

Kapoor et al. found that the SWI/SNF chromatin remodeling complex is capable of regulating the activity of S. cerevisiae checkpoint kinase Mec1. SWI/SNF can activate Mec1 kinase activity in the absence of chromatin or known activators such as Dbp11. The subunit requirement of SWI/SNF-mediated Mec1 regulation differs from that of SWI/SNF-mediated chromatin remodeling. These findings suggest that ATP-dependent chromatin remodeling complexes can regulate non-chromatin substrates such as a checkpoint kinase.
Keywords: chromatin remodeling, checkpoint regulation, Mec1, ATR, SWI/SNF
Abstract
ATP-dependent chromatin remodeling complexes alter chromatin structure through interactions with chromatin substrates such as DNA, histones, and nucleosomes. However, whether chromatin remodeling complexes have the ability to regulate nonchromatin substrates remains unclear. Saccharomyces cerevisiae checkpoint kinase Mec1 (ATR in mammals) is an essential master regulator of genomic integrity. Here we found that the SWI/SNF chromatin remodeling complex is capable of regulating Mec1 kinase activity. In vivo, Mec1 activity is reduced by the deletion of Snf2, the core ATPase subunit of the SWI/SNF complex. SWI/SNF interacts with Mec1, and cross-linking studies revealed that the Snf2 ATPase is the main interaction partner for Mec1. In vitro, SWI/SNF can activate Mec1 kinase activity in the absence of chromatin or known activators such as Dpb11. The subunit requirement of SWI/SNF-mediated Mec1 regulation differs from that of SWI/SNF-mediated chromatin remodeling. Functionally, SWI/SNF-mediated Mec1 regulation specifically occurs in S phase of the cell cycle. Together, these findings identify a novel regulator of Mec1 kinase activity and suggest that ATP-dependent chromatin remodeling complexes can regulate nonchromatin substrates such as a checkpoint kinase.
DNA damage occurs frequently in the cell due to endogenous and exogenous DNA-damaging agents. To deal with DNA damage, eukaryotic cells have evolved complex and specialized mechanisms, including cell cycle checkpoint activation, transcriptional responses to DNA damage, DNA repair, and apoptosis. In yeast, phosphoinositide 3-kinase (PI3K)-related protein kinases (PIKK) Mec1/Tel1 (ATR/ATM in mammals) are the master regulators in response to DNA damage (Gobbini et al. 2013). Although ATM/Tel1 is primarily activated in response to DNA double-strand breaks (DSBs), ATR/Mec1 responds to a broader range of DNA damages as well as replication stress (Sirbu and Cortez 2013). Upon DSB formation, the Mre11–Rad50–Xrs2 (MRX) complex, which contains exonuclease activity, collaborates with other factors to promote the production of ssDNA through resection (Mimitou and Symington 2008). Processive resection coincides with the dissociation of MRX from the DSB and concomitant binding of RPA to ssDNA tails (Lisby et al. 2004). The appearance of RPA-coated ssDNA with extensive resection of the DSB provides increased binding sites for Mec1 through Ddc2 (Cortez et al. 2001), which likely promotes the transition from Tel1/ATM-dependent to robust Mec1/ATR-dependent checkpoint signaling (Shiotani and Zou 2009).
ATM/ATR promotes the activation of downstream effector kinases such as Saccharomyces cerevisiae Chk1 and Rad53 (CHK1 and CHK2 in mammals), which function to target downstream components of the DNA damage response pathways as well as amplify the initial DNA damage response signal (Stracker et al. 2009). The activation of Mec1–Ddc2 (ATR–ATRIP in mammals) is regulated by a number of factors: First, Mec1 signaling is dependent on colocalization of the Mec1–Ddc2 with the PCNA-like 9-1-1 Ddc1–Rad17–Mec3 complex (RAD9–RAD1–HUS1, in mammals) (Parrilla-Castellar et al. 2004; Bonilla et al. 2008). Second, it has been reported that the yeast 9-1-1 complex can activate Mec1 directly in vitro (Majka et al. 2006). Moreover, topoisomerase-binding protein 1 (TopBP1; Dpb11 in budding yeast), which is recruited by the 9-1-1 complex, can activate ATR directly through protein–protein interactions (Kumagai et al. 2006; Mordes et al. 2008). Finally, a recent study suggested that yeast Dna2 is used specifically during S phase to stimulate Mec1 kinase (Kumar and Burgers 2013).
One of the earliest events at a DSB is the phosphorylation of S129 of H2A (γ-H2AX) by Mec1/Tel1 (Rogakou et al. 1998; Dubrana et al. 2007). Recruitment of ATP-dependent chromatin remodeling complexes to DSBs (Ray et al. 2009; Lee et al. 2010; Qi et al. 2015) and the remodeling of the surrounding chromatin occur on a timescale similar to that of H2A phosphorylation. Moreover, it has been shown that the phosphorylation of Ies4 (a subunit of the INO80 chromatin remodeling complex) by Mec1/Tel1 in response to DNA damage directs INO80 function toward checkpoint regulation (Morrison et al. 2007). The timing and close proximity of chromatin remodeling complexes with key checkpoint kinases at damaged sites raise the possibility that ATP-dependent chromatin remodeling complexes may participate in the regulation of checkpoint kinases.
In this study, using genetic and biochemical approaches, we identified SWI/SNF (a chromatin remodeling complex known to be involved in transcriptional regulation and DNA repair) as a novel regulator of Mec1 kinase activity, particularly in the S phase of the cell cycle in response to DNA damage. We found that the optimal Mec1 activation requires Snf2, the core ATPase subunit of the SWI/SNF complex. In vitro, SWI/SNF can activate Mec1 independently of chromatin or other known activators. Mechanistically, the Snf2 ATPase is the main subunit responsible for Mec1 activation, and the Snf2 subunit has multiple direct interactions with Mec1, as shown by cross-linking studies. Given that ATP-dependent chromatin remodeling complexes have been known to regulate chromatin substrates, our findings suggest that nonchromatin substrates such as Mec1 checkpoint kinase are also targeted by ATP-dependent chromatin remodeling complexes.
Results
Snf2 is required for Mec1 kinase activity in vivo
To assess whether ATP-dependent chromatin remodeling complexes are involved in regulating Mec1 or Tel1 activity, we first dissected the pathways of Mec1 and Tel1 activation genetically to determine whether additional pathways might exist. Since Mre11 is a key factor in Tel1 activation and is involved in the initial generation of ssDNA (Nakada et al. 2004), we analyzed the contributions of Mre11 to Tel1 and Mec1 activation. Our results confirmed that although Tel1 activation relies on a single pathway mediated by Mre11, there may be additional pathways of Mec1 activation independent of Mre11 (Supplemental Fig. 1). Experimentally, the Δmre11 mutant provides a background to screen for novel factors required for Mec1 activity, since both Tel1- and Mre11-dependent Mec1 activities are eliminated in this mutant (Fig. 1A).
Figure 1.
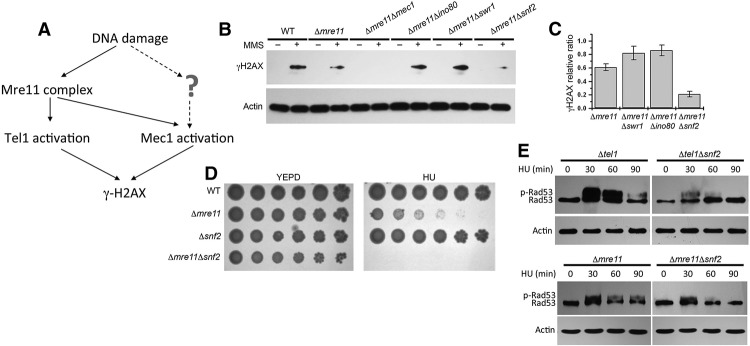
Snf2 is required for Mec1 kinase activity in vivo. (A) The absolute requirement of Mre11 for Tel1 activation in a single pathway contrasts the role of Mre11 in Mec1 activation pathways. Thus, the Δmre11 mutant provides a clean background to screen for novel factors required for Mec1 activity. (B, top panel) Western blot analysis of γ-H2AX induction after MMS treatment for 60 min in different mutants using whole-cell extracts. (Bottom panel) Actin was used as a loading control. (C) Graph showing quantitative analysis of the relative ratio of γ-H2AX in different strains as indicated. The results presented are the mean of five independent experiments ± S.D. (D) Serial dilutions (fivefold) of wild-type (WT) cells. The indicated mutants were tested for sensitivity to 50 mM HU. Plates were incubated for 2 d at 30°C and photographed. (E) Δtel1, Δtel1Δsnf2, Δmre11, and Δmre11Δsnf2 cells were treated with 200 mM HU, and the same amount of cells was collected at 30, 60, and 90 min. (Top panel) The Western blot analysis of Rad53 phosphorylation was carried out as described in the Materials and Methods. (Bottom panel) Actin was used as a loading control.
To analyze the contribution of chromatin remodeling complexes to Mec1 activity, we deleted the core ATPase subunits of different chromatin remodeling complexes in the Δmre11 background and examined the level of γ-H2AX as an indicator of Mec1 activity. Interestingly, among the remodeling complexes such as INO80, SWR1, and SWI/SNF, deletion of Ino80 or Swr1 core ATPases led to a slight increase in γ-H2AX levels. In contrast, deletion of the core ATPase Snf2 of SWI/SNF (Δsnf2Δmre11) resulted in a detectable reduction of γ-H2AX levels in response to DNA damage (Fig. 1B,C). These results were consistent with the earlier findings that mammalian SWI/SNF promotes γ-H2AX induction (Park et al. 2006). Importantly, compared with the Δsnf2 or Δmre11 single mutants, the viability of Δsnf2Δmre11 double deletion mutant strains was further reduced in the presence of HU (Fig. 1D), suggesting that SWI/SNF may contribute to Mec1 activity in addition to Mre11.
Given that one of the major functions of activated Mec1 is to activate Rad53 (mammalian Chk2) (Pellicioli and Foiani 2005), which is the effector kinase that initiates the cell cycle checkpoint, we also used the level of Rad53 activity indicated by Rad53 phosphorylation as a measure of Mec1 activation in vivo. Although robust Rad53 phosphorylation was observed in Δtel1 cells in which Mec1 remains active, the Δtel1Δsnf2 double deletion mutant was notably defective in activating Rad53 (Fig. 1E). These results further confirmed the requirement of SWI/SNF for optimal Mec1 activity during DNA damage response in vivo.
SWI/SNF interacts with Mec1 in a DNA damage-dependent manner
To investigate the mechanism of SWI/SNF in Mec1 activation upon DNA damage, we examined the physical interaction between the two complexes. We immunoprecipitated Snf2 from yeast whole-cell extracts (Fig. 2A) and consistently observed a detectable amount of Mec1 in the pull-down complex (Fig. 2A, lane 4). In contrast, there was no detectable Tel1 in Snf2 coimmunoprecipitates under similar conditions (Fig. 2A, lane 3), although Mec1 and Tel1 protein levels were similar. Moreover, the interaction between Snf2 and Mec1 was not dependent on the presence of DNA, since the interaction remained either after DNase treatment or in the presence of ethidium bromide that disrupted DNA–protein interactions (data not shown). Given that Snf2 has not been found as a single entity and is always associated with the SWI/SNF complex, these results suggest a physical interaction between the SWI/SNF complex and the Mec1 complexes (the Mec1 complex consists of Mec1 and Ddc2). The SWI/SNF complex has multiple functions in the genome, including regulation of numerous genes; it is likely that only a fraction of SWI/SNF is associated with Mec1 outside of transcriptional regulation. Similarly, since Mec1 interacts with multiple factors such as Dpb11 during replication and DNA damage response, it is likely that only a fraction of SWI/SNF and Mec1 interaction is detected by coimmunoprecipitations (co-IPs).
Figure 2.

SWI/SNF interacts with Mec1 in vivo. (A) HA-tagged Snf2 was transformed into cells containing either Flag-tagged Mec1 or Flag-tagged Tel1. Co-IP was carried out with anti-HA magnetic beads. Western blot using anti-Flag antibody shows Snf2-HA specifically coimmunoprecipitated with Mec1-Flag. (Bottom panel) Western blot using anti-HA antibody was used as a loading control. (B) Cells containing Snf2-HA and Mec1-Flag were grown to log phase and then treated with 0.05% MMS. Thirty minutes later, the same amount of treated and untreated cells was collected for co-IP assay carried out with anti-Flag magnetic beads. (Top panel) Western blot using anti-HA antibody shows coimmunoprecipitated Snf2-HA. (Middle panel) Western blot using anti-Flag antibody shows immunoprecipitated Mec1-Flag. (Bottom panel) Western blot analysis of γ-H2AX induction. (C) Graph showing the quantitative analysis of the Western blot analysis in B. The results presented are the mean of five independent experiments ± S.D. (D) Cross-linking map for Mec1, Ddc2 (Lcd1), Snf2, and Snf5. Structural domains were predicted by RaptorX. Red dots represent lysines that were identified in a cross-linked peptide pair. Green dots represent lysine residues that were not identified in a cross-linked peptide pair. Interprotein and intraprotein cross-links are shown as red and blue lines, respectively.
Next, to determine whether Mec1 also coimmunoprecipitates with Snf2 and whether the interaction between SWI/SNF and Mec1 is stimulated by DNA damage, we performed reciprocal co-IP of Mec1 in the presence of DNA-damaging agent MMS. While the level of Mec1 remained stable before and after DNA damage, we observed that the interaction between Snf2 and Mec1 was increased 30 min after MMS treatment (Fig. 2B,C). Furthermore, the level of γ-H2AX was also increased in the MMS-treated cells within 30 min (Fig. 2B,C). The basal level of Mec1 interaction with SWI/SNF is likely due to replication stress encountered during normal DNA replication. These results suggest that the SWI/SNF and Mec1 interaction increases after DNA damage. Moreover, the increased SWI/SNF and Mec1 interaction is also consistent with increased Mec1 activity in response to DNA damage.
Mec1 copurifies with SWI/SNF and directly interacts with the Snf2 ATPase subunit at multiple sites
To further understand how Mec1 interacts with the SWI/SNF complex, we used a cross-linking and mass spectrometry approach to identify protein interactions. We purified SWI/SNF from a Flag-tagged SNF2 strain followed by cross-linking using Bis[sulfosuccinimidyl] suberate (BS3). The cross-linked product was digested with trypsin, and the resulting peptides were analyzed by mass spectrometry. All of the SWI/SNF subunits were identified with high protein coverage (37%–80% coverage). Mec1 and Ddc2 (also called Lcd1), a known Mec1 interacting protein, were also identified with 32% and 37% protein coverage, respectively (Supplemental Table 2). These results further confirm that Mec1 together with Ddc2 interacts with the SWI/SNF complex. Moreover, besides detecting a large number of Mec1 and Lcd1 spectra associated with SWI/SNF (Supplemental Table 2), we did not detect any spectra for known Mec1 regulators such as Dpb11, Dna2, or the 9-1-1 complex in the mass spectrometry analysis, suggesting that the SWI/SNF complex is uniquely associated with the Mec1 complex independent of other Mec1 regulators.
Importantly, we identified 12 interprotein cross-links between Mec1, Ddc2 (Lcd1), and SWI/SNF subunits and 40 intraprotein cross-links for Mec1 and Lcd1 (Fig. 2D; Supplemental Table 3). The BS3 cross-linker has a linker arm of 11.4 Å when fully extended and can cross-link two lysine residues whose Cα atoms are up to 30 Å apart (Merkley et al. 2014). BS3 cross-links has been successfully used in many studies to map domain–domain interactions in protein complexes (Han et al. 2014; Knutson et al. 2014).
Our data allowed us to pinpoint the protein interaction sites at single amino acid levels. The interactions were mapped onto predicted structural domains within these proteins (Fig. 2D). Mec1 has a HEAT repeat and an ARM repeat domain at its N terminus and an mTOR-like kinase domain at its C terminus. Ddc2 (Lcd1) has a HEAT repeat domain. Interestingly, the identified cross-links between the Lcd1 and Mec1 HEAT repeats suggest that these two proteins interact through the HEAT repeats, revealing an important mechanism for how Ddc2 (ATRIP) can interact with Mec1 (ATR).
Importantly, several cross-links were identified between Mec1 and the Snf2 ATPase. Interestingly, Mec1 and Snf2 interactions appear to occur between several functional domains of these proteins. For example, we discovered interactions between the kinase domain of Mec1 and the SnAC domain of Snf2 that regulates the ATPase domain of Snf2 and interacts with histones (Sen et al. 2011, 2013). In fact, the cross-link between K1306 of Snf2 (SnAC domain) and K2318 of Mec1 is close to the active center of the Mec1 kinase domain, suggesting that the Snf2 SnAC domain physically interacts with the Mec1 kinase active center and could potentially regulate its activity. Moreover, several other direct interactions between the HSA, helicase (HELIC), and AT-hook domains of Snf2 and other nonkinase domains of Mec1 were identified (Fig. 2D). Curiously, most interactions were found between Snf2 and Mec1, and the only additional interactions are between Snf5 and Mec1/Lcd1. These results, together with our other biochemical results (Fig. 2A–C), support the Snf2 subunit of the SWI/SNF complex directly interacting with Mec1. The multiple interactions between Snf2 and Mec1 at key functional domains suggest that the SWI/SNF complex may be capable of regulating Mec1 kinase activity.
SWI/SNF stimulates Mec1 kinase activity in a cell-free extract
Given the involvement of SNF2 in Mec1 activation in vivo and the interactions detected by protein–protein cross-linking, we investigated whether SWI/SNF plays a direct role in Mec1 activation. Since SWI/SNF is known to be an important transcriptional regulator (Clapier and Cairns 2009), it is possible that the effect of SWI/SNF on Mec1 activation is indirect through down-regulation of the protein level of Mec1. However, in the absence of SWI/SNF, the level of Mec1 remained unaffected (Supplemental Fig. 2). To further test the contribution of SWI/SNF to Mec1 regulation, we developed a cell-free extract in yeast that allows Mec1/Tel1 activation. The levels of Mec1/Tel1 activities, as indicated by p53 phosphorylation in mutant cell extracts, reflect the Mec1/Tel1 activation defects in specific mutants such as the Δtel1Δmec1 double mutant (R561) (Supplemental Fig. 3A). In this cell-free system, defects in Mec1/Tel1 activities in the mutant extracts can be rescued by adding back the missing factors. This cell-free extract system allows us to address the contribution of specific factors in Mec1/Tel1 activation in the absence of transcription.
To determine the role of SWI/SNF in the cell-free extract system, we first analyzed the level of Mec1/Tel1 activity in the Δsnf2 mutant. Consistent with the in vivo results, the Δsnf2 mutant extract showed a reduced level of Mec1/Tel1 activity (Supplemental Fig. 3A). Given the association of SWI/SNF with Mec1, SWI/SNF purified from wild-type cells may contain contaminating Mec1 activity. The purification of the SWI/SNF complex from the Δtel1Δmec1 cells circumvents this problem, and the purified SWI/SNF is devoid of Mec1/Tel1 kinase activities (Supplemental Fig. 3B, lane 2). We incubated the Δsnf2 cell-free extract with increasing amounts of purified SWI/SNF in the presence of an exogenous substrate, GST-hChk22–107 (Supplemental Fig. 4A). Human Chk2 is the homolog of yeast Rad53 and has been used as a substrate for ATM activity (Lee and Paull 2005). The use of nonyeast substrates of Mec1/ATR kinases, such as human Chk2 and p53, helps to reveal the role of SWI/SNF in the regulation of Mec1 intrinsic kinase activity independent of specific substrates. Phosphorylation of GST-hChk22–107 was monitored by Western blot analysis using a phospho-specific antibody. Addition of purified SWI/SNF recovered the Mec1/Tel1 kinase activity in the Δsnf2 cell extracts. Given the relatively minor contribution of Tel1 to the kinase activities in the extract (data not shown), Mec1 appears to be the main target of SWI/SNF-mediated recovery. Moreover, the SWI/SNF-mediated recovery was dosage-dependent, which is consistent with the correlation between increased SWI/SNF and Mec1 interactions and increased Mec1 activity (Fig. 2B,C). Although the indirect transcriptional effects of SWI/SNF cannot be ruled out, the “add-back” experiments using cell extracts further suggest that SWI/SNF could directly contribute to Mec1 activation.
SWI/SNF activates Mec1 kinase activity in the absence of chromatin and known activators in vitro
To delineate the mechanisms of SWI/SNF-dependent Mec1 activation, we sought to develop in vitro Mec1 activation assays using defined components (Fig. 3A; Supplemental Fig. 4A). We used DNase I and RNase A to remove contaminating DNA or RNA from the purified Mec1 (Supplemental Fig. 4B,C) and other protein complexes used in the assays. The SWI/SNF complex was purified from the Δtel1Δmec1 double deletion mutant, which is devoid of potentially contaminating Mec1. In the purified Mec1 complex, we observed Mec1 and Ddc2 bands as expected but not other Mec1 activators such as Dpb11 (Supplemental Fig. 4C,D). The in vitro kinase assay system was first validated with purified Dpb11, which is a known activator of Mec1 (Fig. 3B; Navadgi-Patil and Burgers 2008). We then incubated purified Mec1 with increasing amounts of purified SWI/SNF or mock purification in the presence of GST-hChk22–107. The phosphorylation of GST-hChk22–107 by Mec1 was detected using a phospho-specific antibody. Compared with the mock purification, addition of purified SWI/SNF induced an increase in the kinase activity of Mec1 (Fig. 3C). Moreover, we used another substrate, a p53 fragment containing ATM/ATR phosphorylation sites in the kinase assay, and the results were comparable with the Chk2 substrate (Supplemental Fig. 3A). Moreover, similar to Dpb11, SWI/SNF has no effect on the kinase-dead Mec1 mutant protein (Fig. 3B; data not shown).
Figure 3.

SWI/SNF stimulates Mec1 kinase activity in vitro. (A) SDS PAGE analysis of purified SWI/SNF and the Mec1–Ddc2 complex as indicated at the top of the gel. Gel was silver-stained and photographed. (B) Western blot analysis of the final product of kinase assay as described in the Materials and Methods; 100 nM GST-hChk22–107 was used as a substrate. The kinase assay contained 5 nM Mec1 purified from strains containing chromosomally tagged Mec1 (lanes 1,3), 5 nM Mec1kd purified from strains containing tagged Mec1kd plasmid (lanes 4,5), or 5 nM Mec1 purified from strains containing tagged Mec1 plasmid (lanes 6,7). Dpb11 (5 nM) was added in lanes 2, 3, 5, and 7. (C) The kinase assay contained 5 nM Mec1, 100 nM GST-hChk22–107, SWI/SNF complex, mock purification of SWI/SNF, and buffer containing Flag peptides as indicated. Concentrations of SWI/SNF were 5 and 10 nM in lanes 3 and 4, respectively. (D) Kinase assay as in B except with 5 nM SWI/SNF, INO80, and Dpb11 in lanes 3, 5, and 7, respectively. (E) Kinase assay as in C except with 5, 10, 15, 20 nM Ipk2 in lanes 5–8, respectively.
In order to determine the specificity of SWI/SNF-dependent Mec1 activation, another chromatin remodeling complex, INO80, and an unrelated nuclear kinase, Ipk2, were purified and used in Mec1 activation experiments. We observed stronger Mec1 kinase activity in the presence of SWI/SNF than with INO80 and Ipk2 (Fig. 3D,E). Furthermore, to test whether contaminating Dpb11 would potentially affect the results, we purified SWI/SNF from a dpb11-1 mutant, which also showed the activation of Mec1 in the absence of functional Dpb11 (Supplemental Fig. 5). Taken together, these in vitro assays using defined components suggest that SWI/SNF can directly stimulate Mec1 kinase activity independently of either chromatin or Mec1 activators such as Dpb11.
SWI/SNF synergize with known Mec1 activation pathways
The activation of Mec1 is regulated by several factors, including ssDNA coated with RPA, the 9-1-1 complex (Ddc1–Rad17–Mec3), Dpb11, and Dna2 (Zou 2013). Furthermore, a number of DNA nucleases or helicases (Mre11, Exo1, and Sgs1) have been implicated in Mec1 activation because of their functions in generating ssDNA during DNA damage response (Nakada et al. 2004; Mimitou and Symington 2008; Zhu et al. 2008). We first tested whether Snf2 functions in the same pathways with these enzymes to facilitate ssDNA formation. Interestingly, we observed synergistic defects of Δsnf2 and Δexo1 mutations in γ-H2AX induction in response to DNA damage (Fig. 4A). Moreover, in the Δmre11 mutant, the Tel1 pathway that compensates for the Mec1 activation defect is eliminated, and the ssDNA-based Mec1 activation is also reduced due to the reduction of Mre11-mediated DNA resection. In the Δmre11Δsnf2 double deletion mutant, a synergistic reduction in γ-H2AX levels was also observed. These results are consistent with the additional roles of SWI/SNF outside Exo1 and Mre11 pathways.
Figure 4.
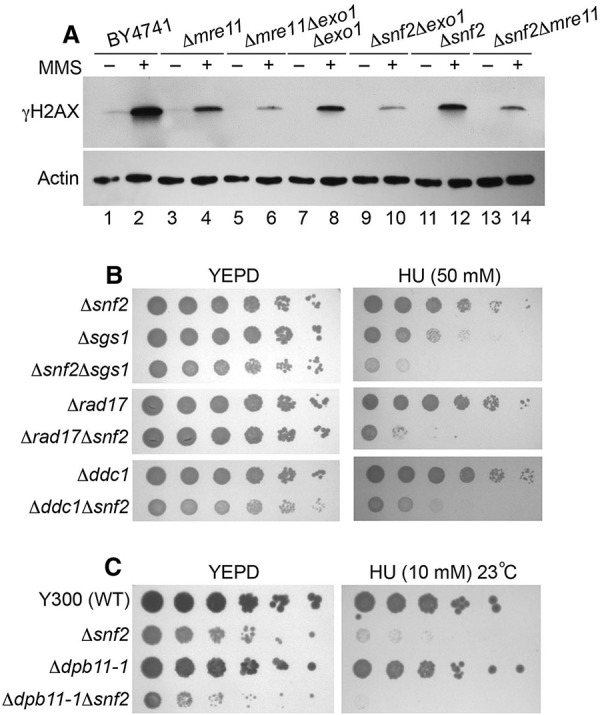
SWI/SNF genetically interact with Mec1 activation pathways. (A, top panel) Western blot analysis of γ-H2AX induction after MMS treatment in different mutants using whole-cell extracts. (Bottom panel) Actin was used as a loading control. (B) Serial dilutions (fivefold) of the indicated single and double mutants were tested for sensitivity to 50 mM HU. Plates were incubated for 2 d at 30°C and photographed. (C) Serial dilutions (fivefold) of the indicated single and double mutants were tested for sensitivity to 10 mM HU. Note that the strain background W303 in C is different from BY4741 in B, and the mutants show higher sensitivity to HU. Plates were incubated for 3 d at 23°C and photographed.
We further dissected the genetic interactions using a phenotypic test, and the Δsgs1Δsnf2 double deletion mutant was more hypersensitive to the DNA damage reagent than the single mutants (Fig. 4B). Although these results do not rule out participation of SWI/SNF in ssDNA generation, they are consistent with SWI/SNF having additional functions in Mec1 activation outside the pathways of Exo1 or Sgs1. Next, we assessed whether SWI/SNF functions in the 9-1-1 (Ddc1–Rad17–Mec3) pathway, which is critical to Mec1 activation after ssDNA generation. Both the Δrad17Δsnf2 and the Δddc1Δsnf2 double deletion mutants were more hypersensitive to the DNA damage reagent than the single mutants (Fig. 4B), suggesting that SWI/SNF has functions distinct from the 9-1-1 complex in response to DNA damage. Finally, we tested whether SWI/SNF is involved in the Mec1 activation pathway mediated by the conserved Mec1/ATR activator Dpb11 (TopBP1 in human), which activates Mec1 kinase activity through protein–protein interactions. Since DPB11 is an essential gene, a temperature-sensitive mutant dpb11-1 in W303 background (Wang and Elledge 2002) was used in our assay. Although the deletion of SNF2 reduced the viability significantly in the presence of HU, the dpb11-1Δsnf2 double mutant showed even lower viability than that of the single mutants (Fig. 4C). This result is consistent with SWI/SNF regulating Mec1 activity independently of Dpb11. Although genetic evidence is indirect, the results are consistent with an additional role of SWI/SNF in Mec1 activation (Fig. 4).
The Snf2 ATPase is required in the SWI/SNF complex for Mec1 activation
SWI/SNF is a large protein complex with 12 subunits, and many of the subunits are required for SWI/SNF chromatin remodeling activities (Cairns et al. 1998; Yang et al. 2007). To determine whether the mechanism of SWI/SNF-mediated Mec1 activation might be similar to or distinct from the SWI/SNF-mediated chromatin remodeling mechanism, we examined the subunit requirement in SWI/SNF for Mec1 activation during DNA damage response. We treated several deletion mutants of SWI/SNF subunits with MMS and detected the level of γ-H2AX induction as a measure of Mec1 activation. Deletion of Snf5, Snf6, Snf11, or Swi3 did not affect γ-H2AX induction in response to DNA damage, suggesting normal Mec1 activities in the absence of these SWI/SNF subunits. In contrast, as shown previously, deletion of the Snf2 ATPase led to a defect in γ-H2AX formation after MMS treatment (Fig. 5A,B; Supplemental Fig. 6). Therefore, except for the essential subunits (Swi1, Arp7, and Arp9) that were not tested in this experiment, Snf2 appeared to be the main subunit in SWI/SNF that was required for Mec1 activation. In contrast, the role of SWI/SNF in chromatin remodeling and transcriptional regulation depends on many of its subunits, including Snf5 and Snf6 (Peterson and Herskowitz 1992).
Figure 5.
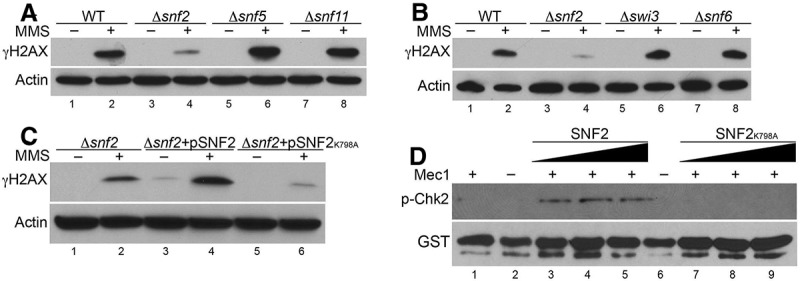
Snf2 is the essential subunit in the SWI/SNF complex for Mec1 activation. (A–C, top panels) Western blot analysis of γ-H2AX induction after MMS treatment in different mutants using whole-cell extracts. (Bottom panels) Actin was used as a loading control. (D) The kinase assay was as in Figure 2F except with 5, 10, and 20 nM SWI/SNF complex (lanes 3–5), or 5, 10, and 20 nM SNF2K798A complex (lanes 7–9).
Interestingly, although subunits such as Swi3 have been shown to organize the distinct modules of SWI/SNF, subcomplexes containing Snf2 ATPase still form in these subunit mutants (Yang et al. 2007), which may contribute to Mec1 activation. The requirement of Snf2 ATPase in Mec1 activation is consistent with the extensive direct interactions between Snf2 and Mec1 (Fig. 2D), suggesting that the Snf2 ATPase itself is the main subunit that regulates Mec1 activity. It is possible that other subunits such as Snf5 may also help in regulating the interactions of Mec1 with Snf2. The unique subunit requirements suggest a distinct mechanism of SWI/SNF in Mec1 activation, different from its conventional mechanism in chromatin remodeling and transcriptional regulation, which requires coordination of multiple subunits (Hirschhorn et al. 1992).
Moreover, it is worth noting that the deletion of SNF2 in wild-type cells also results in the reduction of γ-H2AX (Fig. 5A), suggesting that the Mec1 activation defects of the Δsnf2 mutants are not due to the sensitized mre11 or tel1 deletion backgrounds (Fig. 1). In addition, it is possible that the Mec1 activation defect in Δsnf2 cells is caused by additional mutations accumulated in the SNF2 deletion strains. We tested this possibility through a complementation assay. Introduction of the wild-type SNF2 gene into the Δsnf2 mutant fully restored Mec1 activation (Fig. 5C), indicating that the Mec1 activation defect in Δsnf2 cells is linked to the loss of Snf2. Together, these data support a role of Snf2 in the optimal activation of Mec1.
SWI/SNF-mediated Mec1 activation depends on Snf2 ATPase activity
As a member of the ATP-dependent chromatin remodeling complexes, the SWI/SNF ATPase activity is required for chromatin remodeling. We next examined the requirement of the ATPase activity of SWI/SNF for Mec1 activation. To this end, we generated a plasmid with a point mutation in Snf2 (Snf2K798A) that abolishes SWI/SNF ATPase activity (Richmond and Peterson 1996). We expressed this ATPase-dead mutant of Snf2 in a Δsnf2 background and detected the level of γ-H2AX after DNA damage. The ATPase-dead mutant of Snf2 showed no recovery of γ-H2AX levels after DNA damage in the Δsnf2 mutant. In contrast, the introduction of wild-type Snf2 in a Δsnf2 background rescued the reduction of γ-H2AX levels under similar conditions (Fig. 5C). The detectable level of γ-H2AX in the undamaged cells may be due to a slight overexpression of wild-type Snf2 (Fig. 5C). These results suggest that the ATPase activity of Snf2 is required in vivo for Mec1 activation.
To determine the effect of mutant Snf2K798A in the activation of Mec1 under in vitro conditions, we purified the ATPase-dead SNF2K798A SWI/SNF complex. The SNF2K798A SWI/SNF complex retains all subunits as wild type (data not shown). In the in vitro Mec1 activation assay, we observed a reduction in the phosphorylation of Chk2 by Mec1 in the presence of the mutant Snf2K798A SWI/SNF complex compared with the wild-type SWI/SNF complex (Fig. 5D). This result suggests that the ATPase activity of Snf2 is required for Mec1 activation in vitro. Given that Snf2 has multiple direct interactions with Mec1, especially through the SnAC domain that regulates Snf2 ATPase activity, these results suggest that the activation of Mec1 by SWI/SNF may be driven by ATP-dependent conformational changes in Snf2 and/or Mec1. As such, SWI/SNF appears to use a novel mechanism to regulate nonchromatin substrates such as the Mec1 kinase.
SWI/SNF specifically functions in S phase to promote Mec1 activity
To understand the biological role of SWI/SNF-dependent Mec1 activation, we examined the role of SWI/SNF in cell cycle checkpoints. Under nondamaging conditions, Δsnf2 cells have a normal cell cycle profile, as indicated by the budding index. We then examined the role of Snf2 in Mec1-mediated checkpoint activation in different stages of the cell cycle. Cells were arrested in G1 phase with α factor or arrested in G2/M phase with nocodazole and then treated with 4NQO (4-nitroquinoline 1-oxide), which is a UV-mimetic drug known to induce checkpoint response in G1. Phosphorylation of Rad53 was monitored as a measure of checkpoint activation by Western blot analysis. Similar to wild-type, the Δsnf2 cells showed intact checkpoint activation in both G1 and G2/M phases (Fig. 6A,B; Supplemental Fig. 7), suggesting that SWI/SNF is not required to contribute to Mec1 activation in these two phases of the cell cycle.
Figure 6.
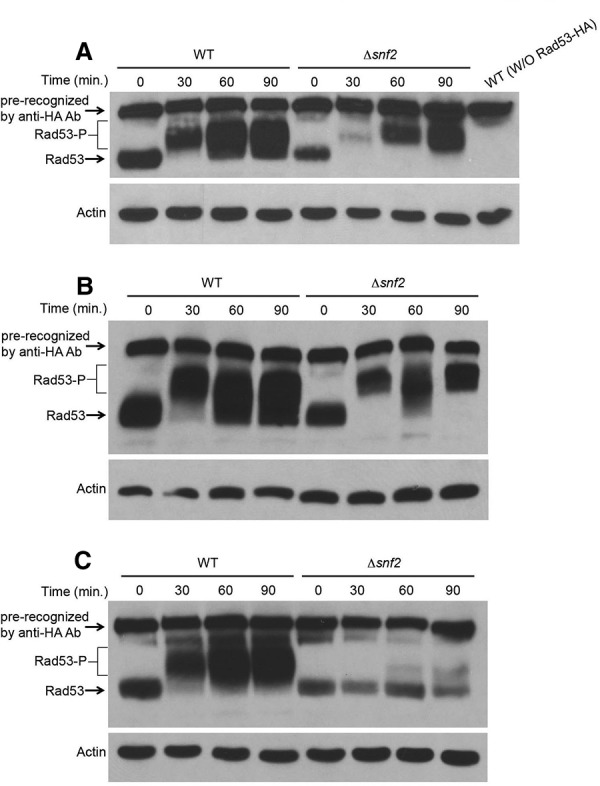
SWI/SNF specifically functions in S phase to promote Mec1 activity. (A) Western blot analysis of Rad53 phosphorylation in G1 cells. Wild-type (WT) and Δsnf2 cells were arrested in G1 phase with α factor and treated with 4NQO. (B) Western analysis of Rad53 phosphorylation in G2 cells. Wild-type and Δsnf2 cells were arrested in G2 phase with nocodazole and treated with 4NQO. (C) Western analysis of Rad53 phosphorylation in S phase. Wild-type and Δsnf2 cells were arrested in G1 and then released into medium containing 200 mM HU.
To analyze the role of SWI/SNF in checkpoint activation during S phase, we synchronized the cells in G1 phase with α factor and released cells into medium with HU, which allows cells to progress into early S phase and induces replication stress. Compared with the wild type, the Δsnf2 cells showed a slightly decreased level of Rad53 protein; however, there is a marked reduction in Rad53 activation in S phase, as indicated by Rad53 phosphorylation in the Δsnf2 mutant, even with extended time points (Fig. 6C). These results suggest that SWI/SNF-dependent Mec1 activation specifically functions in the S phase of the cell cycle to promote optimal Mec1-dependent checkpoint activity in response to replication stress. Taken together, our results suggest that SWI/SNF is part of a novel mechanism of Mec1 activation specifically in S phase of the cell cycle.
Given that Mec1 is still activated during S phase in a strain lacking both Ddc1 and a functional Dpb11 (Navadgi-Patil and Burgers 2009), it is likely that the other pathways for Mec1 activation exist in S phase. A recent study identified Dna2 and Tel1 as additional factors contributing to Mec1 activation in S phase. When these known pathways are eliminated, there is still residual S-phase Rad53 activation (Kumar and Burgers 2013), suggesting that additional factors such as SWI/SNF can contribute to Mec1 activation in S phase. The multiple pathways that regulate S-phase Mec1 activation suggest that the cell requires robust and redundant checkpoint response in S phase, and multiple mechanisms must exist to ensure optimal Mec1 activation (Zou 2013). The SWI/SNF-mediated Mec1 activation may serve to further amplify existing Mec1 activation or function as a backup mechanism for other Mec1 activation pathways. Taken together, our study provides new insights into the mechanisms of checkpoint regulation and reveals that ATP-dependent chromatin remodeling complexes can interact with nonchromatin substrates such as the Mec1 kinase.
Discussion
A novel mechanism of checkpoint regulation through ATP-dependent chromatin remodeling complexes
The functions of chromatin remodeling complexes in transcription have been well established, and recent studies have also begun to reveal links between chromatin remodeling complexes and other nuclear events such as DNA repair and checkpoint regulation (Bao and Shen 2007; Morrison and Shen 2009; van Attikum and Gasser 2009). ATP-dependent chromatin remodeling complexes such as SWI/SNF have been shown to be involved in DNA repair and are also implicated in human cancer (Chai et al. 2005; Wilson and Roberts 2011). The INO80 complex has also been shown to be involved in regulation of cell cycle checkpoints (Morrison et al. 2007). Despite these emerging links, how chromatin remodeling complexes regulate the checkpoint response mechanistically remains unknown.
Given that chromatin remodeling complexes regulate transcription, it is plausible that checkpoint regulation by chromatin remodeling complexes is indirect. However, our study provides evidence for a surprisingly direct role of the SWI/SNF chromatin remodeling complexes in the regulation of the key checkpoint kinase Mec1. Together, our results suggest a novel S-phase checkpoint activation mechanism in which the SWI/SNF complex interacts with the sensor kinase Mec1 in response to DNA damage or replication stress and helps stimulate Mec1 kinase activity together with other Mec1 activators such as Dpb11, 9-1-1, Tel1, and Dna2 (Fig. 7). These emerging studies reveal a new network of regulations in which not only do the checkpoint kinases such as Mec1 regulate ATP-dependent chromatin remodeling complexes such as INO80 (Morrison et al. 2007), but the chromatin remodeling complexes such as SWI/SNF also regulate the checkpoint kinases (Fig. 7).
Figure 7.
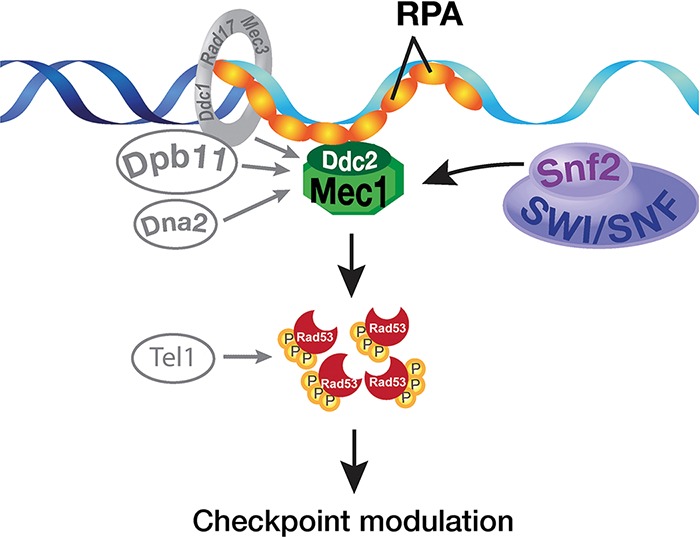
Proposed model for the regulation of Mec1 by the SWI/SNF chromatin remodeling complex in S phase. (Left) The known Mec1 activators such as the 9-1-1 complex, Dpb11, Dna2, and Tel1 are shown in S phase. (Right) SWI/SNF can regulate Mec1 kinase activity through direct interaction between Snf2 ATPase and Mec1. Together, robust S-phase activation of Mec1 is achieved.
Regulating Mec1 kinase activity through the SWI/SNF chromatin remodeling complex
The SWI/SNF complex is a highly conserved multisubunit transcriptional regulator that also plays a role in DSB repair (Chai et al. 2005). In our genetic screens, only SWI/SNF was observed to be required for the optimal activity of Mec1 (Fig. 1), suggesting a role in Mec1 activation distinct from other chromatin remodeling complexes. Although our genetic interaction studies do not rule out the participation of SWI/SNF in established Mec1 activation pathways such as ssDNA generation or Dna2, they are consistent with SWI/SNF having a unique contribution to Mec1 activity. More direct evidence came from cell extract-based and in vitro kinase assays. Using a co-IP assay, we detected that a fraction of Snf2 interacts with Mec1, and the interaction is stimulated by DNA damage (Fig. 2A–C). This interaction was further supported by the cross-linking experiments (Fig. 2D). Furthermore, the cell extract “add-back” experiment suggests a direct stimulation of Mec1 activity by SWI/SNF (Supplemental Fig. 3). Importantly, our in vitro biochemical studies showed that SWI/SNF stimulates Mec1 kinase activity in the absence of chromatin and other Mec1 activators. These biochemical studies suggest a novel mode of Mec1 activation in which SWI/SNF directly interacts with Mec1/Ddc2 to regulate its kinase activity rather than indirectly regulating Mec1 activity through transcription. We noticed a reduction in the protein level of Rad53 in the Δsnf2 mutant cells; it is unclear whether this reduction is due to transcriptional changes or protein stability (Fig. 6). Even with reduced levels of Rad53 protein, the defects of its activation, as indicated by Rad53 phosphorylation, are evident in the Δsnf2 cells.
It has been shown that the Mec1 kinase activity can be activated through direct protein–protein interaction with a bipartite Mec1 activation motif in the unstructured C-terminal tails of Dpb11 and Ddc1 (Navadgi-Patil and Burgers 2008, 2009) and also through the unstructured N-terminal region of Dna2 (Kumar and Burgers 2013). Although our results show that SWI/SNF can directly activate Mec1 independently of the known Mec1 activators such as Dpb11 (Supplemental Fig. 5), we do not yet understand how SWI/SNF can activate Mec1 at the molecular level. Our co-IP and cross-linking results suggest that SWI/SNF can directly bind to Mec1/Ddc2. It is possible that SWI/SNF can induce protein conformational changes in Mec1/Ddc2 in an ATP-dependent manner to facilitate its activation. Further studies will be needed to address the molecular mechanisms of Mec1 activation by SWI/SNF.
Functionally, SWI/SNF regulation of Mec1 activation appears to operate specifically in the S phase during checkpoint activation. Under replication stress, Mec1 is recruited to stalled replication forks. Interestingly, consistent with our model, SWI/SNF has been implicated in both replication and DNA repair (Flanagan and Peterson 1999; Chai et al. 2005). Moreover, the BRG1 catalytic subunit of mammalian SWI/SNF has been shown to colocalize with replication factors, and the loss of human SWI/SNF results in reduction of γ-H2AX levels (Cohen et al. 2010). Thus, the role of SWI/SNF in Mec1 activation may be evolutionarily conserved. Indeed, there is evidence that the human SWI/SNF complex can also interact with ATR and facilitate its activation (G Peng, pers. comm.). Although our in vivo and in vitro results are consistent with a novel mode of action by SWI/SNF, they do not exclude the potential roles of SWI/SNF in chromatin remodeling during checkpoint activation through affecting local chromatin structure. It is likely that both the chromatin-dependent and chromatin-independent function of SWI/SNF work in concert to promote optimal Mec1 activity during checkpoint activation.
Nonchromatin substrates for ATP-dependent chromatin remodeling complexes
Eukaryotic cells use two major mechanisms to regulate chromatin structure: histone post-translational modification and ATP-dependent chromatin remodeling. Although histones are major substrates for histone-modifying enzymes such as HATs and HMTs, it is widely known that histone-modifying enzymes have nonhistone substrates, such as p53 (Shi et al. 2007). In contrast, ATP-dependent chromatin remodeling complexes have only been shown to remodel nucleosomes through their ability to disrupt DNA–histone interactions. To date, there is no clear evidence that nonchromatin substrates existed for ATP-dependent chromatin remodeling complexes. Our studies of SWI/SNF-mediated Mec1 activation provide initial evidence of nonchromatin substrates for ATP-dependent chromatin remodeling complexes. Given the potential of ATP-dependent chromatin remodeling complexes to alter protein–protein interactions, it is possible that other nonchromatin substrates such as enzymes may use chromatin remodeling complexes as cofactors or chaperones. Taken together, our study opens up the possibility of additional roles of chromatin remodeling complexes outside of chromatin modification.
Our genetic and biochemical studies provide initial answers to how chromatin remodeling complexes regulate checkpoint response, and the mechanism is surprisingly direct through physical interactions of the two pathways. SWI/SNF-mediated Mec1 regulation may serve to further amplify existing Mec1 activation or function as backup mechanisms for other known checkpoint activation mechanisms. Our studies highlight the new theme that ATP-dependent chromatin remodeling complexes are emerging players in the complex network of DNA damage response (Fig. 7), forming novel interactions with the established factors. Importantly, research to date on chromatin remodeling complexes has been focused on how these complexes remodel chromatin using nucleosome as substrates; our studies provide a new platform to investigate how ATP-dependent chromatin remodeling complexes might interact with and regulate nonchromatin substrates such as checkpoint kinases.
Materials and methods
Strains and plasmids
All strains were in the S288C genetic background, except for the dpb11-1 mutant (Supplemental Table 1). Standard yeast genetic techniques were used to create gene deletions and epitope-tagged strains. Double Flag tag was inserted into the chromosome of the BY4741 strain at the C terminus of MEC1 or TEL1 as described previously (Morrison et al. 2007). The R561 (Δtel1Δmec1) strain was obtained from Elizabeth Blackburn. Constructions of the R561 strain encoding INO80 with a chromosomal double Flag tag was previously described (Morrison et al. 2007). Y300 (wild-type) and Y1185 (dpb11-1) strains were obtained from Stephen Elledge (Wang and Elledge 2002). Plasmid expressing GST-Chk22–207 was a kind gift from Tanya Paull (Lee and Paull 2005). Plasmid expressing Mec1 (DD513) and plasmid expressing Mec1kd (DD514) were kind gifts from Daniel Durocher (Sweeney et al. 2005). SNF2 was cloned under the control of its native promoter into pRS415 (LEU2) and pRS416 (URA3) vectors containing a double Flag or triple HA sequence, as previously described (Shen et al. 2003). Point mutation (K798A) was generated by site-directed mutagenesis (Stratagene).
Protein purification and analysis
Standard protein techniques such as SDS-PAGE, Western blotting, and silver staining were followed. Phospho-H2AX antibody (Abcam), Rad53 antibody (Santa Cruz Biotechnology), and Actin antibody (Milipore) were used in immunoblotting. INO80, SWI/SNF, and SWI/SNFK798A complexes were purified using the Δmec1Δtel1 strain in the BY4705 background transformed with a Flag-tagged Ino80, Snf2, or Snf2K798A plasmid. Preparation of whole-cell extracts and Flag immunoaffinity purification were described in detail previously (Shen 2004), except for the eluted complexes that were further dialyzed against H-0.1 buffer (25 mM HEPES-KOH at pH 7.6, 1 mM EDTA, 10% glycerol, 0.1 mM KCl, 2.5 mM DTT, 2 mM MgCl2, protease inhibitors). Finally, small aliquots (11 μL) of complex were flash-frozen in liquid nitrogen and stored at −80°C.
Purification of Mec1
Chromosomal MEC1 gene was tagged with Flag using the two-step gene replacement method as described previously (Morrison et al. 2007). Whole-cell extracts were prepared as 100,000g supernatants according to the online protocol from the Steve Hahn laboratory (http://labs.fhcrc.org/hahn/Methods/biochem_meth/polIII_WCE_pavel.html). Dynabeads Protein G and Sigma anti-Flag M2 were used in Flag immunoaffinity purification of the Mec1 complex. For 50 μL of Dynabeads Protein G, 2.2 μg of Sigma anti-Flag M2 and 500 μL of whole-cell extract (∼20 mg total protein) were used. The Dynabeads–M2–Flag-Mec1 complex was washed twice using 500 μL of H-0.5 (0.5 M KCl instead of 0.1 M KCl) for each wash, treated with 500 μL of H-0.5 containing DNase I and RNase A for 30 min at room temperature, washed five times using 500 μL H-0.5 for each wash, and washed three times using 500 μL of H-0.1 for each wash. Next, the Dynabeads–M2–Flag-Mec1 complex was washed once using 500 μL of kinase buffer (25 mM HEPES at pH 7.3, 400 mM NaCl, 20 mM MnCl2, 1 mM DTT, 5 μg/mL EB). Finally, the Mec1 beads was resuspended in 100 μL of kinase buffer and transferred to a tube.
Viability measurements
Cells were grown to mid-log phase in YEPD, and fivefold serial dilutions of cultures were plated onto YEPD or HU plates. Plates were incubated for 2–4 d.
Co-IP assay
Plasmid containing HA-tagged Snf2 was transformed into cells containing either Flag-tagged Mec1 or Flag-tagged Tel1. Cells were grown to log phase and then treated with or without 0.05% MMS for 30 min. The same amount of treated and untreated cells was collected and lysed by glass beads. Standard co-IP protocol was followed using Dynabeads Protein G bound with anti-HA or anti-Flag antibodies. Coimmunoprecipitated Snf2-HA or Mec1-Flag was detected by Western blot using anti-HA or anti-Flag antibodies.
Cross-linking, mass spectrometry analysis, and cross-linked peptide identification
SWI/SNF was purified from a 15-L harvest of the BY4741 Flag-tagged SNF2 strain. The protein complex was first subjected to immunoaffinity purification chromatography using anti-Flag M2 affinity gel (Sigma-Aldrich) and eluted in a buffer containing 1 mg/mL Flag peptide (United Biochemical Research), 20 mM potassium HEPES, 200 mM potassium chloride, 20% glycerol, 0.01% NP40, and 1 mM PMSF. The eluted protein complex was diluted by half and loaded onto a 1-mL SP Sepharose cation exchange column (Sigma-Aldrich) and eluted with a linear gradient from 100 mM to 1 M potassium chloride. Final protein storage conditions were 300 mM potassium chloride, 20 mM potassium HEPES (pH 7.8), 10% glycerol, 0.01% NP40, and 1 mM PMSF. Protein concentration was 0.2 mg/mL. Each series of experiments used 1.5 mL of this purified complex. BS3 (Thermo Scientific) cross-linking was done with 500 μL of purified SWI/SNF (174 fmol/μL) to a final concentration of 2 mM or 5 mM as described earlier (Han et al. 2014; Knutson et al. 2014). The reactions were quenched by addition of 20 μL of 1 M ammonium bicarbonate, and the samples were processed for trypsin digestion followed by mass spectrometry analyses. Peptides were analyzed on a Thermo Scientific Orbitrap Elite with HCD fragmentation and serial mass spectrometry events that included one FTMS1 event at 30,000 resolution followed by 10 FTMS2 events at 15,000 resolution. The RAW files were converted to mzXML files. We used the Comet search engine (Eng et al. 2013) and the Trans-Proteomic Pipline (TPP; http://tools.proteomecenter.org/wiki/index.php?title=Software:TPP) for the identification of unmodified and BS3-monomodified (monolinks) peptides. For cross-linked peptide searches, we used two different cross-link database search algorithms: pLink (Yang et al. 2012) and in-house-designed Nexus with default settings (Knutson et al. 2014).
After performing the pLink and Nexus searches, the results were combined, and each spectrum was manually evaluated for the quality of the match to each peptide using the COMET/Lorikeet spectrum viewer (TPP). Cross-linked peptides were considered confidently identified if at least four consecutive b or y ions for each peptide were observed and the majority of the observed ions were accounted for. The structural domains of Lcd1 and Mec1were predicted by RatorX (Kallberg et al. 2012).
Kinase assay
Standard Mec1 kinase assays (15 μL) contained 25 mM HEPES (pH 7.8), 125 mM NaCl, 8 mM MgCl2, 100 µg/mL BSA, 5 µg/mL ethidium bromide, 1 mM DTT, 100 μM ATP, 100 nM GST-hChk22–107 (used as substrate), and 1 μL of Mec1 beads (∼5 nM Mec1) or whole-cell extracts as indicated. Different amounts (5, 10, or 20 nM) of SWI/SNF or INO80 complex were added, and the assay was incubated for 1 h at 30°C. The reaction was terminated by addition of 5 μL of 4× SDS-PAGE loading buffer. The phosphorylated GST-Chk22–107 was detected by Western blot using phospho-Chk2 (Thr68) antibody (Cell Signaling).
Cell cycle and Western analysis of Rad53 phosphorylation
Cells with HA-tagged Rad53 were grown to OD660 = 0.25 and then arrested in G1 phase by adding 10 µg/mL α factor to the medium twice for 1 h per treatment or in G2 phase with 20 μg/mL nocodazole for 3 h and treated with 2 μg/mL 4NQO for 60–90 min at 30°C. For S-phase analysis, cells were arrested in G1 phase, washed with medium without α factor, and released into medium containing 200 mM HU for 2 h at 30°C. After exposure to the indicated DNA-damaging agents, protein extracts were prepared, and the HA.11 antibody (Covance) was used in the Western blot.
Supplementary Material
Acknowledgments
We thank Elizabeth Blackburn for the R561 strain, Stephen Elledge for the Y1185 and Y300 strains, Tanya Paull for the GST-Chk22–207 plasmid, Mark Bedford for the GST-p53 plasmid, and Laura Denton and Sarah Adai for reading the manuscript. P.K. is supported by the Odyssey post-doctoral program and the Theodore N. Law Endowment for Scientific achievements at The University of Texas M.D. Anderson Cancer Center. Funding to Y.B. is provided by an Odyssey fellowship and Epigenetic scholarship at The University of Texas M.D. Anderson Cancer Center. J.R. is supported by funds and grants from National Institutes of Health (NIH; 2P50 GM076547 and R21CA175849). B.B. is supported by a grant from the National Institute of General Medical Sciences (NIGMS; R01GM48413); X.S. is supported by funds and grants from the U.S. National Cancer Institute (K22CA100017), the American Cancer Society, the U.S. National Institute of General Medical Sciences (R01GM093104), and the Center for Cancer Epigenetics and the IRG program at M.D. Anderson Cancer Center.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.257626.114.
References
- Bao Y, Shen X. 2007. Chromatin remodeling in DNA double-strand break repair. Curr Opin Genet Dev 17: 126–131. [DOI] [PubMed] [Google Scholar]
- Bonilla CY, Melo JA, Toczyski DP. 2008. Colocalization of sensors is sufficient to activate the DNA damage checkpoint in the absence of damage. Mol Cell 30: 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns BR, Erdjument-Bromage H, Tempst P, Winston F, Kornberg RD. 1998. Two actin-related proteins are shared functional components of the chromatin-remodeling complexes RSC and SWI/SNF. Mol Cell 2: 639–651. [DOI] [PubMed] [Google Scholar]
- Chai B, Huang J, Cairns BR, Laurent BC. 2005. Distinct roles for the RSC and Swi/Snf ATP-dependent chromatin remodelers in DNA double-strand break repair. Genes Dev 19: 1656–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier CR, Cairns BR. 2009. The biology of chromatin remodeling complexes. Annu Rev Biochem 78: 273–304. [DOI] [PubMed] [Google Scholar]
- Cohen SM, Chastain PD II, Rosson GB, Groh BS, Weissman BE, Kaufman DG, Bultman SJ. 2010. BRG1 co-localizes with DNA replication factors and is required for efficient replication fork progression. Nucleic Acids Res 38: 6906–6919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez D, Guntuku S, Qin J, Elledge SJ. 2001. ATR and ATRIP: partners in checkpoint signaling. Science 294: 1713–1716. [DOI] [PubMed] [Google Scholar]
- Dubrana K, van Attikum H, Hediger F, Gasser SM. 2007. The processing of double-strand breaks and binding of single-strand-binding proteins RPA and Rad51 modulate the formation of ATR-kinase foci in yeast. J Cell Sci 120: 4209–4220. [DOI] [PubMed] [Google Scholar]
- Eng JK, Jahan TA, Hoopmann MR. 2013. Comet: an open-source MS/MS sequence database search tool. Proteomics 13: 22–24. [DOI] [PubMed] [Google Scholar]
- Flanagan JF, Peterson CL. 1999. A role for the yeast SWI/SNF complex in DNA replication. Nucleic Acids Res 27: 2022–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobbini E, Cesena D, Galbiati A, Lockhart A, Longhese MP. 2013. Interplays between ATM/Tel1 and ATR/Mec1 in sensing and signaling DNA double-strand breaks. DNA Repair 12: 791–799. [DOI] [PubMed] [Google Scholar]
- Han Y, Luo J, Ranish J, Hahn S. 2014. Architecture of the Saccharomyces cerevisiae SAGA transcription coactivator complex. EMBO J 33: 2534–2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschhorn JN, Brown SA, Clark CD, Winston F. 1992. Evidence that SNF2/SWI2 and SNF5 activate transcription in yeast by altering chromatin structure. Genes Dev 6: 2288–2298. [DOI] [PubMed] [Google Scholar]
- Kallberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J. 2012. Template-based protein structure modeling using the RaptorX web server. Nat Protoc 7: 1511–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson BA, Luo J, Ranish J, Hahn S. 2014. Architecture of the Saccharomyces cerevisiae RNA polymerase I Core Factor complex. Nat Struct Mol Biol 21: 810–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Lee J, Yoo HY, Dunphy WG. 2006. TopBP1 activates the ATR–ATRIP complex. Cell 124: 943–955. [DOI] [PubMed] [Google Scholar]
- Kumar S, Burgers PM. 2013. Lagging strand maturation factor Dna2 is a component of the replication checkpoint initiation machinery. Genes Dev 27: 313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Paull TT. 2005. ATM activation by DNA double-strand breaks through the Mre11–Rad50–Nbs1 complex. Science 308: 551–554. [DOI] [PubMed] [Google Scholar]
- Lee HS, Park JH, Kim SJ, Kwon SJ, Kwon J. 2010. A cooperative activation loop among SWI/SNF, γ-H2AX and H3 acetylation for DNA double-strand break repair. EMBO J 29: 1434–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Barlow JH, Burgess RC, Rothstein R. 2004. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 118: 699–713. [DOI] [PubMed] [Google Scholar]
- Majka J, Niedziela-Majka A, Burgers PM. 2006. The checkpoint clamp activates Mec1 kinase during initiation of the DNA damage checkpoint. Mol Cell 24: 891–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkley ED, Rysavy S, Kahraman A, Hafen RP, Daggett V, Adkins JN. 2014. Distance restraints from crosslinking mass spectrometry: mining a molecular dynamics simulation database to evaluate lysine-lysine distances. Protein Sci 23: 747–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimitou EP, Symington LS. 2008. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455: 770–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordes DA, Glick GG, Zhao R, Cortez D. 2008. TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev 22: 1478–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison AJ, Shen X. 2009. Chromatin remodelling beyond transcription: the INO80 and SWR1 complexes. Nat Rev Mol Cell Biol 10: 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison AJ, Kim JA, Person MD, Highland J, Xiao J, Wehr TS, Hensley S, Bao Y, Shen J, Collins SR, et al. 2007. Mec1/Tel1 phosphorylation of the INO80 chromatin remodeling complex influences DNA damage checkpoint responses. Cell 130: 499–511. [DOI] [PubMed] [Google Scholar]
- Nakada D, Hirano Y, Sugimoto K. 2004. Requirement of the Mre11 complex and exonuclease 1 for activation of the Mec1 signaling pathway. Mol Cell Biol 24: 10016–10025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navadgi-Patil VM, Burgers PM. 2008. Yeast DNA replication protein Dpb11 activates the Mec1/ATR checkpoint kinase. J Biol Chem 283: 35853–35859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navadgi-Patil VM, Burgers PM. 2009. The unstructured C-terminal tail of the 9-1-1 clamp subunit Ddc1 activates Mec1/ATR via two distinct mechanisms. Mol Cell 36: 743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Park EJ, Lee HS, Kim SJ, Hur SK, Imbalzano AN, Kwon J. 2006. Mammalian SWI/SNF complexes facilitate DNA double-strand break repair by promoting γ-H2AX induction. EMBO J 25: 3986–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrilla-Castellar ER, Arlander SJ, Karnitz L. 2004. Dial 9-1-1 for DNA damage: the Rad9–Hus1–Rad1 (9-1-1) clamp complex. DNA Repair (Amst) 3: 1009–1014. [DOI] [PubMed] [Google Scholar]
- Pellicioli A, Foiani M. 2005. Signal transduction: how rad53 kinase is activated. Curr Biol 15: R769–R771. [DOI] [PubMed] [Google Scholar]
- Peterson CL, Herskowitz I. 1992. Characterization of the yeast SWI1, SWI2, and SWI3 genes, which encode a global activator of transcription. Cell 68: 573–583. [DOI] [PubMed] [Google Scholar]
- Qi W, Wang R, Chen H, Wang X, Xiao T, Boldogh I, Ba X, Han L, Zeng X. 2015. BRG1 promotes the repair of DNA double-strand breaks by facilitating the replacement of RPA with RAD51. J Cell Sci 128: 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray A, Mir SN, Wani G, Zhao Q, Battu A, Zhu Q, Wang QE, Wani AA. 2009. Human SNF5/INI1, a component of the human SWI/SNF chromatin remodeling complex, promotes nucleotide excision repair by influencing ATM recruitment and downstream H2AX phosphorylation. Mol Cell Biol 29: 6206–6219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond E, Peterson CL. 1996. Functional analysis of the DNA-stimulated ATPase domain of yeast SWI2/SNF2. Nucleic Acids Res 24: 3685–3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 273: 5858–5868. [DOI] [PubMed] [Google Scholar]
- Sen P, Ghosh S, Pugh BF, Bartholomew B. 2011. A new, highly conserved domain in Swi2/Snf2 is required for SWI/SNF remodeling. Nucleic Acids Res 39: 9155–9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen P, Vivas P, Dechassa ML, Mooney AM, Poirier MG, Bartholomew B. 2013. The SnAC domain of SWI/SNF is a histone anchor required for remodeling. Mol Cell Biol 33: 360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X. 2004. Preparation and analysis of the INO80 complex. Methods Enzymol 377: 401–412. [DOI] [PubMed] [Google Scholar]
- Shen X, Xiao H, Ranallo R, Wu WH, Wu C. 2003. Modulation of ATP-dependent chromatin-remodeling complexes by inositol polyphosphates. Science 299: 112–114. [DOI] [PubMed] [Google Scholar]
- Shi X, Kachirskaia I, Yamaguchi H, West LE, Wen H, Wang EW, Dutta S, Appella E, Gozani O. 2007. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol Cell 27: 636–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiotani B, Zou L. 2009. Single-stranded DNA orchestrates an ATM-to-ATR switch at DNA breaks. Mol Cell 33: 547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirbu BM, Cortez D. 2013. DNA damage response: three levels of DNA repair regulation. Cold Spring Harb Perspect Biol 5: a012724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stracker TH, Usui T, Petrini JH. 2009. Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair 8: 1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney FD, Yang F, Chi A, Shabanowitz J, Hunt DF, Durocher D. 2005. Saccharomyces cerevisiae Rad9 acts as a Mec1 adaptor to allow Rad53 activation. Curr Biol 15: 1364–1375. [DOI] [PubMed] [Google Scholar]
- van Attikum H, Gasser SM. 2009. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol 19: 207–217. [DOI] [PubMed] [Google Scholar]
- Wang H, Elledge SJ. 2002. Genetic and physical interactions between DPB11 and DDC1 in the yeast DNA damage response pathway. Genetics 160: 1295–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BG, Roberts CW. 2011. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer 11: 481–492. [DOI] [PubMed] [Google Scholar]
- Yang X, Zaurin R, Beato M, Peterson CL. 2007. Swi3p controls SWI/SNF assembly and ATP-dependent H2A–H2B displacement. Nat Struct Mol Biol 14: 540–547. [DOI] [PubMed] [Google Scholar]
- Yang B, Wu YJ, Zhu M, Fan SB, Lin J, Zhang K, Li S, Chi H, Li YX, Chen HF, et al. 2012. Identification of cross-linked peptides from complex samples. Nat Methods 9: 904–906. [DOI] [PubMed] [Google Scholar]
- Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. 2008. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134: 981–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L. 2013. Four pillars of the S-phase checkpoint. Genes Dev 27: 227–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.