

Abstract
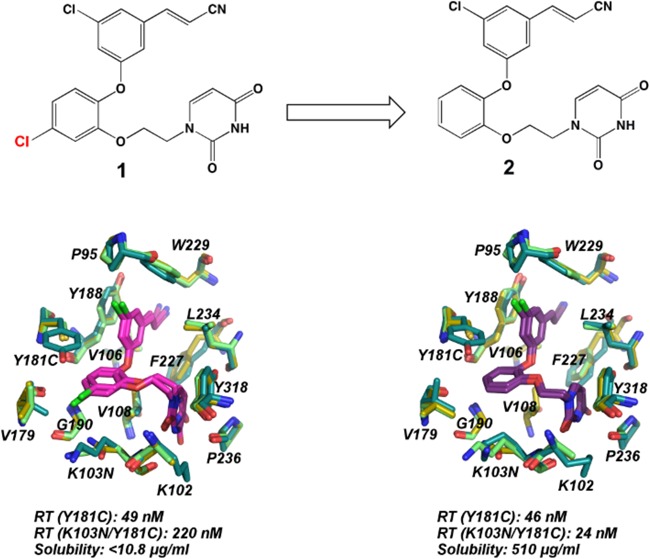
The development of novel non-nucleoside inhibitors (NNRTIs) with activity against variants of HIV reverse transcriptase (RT) is crucial for overcoming treatment failure. The NNRTIs bind in an allosteric pocket in RT ∼10 Å away from the active site. Earlier analogues of the catechol diether compound series have picomolar activity against HIV strains with wild-type RT but lose potency against variants with single Y181C and double K103N/Y181C mutations. As guided by structure-based and computational studies, removal of the 5-Cl substitution of compound 1 on the catechol aryl ring system led to a new analogue compound 2 that maintains greater potency against Y181C and K103N/Y181C variants and better solubility (510 μg/mL). Crystal structures were determined for wild-type, Y181C, and K103N/Y181C RT in complex with both compounds 1 and 2 to understand the structural basis for these findings. Comparison of the structures reveals that the Y181C mutation destabilizes the binding mode of compound 1 and disrupts the interactions with residues in the pocket. Compound 2 maintains the same conformation in wild-type and mutant structures, in addition to several interactions with the NNRTI binding pocket. Comparison of the six crystal structures will assist in the understanding of compound binding modes and future optimization of the catechol diether series.
Introduction
Non-nucleoside reverse transcriptase inhibitors (NNRTIs) are vital components of highly active antiretroviral therapy (HAART) for the treatment of HIV.1−3 Currently, there are five FDA-approved NNRTIs coadministered as a combination therapy with nucleoside reverse transcriptase inhibitors (NRTIs) or HIV protease inhibitors. Among the most successful combination therapies used for the treatment of HIV are Atripla and Complera.4,5 Although HAART has been effective in suppressing viral loads in patients,6 drug resistance continues to be a major cause of treatment failure.1,7 The predominant mechanism of resistance involves the selection of mutations in target enzymes reverse transcriptase (RT), HIV protease, and integrase. In RT, several mutations have been identified that confer resistance to both NRTI and NNRTI classes of antiretroviral drugs.7,8 Specifically, mutations conferring resistance to NNRTIs are located within the non-nucleoside binding pocket (NNBP) located ∼10 Å away from the active site. These mutations often eliminate a key interaction with the inhibitor or induce steric penalties on inhibitor binding by restricting space in the pocket.7,9
Among several variants identified in the clinic, mutations at the Y181 position are highly prevalent and exist as single variants, such as RT (Y181I), RT (Y181V), and RT (Y181C),10,11 as well as the double variant RT (K103N/Y181C).12 Although flexible diarylpyrimidine inhibitors (DAPYs) etravirine and rilpivirine maintain potency over Y181C variants, several first-generation inhibitors, such as nevirapine and efavirenz, suffer from ∼63- and 12-fold changes in potency against RT (Y181C) compared with RT (WT).12,13 Changes in potency against the RT (K103N/Y181C) variant are dramatic as well for nevirapine and efavirenz, as observed in the decrease in potency by 625- and 1176-fold, respectively.12 The rapid selection of resistance mutations necessitates the development of new, chemically diverse inhibitors that are effective against multiple-variants of RT.
Despite the challenge of developing inhibitors with activity for mutant variants of RT, efforts to design novel NNRTIs using computer-aided and structure-based drug design are promising. There are several research groups that use a multidisciplinary approach in designing new NNRTIs with better pharmacological and resistance profiles.14−17 Previously, we have reported the computational design, synthesis, antiviral activity, and wild-type crystal structures for potent analogues of wild-type RT known as the catechol diethers.18−21 Although our leading catechol diether derivative compound 1 has picomolar potency against the wild-type RT enzyme, potency is lost for the single Y181C and K103N/Y181C variants. In antiviral assays, EC50 values increase from 55 pM to 49 nM for viral strains containing RT with the single Y181C mutation and 220 nM for viral strains containing double mutation K103N/Y181C.18 This dramatic change in potency between wild-type and mutant forms of the RT enzyme warrants the investigation of RT (Y181C) and RT (K103N/Y181C) structures in complex with our leading catechol diether compound. Such structural efforts will assist in the identification of new areas for targeting in the binding pocket.
In parallel with the structural efforts, computational methods predicted that a modified analogue of the catechol diether series lacking the 5-Cl substituent on the catechol ring (compound 2) would have good solubility while retaining potency against the RT (WT) enzyme. This analogue was synthesized and evaluated for solubility and activity against HIV-1 virus containing wild-type, Y181C, and K103N/Y181C variants of RT. In addition to being highly soluble,22 the compound retains better potency against both the Y181C and K103N/Y181C variants of RT. In contrast to compound 1, the modified analogue maintains activity for wild-type, Y181C, and K103N/Y181C RT variants in the low- to midnanomolar range.
To elucidate the interactions maintained or lost with the mutant enzymes, we determined additional crystal structures of RT (WT), RT (Y181C), and RT (K103N/Y181C) in complex with compound 2 as well as crystal structures of RT (Y181C) and RT (K103N/Y181C) in complex with compound 1. In comparing the RT complexes with 1, it is apparent that the conformation of the inhibitor varies in the wild-type and mutant structures. In all three structures with compound 1, the ethoxy uracil side chain is in the syn-antigauche (sag) conformation previously discovered in crystal structures of RT in complex with various C5 analogues of the catechol diether series.20 In the mutant structures, the sag conformation seems unfavorable, causing the destabilization of the former binding mode observed in the RT (WT):1 crystal structure. Noticeably, 1 shifts in the RT (Y181C) and RT (K103N/Y181C) structures because of the elimination of a π–π stacking interaction between Tyr181 (replaced with Cys181) and the catechol aryl ring. The 5-Cl substitution on the catechol ring of 1 restricts the conformational flexibility of the ethoxy uracil side chain. In addition, the uracil ring of 1 in the RT (Y181C) and RT (K103N/Y181C) structures is slightly rotated and prevents the formation of key hydrogen bonds that were previously formed in the RT (WT):1 crystal structure. Despite the loss of a π–π stacking interaction caused by the Y181C mutation, crystal structures of RT (WT), RT (Y181C), and RT (K103N/Y181C) in complex with 2 reveal that the inhibitor maintains the same conformation. Compound 2 maintains the anti-antigauche (aag) conformation in all three complexes, allowing the maintenance of two hydrogen bonds with the uracil ring.
The hydrogen bonds maintained or lost in the mutant RT pockets correlate with the EC50 values observed for the two compounds. Interestingly, this phenomenon, in which the hydrogen bonds correlate with the EC50 values, has been observed previously in RT (WT) structures in complex with catechol diether derivatives with varying halogen substituents at the C5 position of the cyanovinyl aryl ring.20 As observed in all six structures, the protein backbone interaction with Lys/Asn103 is retained with the uracil. Encouragingly, the elimination of the 5-Cl from the catechol ring significantly improves both compound solubility and performance against Y181C and K103N/Y181C resistant variants of RT. Future derivatives retaining the undecorated catechol ring may be optimized to gain activity for additional variants of RT while maintaining high solubility.
Results
Antiviral Activity and Solubility Measurements for Compounds 1 and 2
Previously, we reported the antiviral activity for compounds 1 and 2 in MT-2 cells infected with HIV-1 virus containing RT (WT), RT (Y181C), and RT (K103N/Y181C) variants.18,19,22 As observed in the assay data for compound 1, an 890-fold decrease in activity was observed for the RT (Y181C) variant compared with RT (WT), and an even greater 4000-fold decrease in activity was observed for the RT (K103N/Y181C) variant compared with RT (WT).18 The elimination of the 5-Cl from the catechol ring led to compound 2, an analogue that maintains significantly better potency against RT (Y181C) and RT (K103N/Y181C) variants. As summarized in Table 1, compound 2 has an EC50 of 0.310 nM for RT (WT), 46 nM for RT (Y181C), and 24 nM for RT (K103N/Y181C). In addition to maintaining nanomolar–picomolar activity for three variants of RT, compound 2 has a solubility of 510 μg/mL, a considerable improvement compared with the difluoro version of compound 1 (compound 3), which has a solubility of 10.8 μg/mL (Table 1). Most strikingly, compound 2 has a better solubility profile than current FDA-approved NNRTIs nevirapine, efavirenz, and rilpivirine (Table 1). Balancing both optimal activity for RT variants and solubility, complexes with both compounds 1 and 2 were determined to understand how removal of the 5-Cl group allowed for the maintenance of potency with RT (Y181C) and RT (K103N/Y181C).
Table 1. Antiviral Activity and Solubility Data for Compounds 1-3 and FDA-Approved NNRTIs.
| compd | WT EC50 (nM) | Y181C EC50 (nM) | K103N/Y181C EC50 (nM) | solubility (μg/mL) |
|---|---|---|---|---|
| 1 | 0.055 | 49 | 220 | NA |
| 2 | 0.31 | 46 | 24 | 510 |
| 3 | 0.32 | 16 | 85 | 10.8 |
| rilpivirine | 0.67 | 0.65 | 2 | 0.02–0.24 |
| nevirapine | 110 | NA | NA | 167 |
| efavirenz | 2 | 10 | 30 | 68 |
General Structure Details for RT (WT), RT (Y181C), and RT (K103N/Y181C)
To elucidate the effects of K103N and Y181C mutations on inhibitor binding, we determined crystal structures of RT (Y181C) and RT (K103N/Y181C) in complex with compound 1 to compare with the previously determined RT (WT):1 crystal structure (PDB code: 4H4M).19 Similarly, we determined three additional crystal structures of RT (WT), RT (Y181C), and RT (K103N/Y181C) in complex with 2 to compare with the complexes of 1 (Table 2). Initially, we used cocrystallization methods and optimization conditions similar to those reported earlier for RT (WT) crystals19−21,23 to form RT (Y181C) and RT (K103N/Y181C) crystals. Diffraction data for the mutant RT cocrystals diffracted to lower resolution amplitudes (between 3.3 and 3.6 Å) and indexing revealed that the crystals adopted a different space group, P21, instead of the more common RT (WT) space group, C2. On the basis of Matthews coefficient analysis,24 we speculate that an increase in solvent content (to 66–68%) observed in the P21 crystal forms may contribute to the lower resolution data, and moreover, obtaining crystals that adopted the C2 space group as in previous studies,12,19,20,25 would reduce solvent content and yield crystals with better diffraction. To get better diffracting crystals that retained the C2 space group, we used crystal soaking and dehydration techniques to improve the RT mutant crystal complexes. Both techniques improved the resolution of the mutant structures (now ranging from 2.6 to 3.0 Å) and helped retain the C2 space group with reduced solvent content (∼50–53% solvent).
Table 2. Data Collection and Refinement Statistics for RT (Y181C):1, RT (K103N/Y181C):1, RT (WT):2, RT (Y181C):2, and RT (K103N/Y181C):2.
| complex | (Y181C):1 | (K103N/Y181C):1 | (WT):2 | (Y181C):2 | (K103N/Y181C):2 |
|---|---|---|---|---|---|
| PDB code | 4RW6 | 4RW4 | 4RW8 | 4RW9 | 4RW7 |
| resolution limit (Å) | 2.63 | 2.68 | 2.88 | 2.98 | 3.01 |
| X-ray source | NSLS X29A | NSLS X29A | NSLS X29A | NSLS X29A | NSLS X29A |
| wavelength, Å | 1.075 | 1.075 | 1.075 | 1.075 | 1.075 |
| space group | C2 | C2 | C2 | C2 | C2 |
| no. molecules in asymmetric unit | 1 | 1 | 1 | 1 | 1 |
| unit cell, a, b, c in Å (β in deg) | a = 161.3, b = 73.9, c = 107.6, β = 99.8 | a = 163.7, b = 74.1, c = 108.4, β = 100.6 | a = 224.0, b = 69.4, c = 104.4, β = 105.8 | a = 161.4, b = 74.0, c = 108.1, β = 99.7 | a = 162.0, b = 73.9, c = 108.5, β = 99.9 |
| resolution range, Å | 50.0–2.63 | 50.0–2.68 | 50.0–2.81 | 50.0–2.98 | 50.0–3.01 |
| last shell, Å | 2.68–2.63 | 2.73–2.68 | 2.86–2.81 | 3.03–2.98 | 3.06–3.01 |
| R sym (last shell) | 0.055 (0.543) | 0.086 (0.425) | 0.088 (0.527) | 0.113 (0.566) | 0.122 (0.488) |
| completeness, % (last shell, %) | 99.6 (100.0) | 98.5 (99.0) | 99.3 (92.0) | 97.1 (98.7) | 99.6 (100.0) |
| no. of reflections (unique reflections) | 139409 (37152) | 132864 (35458) | 140075 (37560) | 91067 (24980) | 93383 (25032) |
| redundancy (last shell) | 3.8 (3.7) | 3.8 (3.8) | 3.7 (3.2) | 3.7 (3.7) | 3.7 (3.8) |
| av I/σ (last shell) | 31.7 (2.1) | 35.3 (3.4) | 25.8 (1.8) | 25.0 (2.7) | 32.2 (4.1) |
| total number of atoms (protein/inhibitor/solvent/ions) | 7716, 30, 29, N/A | 7724, 30, 12, N/A | 7882, 29, 19, N/A | 7716, 29, N/A, N/A | 7715, 29, N/A, N/A |
| R free, R factor | 0.2778, 0.2338 | 0.2777, 0.2309 | 0.2820, 0.2422 | 0.2804, 0.2318 | 0.2657, 0.2289 |
| RMS deviation bond lengths (Å), angles (deg) | 0.003, 0.726 | 0.003, 0.726 | 0.003, 0.730 | 0.003, 0.711 | 0.003, 0.724 |
| av B factor (protein/inhibitor/solvent, ions) | 68.49, 55.34, 42.38, N/A | 67.74, 55.91, 42.88, N/A | 74.16, 62.94, 51.83, N/A | 56.90, 36.17, N/A, N/A | 60.89, 44.40, N/A, N/A |
| Ramachandran favored, allowed, outliers (%) [MolProbity] | 97.31, 2.69, 0 | 98.18, 1.82, 0 | 95.51, 4.49, 0 | 96.99, 3.01, 0 | 97.64, 2.36, 0 |
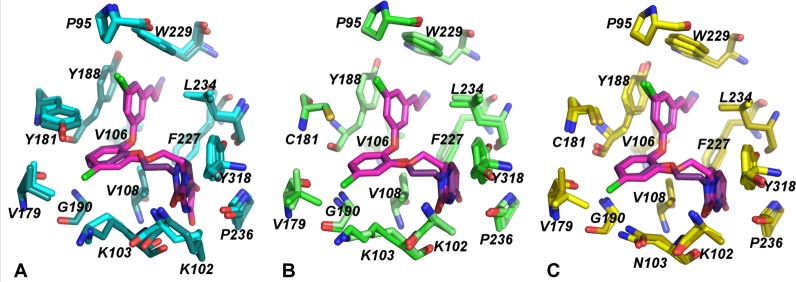
The omit electron density clearly defines the binding site residues for all five structures and compounds 1 and 2 (Figure S3). All atom alignments of the inhibitor and binding pockets for wild-type and mutant-inhibitor complexes (Figure 1) reveal the lack of global conformational changes imparted by the resistance mutations K103N and Y181C, which was also observed in earlier complexes determined for RT (K103N/Y181C):rilpivirine (PDB code: 3BGR), RT (Y181C):nevirapine (PDB code: 1JLB), RT (K103N):rilpivirine (PDB code: 3MEG), and RT(K103N):etravirine (PDB code: 3MED).11,12,15 In addition, Cα backbone traces (Figure S1) and root-mean-square deviations (rmsds) for the alignments (Table S1) reveal that the wild-type and mutant complexes are similar and adopt the usual “open” conformation observed in RT (WT) complexes (Figures 1, S1).12,19,20 Despite the dramatic change in EC50 values, both compounds 1 and 2 retain several van der Waals and hydrophobic interactions in the RT (Y181C) and RT (K103N/Y181C) binding pockets. Specifically, residues Leu100, Val106, Val108, Val179, Tyr188, Gly190, Phe227, Trp229, Leu234, Pro236, and Tyr188 interact with both compounds, despite the presence of Cys181 or Asn103 mutations (Figure 1). In all complexes, the planar aromatic ring moieties of compounds 1 and 2 arrange in a similar orientation, which seems influenced by the arrangement of hydrophobic residues in the binding pocket. Slight changes in rotamer conformation are observed in the mutant pockets most likely as a result of the changing spatial environment imparted by the K103N and Y181C mutations. Specifically, the absence of Tyr181 creates an opening in the pocket near Val179, which consequently affects the binding orientation of compound 1 (Figure 2, Figure S2). The overall surface area for this region is reduced by 41–58 Å2 (Table S2) in the RT (Y181C) and RT (K103N/Y181C) structures as compared with the RT (WT) structures, where Tyr181 is present (Table S2).
Figure 1.

Stereo view of superimposed wild-type and mutant RT complexes bound to 1 (pink) and 2 (purple). RT (WT) structures are illustrated in teal, RT (Y181C) structures are illustrated in light green, and RT (K103N/Y181C) structures are illustrated in gold. The orientation of compound 1 shifts in the various wild-type and mutant RT binding pockets; compound 2 maintains the same conformation.
Figure 2.

RT non-nucleoside binding pockets in surface representation with key residues and compounds in stick representation. (A) RT (WT):1; (B) RT (Y181C):1; (C) RT (K103N/Y181C):l; (D) RT (WT):2; (E) RT (Y181C):2; and (F) RT (K103N/Y181C):2.
Comparison of Binding Modes for Compounds 1 and 2
Previously, we identified variations in the ethoxy uracil conformation in various structures of RT (WT) in complex with several analogues of the catechol diether series, which seem influenced by the C5 substitution on the cyanovinyl aryl ring.20 Crystal structures from the analysis revealed two unique conformations of the ethoxy uracil side chain characterized by unique torsion angles: syn-anti-gauche (sag) and anti-anti-gauche (aag) conformations.20 Alignments for compounds 1 and 2 in the RT (WT), RT (Y181C), and RT (K103N/Y181C) structures show the differences between the ethoxy uracil side chain for the sag and aag conformations (Figure 3). Although the sag conformation seems favorable in the RT (WT):1 structure, as observed by the multiple hydrogen bonds formed between the uracil ring and Lys102, Lys103, and Pro236 (Figure 4), it seems that this conformation is unfavorable in the RT (Y181C):1 and RT (K103N/Y181C):1 structure complexes. In comparing the three binding modes of 1 in the structures, in which the ethoxy uracil is in the sag conformation, the compound seems to shift in the RT (Y181C) and RT (K103N/Y181C) complexes. An all atom superposition of the three binding pockets in complex with 1 (Figure 1A) reveals slight differences in binding conformations in which the compound shifts toward the pocket opening created by the Cys181 mutation in RT (Y181C):1 and RT (K103N/Y181C):1 structures (Figure 2B,C). Observed only in the mutant complexes with compound 1, the uracil ring rotates ∼40° in the RT (Y181C):1 and RT (K103N/Y181C):1 complexes (Figure 1A, Figure S2A) in which the O2 of the uracil carbonyl in RT (WT) and RT (Y181C)/RT (K103N/Y181C) structures serves as a reference point. Elimination of a π–π stacking interaction between Tyr181 and the catechol aryl may contribute to the observed shift of compound 1 in the mutant structures.
Figure 3.
Comparison of binding modes for compounds 1 and 2. (A) RT (WT) (dark teal) in complex with 1 (pink) aligned with RT (WT) (light teal) in complex with 2 (purple). (B) RT (Y181C) (light green) in complex with 1 (pink) aligned with RT (Y181C) (darker green) in complex with 2 (purple). (C) RT (K103N/Y181C) (gold) in complex with 1 (pink) aligned with RT (K103N/Y181C) (yellow) in complex with 2 (purple). Superposition of compounds 1 (sag orientation) and 2 (aag orientation) clearly shows the difference in ethoxy linker orientation.
Figure 4.

Ethoxy uracil conformation of compounds 1 and 2 in the RT (WT) [teal], RT (Y181C) [light green], and RT (K103N/Y181C) [gold] crystal structures. The sag conformation of compound 1 differs from the aag of compound 2. The sag conformation and rotation of the uracil in the RT (Y181C):1 and RT (K103N/Y181C):1 causes the loss of hydrogen bonds in the mutant structures. Compound 2 maintains the same aag conformation in all of the structures; two hydrogen bonds with residues Lys103 and Pro236 are maintained in the RT (Y181C):2 and RT (K103N/Y181C:2 structures.
The 5-Cl on the catechol ring is the only structural difference between compound 1 and compound 2. Although the difference of a single Cl atom may seem like a minor change, it has major consequences on how compound 1 adapts in the Y181C and K103N/Y181C binding pockets. Noticeably, the compound shift causes the 5-Cl on the catechol ring to move closer toward the side chains of Lys103/Asn103 and Val179 (Figure 1A, Figure S2A). To avoid steric clash with the 5-Cl, Val179 adopts a different rotamer conformation in the RT (Y181C) and RT (K103N/Y181C) structures in which the CG1 and CG2 atoms in the side chain rotate ∼180° compared with the original orientation observed in the RT (WT) structure (Figure S2A). In the RT (WT) structure, the rotamer of Val179 is stabilized by a VDW interaction with Y181, and the 5-Cl can accommodate the limited space below the phenol side chain. This Val179 interaction is lost with Cys181, and concurrently, the VDW interaction between the 5-Cl of compound 1 and the CG1 atom of Val179 (which is ∼3.3 Å) in the RT (WT) structures is lost in the RT (Y181C) and RT (K103N/Y181C) structure (distance is now 3.9 and 4.1 Å, respectively). Compound shifting observed in the mutant structures with 1 is most likely attributed to destabilization of the complex from lost interactions with Cys181 and Val179.
Despite the elimination of crucial π–π stacking between the catechol aryl and Tyr181, compound 2 maintains a similar conformation in the RT (WT), RT (Y181C), and RT (K103N/Y181C) structures (Figure 1B, 3,4). In both wild-type and mutant structures in complex with 2, there is no apparent shift in the compound, variation in the ethoxy linker, or rotation of the uracil ring. As observed in our previous work, the ethoxy uracil side chain of compound 2 adopts the aag conformation instead of the sag conformation observed in the structure complexes of 1. Unlike compound 1, absence of the 5-Cl provides more space in the pocket and reduces steric conflict with the side chain of Val179 (Figure 2). Instead, Val179 in both RT (Y181C):2 and RT (K103N/Y181C):2 structures adopts a rotamer conformation amenable for a VDW interaction with the catechol aryl. The interaction with Val179 and catechol ring identified in the mutant complexes with 2 may help stabilize the aag conformation, which seems to maintain better activity for RT (WT), RT (Y181C) and RT (K103N/Y181C) variants.
Additional differences observed among mutant complexes for 1 and 2 include the number of hydrogen bonding interactions with the uracil as guided by the compound orientation in the structures. In the RT (WT):1 crystal structure, four possible hydrogen bonds are observed between the uracil ring and residues Lys102, Lys103, and Pro236. The sag conformation of 1 is compromised by the Cys181 mutation, in which only 2 hydrogen bonds are observed in the RT (Y181C):1 structure, and only 1 hydrogen bond is observed in the RT (K103N/Y181C):1 structure (Figure 4). Although compound 2 can form only three hydrogen bonds in the RT (WT):2 structure, as compared with the possible four hydrogen bonds formed in the RT (WT):1 structure, the aag conformation allows for the maintenance of two hydrogen bonds in RT (Y181C) and RT (K103N/Y181C) structures. In comparing the RT (WT), RT (Y181C), and RT (K103N/Y181C) structures with 2, the interaction between the NH3+ side chain of K102 and O4 of the uracil carbonyl is not identified in the crystal structures (Figure 4). This interaction is not present because of disordered electron density for the side chain, and only the Cβ of the side chain is modeled in all of the mutant structures (Figure 1). The hydrogen bonding analysis extends to only the crystal structures, and these interactions with solvent-exposed residues Lys102, Lys103, and Pro236 may vary significantly in solution. However, the structures interpreted here represent low energy conformations that may assist in the understanding of which compound orientation is favorable against the RT (Y181C) and RT (K103N/Y181C) binding pockets.
The interaction between Tyr181 and the CG of Pro95 observed in the RT (WT):1 and RT (WT):2 structures is lost, with the Cys181 mutation causing a rotamer change from CG endo (WT) to CG exo as observed in the RT (Y181C) and RT (K103N/Y181C) structures. For both compounds 1 and 2, the distance between the CG atom of Pro95 and the 5-Cl of the cyanovinyl aryl ring is 3.4 Å. The opening in the pocket caused by the Cys181 mutation weakens the interaction between the 5-Cl and CG of Pro95; the distances between these atoms in the RT (Y181C):1 and RT (K103N/Y181C):1 complexes are now 3.8 and 3.9 Å, respectively. Similarly, this same interaction is farther apart, with distances of 3.9 Å for RT (Y181C):2 and 3.8 Å RT (K103N/Y181C):2. The effect of Cys181 on the rotamer conformation of Pro95 has negative consequences on the interaction between the 5-Cl on the cyanovinyl aryl ring and the CG of Pro95, an interaction that seems crucial for optimal compound placement in the binding pocket.20
Discussion and Conclusions
Mutations at the Y181 position of HIV-1 RT are known to reduce interactions with the NNRTI by eliminating key interactions or inducing steric penalties on the inhibitor binding conformation.7,9,11,26 Earlier comparisons of the RT (WT) and RT (Y181C) complexes with nevirapine revealed that the lost interaction between Y181 and the dipyrido-diazepine ring contributes to the 100-fold reduction in potency.11 For the catechol diethers, Tyr181 remains a key residue required for picomolar potency with wild-type RT, and the Cys181 mutation disrupts the parallel-displaced stacking interaction with the catechol aryl ring. Analysis of the six crystal structures reveals that the Y181C mutation indirectly affects compound binding by affecting the orientation of the ethoxy linked uracil side chain. Unlike nevirapine, the catechol diether inhibitors contain the flexible ethoxy uracil side chain that can potentially adapt and gain new interactions with residues in the region of the pocket near Lys102, Lys103, and Pro236. Although the sag conformation of 1 is optimal for RT (WT), this conformation is suboptimal for adapting to the Y181C and K103N/Y181C binding pockets by preventing hydrogen bonds with the uracil. The conformation of compound 2 remains unchanged in the wild-type and mutant structures, and the aag conformation of the ethoxy uracil may be an advantage in maintaining hydrogen bonding interactions. The 5-Cl substitution on the catechol aryl ring may influence the sag conformation of 1, and restrict the space required in the pocket for the change to the aag conformation, as in 2.
Unlike the Y181 mutation, the crystal structures reveal that the K103N mutation may have less of an effect on catechol diether binding. Previous structural studies comparing RT (WT) and RT (K103N) enzymes postulated that the K103N mutation could restrict the entry of NNRTIs and, thus, the formation of the NNBP.27 We speculate that the K103N mutation does not prevent the entry of the catechol diethers because crystallization of the RT (K103N/Y181C) complexes was possible, and both compounds 1 and 2 were present at full occupancy in the refined structures. In fact, the opening of the pocket entrance created by the Cys181 mutation near Lys/Asn103 may actually enhance compound entry.
The uracil rotation observed in the RT (Y181C):1 and RT (K103N/Y181C):1 structures seems to influence the hydrogen bonding interactions with the uracil ring. In the RT (WT):1 structure, the backbone of K103 can hydrogen-bond with the C2 carbonyl and NH of the uracil. Only the hydrogen bond between the C2 carbonyl and NH of Lys103 is observed in the RT (WT):2 structure. In both RT (K103N/Y181C):1 and RT (K103N/Y181C):2 structures, the interaction between the NH of Asn103 and the C2 carbonyl is retained, but the interaction between the backbone carbonyl of Asn103 observed in the RT (WT):1 complex is lost. This interaction is also lost in the RT (Y181C):1 structure where Lys103 is still present. Although the Asn103 and Lys103 backbone conformations are similar, it is apparent that the rotation of the uracil ring observed in the RT (Y181C):1 and RT (K103N/Y181C):1 structures prevents the formation of the second hydrogen bond with the backbone of Lys103/Asn103. For compound 2, the single backbone interaction with Lys103/Asn103 is retained in the RT (WT), RT (Y181C), and RT (K103N/Y181C) structures. Thus, it appears that the K103N mutation is not causing structural changes in the binding pocket that prevent hydrogen bonds with the uracil ring. Instead, the uracil shift induced by the structural consequences of the Cys181 mutation prevents backbone interactions with Asn103 indirectly.
The reliance on the aromatic side chain of Tyr181 is problematic for the catechol diethers, especially for compound 1, because of the destabilization of the binding complex. Interestingly, the interaction between the Lys103/Asn103 backbone NH and C2 carbonyl of the uracil is conserved in the six crystal structures, whether the ethoxy uracil side chain is in the sag or aag conformation. Previously, Ren et al. proposed the targeting of backbone or “main-chain” interactions with binding pocket residues as an effective strategy to design better inhibitors resilient to resistance mutations.11 Thus, it seems beneficial to target backbone hydrogen bond donors and acceptors to maintain interactions with residues prone to mutation, especially mutations that confer significant changes in the properties of the amino acid side chain.
Development of therapeutics targeting multiple variants of RT is a crucial strategy in overcoming treatment failure due to resistance. To that end, we have investigated the crystal of structures Y181C and K103N/Y181C RT variants in complex with novel catechol diether NNRTIs to understand the structural impact of these mutations on inhibitor binding and potency. By comparing crystal structures of wild-type and mutant RT variants in complex with catechol diether compounds 1 and 2, it is apparent that the aag conformation of 2 is optimal for adapting in both RT (Y181C) and RT (K103N/Y181C) binding pockets. Conversely, the sag conformation of 1 is less optimal and seems influenced by the steric limitations from the 5-Cl substitution on the catechol ring. On the basis of the structures, it appears that the Y181C mutation has an impact on inhibitor binding that may disrupt hydrogen bonding with the backbone of Lys103/Asn103. On the basis of knowledge gained from the comparison of the wild-type and mutant complexes of 1 and 2, further optimization of the compound series will focus on targeting backbone interactions and eliminating the 5-Cl from the catechol ring because both solubility and potency are improved against the RT mutants.
Experimental Section
Synthesis of Compounds
Synthesis of compounds 1 and 2 have been reported previously.18,22 All compounds have >95% purity as verified by HPLC analysis.
Cloning, Expression, and Purification of RT Constructs
The RT52A construct, referred here as RT (WT) construct, was used as the template for PCR amplification of the region encoding the p66 subunit. The PCR-amplified insert of the p66 subunit containing NdeI forward and XhoI reverse cut sites was inserted into the pCR 2.1-topo vector, and the resulting construct was used as the template for site-directed mutagenesis. The first cycle of mutagenesis was used to create the single (Y181C) mutation, and a second cycle of mutagenesis was used to create the double (K103N/Y181C) mutation. The pCR 2.1-topo vector containing the single Y181C p66 insert and K103N/Y181C p66 insert were digested with NdeI and XhoI, then ligated back into the original RT52A vector. Final clones for the RT (Y181C) and RT (K103N/Y181C) were verified by DNA sequencing. Recombinant RT (WT), RT (Y181C), and RT (K103N) enzymes were expressed and purified to homogeneity using methods described previously.19−21,23
Crystallization
Crystals of RT (WT):2 were prepared using cocrystallization methods described previously.19−21 The final optimized condition for crystal growth consisted of 18% (w/v) PEG 8000, 100 mM ammonium sulfate, 15 mM magnesium sulfate, 5 mM spermine, and 50 mM citric acid, pH 7.5. Crystals were transferred to a cryo-solution containing 27% (v/v) ethylene glycol and flash-cooled with liquid nitrogen. For the RT (Y181C):1, RT (Y181C):2, and RT (K103N/Y181C):2 complexes, crystal soaking techniques were used to obtain the C2 crystal forms. Initially, the RT (Y181C) and RT (K103N/Y181C) enzymes were cocrystalized with low concentrations of rilpivirine (1–5 μM). The mutant/rilpivirine crystals grew in 16–20% (w/v) PEG 8000, 100 mM ammonium sulfate, 15 mM magnesium sulfate, 5 mM spermine, and 50 mM citric acid pH 6.0–6.5, conditions similar to the cocrystallized complexes with RT (WT). The RT mutant/rilpivirine crystals were first transferred to a stabilizing solution containing 2% higher precipitating agent, then soaked with excess compound 1 or 2 for 16–18 h overnight. Following the overnight incubation, crystals were transferred to a cryo solution containing 27% (w/v) ethylene glycol, then flash-cooled with liquid nitrogen.
For the RT (K103N/Y181C):1 complexes, crystals were first obtained using cocrystallization then dehydrated using similar methods described previously.28,29 Crystals were first grown in an optimized condition of 16–20% (w/v) PEG 8000, 100 mM ammonium sulfate, 15 mM magnesium sulfate, 5 mM spermine, and 50 mM citric acid pH 7.0–7.5. The RT (K103N/Y181C):1 crystals were then dehydrated to reduce solvent content. Following growth, crystals were transferred to a stabilizing solution containing 2% higher precipitating agent. For dehydration, pooled crystals were transferred to solutions containing 25, 30, or 35 (w/v) % PEG 8000, at which crystals were incubated in each of the dehydration solutions for 8–12 h, at 4 °C. After the final incubation at 35% (w/v), crystals were flash-cooled with liquid nitrogen in a cryo solution containing 35% (w/v) PEG 8000 and 27% (w/v) ethylene glycol.
Diffraction data for all crystals were collected at Brookhaven NSLS on beamline X29A. High-resolution data sets for the best diffracting crystals were scaled and merged in space group C2 using HKL2000.30 Phases were determined using either Difference Fourier Methods or Molecular Replacement with Phaser31 using methods described previously. The program Coot32 was used for model building into the electron density. Maximum-likelihood restrained refinement in Phenix33 was used to refine the structure after each cycle of model building until acceptable R factors, geometry statistics (ideal rmsd for bonds and angles), and Ramachandran statistics were achieved (Table 2). PyMOL molecular viewer was used to visualize and analyze the structures.34 Iterative build omit σA-weighted 2mF0 – Fc electron density maps were generated using Phenix Autobuild.35
Acknowledgments
Gratitude is expressed to the National Institutes of Health (AI44616 and GM49551) for research support and for fellowship of K.M.F. (AI104334) and training support for D.E.P. (GM007324). We also thank Brookhaven National Synchrotron Light Source (NSLS) for beam time on X29A and assistance with data collection.
Glossary
Abbreviations Used
- HIV
human immunodeficiency virus
- RT
reverse transcriptase
- NNRTI
non-nucleoside reverse transcriptase inhibitor
- NNBP
non-nucleoside binding pocket
- WT
wild-type
- aag
anti-antigauche
- sag
syn-antigauche
- PEG
polyethylene glycol
- DAPY
diarylpyrimidines
Supporting Information Available
Additional information and figures used for the crystal structure analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
All crystal structures are deposited to the Protein Data Bank (PDB) with the following accession codes: 4RW4, 4RW6, 4RW7, 4RW8, and 4RW9.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Asahchop E. L.; Wainberg M. A.; Sloan R. D.; Tremblay C. L. Antiviral drug resistance and the need for development of new HIV-1 reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 2012, 56, 5000–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bethune M. P. Non-nucleoside reverse transcriptase inhibitors (NNRTIs), their discovery, development, and use in the treatment of HIV-1 infection: A review of the last 20 years (1989–2009). Antiviral Res. 2010, 85, 75–90. [DOI] [PubMed] [Google Scholar]
- De Clercq E. Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int. J. Antimicrob Ag 2009, 33, 307–320. [DOI] [PubMed] [Google Scholar]
- James C.; Preininger L.; Sweet M. Rilpivirine: A second-generation nonnucleoside reverse transcriptase inhibitor. Am. J. Health-Syst. Pharm. 2012, 69, 857–861. [DOI] [PubMed] [Google Scholar]
- Permpalung N.; Putcharoen O.; Avihingsanon A.; Ruxrungtham K. Treatment of HIV infection with once-daily regimens. Expert Opin. Pharmacother. 2012, 13, 2301–2317. [DOI] [PubMed] [Google Scholar]
- Engelman A.; Cherepanov P. The structural biology of HIV-1: Mechanistic and therapeutic insights. Nat. Rev. Microbiol 2012, 10, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das K.; Arnold E. HIV-1 reverse transcriptase and antiviral drug resistance. Part 2. Curr. Opin. Virol. 2013, 3, 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das K.; Arnold E. HIV-1 reverse transcriptase and antiviral drug resistance. Part 1. Curr. Opin. Virol. 2013, 3, 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson A. C. Winning the arms race by improving drug discovery against mutating targets. ACS Chem. Biol. 2012, 7, 278–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccherini-Silberstein F.; Gago F.; Santoro M.; Gori C.; Svicher V.; Rodriguez-Barrios F.; d’Arrigo R.; Ciccozzi M.; Bertoli A.; d’Arminio Monforte A.; Balzarini J.; Antinori A.; Perno C. F. High sequence conservation of human immunodeficiency virus type 1 reverse transcriptase under drug pressure despite the continuous appearance of mutations. J. Virol. 2005, 79, 10718–10729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J.; Nichols C.; Bird L.; Chamberlain P.; Weaver K.; Short S.; Stuart D. I.; Stammers D. K. Structural mechanisms of drug resistance for mutations at codons 181 and 188 in HIV-1 reverse transcriptase and the improved resilience of second generation non-nucleoside inhibitors. J. Mol. Biol. 2001, 312, 795–805. [DOI] [PubMed] [Google Scholar]
- Das K.; Bauman J. D.; Clark A. D. Jr.; Frenkel Y. V.; Lewi P. J.; Shatkin A. J.; Hughes S. H.; Arnold E. High-resolution structures of HIV-1 reverse transcriptase/TMC278 complexes: Strategic flexibility explains potency against resistance mutations. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 1466–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das K.; Clark A. D. Jr.; Lewi P. J.; Heeres J.; De Jonge M. R.; Koymans L. M.; Vinkers H. M.; Daeyaert F.; Ludovici D. W.; Kukla M. J.; De Corte B.; Kavash R. W.; Ho C. Y.; Ye H.; Lichtenstein M. A.; Andries K.; Pauwels R.; De Bethune M. P.; Boyer P. L.; Clark P.; Hughes S. H.; Janssen P. A.; Arnold E. Roles of conformational and positional adaptability in structure-based design of TMC125-R165335 (etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant HIV-1 variants. J. Med. Chem. 2004, 47, 2550–2560. [DOI] [PubMed] [Google Scholar]
- Johnson B. C.; Pauly G. T.; Rai G.; Patel D.; Bauman J. D.; Baker H. L.; Das K.; Schneider J. P.; Maloney D. J.; Arnold E.; Thomas C. J.; Hughes S. H. A comparison of the ability of rilpivirine (TMC278) and selected analogues to inhibit clinically relevant HIV-1 reverse transcriptase mutants. Retrovirology 2012, 9, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansdon E. B.; Brendza K. M.; Hung M.; Wang R.; Mukund S.; Jin D.; Birkus G.; Kutty N.; Liu X. Crystal structures of HIV-1 reverse transcriptase with etravirine (TMC125) and rilpivirine (TMC278): implications for drug design. J. Med. Chem. 2010, 53, 4295–4299. [DOI] [PubMed] [Google Scholar]
- Ren J.; Chamberlain P. P.; Stamp A.; Short S. A.; Weaver K. L.; Romines K. R.; Hazen R.; Freeman A.; Ferris R. G.; Andrews C. W.; Boone L.; Chan J. H.; Stammers D. K. Structural basis for the improved drug resistance profile of new generation benzophenone non-nucleoside HIV-1 reverse transcriptase inhibitors. J. Med. Chem. 2008, 51, 5000–5008. [DOI] [PubMed] [Google Scholar]
- Tucker T. J.; Saggar S.; Sisko J. T.; Tynebor R. M.; Williams T. M.; Felock P. J.; Flynn J. A.; Lai M. T.; Liang Y.; McGaughey G.; Liu M.; Miller M.; Moyer G.; Munshi V.; Perlow-Poehnelt R.; Prasad S.; Sanchez R.; Torrent M.; Vacca J. P.; Wan B. L.; Yan Y. The design and synthesis of diaryl ether second generation HIV-1 non-nucleoside reverse transcriptase inhibitors (NNRTIs) with enhanced potency versus key clinical mutations. Bioorg. Med. Chem. Lett. 2008, 18, 2959–2966. [DOI] [PubMed] [Google Scholar]
- Bollini M.; Domaoal R. A.; Thakur V. V.; Gallardo-Macias R.; Spasov K. A.; Anderson K. S.; Jorgensen W. L. Computationally guided optimization of a docking hit to yield catechol diethers as potent anti-HIV agents. J. Med. Chem. 2011, 54, 8582–8591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey K. M.; Bollini M.; Mislak A. C.; Cisneros J. A.; Gallardo-Macias R.; Jorgensen W. L.; Anderson K. S. Crystal structures of HIV-1 reverse transcriptase with picomolar inhibitors reveal key interactions for drug design. J. Am. Chem. Soc. 2012, 134, 19501–19503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey K. M.; Gray W. T.; Spasov K. A.; Bollini M.; Gallardo-Macias R.; Jorgensen W. L.; Anderson K. S. Structure-Based Evaluation of C5 Derivatives in the Catechol Diether Series Targeting HIV-1 Reverse Transcriptase. Chem. Biol. Drug Des 2014, 83, 541–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W. G.; Gallardo-Macias R.; Frey K. M.; Spasov K. A.; Bollini M.; Anderson K. S.; Jorgensen W. L. Picomolar inhibitors of HIV reverse transcriptase featuring bicyclic replacement of a cyanovinylphenyl group. J. Am. Chem. Soc. 2013, 135, 16705–16713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W. G.; Frey K. M.; Gallardo-Macias R.; Spasov K. A.; Bollini M.; Anderson K. S.; Jorgensen W. L. Picomolar inhibitors of HIV-1 reverse transcriptase: design and crystallography of naphthyl phenyl ethers. ACS Med. Chem. Lett. 2014, 5, 1259–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollini M.; Frey K. M.; Cisneros J. A.; Spasov K. A.; Das K.; Bauman J. D.; Arnold E.; Anderson K. S.; Jorgensen W. L. Extension into the entrance channel of HIV-1 reverse transcriptase—Crystallography and enhanced solubility. Bioorg. Med. Chem. Lett. 2013, 23, 5209–5212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews B. W. Solvent content of protein crystals. J. Mol. Biol. 1968, 33, 491–497. [DOI] [PubMed] [Google Scholar]
- Bauman J. D.; Das K.; Ho W. C.; Baweja M.; Himmel D. M.; Clark A. D. Jr.; Oren D. A.; Boyer P. L.; Hughes S. H.; Shatkin A. J.; Arnold E. Crystal engineering of HIV-1 reverse transcriptase for structure-based drug design. Nucleic Acids Res. 2008, 36, 5083–5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das K.; Ding J.; Hsiou Y.; Clark A. D. Jr.; Moereels H.; Koymans L.; Andries K.; Pauwels R.; Janssen P. A.; Boyer P. L.; Clark P.; Smith R. H. Jr.; Kroeger Smith M. B.; Michejda C. J.; Hughes S. H.; Arnold E. Crystal structures of 8-Cl and 9-Cl TIBO complexed with wild-type HIV-1 RT and 8-Cl TIBO complexed with the Tyr181Cys HIV-1 RT drug-resistant mutant. J. Mol. Biol. 1996, 264, 1085–1100. [DOI] [PubMed] [Google Scholar]
- Hsiou Y.; Ding J.; Das K.; Clark A. D. Jr.; Boyer P. L.; Lewi P.; Janssen P. A.; Kleim J. P.; Rosner M.; Hughes S. H.; Arnold E. The Lys103Asn mutation of HIV-1 RT: a novel mechanism of drug resistance. J. Mol. Biol. 2001, 309, 437–445. [DOI] [PubMed] [Google Scholar]
- Esnouf R. M.; Ren J.; Garman E. F.; Somers D. O.; Ross C. K.; Jones E. Y.; Stammers D. K.; Stuart D. I. Continuous and discontinuous changes in the unit cell of HIV-1 reverse transcriptase crystals on dehydration. Acta Crystallogr., D 1998, 54, 938–953. [DOI] [PubMed] [Google Scholar]
- Heras B.; Martin J. L. Post-crystallization treatments for improving diffraction quality of protein crystals. Acta Crystallogr., D 2005, 61, 1173–1180. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Lohkamp B.; Scott W. G.; Cowtan K. Features and development of Coot. Acta Crystallogr., D 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: A comprehensive python-based system for macromolecular structure solution. Acta Crystallogr., D 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W. L. PyMOL molecular viewer: Updates and refinements. Abstr. Pap. Am. Chem. Soc. 2009, 238. [Google Scholar]
- Terwilliger T. C.; Grosse-Kunstleve R. W.; Afonine P. V.; Moriarty N. W.; Adams P. D.; Read R. J.; Zwart P. H.; Hung L. W. Iterative-build OMIT maps: Map improvement by iterative model building and refinement without model bias. Acta Crystallogr., D 2008, 64, 515–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.