators of translation of interferon-up-regulated mRNAs (Bidet et al. 2014). The RNAi inhibitory activity of sfRNA appears to be especially important for counteracting a potent antiviral RNAi response in the insect phase of the Flavivirus life cycle.
Many negative-strand RNA viruses also produce subgenomic ncRNAs. It has been known for >35 years that those viruses with nonsegmented genomes (e.g., vesicular stomatitis virus [VSV] as well as rabies and Sendai viruses) synthesize an ∼50-nt-long plus-strand leader RNA (leRNA) (Leppert et al. 1979; Kurilla et al. 1984). In VSV, similarly sized minus-strand leRNAs have also been reported (Leppert et al. 1979). These VSV RNAs accumulate to ∼300 copies per infected cell as a result of premature termination by the viral RNA polymerase. Despite their relatively low abundance, leRNAs have been implicated in controlling viral genome replication (Blumberg et al. 1981).
Recently, minus-strand leRNAs, also known as small viral RNAs (svRNAs), have also been detected in cells infected with influenza A virus, which has a segmented negative-strand genome (Perez et al. 2010; Umbach et al. 2010). Here, 18- to 27-nt-long RNAs that are colinear with the 5′-most sequences of each of the eight genomic segments accumulate to very high levels (>100,000 copies per cell). Although not yet definitively proven, the influenza leRNAs are believed to originate from premature termination of genomic RNA synthesis. They were shown to interact with the viral RNA polymerase and proposed to regulate the switch from mRNA synthesis to viral genome replication (Perez et al. 2010, 2012).
Prospects
Although we have learned much, functions for many of the viral ncRNAs described here remain to be determined, particularly at the mechanistic level. In general, as for their cellular counterparts, the roles of viral small ncRNAs are better understood than those of lncRNAs. Elucidating lncRNA functions is only beginning and will undoubtedly be an area of active investigation for years to come. Meanwhile, the rate of discovery of new viral ncRNAs will continue to increase and bring new surprises. The challenges posed will no doubt be met by RNA biologists armed with improved technologies that continue to emerge. Additional unexpected insights into host ncRNAs can be expected to emerge from examination of their viral counterparts.
Acknowledgments
We thank Jess Brown, Johanna Withers, and Seyed Torabi for comments on the manuscript; Angie Miccinello for expert editing; and all other past and present members of the Steitz laboratory for stimulating discussions. Our work was supported by National Institutes of Health grant CA16038. W.N.M. holds a post-doctoral fellowship from the American Cancer Society, and M.X. is supported by a K99 grant (CA190886) from the National Cancer Institute. J.A.S. is an investigator of the Howard Hughes Medical Institute.
Note added in proof
The van Dyk group (Diebel et al. 2015) has recently generated and characterized a recombinant MHV68 strain devoid of all tRNA–pre-miRNA chimeric genes. They found that this strain has wild-type levels of lytic replication in vitro and normal establishment of latency in B cells upon acute infection in vivo. However, in acute infection of immunocompromized mice, tRNA–pre-miRNA-deficient MHV68 exhibits reduced virulence in a model of viral pneumonia. It appears that the tRNAs contribute most importantly to this phenotype.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.259077.115.
References
- Abend JR, Uldrick T, Ziegelbauer JM. 2010. Regulation of tumor necrosis factor-like weak inducer of apoptosis receptor protein (TWEAKR) expression by Kaposi's sarcoma-associated herpesvirus microRNA prevents TWEAK-induced apoptosis and inflammatory cytokine expression. J Virol 84: 12139–12151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abend JR, Ramalingam D, Kieffer-Kwon P, Uldrick TS, Yarchoan R, Ziegelbauer JM. 2012. Kaposi's sarcoma-associated herpesvirus microRNAs target IRAK1 and MYD88, two components of the toll-like receptor/interleukin-1R signaling cascade, to reduce inflammatory-cytokine expression. J Virol 86: 11663–11674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed W, Philip PS, Tariq S, Khan G. 2014. Epstein-Barr virus-encoded small RNAs (EBERs) are present in fractions related to exosomes released by EBV-transformed cells. PLoS One 9: e99163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht JC, Fleckenstein B. 1992. Nucleotide sequence of HSUR 6 and HSUR 7, two small RNAs of Herpesvirus saimiri. Nucleic Acids Res 20: 1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson MG, Haasnoot PC, Xu N, Berenjian S, Berkhout B, Akusjarvi G. 2005. Suppression of RNA interference by adenovirus virus-associated RNA. J Virol 79: 9556–9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio O, Razquin N, Zaratiegui M, Narvaiza I, Fortes P. 2006. Adenovirus virus-associated RNA is processed to functional interfering RNAs involved in virus production. J Virol 80: 1376–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio O, Carnero E, Abad X, Razquin N, Guruceaga E, Segura V, Fortes P. 2010. Adenovirus VA RNA-derived miRNAs target cellular genes involved in cell growth, gene expression and DNA repair. Nucleic Acids Res 38: 750–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvey A, Tempera I, Tsai K, Chen HS, Tikhmyanova N, Klichinsky M, Leslie C, Lieberman PM. 2012. An atlas of the Epstein-Barr virus transcriptome and epigenome reveals host-virus regulatory interactions. Cell Host Microbe 12: 233–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiarz JE, Ruby JG, Wang Y, Bartel DP, Blelloch R. 2008. Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes Dev 22: 2773–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baillat D, Hakimi MA, Naar AM, Shilatifard A, Cooch N, Shiekhattar R. 2005. Integrator, a multiprotein mediator of small nuclear RNA processing, associates with the C-terminal repeat of RNA polymerase II. Cell 123: 265–276. [DOI] [PubMed] [Google Scholar]
- Bekerman E, Jeon D, Ardolino M, Coscoy L. 2013. A role for host activation-induced cytidine deaminase in innate immune defense against KSHV. PLoS Pathog 9: e1003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellare P, Ganem D. 2009. Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: an evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe 6: 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender BJ, Coen DM, Strang BL. 2014. Dynamic and nucleolin-dependent localization of human cytomegalovirus UL84 to the periphery of viral replication compartments and nucleoli. J Virol 88: 11738–11747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat RA, Thimmappaya B. 1983. Two small RNAs encoded by Epstein-Barr virus can functionally substitute for the virus-associated RNAs in the lytic growth of adenovirus 5. Proc Natl Acad Sci 80: 4789–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidet K, Garcia-Blanco MA. 2014. Flaviviral RNAs: weapons and targets in the war between virus and host. Biochem J 462: 215–230. [DOI] [PubMed] [Google Scholar]
- Bidet K, Dadlani D, Garcia-Blanco MA. 2014. G3BP1, G3BP2 and CAPRIN1 are required for translation of interferon stimulated mRNAs and are targeted by a dengue virus non-coding RNA. PLoS Pathog 10: e1004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumberg BM, Leppert M, Kolakofsky D. 1981. Interaction of VSV leader RNA and nucleocapsid protein may control VSV genome replication. Cell 23: 837–845. [DOI] [PubMed] [Google Scholar]
- Bogerd HP, Karnowski HW, Cai X, Shin J, Pohlers M, Cullen BR. 2010. A mammalian herpesvirus uses noncanonical expression and processing mechanisms to generate viral MicroRNAs. Mol Cell 37: 135–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borah S, Darricarrere N, Darnell A, Myoung J, Steitz JA. 2011. A viral nuclear noncoding RNA binds re-localized poly(A) binding protein and is required for late KSHV gene expression. PLoS Pathog 7: e1002300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowden RJ, Simas JP, Davis AJ, Efstathiou S. 1997. Murine γ-herpesvirus 68 encodes tRNA-like sequences which are expressed during latency. J Gen Virol 78: 1675–1687. [DOI] [PubMed] [Google Scholar]
- Brown JA, Valenstein ML, Yario TA, Tycowski KT, Steitz JA. 2012. Formation of triple-helical structures by the 3′-end sequences of MALAT1 and MENβ noncoding RNAs. Proc Natl Acad Sci 109: 19202–19207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA, Bulkley D, Wang J, Valenstein ML, Yario TA, Steitz TA, Steitz JA. 2014. Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix. Nat Struct Mol Biol 21: 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck AH, Perot J, Chisholm MA, Kumar DS, Tuddenham L, Cognat V, Marcinowski L, Dolken L, Pfeffer S. 2010. Post-transcriptional regulation of miR-27 in murine cytomegalovirus infection. RNA 16: 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JM, Bass CR, Kincaid RP, Sullivan CS. 2014. Identification of tri-phosphatase activity in the biogenesis of retroviral microRNAs and RNAP III-generated shRNAs. Nucleic Acids Res 42: 13949–13962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell M, Kim KY, Chang PC, Huerta S, Shevchenko B, Wang DH, Izumiya C, Kung HJ, Izumiya Y. 2014. A lytic viral long noncoding RNA modulates the function of a latent protein. J Virol 88: 1843–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao S, Strong MJ, Wang X, Moss WN, Concha M, Lin Z, O'Grady T, Baddoo M, Fewell C, Renne R, et al. 2015. High-throughput RNA sequencing based virome analysis of 50 lymphoma cell lines from the Cancer Cell Line Encyclopedia project. J Virol 89: 713–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazalla D, Yario T, Steitz JA. 2010. Down-regulation of a host microRNA by a Herpesvirus saimiri noncoding RNA. Science 328: 1563–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazalla D, Xie M, Steitz JA. 2011. A primate herpesvirus uses the integrator complex to generate viral microRNAs. Mol Cell 43: 982–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman EG, Costantino DA, Rabe JL, Moon SL, Wilusz J, Nix JC, Kieft JS. 2014a. The structural basis of pathogenic subgenomic flavivirus RNA (sfRNA) production. Science 344: 307–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman EG, Moon SL, Wilusz J, Kieft JS. 2014b. RNA structures that resist degradation by Xrn1 produce a pathogenic Dengue virus RNA. eLife 3: e01892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ. 2010. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 465: 584–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Zang JB, Mele A, Darnell RB. 2009. Argonaute HITS-CLIP decodes microRNA–mRNA interaction maps. Nature 460: 479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy EY, Siu KL, Kok KH, Lung RW, Tsang CM, To KF, Kwong DL, Tsao SW, Jin DY. 2008. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J Exp Med 205: 2551–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. 2011. Genomic maps of long noncoding RNA occupancy reveal principles of RNA–chromatin interactions. Mol Cell 44: 667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, Ma E, Mane S, Hannon GJ, Lawson ND, et al. 2010. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science 328: 1694–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke PA, Sharp NA, Clemens MJ. 1990. Translational control by the Epstein-Barr virus small RNA EBER-1. Reversal of the double-stranded RNA-induced inhibition of protein synthesis in reticulocyte lysates. Eur J Biochem 193: 635–641. [DOI] [PubMed] [Google Scholar]
- Cliffe AR, Garber DA, Knipe DM. 2009. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J Virol 83: 8182–8190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad NK, Steitz JA. 2005. A Kaposi's sarcoma virus RNA element that increases the nuclear abundance of intronless transcripts. EMBO J 24: 1831–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook HL, Mischo HE, Steitz JA. 2004. The Herpesvirus saimiri small nuclear RNAs recruit AU-rich element-binding proteins but do not alter host AU-rich element-containing mRNA levels in virally transformed T cells. Mol Cell Biol 24: 4522–4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook HL, Lytle JR, Mischo HE, Li MJ, Rossi JJ, Silva DP, Desrosiers RC, Steitz JA. 2005. Small nuclear RNAs encoded by Herpesvirus saimiri upregulate the expression of genes linked to T cell activation in virally transformed T cells. Curr Biol 15: 974–979. [DOI] [PubMed] [Google Scholar]
- de Haro C, Mendez R, Santoyo J. 1996. The eIF-2α kinases and the control of protein synthesis. FASEB J 10: 1378–1387. [DOI] [PubMed] [Google Scholar]
- Diebel KW, Smith AL, van Dyk LF. 2010. Mature and functional viral miRNAs transcribed from novel RNA polymerase III promoters. RNA 16: 170–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebel KW, Oko LM, Medina EM, Niemeyer BF, Warren CJ, Claypool DJ, Tibbetts SA, Cool CD, Clambey ET, van Dyk LF. 2015. Gammaherpesvirus small noncoding RNAs are bifunctional elements that regulate infection and contribute to virulence in vivo. MBio 6: e01670-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominski Z, Yang XC, Marzluff WF. 2005. The polyadenylation factor CPSF-73 is involved in histone-pre-mRNA processing. Cell 123: 37–48. [DOI] [PubMed] [Google Scholar]
- Edwards RH, Marquitz AR, Raab-Traub N. 2008. Epstein-Barr virus BART microRNAs are produced from a large intron prior to splicing. J Virol 82: 9094–9106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggington JM, Greene T, Bass BL. 2011. Predicting sites of ADAR editing in double-stranded RNA. Nat Commun 2: 319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ensser A, Fleckenstein B. 2005. T-cell transformation and oncogenesis by γ2-herpesviruses. Adv Cancer Res 93: 91–128. [DOI] [PubMed] [Google Scholar]
- Fan XC, Myer VE, Steitz JA. 1997. AU-rich elements target small nuclear RNAs as well as mRNAs for rapid degradation. Genes Dev 11: 2557–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell MJ, Dobson AT, Feldman LT. 1991. Herpes simplex virus latency-associated transcript is a stable intron. Proc Natl Acad Sci 88: 790–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feederle R, Linnstaedt SD, Bannert H, Lips H, Bencun M, Cullen BR, Delecluse HJ. 2011. A viral microRNA cluster strongly potentiates the transforming properties of a human herpesvirus. PLoS Pathog 7: e1001294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fok V, Friend K, Steitz JA. 2006. Epstein-Barr virus noncoding RNAs are confined to the nucleus, whereas their partner, the human La protein, undergoes nucleocytoplasmic shuttling. J Cell Biol 173: 319–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francoeur AM, Mathews MB. 1982. Interaction between VA RNA and the lupus antigen La: formation of a ribonucleoprotein particle in vitro. Proc Natl Acad Sci 79: 6772–6776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem D. 2006. KSHV infection and the pathogenesis of Kaposi's sarcoma. Annu Rev Pathol 1: 273–296. [DOI] [PubMed] [Google Scholar]
- Gardner EJ, Nizami ZF, Talbot CC Jr, Gall JG. 2012. Stable intronic sequence RNA (sisRNA), a new class of noncoding RNA from the oocyte nucleus of Xenopus tropicalis. Genes Dev 26: 2550–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golembe TJ, Yong J, Battle DJ, Feng W, Wan L, Dreyfuss G. 2005. Lymphotropic Herpesvirus saimiri uses the SMN complex to assemble Sm cores on its small RNAs. Mol Cell Biol 25: 602–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottwein E, Cullen BR. 2010. A human herpesvirus microRNA inhibits p21 expression and attenuates p21-mediated cell cycle arrest. J Virol 84: 5229–5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottwein E, Corcoran DL, Mukherjee N, Skalsky RL, Hafner M, Nusbaum JD, Shamulailatpam P, Love CL, Dave SS, Tuschl T, et al. 2011. Viral microRNA targetome of KSHV-infected primary effusion lymphoma cell lines. Cell Host Microbe 10: 515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenaway PJ, Wilkinson GW. 1987. Nucleotide sequence of the most abundantly transcribed early gene of human cytomegalovirus strain AD169. Virus Res 7: 17–31. [DOI] [PubMed] [Google Scholar]
- Gregorovic G, Bosshard R, Karstegl CE, White RE, Pattle S, Chiang AK, Dittrich-Breiholz O, Kracht M, Russ R, Farrell PJ. 2011. Cellular gene expression that correlates with EBER expression in Epstein-Barr Virus-infected lymphoblastoid cell lines. J Virol 85: 3535–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grey F. 2014. The role of microRNAs in herpesvirus latency and persistence. J Gen Virol 10.1099/vir.1090.070862-070860. [DOI] [PubMed] [Google Scholar]
- Grey F, Meyers H, White EA, Spector DH, Nelson J. 2007. A human cytomegalovirus-encoded microRNA regulates expression of multiple viral genes involved in replication. PLoS Pathog 3: e163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo YE, Steitz JA. 2014a. 3′-biotin-tagged microRNA-27 does not associate with Argonaute proteins in cells. RNA 20: 985–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo YE, Steitz JA. 2014b. Virus meets host microRNA: the destroyer, the booster, the hijacker. Mol Cell Biol 34: 3780–3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo YE, Riley KJ, Iwasaki A, Steitz JA. 2014. Alternative capture of noncoding RNAs or protein-coding genes by herpesviruses to alter host T cell function. Mol Cell 54: 67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha M, Kim VN. 2014. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 15: 509–524. [DOI] [PubMed] [Google Scholar]
- Haecker I, Gay LA, Yang Y, Hu J, Morse AM, McIntyre LM, Renne R. 2012. Ago HITS-CLIP expands understanding of Kaposi's sarcoma-associated herpesvirus miRNA function in primary effusion lymphomas. PLoS Pathog 8: e1002884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jungkamp AC, Munschauer M, et al. 2010. PAR-CliP - a method to identify transcriptome-wide the binding sites of RNA binding proteins. J Vis Exp 10.3791/2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselberth JR. 2013. Lives that introns lead after splicing. Wiley Interdiscip Rev RNA 4: 677–691. [DOI] [PubMed] [Google Scholar]
- Hook LM, Grey F, Grabski R, Tirabassi R, Doyle T, Hancock M, Landais I, Jeng S, McWeeney S, Britt W, et al. 2014. Cytomegalovirus miRNAs target secretory pathway genes to facilitate formation of the virion assembly compartment and reduce cytokine secretion. Cell Host Microbe 15: 363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houmani JL, Davis CI, Ruf IK. 2009. Growth-promoting properties of Epstein-Barr virus EBER-1 RNA correlate with ribosomal protein L22 binding. J Virol 83: 9844–9853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe JG, Steitz JA. 1986. Localization of Epstein-Barr virus-encoded small RNAs by in situ hybridization. Proc Natl Acad Sci 83: 9006–9010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutzinger R, Feederle R, Mrazek J, Schiefermeier N, Balwierz PJ, Zavolan M, Polacek N, Delecluse HJ, Huttenhofer A. 2009. Expression and processing of a small nucleolar RNA from the Epstein-Barr virus genome. PLoS Pathog 5: e1000547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imig J, Brunschweiger A, Brummer A, Guennewig B, Mittal N, Kishore S, Tsikrika P, Gerber AP, Zavolan M, Hall J. 2015. miR-CLIP capture of a miRNA targetome uncovers a lincRNA H19-miR-106a interaction. Nat Chem Biol 11: 107–114. [DOI] [PubMed] [Google Scholar]
- Inman M, Perng GC, Henderson G, Ghiasi H, Nesburn AB, Wechsler SL, Jones C. 2001. Region of herpes simplex virus type 1 latency-associated transcript sufficient for wild-type spontaneous reactivation promotes cell survival in tissue culture. J Virol 75: 3636–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakiri D, Sheen TS, Chen JY, Huang DP, Takada K. 2005. Epstein-Barr virus-encoded small RNA induces insulin-like growth factor 1 and supports growth of nasopharyngeal carcinoma-derived cell lines. Oncogene 24: 1767–1773. [DOI] [PubMed] [Google Scholar]
- Iwakiri D, Zhou L, Samanta M, Matsumoto M, Ebihara T, Seya T, Imai S, Fujieda M, Kawa K, Takada K. 2009. Epstein-Barr virus (EBV)-encoded small RNA is released from EBV-infected cells and activates signaling from Toll-like receptor 3. J Exp Med 206: 2091–2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeang KT, Hayward SD. 1983. Organization of the Epstein-Barr virus DNA molecule. III. Location of the P3HR-1 deletion junction and characterization of the NotI repeat units that form part of the template for an abundant 12-O-tetradecanoylphorbol-13-acetate-induced mRNA transcript. J Virol 48: 135–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung YJ, Choi H, Kim H, Lee SK. 2014. MicroRNA miR-BART20-5p stabilizes Epstein-Barr virus latency by directly targeting BZLF1 and BRLF1. J Virol 88: 9027–9037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamel W, Segerman B, Oberg D, Punga T, Akusjarvi G. 2013. The adenovirus VA RNA-derived miRNAs are not essential for lytic virus growth in tissue culture cells. Nucleic Acids Res 41: 4802–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang W, Mukerjee R, Gartner JJ, Hatzigeorgiou AG, Sandri-Goldin RM, Fraser NW. 2006. Characterization of a spliced exon product of herpes simplex type-1 latency-associated transcript in productively infected cells. Virology 356: 106–114. [DOI] [PubMed] [Google Scholar]
- Katsumura KR, Maruo S, Takada K. 2011. EBV lytic infection enhances transformation of B-lymphocytes infected with EBV in the presence of T-lymphocytes. J Med Virol 84: 504–510. [DOI] [PubMed] [Google Scholar]
- Kelly GL, Long HM, Stylianou J, Thomas WA, Leese A, Bell AI, Bornkamm GW, Mautner J, Rickinson AB, Rowe M. 2009. An Epstein-Barr virus anti-apoptotic protein constitutively expressed in transformed cells and implicated in Burkitt lymphomagenesis: the Wp/BHRF1 link. PLoS Pathog 5: e1000341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Lee S, Shin J, Kim Y, Evnouchidou I, Kim D, Kim YK, Kim YE, Ahn JH, Riddell SR, et al. 2011. Human cytomegalovirus microRNA miR-US4-1 inhibits CD8+ T cell responses by targeting the aminopeptidase ERAP1. Nat Immunol 12: 984–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Lee S, Kim S, Kim D, Ahn JH, Ahn K. 2012. Human cytomegalovirus clinical strain-specific microRNA miR-UL148D targets the human chemokine RANTES during infection. PLoS Pathog 8: e1002577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kincaid RP, Burke JM, Sullivan CS. 2012. RNA virus microRNA that mimics a B-cell oncomiR. Proc Natl Acad Sci 109: 3077–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kincaid RP, Chen Y, Cox JE, Rethwilm A, Sullivan CS. 2014. Noncanonical microRNA (miRNA) biogenesis gives rise to retroviral mimics of lymphoproliferative and immunosuppressive host miRNAs. mBio 5: e00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa N, Goto M, Kurozumi K, Maruo S, Fukayama M, Naoe T, Yasukawa M, Hino K, Suzuki T, Todo S, et al. 2000. Epstein-Barr virus-encoded poly(A)− RNA supports Burkitt's lymphoma growth through interleukin-10 induction. EMBO J 19: 6742–6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komano J, Maruo S, Kurozumi K, Oda T, Takada K. 1999. Oncogenic role of Epstein-Barr virus-encoded RNAs in Burkitt's lymphoma cell line Akata. J Virol 73: 9827–9831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummenacher C, Zabolotny JM, Fraser NW. 1997. Selection of a nonconsensus branch point is influenced by an RNA stem-loop structure and is important to confer stability to the herpes simplex virus 2-kilobase latency-associated transcript. J Virol 71: 5849–5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulesza CA, Shenk T. 2004. Human cytomegalovirus 5-kilobase immediate–early RNA is a stable intron. J Virol 78: 13182–13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulesza CA, Shenk T. 2006. Murine cytomegalovirus encodes a stable intron that facilitates persistent replication in the mouse. Proc Natl Acad Sci 103: 18302–18307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurilla MG, Cabradilla CD, Holloway BP, Keene JD. 1984. Nucleotide sequence and host La protein interactions of rabies virus leader RNA. J Virol 50: 773–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski DL, Thompson HW, Bloom DC. 2009. The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J Virol 83: 8173–8181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SI, Murthy SC, Trimble JJ, Desrosiers RC, Steitz JA. 1988. Four novel U RNAs are encoded by a herpesvirus. Cell 54: 599–607. [DOI] [PubMed] [Google Scholar]
- Lee N, Pimienta G, Steitz JA. 2012. AUF1/hnRNP D is a novel protein partner of the EBER1 noncoding RNA of Epstein-Barr virus. RNA 18: 2073–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N, Moss WN, Yario TA, Steitz JA. 2015. An EBV noncoding RNA base pairs with nascent RNA to enable PAX5 transcription factor binding to viral DNA. Cell 160: 607–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei X, Bai Z, Ye F, Xie J, Kim CG, Huang Y, Gao SJ. 2010. Regulation of NF-κB inhibitor IκBα and viral replication by a KSHV microRNA. Nat Cell Biol 12: 193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppert M, Rittenhouse L, Perrault J, Summers DF, Kolakofsky D. 1979. Plus and minus strand leader RNAs in negative strand virus-infected cells. Cell 18: 735–747. [DOI] [PubMed] [Google Scholar]
- Lerner MR, Steitz JA. 1981. Snurps and scyrps. Cell 25: 298–300. [DOI] [PubMed] [Google Scholar]
- Lerner MR, Andrews NC, Miller G, Steitz JA. 1981. Two small RNAs encoded by Epstein-Barr virus and complexed with protein are precipitated by antibodies from patients with systemic lupus erythematosus. Proc Natl Acad Sci 78: 805–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libri V, Helwak A, Miesen P, Santhakumar D, Borger JG, Kudla G, Grey F, Tollervey D, Buck AH. 2012. Murine cytomegalovirus encodes a miR-27 inhibitor disguised as a target. Proc Natl Acad Sci 109: 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Yue L, Li X, Yu X, Zhao H, Jiang Z, Qin E, Qin C. 2010. Identification and characterization of small sub-genomic RNAs in dengue 1-4 virus-infected cell cultures and tissues. Biochem Biophys Res Commun 391: 1099–1103. [DOI] [PubMed] [Google Scholar]
- Lord PC, Rothschild CB, DeRose RT, Kilpatrick BA. 1989. Human cytomegalovirus RNAs immunoprecipitated by multiple systemic lupus erythematosus antisera. J Gen Virol 70: 2383–2396. [DOI] [PubMed] [Google Scholar]
- Lu S, Cullen BR. 2004. Adenovirus VA1 noncoding RNA can inhibit small interfering RNA and MicroRNA biogenesis. J Virol 78: 12868–12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Stedman W, Yousef M, Renne R, Lieberman PM. 2010. Epigenetic regulation of Kaposi's sarcoma-associated herpesvirus latency by virus-encoded microRNAs that target Rta and the cellular Rbl2-DNMT pathway. J Virol 84: 2697–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrae R. 2014. Uninvited guests. Cell 157: 761. [Google Scholar]
- Marcinowski L, Tanguy M, Krmpotic A, Radle B, Lisnic VJ, Tuddenham L, Chane-Woon-Ming B, Ruzsics Z, Erhard F, Benkartek C, et al. 2012. Degradation of cellular mir-27 by a novel, highly abundant viral transcript is important for efficient virus replication in vivo. PLoS Pathog 8: e1002510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews MB, Shenk T. 1991. Adenovirus virus-associated RNA and translation control. J Virol 65: 5657–5662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina MF, Joshi S. 1999. RNA-polymerase III-driven expression cassettes in human gene therapy. Curr Opin Mol Ther 1: 580–594. [PubMed] [Google Scholar]
- Medvedovic J, Ebert A, Tagoh H, Busslinger M. 2011. Pax5: a master regulator of B cell development and leukemogenesis. Adv Immunol 111: 179–206. [DOI] [PubMed] [Google Scholar]
- Mitchell WJ, Deshmane SL, Dolan A, McGeoch DJ, Fraser NW. 1990. Characterization of herpes simplex virus type 2 transcription during latent infection of mouse trigeminal ganglia. J Virol 64: 5342–5348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitton-Fry RM, DeGregorio SJ, Wang J, Steitz TA, Steitz JA. 2010. Poly(A) tail recognition by a viral RNA element through assembly of a triple helix. Science 330: 1244–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody R, Zhu Y, Huang Y, Cui X, Jones T, Bedolla R, Lei X, Bai Z, Gao SJ. 2013. KSHV microRNAs mediate cellular transformation and tumorigenesis by redundantly targeting cell growth and survival pathways. PLoS Pathog 9: e1003857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon SL, Anderson JR, Kumagai Y, Wilusz CJ, Akira S, Khromykh AA, Wilusz J. 2012. A noncoding RNA produced by arthropod-borne flaviviruses inhibits the cellular exoribonuclease XRN1 and alters host mRNA stability. RNA 18: 2029–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss WN, Steitz JA. 2013. Genome-wide analyses of Epstein-Barr virus reveal conserved RNA structures and a novel stable intronic sequence RNA. BMC Genomics 14: 543–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss WN, Lee N, Pimienta G, Steitz JA. 2014. RNA families in Epstein-Barr virus. RNA Biol 11: 10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukerjee R, Kang W, Suri V, Fraser NW. 2004. A non-consensus branch point plays an important role in determining the stability of the 2-kb LAT intron during acute and latent infections of herpes simplex virus type-1. Virology 324: 340–349. [DOI] [PubMed] [Google Scholar]
- Murphy E, Vanicek J, Robins H, Shenk T, Levine AJ. 2008. Suppression of immediate–early viral gene expression by herpesvirus-coded microRNAs: implications for latency. Proc Natl Acad Sci 105: 5453–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy SC, Trimble JJ, Desrosiers RC. 1989. Deletion mutants of Herpesvirus saimiri define an open reading frame necessary for transformation. J Virol 63: 3307–3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myer VE, Lee SI, Steitz JA. 1992. Viral small nuclear ribonucleoproteins bind a protein implicated in messenger RNA destabilization. Proc Natl Acad Sci 89: 1296–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachmani D, Stern-Ginossar N, Sarid R, Mandelboim O. 2009. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe 5: 376–385. [DOI] [PubMed] [Google Scholar]
- Nachmani D, Lankry D, Wolf DG, Mandelboim O. 2010. The human cytomegalovirus microRNA miR-UL112 acts synergistically with a cellular microRNA to escape immune elimination. Nat Immunol 11: 806–813. [DOI] [PubMed] [Google Scholar]
- Nikitin PA, Yan CM, Forte E, Bocedi A, Tourigny JP, White RE, Allday MJ, Patel A, Dave SS, Kim W, et al. 2010. An ATM/Chk2-mediated DNA damage-responsive signaling pathway suppresses Epstein-Barr virus transformation of primary human B cells. Cell Host Microbe 8: 510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, Chung WJ, Ruby JG, Guo H, Bartel DP, Lai EC. 2008. The Drosophila hairpin RNA pathway generates endogenous short interfering RNAs. Nature 453: 803–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ou Y, Davis KA, Traina-Dorge V, Gray WL. 2007. Simian varicella virus expresses a latency-associated transcript that is antisense to open reading frame 61 (ICP0) mRNA in neural ganglia of latently infected monkeys. J Virol 81: 8149–8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Vitvitskaia O, Carpenter D, Wechsler SL, Jones C. 2008. Identification of two small RNAs within the first 1.5-kb of the herpes simplex virus type 1-encoded latency-associated transcript. J Neurovirol 14: 41–52. [DOI] [PubMed] [Google Scholar]
- Perez JT, Varble A, Sachidanandam R, Zlatev I, Manoharan M, Garcia-Sastre A, tenOever BR. 2010. Influenza A virus-generated small RNAs regulate the switch from transcription to replication. Proc Natl Acad Sci 107: 11525–11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez JT, Zlatev I, Aggarwal S, Subramanian S, Sachidanandam R, Kim B, Manoharan M, tenOever BR. 2012. A small-RNA enhancer of viral polymerase activity. J Virol 86: 13475–13485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perng GC, Jones C, Ciacci-Zanella J, Stone M, Henderson G, Yukht A, Slanina SM, Hofman FM, Ghiasi H, Nesburn AB, et al. 2000. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 287: 1500–1503. [DOI] [PubMed] [Google Scholar]
- Pfeffer S, Zavolan M, Grasser FA, Chien M, Russo JJ, Ju J, John B, Enright AJ, Marks D, Sander C, et al. 2004. Identification of virus-encoded microRNAs. Science 304: 734–736. [DOI] [PubMed] [Google Scholar]
- Pijlman GP, Funk A, Kondratieva N, Leung J, Torres S, van der Aa L, Liu WJ, Palmenberg AC, Shi PY, Hall RA, et al. 2008. A highly structured, nuclease-resistant, noncoding RNA produced by flaviviruses is required for pathogenicity. Cell Host Microbe 4: 579–591. [DOI] [PubMed] [Google Scholar]
- Pont AR, Sadri N, Hsiao SJ, Smith S, Schneider RJ. 2012. mRNA decay factor AUF1 maintains normal aging, telomere maintenance, and suppression of senescence by activation of telomerase transcription. Mol Cell 47: 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Thorley-Lawson DA. 2014. EBV microRNA BART 18-5p targets MAP3K2 to facilitate persistence in vivo by inhibiting viral replication in B cells. Proc Natl Acad Sci 111: 11157–11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese TA, Xia J, Johnson LS, Zhou X, Zhang W, Virgin HW. 2010. Identification of novel microRNA-like molecules generated from herpesvirus and host tRNA transcripts. J Virol 84: 10344–10353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves MB, Davies AA, McSharry BP, Wilkinson GW, Sinclair JH. 2007. Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science 316: 1345–1348. [DOI] [PubMed] [Google Scholar]
- Reich PR, Forget BG, Weissman SM. 1966. RNA of low molecular weight in KB cells infected with adenovirus type 2. J Mol Biol 17: 428–439. [DOI] [PubMed] [Google Scholar]
- Rennekamp AJ, Lieberman PM. 2011. Initiation of Epstein-Barr virus lytic replication requires transcription and the formation of a stable RNA–DNA hybrid molecule at OriLyt. J Virol 85: 2837–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riaz A, Dry I, Levy CS, Hopkins J, Grey F, Shaw DJ, Dalziel RG. 2014. Ovine herpesvirus-2-encoded microRNAs target virus genes involved in virus latency. J Gen Virol 95: 472–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley KJ, Rabinowitz GS, Steitz JA. 2010. Comprehensive analysis of Rhesus lymphocryptovirus microRNA expression. J Virol 84: 5148–5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley KJ, Rabinowitz GS, Yario TA, Luna JM, Darnell RB, Steitz JA. 2012. EBV and human microRNAs co-target oncogenic and apoptotic viral and human genes during latency. EMBO J 31: 2207–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roby JA, Pijlman GP, Wilusz J, Khromykh AA. 2014. Noncoding subgenomic flavivirus RNA: multiple functions in West Nile virus pathogenesis and modulation of host responses. Viruses 6: 404–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock DL, Beam SL, Mayfield JE. 1987. Mapping bovine herpesvirus type 1 latency-related RNA in trigeminal ganglia of latently infected rabbits. J Virol 61: 3827–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodahl E, Haarr L. 1997. Analysis of the 2-kilobase latency-associated transcript expressed in PC12 cells productively infected with herpes simplex virus type 1: evidence for a stable, nonlinear structure. J Virol 71: 1703–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosa MD, Gottlieb E, Lerner MR, Steitz JA. 1981. Striking similarities are exhibited by two small Epstein-Barr virus-encoded ribonucleic acids and the adenovirus-associated ribonucleic acids VAI and VAII. Mol Cell Biol 1: 785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetto CC, Pari G. 2012. KSHV PAN RNA associates with demethylases UTX and JMJD3 to activate lytic replication through a physical interaction with the virus genome. PLoS Pathog 8: e1002680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossetto CC, Tarrant-Elorza M, Verma S, Purushothaman P, Pari GS. 2013. Regulation of viral and cellular gene expression by Kaposi's sarcoma-associated herpesvirus polyadenylated nuclear RNA. J Virol 87: 5540–5553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouha H, Thurner C, Mandl CW. 2010. Functional microRNA generated from a cytoplasmic RNA virus. Nucleic Acids Res 38: 8328–8337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby JG, Jan CH, Bartel DP. 2007. Intronic microRNA precursors that bypass Drosha processing. Nature 448: 83–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin BB, Patel D, Conrad NK. 2010. Kaposi's sarcoma-associated herpesvirus ORF57 protein binds and protects a nuclear noncoding RNA from cellular RNA decay pathways. PLoS Pathog 6: e1000799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnettler E, Sterken MG, Leung JY, Metz SW, Geertsema C, Goldbach RW, Vlak JM, Kohl A, Khromykh AA, Pijlman GP. 2012. Noncoding flavivirus RNA displays RNA interference suppressor activity in insect and mammalian cells. J Virol 86: 13486–13500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuessler A, Funk A, Lazear HM, Cooper DA, Torres S, Daffis S, Jha BK, Kumagai Y, Takeuchi O, Hertzog P, et al. 2012. West Nile virus noncoding subgenomic RNA contributes to viral evasion of the type I interferon-mediated antiviral response. J Virol 86: 5708–5718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz TM, Kulesza CA. 2014. Stability determinants of murine cytomegalovirus long noncoding RNA7.2. J Virol 88: 11630–11633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seto E, Moosmann A, Gromminger S, Walz N, Grundhoff A, Hammerschmidt W. 2010. Micro RNAs of Epstein-Barr virus promote cell cycle progression and prevent apoptosis of primary human B cells. PLoS Pathog 6: e1001063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro JS, Varble A, Pham AM, Tenoever BR. 2010. Noncanonical cytoplasmic processing of viral microRNAs. RNA 16: 2068–2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Sa e Silva M, Jaber T, Vitvitskaia O, Li S, Henderson G, Jones C. 2009. Two small RNAs encoded within the first 1.5 kilobases of the herpes simplex virus type 1 latency-associated transcript can inhibit productive infection and cooperate to inhibit apoptosis. J Virol 83: 9131–9139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon MD, Wang CI, Kharchenko PV, West JA, Chapman BA, Alekseyenko AA, Borowsky ML, Kuroda MI, Kingston RE. 2011. The genomic binding sites of a noncoding RNA. Proc Natl Acad Sci 108: 20497–20502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skalsky RL, Corcoran DL, Gottwein E, Frank CL, Kang D, Hafner M, Nusbaum JD, Feederle R, Delecluse HJ, Luftig MA, et al. 2012. The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog 8: e1002484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderlund H, Pettersson U, Vennstrom B, Philipson L, Mathews MB. 1976. A new species of virus-coded low molecular weight RNA from cells infected with adenovirus type 2. Cell 7: 585–593. [DOI] [PubMed] [Google Scholar]
- Spector DH. 1996. Activation and regulation of human cytomegalovirus early genes. Intervirology 39: 361–377. [DOI] [PubMed] [Google Scholar]
- Steitz J, Borah S, Cazalla D, Fok V, Lytle R, Mitton-Fry R, Riley K, Samji T. 2010. Noncoding RNPs of viral origin. Cold Spring Harb Perspect Biol 3: 165–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern-Ginossar N, Elefant N, Zimmermann A, Wolf DG, Saleh N, Biton M, Horwitz E, Prokocimer Z, Prichard M, Hahn G, et al. 2007. Host immune system gene targeting by a viral miRNA. Science 317: 376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern-Ginossar N, Weisburd B, Michalski A, Le VT, Hein MY, Huang SX, Ma M, Shen B, Qian SB, Hengel H, et al. 2012. Decoding human cytomegalovirus. Science 338: 1088–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens JG, Wagner EK, Devi-Rao GB, Cook ML, Feldman LT. 1987. RNA complementary to a herpesvirus α gene mRNA is prominent in latently infected neurons. Science 235: 1056–1059. [DOI] [PubMed] [Google Scholar]
- Suffert G, Malterer G, Hausser J, Viiliainen J, Fender A, Contrant M, Ivacevic T, Benes V, Gros F, Voinnet O, et al. 2011. Kaposi's sarcoma herpesvirus microRNAs target caspase 3 and regulate apoptosis. PLoS Pathog 7: e1002405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan CS, Grundhoff AT, Tevethia S, Pipas JM, Ganem D. 2005. SV40-encoded microRNAs regulate viral gene expression and reduce susceptibility to cytotoxic T cells. Nature 435: 682–686. [DOI] [PubMed] [Google Scholar]
- Sun R, Lin SF, Gradoville L, Miller G. 1996. Polyadenylylated nuclear RNA encoded by Kaposi sarcoma-associated herpesvirus. Proc Natl Acad Sci 93: 11883–11888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan S, Tomkinson B, Kieff E. 1991. Recombinant Epstein-Barr virus with small RNA (EBER) genes deleted transforms lymphocytes and replicates in vitro. Proc Natl Acad Sci 88: 1546–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SM, Kirchner R, Jin J, Hofmann O, McReynolds L, Hide W, Lieberman J. 2014. Sequencing of captive target transcripts identifies the network of regulated genes and functions of primate-specific miR-522. Cell Rep 8: 1225–1239. [DOI] [PubMed] [Google Scholar]
- Tierney RJ, Kao KY, Nagra JK, Rickinson AB. 2011. Epstein-Barr virus BamHI W repeat number limits EBNA2/EBNA-LP coexpression in newly infected B cells and the efficiency of B-cell transformation: a rationale for the multiple W repeats in wild-type virus strains. J Virol 85: 12362–12375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toczyski DP, Steitz JA. 1991. EAP, a highly conserved cellular protein associated with Epstein-Barr virus small RNAs (EBERs). EMBO J 10: 459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toczyski DP, Matera AG, Ward DC, Steitz JA. 1994. The Epstein-Barr virus (EBV) small RNA EBER1 binds and relocalizes ribosomal protein L22 in EBV-infected human B lymphocytes. Proc Natl Acad Sci 91: 3463–3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai MC, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. 2010. Long noncoding RNA as modular scaffold of histone modification complexes. Science 329: 689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tycowski KT, Shu MD, Borah S, Shi M, Steitz JA. 2012. Conservation of a triple-helix-forming RNA stability element in noncoding and genomic RNAs of diverse viruses. Cell Rep 2: 26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. 2008. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 454: 780–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach JL, Yen HL, Poon LL, Cullen BR. 2010. Influenza A virus expresses high levels of an unusual class of small viral leader RNAs in infected cells. mBio 1: e00204–e00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vereide DT, Seto E, Chiu YF, Hayes M, Tagawa T, Grundhoff A, Hammerschmidt W, Sugden B. 2014. Epstein-Barr virus maintains lymphomas via its miRNAs. Oncogene 33: 1258–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassarman DA, Lee SI, Steitz JA. 1989. Nucleotide sequence of HSUR 5 RNA from Herpesvirus saimiri. Nucleic Acids Res 17: 1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinmann R, Brendler TG, Raskas HJ, Roeder RG. 1976. Low molecular weight viral RNAs transcribed by RNA polymerase III during adenovirus 2 infection. Cell 7: 557–566. [DOI] [PubMed] [Google Scholar]
- Whisnant AW, Kehl T, Bao Q, Materniak M, Kuzmak J, Lochelt M, Cullen BR. 2014. Identification of novel, highly expressed retroviral microRNAs in cells infected by bovine foamy virus. J Virol 88: 4679–4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White EA, Spector DH. 2007. Early viral gene expression and function. In Human herpesviruses: biology, therapy, and immunoprophylaxis (ed. Arvin A, et al. ), pp. 264–294 Cambridge University Press, Cambridge, UK. [PubMed] [Google Scholar]
- Wilson JL, Vachon VK, Sunita S, Schwartz SL, Conn GL. 2014. Dissection of the adenoviral VA RNAI central domain structure reveals minimum requirements for RNA-mediated inhibition of PKR. J Biol Chem 289: 23233–23245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilusz JE, JnBaptiste CK, Lu LY, Kuhn CD, Joshua-Tor L, Sharp PA. 2012. A triple helix stabilizes the 3′ ends of long noncoding RNAs that lack poly(A) tails. Genes Dev 26: 2392–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolin SL, Cedervall T. 2002. The La protein. Annu Rev Biochem 71: 375–403. [DOI] [PubMed] [Google Scholar]
- Wu TT, Su YH, Block TM, Taylor JM. 1996. Evidence that two latency-associated transcripts of herpes simplex virus type 1 are nonlinear. J Virol 70: 5962–5967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Steitz JA. 2014. Versatile microRNA biogenesis in animals and their viruses. RNA Biol 11: 673–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie M, Li M, Vilborg A, Lee N, Shu MD, Yartseva V, Sestan N, Steitz JA. 2013. Mammalian 5′-capped microRNA precursors that generate a single microRNA. Cell 155: 1568–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima M, Kanda T, Takada K. 2005. Critical role of Epstein-Barr virus (EBV)-encoded RNA in efficient EBV-induced B-lymphocyte growth transformation. J Virol 79: 4298–4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Aozasa K, Oshimi K, Takada K. 2004. Epstein-Barr virus (EBV)-encoded RNA promotes growth of EBV-infected T cells through interleukin-9 induction. Cancer Res 64: 5332–5337. [DOI] [PubMed] [Google Scholar]
- Yao Y, Smith LP, Nair V, Watson M. 2014. An avian retrovirus uses canonical expression and processing mechanisms to generate viral microRNA. J Virol 88: 2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Harris SL, Levine AJ. 2006. The regulation of exosome secretion: a novel function of the p53 protein. Cancer Res 66: 4795–4801. [DOI] [PubMed] [Google Scholar]
- Zabolotny JM, Krummenacher C, Fraser NW. 1997. The herpes simplex virus type 1 2.0-kilobase latency-associated transcript is a stable intron which branches at a guanosine. J Virol 71: 4199–4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Sroller V, Zanwar P, Chen CJ, Halvorson SJ, Ajami NJ, Hecksel CW, Swain JL, Wong C, Sullivan CS, et al. 2014. Viral microRNA effects on pathogenesis of polyomavirus SV40 infections in syrian golden hamsters. PLoS Pathog 10: e1003912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Xu H, Yao Y, Smith LP, Kgosana L, Green J, Petherbridge L, Baigent SJ, Nair V. 2011. Critical role of the virus-encoded microRNA-155 ortholog in the induction of Marek's disease lymphomas. PLoS Pathog 7: e1001305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong W, Ganem D. 1997. Characterization of ribonucleoprotein complexes containing an abundant polyadenylated nuclear RNA encoded by Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8). J Virol 71: 1207–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegelbauer JM, Sullivan CS, Ganem D. 2009. Tandem array-based expression screens identify host mRNA targets of virus-encoded microRNAs. Nat Genet 41: 130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]