

Abstract
The activity-dependent refinement of neural circuit connectivity during critical periods of brain development is essential for optimized behavioral performance. We hypothesize that this mechanism is defective in fragile X syndrome (FXS), the leading heritable cause of intellectual disability and autism spectrum disorders. Here, we use optogenetic tools in the Drosophila FXS disease model to test activity-dependent dendritogenesis in two extrinsic neurons of the mushroom body (MB) learning and memory brain center: (1) the input projection neuron (PN) innervating Kenyon cells (KCs) in the MB calyx microglomeruli and (2) the output MVP2 neuron innervated by KCs in the MB peduncle. Both input and output neuron classes exhibit distinctive activity-dependent critical period dendritic remodeling. MVP2 arbors expand in Drosophila mutants null for fragile X mental retardation 1 (dfmr1), as well as following channelrhodopsin-driven depolarization during critical period development, but are reduced by halorhodopsin-driven hyperpolarization. Optogenetic manipulation of PNs causes the opposite outcome – reduced dendritic arbors following channelrhodopsin depolarization and expanded arbors following halorhodopsin hyperpolarization during development. Importantly, activity-dependent dendritogenesis in both neuron classes absolutely requires dfmr1 during one developmental window. These results show that dfmr1 acts in a neuron type-specific activity-dependent manner for sculpting dendritic arbors during early-use, critical period development of learning and memory circuitry in the Drosophila brain.
Keywords: Fragile X syndrome, Critical period, Dendrite, Synapse, Channelrhodopsin, Halorhodopsin, Mushroom body, Drosophila, Fmr1
Summary: In the Drosophila brain, the fragile X mental retardation protein mediates neuron type-specific and activity-dependent changes in dendritic synaptic connectivity within the learning/memory center.
INTRODUCTION
Activity-dependent remodeling of neural connectivity is a hallmark of late critical period brain development (Doll and Broadie, 2014). Following early overproduction, synapses undergo activity-dependent pruning to achieve the mature architecture, optimizing circuit function and behavioral output (Katz and Shatz, 1996; West and Greenberg, 2011). The critical period defines a transient developmental window of heightened sensitivity to sensory input that drives changes in synaptic connectivity (Holtmaat and Svoboda, 2009). Defects in activity-dependent remodeling of neural circuitry have been linked to numerous neurological diseases, primarily manifesting as immature, structurally abnormal dendrites in patient post-mortem examinations, including schizophrenia (Penzes et al., 2011), autism spectrum disorders (Hutsler and Zhang, 2010) and fragile X syndrome (FXS) (Irwin et al., 2001). Dysmorphic dendritic arbors might indicate abnormal development or compensation for lack of proper synaptic connectivity.
FXS is typically caused by trinucleotide repeat expansion in the fragile X mental retardation 1 (FMR1) 5′UTR, causing hypermethylation and transcriptional silencing of fragile X mental retardation protein (FMRP) expression (Kelleher and Bear, 2008). FXS is characterized by a host of behavioral symptoms, prominently including cognitive impairment (Hagerman et al., 2009). The FXS structural hallmark is structurally abnormal and immature dendritic spines (Irwin et al., 2001), suggestive of improper synaptic development. Therefore, defects in critical period synaptogenesis and refinement have been hypothesized to underlie FXS (Belmonte et al., 2004; Geschwind and Levitt, 2007). Consistently, Fmr1-knockout mice exhibit dendritic abnormalities (Kooy, 2003), associated with brain region-specific defects in activity-dependent critical periods (Cruz-Martin et al., 2010). Synaptically localized FMRP is upregulated by activity, and in turn modulates activity and controls activity-dependent processes including synapse elimination (Antar et al., 2004; Gabel et al., 2004; Pfeiffer and Huber, 2007; Bureau et al., 2008; Tessier and Broadie, 2008; Pfeiffer et al., 2010).
In the Drosophila FXS disease model, Drosophila (d) FMRP is activity-regulated by sensory input, with peak expression during early post-eclosion development (Gatto and Broadie, 2008; Tessier and Broadie, 2008). In the mushroom body (MB) learning and memory center, Kenyon cells (KCs) exhibit defects in both dendritic arbors and axonal outputs in null mutants of dfmr1 (Fmr1 – FlyBase) (Pan et al., 2004, 2008). KC synaptic connectivity is activity-regulated during the critical period coinciding with peak dFMRP expression (Tessier and Broadie, 2008, 2012). However, intersecting roles of activity and dFMRP function in MB circuitry have not been explored beyond KCs. MB extrinsic neurons (ENs) synapse with KCs, providing presynaptic input and postsynaptic output (Tanaka et al., 2008). KCs receive input in the MB calyx from projection neurons (PNs), which gather olfactory information via dendritic arbors in antennal lobe (AL) glomeruli (Jefferis et al., 2001; Tanaka et al., 2012). Downstream, MVP2 neurons receive input from KC axons via dendritic arbors at the MB spur (Tanaka et al., 2008).
Optogenetics has enhanced the ability to noninvasively manipulate activity in vivo (Fenno et al., 2011), allowing pioneering work in Drosophila (Tessier and Broadie, 2008; Inada et al., 2011; Honjo et al., 2012; de Vries and Clandinin, 2013). Depolarizing channelrhodopsin (a Na+ channel activated by blue light) and hyperpolarizing halorhodopsin (a Cl− pump activated by amber light) provide bidirectional control over activity (Tye and Deisseroth, 2012). To target MB circuit neurons, we screened the FlyLight Gal4 library for input PN (13-4-4ζ-Gal4) and output MVP2 (Oli-Gal4) EN drivers (Jenett et al., 2012). Surprisingly, we find that dendritic arbors of these two neurons respond to activity in an opposing manner during critical period brain development, with bidirectional responses to channelrhodopsin and halorhodopsin. Neurons that are null for dfmr1 do not respond to activity modulation, and they retain overgrown dendrites despite both types of activity modulation, demonstrating that dFMRP is an essential link in activity-dependent dendritic remodeling. dFMRP is specifically required in the early-use critical period.
RESULTS
Two classes of mushroom body extrinsic input and output neurons
The Drosophila Gal4-UAS system allows genetic targeting of neuron subsets (Jones, 2009), but standard Gal4 drivers express in many neurons. The FlyLight library of tightly limited drivers (Jenett et al., 2012) provides much more restricted targeting, often with resolution down to single individually identifiable neurons. We screened this Gal4 collection for expression in single classes of MB extrinsic neurons (MB-ENs), and selected two lines – R12G04-Gal4 and R65G01-Gal4 (Jenett et al., 2012). R12G04-Gal4 is driven by regulatory sequence from the Olig (Oli) transcription factor gene (Jenett et al., 2012; Oyallon et al., 2012), expressing in the MB pedunculus-medial lobe and vertical lobe arborizing neuron 2 (MB-MVP2). R65G01-Gal4 is driven by regulatory sequence from the Leonardo (14-3-3ζ – FlyBase) gene (Broadie et al., 1997), with tightly restricted expression in just two projection neurons (PNs). The two MB-EN classes revealed with these two transgenic Gal4 drivers are shown in Fig. 1.
Fig. 1.

Two classes of mushroom body extrinsic input and output neurons. Drosophila brains at 1 day post eclosion show MB pedunculus-medial lobe and vertical lobe arborizing neuron 2 (MVP2) and projection neuron (PN) architecture. (A) Schematic illustrating MVP2 (left) and PN (right) classes. ML, medial lobe. (B) Oli-Gal4 driving UAS-DenMark in MVP2 dendrites (green) within the MB spur (SPU), anterior to the distal-most pedunculus (PED), with anti-fasciclin II (FasII, red) labeling Kenyon cell (KC) axons. Insets show MVP2 dendrites at higher magnification receiving an MB spur input, and an MVP2 axon innervating the MB vertical lobe (VL). D, dorsal; V, ventral. (C) 14-3-3ζ-Gal4 driving UAS-mCD8::GFP in PNs (green) deriving presynaptic input from the antennal lobe (AL) VL1 glomerulus, and innervating KCs in the MB calyx (FasII, red). Insets show FasII-labeled AL and phalloidin (F-actin)-labeled MB calyx, with PN dendritic arbors in the AL and axonal presynaptic microglomeruli in the MB calyx. Scale bars: 50 μm in B; 30 μm in C.
MVP2 soma in anterior inferiormedial protocerebrum, anterior to the medial lobe, have processes running posteriorly before bifurcating laterally (Fig. 1A,B) (Tanaka et al., 2008). The medial branch forms a contralateral projection with feedback into the MB vertical lobe (VL; α lobe). MVP2 dendrites form dense reticular arborizations at the MB spur (SPU) (Ito et al., 2014), where KC axons branch in separate lobes (Fig. 1B) (Sinakevitch et al., 2010). MVP2 dendrites are situated to receive postsynaptic input from KC axons, perhaps mediating functional correlations among specific groups (Tanaka et al., 2008). Double labeling with antibodies against Fasciclin II (FasII) to mark KC axons (Gatto et al., 2014) and the specific dendrite reporter UAS-DenMark (DM) (Nicolai et al., 2010) shows MVP2 arbors occupy a large MB spur region (Fig. 1B). The anterior dendritic core is wider in area and tapers at the posterior end to a smaller cross-sectional area, effectually mirroring the morphology of the MB spur and lower peduncle.
PNs provide presynaptic input to KCs (Li et al., 2013). The tiny subset of R65G01-Gal4 PNs have reticular dendritic arbors in the AL, deriving input from the VL1 glomerulus (Fig. 1A,C). This innervation pattern is clearly defined with FasII co-labeling and measurement of the relative depth of the VL1 glomerulus (Laissue et al., 1999; Couto et al., 2005). The paired R65G01-Gal4 PN soma are located at the extreme ventral surface of the brain, with processes running medially to within ∼20 μm of the midline, before bifurcating into dorsolateral and contralateral branches (Fig. 1C). The dorsolateral branch enters the AL, forming a dense reticular dendritic arbor in the VL1 glomerulus (a subcompartment of the AL). The axon continues dorsally and posteriorly to innervate KCs within the MB calyx, with 4-5 microglomeruli (calyx synaptic units; Fig. 1C) (Butcher et al., 2012). Together, these two new transgenic drivers allow labeling and manipulation of two complementary MB-EN classes, thereby permitting investigation of KC input (PN) and output (MVP2) neurons with single-cell resolution.
Null dfmr1 mutants display expanded dendritic arbors during critical period development
Drosophila FXS model studies have focused almost exclusively on axonal processes (Pan et al., 2004; Gatto and Broadie, 2008, 2009). However, the FXS disease state has been most characterized for dendritic defects (Irwin et al., 2000), with the majority of mammalian studies focused on dendritic processes (Portera-Cailliau, 2012). We therefore concentrated on examining dendritic arbors in our two target MB-EN classes, focusing on the well-defined MB critical period 24 h immediately post-eclosion (Tessier and Broadie, 2008, 2012). Newly eclosed flies expressing UAS-DenMark (dendrite marker) and UAS-mCD8::GFP (general membrane marker) driven by our two Gal4s were compared between genetic background w1118, dfmr150M/+ heterozygote controls and homozygous dfmr150M null mutants (Zhang et al., 2001). Dissected brains were double-labeled with antibodies against FasII to mark KC processes and RFP to label the dendritic reporter (Fig. 1), and dendritic arbors were assayed by quantifying cross-sectional area, depth and volume.
In control MVP2 neurons (UAS-DenMark>Oli-Gal4), dendritic arbors are tapered – they are narrower at both anterior and posterior ends, with the maximum cross-sectional area at the center (Fig. 2A). By contrast, dfmr1-null mutants (UAS-DenMark;dfmr150M/TM6>dfmr150M,Oli-Gal4/TM6TbGFP) display a striking expansion of MVP2 dendritic arbors (Fig. 2B). The central core of null arbors is much wider than that of the wild type, with less-pronounced anterior and posterior ends. Owing to the great arbor depth, MVP2 dendrites are also presented in a serial z-stack series (dorsal to ventral) showing the full extent of reticular arborization (Fig. 2A,B, lower images). Mutant arbors occupy much more dorsoventral depth [37.8±1.7 µm (s.e.m.)] compared with those of wild-type controls (29.5±0.5 µm), a highly significant expansion (ANOVA, P<0.001; wt, n=16; null, n=16; Fig. 2A,B). Null dfmr1 mutants similarly show a striking >30% increase in total dendritic arbor volume compared with that of controls (P<0.001; Fig. 2C). On average, the mutants develop 7124±174 μm3 of dendritic volume, compared to 4764±186 μm3 in wild-type and 5274±147 μm3 in dfmr150M/+ heterozygote controls (Fig. 2C). These results show that MVP2 dendritic arbors are enlarged and overelaborated in the FXS disease-state model at day 1 post eclosion.
Fig. 2.

MB extrinsic neuron dendritic arbors expand in dfmr1-null mutants. MVP2 dendritic arbors labeled with Oli-Gal4>UAS-DenMark at day 1 post eclosion. Representative images of (A) w1118 genetic background control (wildtype) and (B) the homozygous dfmr150M/dfmr150M null mutant (dfmr1), showing dorsoventral dendritic arbor expansion in the absence of FMRP. Bottom panels show a dorsoventral optical section series illustrating every third frame of z-stack images. D, dorsal; V, ventral; A, anterior; P, posterior. Scale bars: 10 μm (top), 20 μm (z-series). (C) Quantification of dendritic arbor volume comparing w1118 genetic background control (wildtype), heterozygous control (dfmr150M/+) and homozygous null mutant (dfmr150M/dfmr150M). Data are presented as box and whisker plots showing the minimum, 25th percentile, median, 75th percentile and maximum values. N values are as follows: wildtype, 16; dfmr1/+, 13; null, 16. ***P<0.001 (ANOVA).
PN dendritic arbors are likewise overgrown in dfmr1-null mutants (supplementary material Fig. S1). In control neurons, the densest arbor region lies within the antennal lobe VL1 glomerulus, with reticular arborization demarcated by mCD8::GFP and FasII co-labeling defining glomeruli boundaries (Fig. 1B). Null dfmr1 PN dendritic arbors show a highly significant 23±2% expansion in total dendritic volume compared with that of controls (ANOVA, P<0.001; wt, n=12; null, n=14; supplementary material Fig. S1A,B). These results reveal that dendritic arbors are significantly expanded in both MB-EN classes in dfmr1 mutants at day 1 post eclosion. Importantly, at earlier developmental times up to eclosion and prior to use, dfmr1-null PN and MVP2 dendritic arbors are not significantly different from those of wild type. At pupal day 4 (P4), PN dendritic arbor volumes are 2704±71 µm3 and 2565±68 µm3 (n=8; not significant) and MVP2 arbor volumes are 4801±161 µm3 and 4864±199 µm3 (n=14, 12; not significant) in controls and dfmr1 nulls, respectively. Likewise at <1 h post eclosion, PN dendritic arbor volumes are 2318±48 µm3 and 2404±51 µm3 (n=12, 10; not significant), and MVP2 arbor volumes are 4990±158 µm3 and 5163±341 µm3 (n=17, 16; not significant) in controls and nulls, respectively. Thus, significant differences in dendritic arbors of both neuron classes occur only at day 1 post eclosion, indicating a restricted early-use critical period.
FMRP is required for activity-dependent changes in MVP2 dendritic arborization
Activity-dependent changes in synaptic connectivity are hypothesized to underlie the dysmorphic dendrites characterizing FXS (Doll and Broadie, 2014). We therefore tested target MB-ENs for manifestation of activity-dependent restructuring of dendritic arbors during the 24-h critical period immediately following eclosion (Tessier and Broadie, 2008, 2012). We used new transgenic optogenetic tools to manipulate activity, including a UAS-ChR2(H134R)-mCherry Na+ channel for targeted depolarization (Honjo et al., 2012) and a UAS-eNpHR3.0-EGFP Cl− pump for targeted hyperpolarization (Inada et al., 2011). To make the latter transgenic line, we cloned eNpHR3.0 halorhodopsin into a UAS expression construct, including a 3′ EGFP tag for visualization. Transgenic animals were raised on food supplemented with either ethanol vehicle (control) or all-trans retinal (ATR) co-factor (Schroll et al., 2006; Ataman et al., 2008). ATR is an essential supplement for optogenetic manipulations. Newly eclosed flies were exposed to 24 h of light stimulation (0.5-5 Hz; 20 ms pulses) and then dissected to examine dendritic architecture. Representative images and a summary of the results are shown in Figs 3 and 4 and supplementary material Figs S2-S5.
Fig. 3.

FMRP is required for activity-dependent MVP2 dendritic development. Dorsoventral series of MVP2 dendritic arbors with Oli-Gal4-driven UAS-ChR2(H134R) depolarization during day 1 post eclosion. Representative images show vehicle-fed wild-type control (A), ATR-fed wild-type experimental (B), vehicle-fed dfmr150M null (C) and ATR-fed dfmr150M null experimental (D) conditions following 24 h of 5 Hz blue light stimuli. On the right, dendritic arbors are shown in a dorsoventral series of 3 µm steps through the z-stack images. Scale bars: 10 μm. (E) Quantification of dendritic arbor volume in all four conditions. N values are as follows: wtETOH, 25; wtATR, 31; nullETOH, 32; nullATR, 25. ***P<0.001; n.s., not significant (ANOVA).
Fig. 4.

PN dendrites exhibit the opposite FMRP-dependent activity dependence. PN dendrites with 14-3-3ζ-Gal4-driven UAS-eNpHR3.0 hyperpolarization during day 1 post eclosion. Representative images show flattened 3D representations of z-stacks from anterior (left) and lateral (right) perspectives for vehicle-fed wild-type control (A), ATR-fed wild-type experimental (B), vehicle-fed dfmr150M null (C) and ATR-fed dfmr150M null experimental (D) dendritic arbors following 24 h exposure to 5 Hz amber light. Scale bar: 10 μm. (E) Quantification of dendritic arbor volume in the four conditions. N values are as follows: wtETOH, 21; wtATR, 26; nullETOH, 16; nullATR, 15. ***P<0.001; n.s., not significant (ANOVA).
There is a striking increase in wild-type MVP2 dendritic arbor volume following ChR2 depolarization during critical period development [Oli-Gal4>UAS-ChR2(H134R)-mCherry; Fig. 3A,B], reminiscent of dfmr1 phenotypes (Fig. 2). At 5 Hz stimulation, MVP2 arbors expand to nearly 20% more volume (ATR-fed, 5594±134 µm3) compared to those of controls (4763±187 µm3), a highly significant expansion (P<0.001; n=31, 25, respectively; Fig. 3E). There is a similar ∼20% increase (5282±138 µm3) with 0.5 Hz stimulation compared with control (4427±172 µm3, P<0.001; n=16, 10, respectively). To test our hypothesis that dFMRP is necessary for these activity-dependent developmental changes (Doll and Broadie, 2014), we repeated optogenetic depolarizing manipulations in dfmr1 nulls. We found that dfmr1 mutants [dfmr150M,Oli-Gal4/TM6TbGFP>UAS-ChR2(H134R);dfmr150M/TM6] are completely unable to respond to ChR2 depolarization (Fig. 3C,D). Both vehicle-fed (5316±723 μm3, P<0.001, n=32) and ATR-fed (5309±102 μm3, P<0.001, n=25) mutants develop expanded MVP2 dendritic arbors indistinguishable from those of the dfmr1 null alone (Fig. 3E).
MVP2 hyperpolarization (Oli-Gal4>UAS-eNpHR3.0-EGFP) causes the opposing consequence of dendritic arbor undergrowth in wild-type ATR-fed experimental animals compared with vehicle-fed controls (supplementary material Fig. S2A,B). Although halorhodopsin-mediated inhibition causes less dramatic changes than channelrhodopsin-mediated activation, ATR-fed animals exposed to 5 Hz amber light stimulation during the 24 h critical period clearly develop less expansive MVP2 arbors, with a nearly 15% volume decrease (3782±140 µm3, n=19) compared with those of vehicle-fed controls (4294±139 µm3, P=0.004, n=18; supplementary material Fig. S2E). As with ChR2 depolarization, there are no halorhodopsin-dependent changes in MVP2 dendritic arbors in dfmr1 nulls following amber light inhibition (supplementary material Fig. S2C-E). These results clearly show that dfmr1-null MVP2 neurons are incapable of responding to activity modulation, indicating that dFMRP is required for developmental remodeling of the dendritic arbor. We also tested whether MVP2 neurons can respond to optogenetic excitation at maturity (6-7 days post eclosion; supplementary material Fig. S3). We found no significant difference in either wild-type or dfmr1-null dendritic arbors following 24 h of either ChR2 depolarization (supplementary material Fig. S3A) or eNpHR hyperpolarization (supplementary material Fig. S3B), indicating a restricted critical period. Taken together, these results show that MVP2 dendrites display bimodal activity-dependent remodeling during critical period development, with expanding arbors during ChR2-mediated depolarization. Importantly, dFMRP is required for all activity-dependent changes in dendritic arborization.
PN arborization is inversely regulated by activity during critical period development
To test activity regulation in the PN class, we first used 14-3-3ζ-Gal4 to drive UAS-ChR2(H134R)-mCherry for targeted depolarization during the same 24 h critical period immediately post eclosion, comparing ATR-fed experimental animals to vehicle-fed controls (supplementary material Fig. S4). Using identical paradigms to the above MVP2 experiments, we were surprised to find that PN optogenetic manipulations cause the opposite dendritic arbor changes (compare Fig. 3 and supplementary material Fig. S4). ChR2-driven depolarization causes a clear and obvious decrease in PN dendritic arborization in ATR-fed experimental animals compared with vehicle-fed controls (supplementary material Fig. S4A,B). With 5 Hz stimulation during the critical period, dendritic arbor volumes decrease ∼20% (19±2.8%) in ATR-fed experimental animals compared with those of vehicle-fed controls, a highly significant change (ANOVA, P<0.001, n=16 for both; supplementary material Fig. S4E). Importantly, there are no detectable changes in PN dendritic arborization following channelrhodopsin-mediated depolarization in dfmr1-null mutants (supplementary material Fig. S4C-E).
By contrast, PN hyperpolarization with UAS-eNpHR3.0-EGFP causes dendritic arbor expansion in ATR-fed experimental animals compared with vehicle-fed controls (Fig. 4A,B). Volumetric analyses show a 15±3% increase in arbor volume in activity-inhibited ATR-fed PNs compared with that of vehicle-fed controls, a highly significant expansion (P<0.001, n=26, 21, respectively; Fig. 4E). Importantly, there is again no difference between ATR-fed and vehicle-fed dfmr1-null mutants in response to halorhodopsin-driven hyperpolarization (Fig. 4C,D), showing that activity-dependent changes are entirely dFMRP dependent (Fig. 4E). We next tested whether PNs respond to optogenetic modulation at maturity (6-7 days post eclosion) and found no significant difference in wild-type dendritic arbors following 24 h of stimulation (supplementary material Fig. S5A,B), indicating a restricted critical period. Interestingly, however, dfmr1-null PNs do respond to hyperpolarization with a significant reduction (P<0.001) in dendritic volume in ATR-fed nulls compared with that of vehicle-fed null controls (supplementary material Fig. S5B), which suggests a critical period shifted in an opposing direction compared with that of wild-type PNs. We conclude that PNs providing input to the MB circuit respond to activity in an exactly opposite manner to the MVP2 output neurons, demonstrating neuron type-specific responses to activity during critical period development, and further showing that dfmr1-null neurons are incapable of responding to activity, indicating a tight dFMRP requirement.
Conditional dfmr1 rescue defines the requirement for dfmr1 during critical period development
We hypothesized that the developmental absence of dfmr1 during the early-use post eclosion period results in defects in dendritic arborization during a short critical period window (Doll and Broadie, 2014). To test this hypothesis, we used conditional transgenic induction of targeted wild-type dfmr1 expression in otherwise dfmr1-null mutants employing a Gal80 temperature-sensitive (Gal80ts) approach (McGuire et al., 2003; Tutor et al., 2014). For the MVP2 neuron class, a tubP-Gal80ts/Cyo;dfmr150M,Oli-Gal4/TM6 line was crossed to a UAS-DM/CyO;dfmr150M,UAS-9557-3/TM6 line (a wild-type dfmr1 transgene under UAS control; Gatto and Broadie, 2009), with progeny raised at the permissive temperature (18°C) until the last pupal day of development (pupal day 4, P4). At P4, animals were shifted to the restrictive temperature (29°C) until day 1 post eclosion, thereby releasing Gal80 inhibition and activating dfmr1 expression in targeted neurons. A control lacking dfmr1 (dfmr150M,Oli-Gal4/TM6TbGFP>UAS-DM/CyO,dfmr150M/TM6) was similarly shifted to 29°C at P4 to control for the temperature shift. A summary of these data for both neuron classes is shown in Figs 5 (MVP2) and 6 (PN).
Fig. 5.
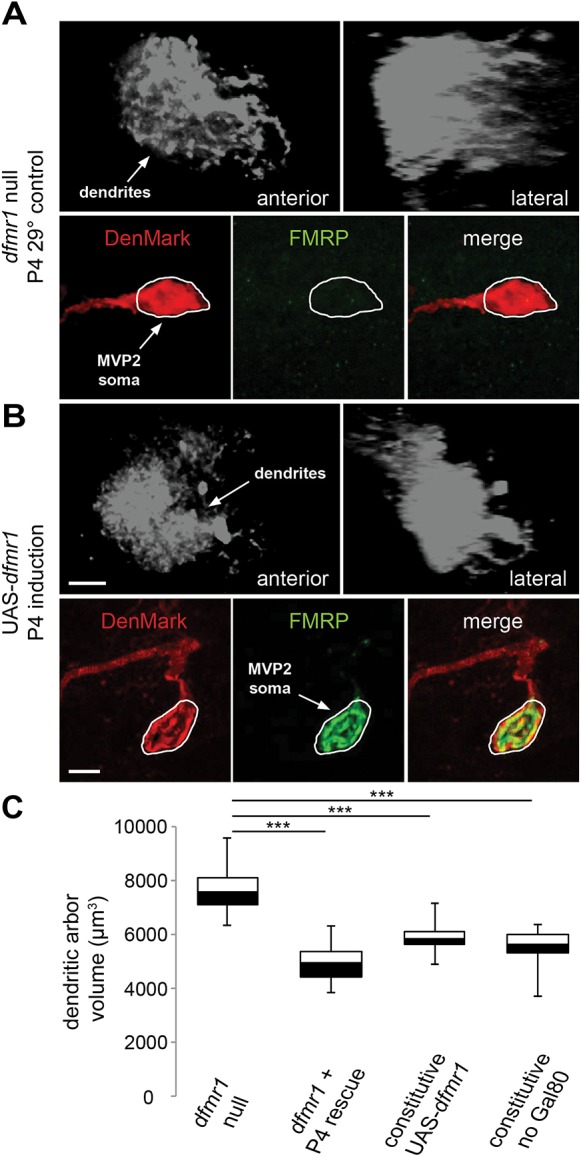
Null MVP2 dendritic arbors rescued with conditional dfmr1 expression. Targeted critical period expression of wild-type dfmr1 in MVP2 neurons in an otherwise dfmr1-null background. Compared with null mutant (dfmr150M,Oli-Gal4/TM6TbGFP>UAS-DM;dfmr150M/TM6) (A), conditional dfmr1 P4 induction (tubP-Gal80ts/CyO;dfmr150M,Oli-Gal4/TM6>UAS-DM/CyO;dfmr150M,UAS-9557-3/TM6) (B) by temperature shift to 29°C causes a reduction in MVP2 dendritic arbor volume. Gray images (top) show z-stack projections from anterior (left) and lateral (right) perspectives. Color images (bottom) show the absence (A) and presence (B) of FMRP (green) in MVP2 soma labeled with DenMark (red) in an otherwise dfmr1-null brain. Scale bars: 10 μm (upper rows), 5 μm (lower rows). (C) Quantification of dendritic arbor volumes compared to those of constitutive dfmr1 expression controls, including constant restrictive temperature with Gal80ts repressor and expression in the absence of Gal80ts. N values are as follows: nullcontrol, 22; nullP4-rescue, 23; Nullconstitutive, 18; Nullconstitutive(-Gal80), 15. ***P<0.001 (ANOVA).
Fig. 6.

Critical period dfmr1 induction rescues mutant PN dendritic arbors. Targeted critical period expression of wild-type dfmr1 in PNs in an otherwise dfmr1-null background. Compared to the dfmr1 null (dfmr150M,14-3-3ζ-Gal4/TM6TbGFP>UAS-DM;dfmr150M/TM6) (A), dfmr1 induction at P4 (tubP-Gal80ts/CyO;dfmr150M,14-3-3ζ-Gal4/TM6>UAS-DM/CyO;dfmr150M,UAS-9557-3/TM6) (B) by temperature shift to the restrictive 29°C reduces PN dendritic arbor volume. Top rows depict PN dendritic arbors within the phalloidin-labeled (purple) AL. FMRP expression (green) occurs only with conditional expression in DenMark-labeled (red) PNs (B, lower row) in brains otherwise completely deficient for FMRP. The far-right column displays two dendritic arbor examples from temperature-shifted null (A) and conditional rescue (B). Scale bars: 10 μm (upper rows), 5 μm (lower rows). (C) Quantification of dendritic arbor volumes compared to two constitutive dfmr1 expression controls, including constant restrictive temperature with the Gal80ts repressor and expression in the absence of the Gal80ts. N values are as follows: nullcontrol, 15; nullP4-rescue, 14; nullconstitutive, 13; nullconstitutive(-Gal80), 13. *P<0.05, **P<0.01 (ANOVA).
Conditional dfmr1 induction drives tight temporal dFMRP expression in MVP2 neurons, which are otherwise completely devoid of the protein (compare Fig. 5A,B). Conditional dfmr1 induction during the critical period causes a very clear reduction in MVP2 dendritic arborization compared with that of nulls (Fig. 5A,B), restoring the normal architecture. There is a very highly significant reduction (P<0.001; n=22, 23, respectively) in MVP2 dendritic arbor volume in dfmr1 nulls with conditional rescue (4910±139 μm3) compared with that of non-rescued controls (7686±184 μm3) at 1 day post-eclosion (Fig. 5C). Constitutive dfmr1 expression in MVP2 neurons also causes a highly significant reduction (P<0.001, n=18) in dendritic arbor volume (5876±147 μm3; Fig. 5C), albeit to a slightly reduced degree compared with temporally targeted rescue. This shows that critical period dfmr1 expression optimally rescues the dendritic arborization defect.
Similarly in the PN class, conditional critical period expression of wild-type dfmr1 reduces dendritic volume to rescue to the wild-type condition (Fig. 6). In this experiment, tubP-Gal80ts/Cyo;dfmr150M,14-3-3ζ-Gal4/TM6 animals were crossed to UAS-DM;dfmr150M,UAS-9557-3/TM6, with progeny likewise raised at permissive temperature (18°C) until P4 and then shifted to the restrictive temperature (29°C). This tight temporal induction of wild-type dfmr1 in PNs in otherwise null brains causes an obvious reduction in dendritic arbors compared with temperature-shifted dfmr1 nulls (dfmr150M,14-3-3ζ-Gal4-Gal4/TM6TbGFP>UAS-DM/CyO,dfmr150M/TM6; compare Fig. 6A,B). Both the targeted critical period-shifted PN dendritic arbors (2700±124 μm3) and constitutive dfmr1-rescued controls (2721±77 μm3) display very significantly reduced (both P<0.001; n=14, 13, respectively) dendritic volume compared with that of the dfmr1-null mutant (3176±102 μm3; n=15; Fig. 6C). Taken together, these results clearly demonstrate that spatiotemporally targeted dfmr1 expression provides complete rescue of dendritic arborization during the early-use period immediately post eclosion.
Targeted conditional dfmr1 knockdown demonstrates an FMRP requirement during the critical period
To further define dfmr1 requirements during the early-use critical period, we used conditional transgenic RNA interference (RNAi) against dfmr1 in wild-type neurons using a Gal80ts strategy (Tutor et al., 2014). For the MVP2 neuron class, tubP-Gal80ts/Cyo;Oli-Gal4/TM6 was crossed to UAS-DM/Cyo;UAS-dfmr1-RNAi/TM6, with progeny raised at the permissive temperature (18°C) until P4, whereupon they were shifted to the restrictive temperature (29°C) to activate dfmr1-RNAi (supplementary material Fig. S6). Controls were similarly shifted to 29°C at P4 to test for effects of the temperature shift. Critical period induction of dfmr1-RNAi led to a rapid temporal loss of dFMRP specifically in targeted MVP2 neurons (compare supplementary material Fig. S6A,B), and a ∼25% increase in dendritic arbor volume with conditional dfmr1 knockdown compared with that of temperature-shifted wild-type controls, a highly significant result (P<0.001; n=27, 22, respectively; supplementary material Fig. S6C). An indistinguishable increase in arborization occurs with two constitutive dfmr1-RNAi controls (P<0.001; n=21; supplementary material Fig. S6C), showing that targeted spatiotemporal dfmr1 knockdown only in MVP2 neurons and solely during the early-use critical period generates the full dfmr1 phenotype.
Similarly, to test PN class requirements, tubP-Gal80ts/Cyo;14-3-3ζ-Gal4/TM6 was crossed to UAS-DM/Cyo;UAS-dfmr1-RNAi/TM6 (supplementary material Fig. S7). As above, the tight conditional induction of dfmr1-RNAi causes both a striking loss of dFMRP only from the targeted PNs (compare supplementary material Fig. S7A,B) and a very clear and obvious expansion of the PN dendritic arbor compared with that of controls (supplementary material Fig. S7A,B). Compared with wild-type control arbors (2801±125 μm3), there is a highly significant expansion (P<0.01) of dendritic arbor volume with conditional RNAi knockdown (3302±194 μm3) during the 1 day post eclosion period (supplementary material Fig. S7C). Constitutive dfmr1 knockdown causes a similar increase in PN arbor volume (3444±155 μm3; P<0.001; supplementary material Fig. S7C), which is similar to conditional dfmr1 knockdown during the critical period. Thus, spatiotemporally targeted dFMRP removal only in the PN class also indicates a highly restricted role in dendritic arborization during the early-use critical period immediately post eclosion.
Temporally targeted dfmr1 rescue restores activity-dependent dendritic development
As a final test of critical period dfmr1 requirements in activity-dependent regulation of dendritic arborization, we combined conditional dfmr1 rescue of otherwise null mutant animals with optogenetic modulation of electrical activity (Fig. 7). As in other conditional experiments, we used the Gal80ts repressive paradigm to restrict wild-type dfmr1 expression to MVP2 neurons during the critical period [tub-PGal80ts/CyO;dfmr150M,Oli-Gal4/TM6>UAS-ChR2(H134R)-mCherry/CyO;dfmr150M,UAS-9557-3/TM6]. Following the shift to the restrictive temperature at P4, animals were exposed to 24 h of depolarization by stimulation with 5 Hz blue light, comparing animals fed vehicle (Fig. 7A, left) or the essential ATR co-factor (Fig. 7A, right). Dendritic arbors in rescued dfmr1-null mutants (without ATR) develop normal arborization, indistinguishable from that of unstimulated wild-type animals (dashed line). However, dendritic arbors in ATR-fed animals dramatically increase in size (Fig. 7A). Quantification of dendritic arbor volume shows a striking increase in ATR-fed optogenetically stimulated animals (6899±208 μm3) compared with vehicle-fed controls (4502±257 μm3), a highly significant elevation (P<0.001; n=18, 20, respectively; Fig. 7B). These data show that spatiotemporal reintroduction of wild-type dfmr1 solely during the early-use critical period fully restores the capacity for activity-dependent regulation of dendritic arborization during development.
Fig. 7.
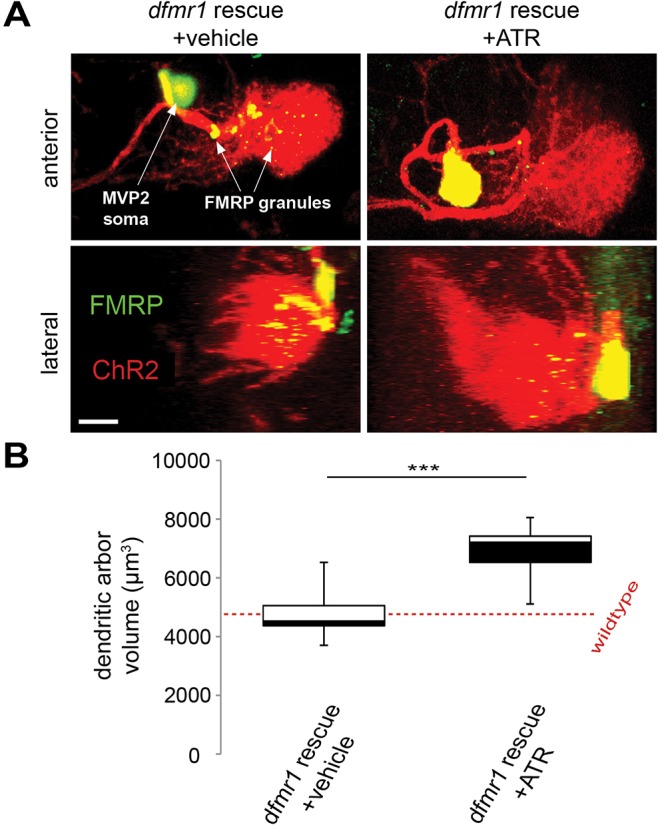
FMRP rescue restores activity-dependent dendritic arbor development. MVP2 dendritic arbors with dfmr1 conditional rescue in an otherwise dfmr1-null background and following ChR2(H134R)-mediated depolarization during the critical period, with and without feeding the essential ATR co-factor. Following dfmr1 P4 conditional induction, tubP-Gal80ts/CyO;dfmr150M,Oli-Gal4/TM6>UAS-ChR2(H134R)-mCherry/CyO;dfmr150M, UAS-9557-3/TM6 animals were exposed to 24 h of 5 Hz blue light stimulation. (A) Representative images of MVP2 dendritic arbors from anterior (top) and lateral (bottom) perspectives from animals fed with either vehicle control (left column) or ATR (right column). Anti-FMRP (green) in UAS-DenMark (red) labeled soma and dendritic arbors (FMRP granules, arrows) in the otherwise dfmr1-null brains. Scale bar: 10 μm. (B) Quantification of dendritic arbor volumes. N values are as follows: nullrescue+vehicle, 18; nullrescue+ATR, 20. The dashed line shows the wild-type MVP2 arbor volume. ***P<0.001.
DISCUSSION
FMRP is hypothesized to act during a critical period of brain development as a mechanistic link between early-use activity and optimized synaptic connectivity (Doll and Broadie, 2014). In direct mRNA-binding mechanisms, FMRP regulates activity-dependent transcript stability, transport and/or translation (Kanai et al., 2004; Antar et al., 2005; Ferrari et al., 2007; Dictenberg et al., 2008; Charalambous et al., 2013). Although the full range of FMRP targets is not known, established targets include microtubule [e.g. futsch (a Drosophila MAP1B homolog)] and actin (e.g. Rac1, Arc, PAK) cytoskeleton modulators, membrane-associated scaffolds (e.g. PSD-95) and other key synaptic regulators (e.g. CaMKII), which are all well positioned to control the choice between activity-dependent synapse pruning versus stabilization (Zhang et al., 2001; Zalfa et al., 2007; Chen et al., 2010; Bongmba et al., 2011; Dolan et al., 2013). FMRP expression is itself positively regulated by activity (Antar et al., 2004; Gabel et al., 2004; Wang et al., 2008), with peak brain expression coincident with critical period synaptic refinement (Lu et al., 2004; Singh et al., 2007; Tessier and Broadie, 2008). Thus, FMRP appears to be perfectly situated to function as the ‘activity sensor’ directly regulating the translation of a specialized cassette of proteins that determine the key synapse stabilization/removal decision during late-stage brain development.
To test this hypothesis, we targeted two classes of MB learning and memory center extrinsic neurons; input PNs and output MVP2 neurons. The dendritic arbors of both neuron types fail to undergo critical period refinement in dfmr1-null mutants, and fail to respond to optogenetic activity modulation in the absence of FMRP during the critical period of development immediately following eclosion. The overgrown dendritic arbors in dfmr1 nulls resemble expanded pyramidal cell dendrites found in mouse FXS models and human FXS patients (Comery et al., 1997; Irwin et al., 2000). The correlation between dendritic arbor volume in dfmr1 nulls and neurons experiencing ChR2 depolarization during early-use critical period development suggests excessive excitation in the mutant condition, consistent with established heightened sensory perception (Tessier and Broadie, 2008) and elevated synaptic excitability (Repicky and Broadie, 2009) in the Drosophila FXS model. This connection is also consistent with increased synaptic drive and altered excitation/inhibition (E/I) synaptic balance in Fmr1 mutant mice (Hays et al., 2011; Patel et al., 2013), as well as increased sensory sensitivity in human FXS patients (Harris et al., 2008).
The differential response to developmental activity modulation between MB input PNs and output MVP2 neurons is particularly fascinating. Both neuron classes display clear activity-dependent bidirectional restructuring of their dendritic arbors during critical period development, but with opposite directionality. There is very little information as yet on MVP2 neurons, other than anatomical nomenclature (Tanaka et al., 2008), but PNs have been intensively studied (Laissue et al., 1999; Couto et al., 2005). The fact that local inhibitory interneurons (LNs) provide AL hyperpolarizing input onto PNs might explain why these neurons are more responsive to halorhodopsin-mediated hyperpolarization during development (Das et al., 2011). Interestingly, dfmr1 PNs acquire late-stage remodeling capabilities, consistent with previous work showing correction of KC axon structure at 4 days post eclosion (Tessier and Broadie, 2008). By contrast, MVP2 neurons form reticular dendritic arbors within the MB spur, where KCs bifurcate into separate axonal output lobes (Tanaka et al., 2008). KC neurotransmitters remain unknown, but are likely to be predominantly excitatory (Yasuyama et al., 2002; Johard et al., 2008; Henry et al., 2012), with a GABAergic component (Gatto et al., 2014). This circuit connectivity might explain why MVP2 neurons are responsive to both depolarization and hyperpolarization during critical period development. In dfmr1 mutants, MVP2 dendrites might have attained a maximum size, preventing further expansion during channelrhodopsin-mediated hyperexcitation. However, FMRP-deficient MVP2 neurons also fail to respond to hyperpolarizing input. Under both conditions, dendritic arbors are always expanded in the absence of FMRP, in a cell-autonomous defect independent of activity directionality.
Importantly, both targeted dfmr1 rescue and knockdown during the critical period of development immediately post eclosion completely accounts for dendritic architecture requirements (McGuire et al., 2003; Tutor et al., 2014). Conditional, spatiotemporal control of dfmr1 expression in both neuron classes shows a cell-autonomous FMRP requirement that is solely restricted to the early-use developmental window coincident with eclosion (Doll and Broadie, 2014). In MVP2 neurons, both FMRP- and activity-dependent requirements are completely transitory in this brief window, with no developmental impact of dfmr1 loss persisting at maturity. Similarly, wild-type PNs normally show no activity-driven changes at maturity, also demonstrating a restricted critical period. Interestingly, however, dfmr1-null PNs appear to exhibit an expanded critical period, with abnormally persistent activity-dependent remodeling. This suggests that loss of FMRP can shift the critical period of synaptic connectivity development and refinement (Doll and Broadie, 2014). Taken together, the results of this study clearly show neuron type-specific responses to activity within a single neural circuit, indicating that differential mechanisms control activity-dependent synaptic pruning versus stabilization during critical period development of dendritic arborization.
Optogenetics has only minimally been used to address developmental questions (Tye and Deisseroth, 2012). However, a recent study showed that optogenetic manipulation can alter axon pathfinding in chick motor neurons (Kastanenka and Landmesser, 2013), revealing that activity can regulate morphological changes during early periods of synaptic development. The present study builds upon our previous work (Tessier and Broadie, 2008), which similarly showed that ChR2 optogenetic manipulation can alter KC axonal development in the Drosophila MB. Here, we use both ChR2 and eNpHR3 tools, target dendritic arbors and take advantage of the essential ATR co-factor requirement in Drosophila (Schroll et al., 2006) to control for both genotype and light stimulation. By contrast, mammalian cells contain ATR (Fenno et al., 2011), preventing application of a similar control. Future studies could use direct focal illumination of neuronal subcompartments (Packer et al., 2012) to assay effects of even more targeted depolarization or hyperpolarization on neural circuit morphogenesis. In addition, it will be particularly interesting to use optogenetics to test the altered E/I synaptic balance hypothesis in the FXS disease state (Gatto and Broadie, 2011; Gatto et al., 2014), including both hyperexcitation and hypoinhibition (Selby et al., 2007; Gibson et al., 2008), by hyperpolarizing MB circuit excitatory neurons or depolarizing MB circuit inhibitory neurons to assay restoration of circuit architecture and learning and memory behavioral outputs.
Drosophila optogenetics is facilitated by the penetration of blue or amber light through a semitransparent cuticle (Honjo et al., 2012). This is particularly true during relatively unpigmented developmental stages but, even at maturity, 100% of flies expressing ChR2(H134R) show behavioral responses to blue light (Inagaki et al., 2013). The fact that MVP2 and PN soma are located in extreme anterior and ventral brain regions, respectively (Fig. 1), further facilitates optogenetic manipulations in these neuronal classes. The vast catalog of new transgenic drivers in the FlyLight Gal4 library (Jenett et al., 2012) provides an unrivaled opportunity for targeted control of individually identifiable neurons. Activity-dependent restructuring underlying refinement of synaptic partnerships could be further pursued in conjunction with new mapping strategies, such as genetic reconstitution across synaptic partners (GRASP) (Feinberg et al., 2008) or the CaLexA system of neural tracing (Masuyama et al., 2012). Recent efforts have generated increasingly precise maps of Drosophila brain circuits, including MB circuitry (Parnas et al., 2013; Perisse et al., 2013) and linked AL circuitry (Tanaka et al., 2012). With new optogenetic tools for activity modulation in the context of such highly defined neural circuits, developmental milestones of activity-dependent mechanisms can now be dissected with unparalleled resolution, to address the basis of the FXS disease state as well as related neurodevelopmental disorders.
MATERIALS AND METHODS
Drosophila genetics
Stocks were reared on standard cornmeal/agar/molasses food at 25°C unless stated otherwise. Transgenic parental lines harboring the dfmr1 null allele dfmr150M (Zhang et al., 2001) were generated with standard genetic techniques in combination with R12G04-Gal4, R65G01-Gal4, UAS-mCD8::GFP, UAS-DenMark (DM) and tubP-Gal80[ts];TM2/TM6, obtained from the Drosophila Stock Center (Bloomington). For optogenetic manipulations, UAS-ChR2(H134R)-mCherry generously provided by Leslie Griffith (Pulver et al., 2009) and UAS-eNpHR3.0-EGFP (this study, see below) were introduced using standard genetic techniques. For conditional dfmr1 manipulation, UAS-dfmr1-RNAi (TRiP, Harvard) and a wild-type dfmr1 rescue construct (UAS-9557-3; Gatto et al., 2009) were crossed into control and mutant backgrounds, with all progeny raised at the permissive temperature (18°C) until pupal day 4 (P4), and then shifted to the restrictive temperature (29°C). These experiments involved the following sets of parental pairings: (1) tubP-Gal80ts/CyO;dfmr150M,Oli-Gal4/TM6>UAS-DM/CyO;dfmr150M, UAS-9557-3/TM6, (2) tubP-Gal80ts/CyO;dfmr150M,14-3-3ζ-Gal4/TM6>UAS-DM/CyO; dfmr150M,UAS-9557-3/TM6, (3) dfmr150M,Oli-Gal4/TM6TbGFP>UAS-DM/CyO;dfmr150M/TM6, (4) dfmr150M,14-3-3ζ-Gal4/TM6TbGFP>UAS-DM/CyO,dfmr150M/TM6, (5) tubP-Gal80ts/CyO;14-3-3ζ-Gal4/TM6>UAS-DM/CyO;UAS-dfmr1-RNAi/TM6, (6) tubP-Gal80ts/CyO;Oli-Gal4/TM6>UAS-DM/CyO;UAS-dfmr1-RNAi/TM6, (7) 14-3-3ζ-Gal4>UAS-DM and (8) Oli-Gal4>UAS-DM.
Halorhodopsin transgenics
A pAAV-CaMKIIa-eNpHR3.0-EYFP construct vector, kindly provided by Karl Deisseroth (Stanford University) was PCR excised and amplified using the following primers: F, 5ʹ-CTCAGAAGCCCCAAGCTCGTC-3ʹ and R, 5ʹ-GCAATAGCATGATACAAAGG-3ʹ. The eNpHR3.0 construct was then subcloned into a pENTR vector through a second PCR amplification using the following primers: F, 5ʹ-CACCATGTCGCGGAGGCCATGGCTTC-3ʹ and R, 5ʹ-ACCACGTTGATGTCGATCTG-3ʹ. The entry vector was then cloned into a pTWG Gateway cassette (5′UAS, 3′EGFP; DGRC). The confirmed plasmid was then finally injected into Drosophila embryos (Genetic Services) for stable genomic integration. Injected adults were crossed to w1118 to identify w+ transgenic carriers using standard genetic techniques. Isolated strains with robust w+ expression were mapped using standard genetic techniques and introduced into the dfmr150M null background.
Optogenetics
Transgenic animals were generated from pairwise crosses with one of two responders [UAS-ChR2(H134R)-mCherry or UAS-eNpHR3.0-EGFP] and one of two drivers (Oli-Gal4 or 14-3-3ζ-Gal4), in both w1118 and dfmr1 null backgrounds: Oli-Gal4>UAS-ChR2(H134R), Oli-Gal4>UAS-eNpHR, 14-3-3ζ-Gal4>UAS-ChR2(H134R), 14-3-3ζ-Gal4>UAS-eNpHR, dfmr150M,Oli-Gal4>UAS-ChR2(H134R);dfmr150M/TM6, dfmr150M,Oli-Gal4>UAS-dfmr150M, eNpHR/TM6TbGFP, dfmr150M,14-3-3ζ-Gal4>UAS-ChR2(H134R);dfmr150M/TM6 and dfmr150M,14-3-3ζ-Gal4>UAS-dfmr150M, eNpHR/TM6TbGFP. Offspring from these eight genotypes were fed from hatching on standard food supplemented with either 10 μl ethanol vehicle (in 10 ml volume; control) or 100 μM all-trans retinal (ATR) (Ataman et al., 2008). Upon either adult eclosion or after 6 days post-eclosion, animals were placed in 30 mm petri dishes with Whatman paper strips saturated with vehicle control or ATR in a 20% sucrose solution. The chambers were placed in a LED exposure chamber with two Luxeon Rebel Endor Star 3X 15-Watt LED arrays (470 nm blue or 590 nm amber light, LED Supply). At 15 volts, the LED arrays generate ∼100 μW/cm2 at a working distance of 2 cm. Animals were exposed to 24 h of 20 ms light pulses at either 0.5 or 5 Hz frequency, as indicated. Brains from light-exposed animals were immediately dissected and immunolabeled for imaging. For conditional UAS-dfmr1 rescue experiments with channelrhodopsin stimulation, staged tubP-Gal80ts/CyO;dfmr150M,Oli-Gal4/TM6>UAS-ChR2(H134R)/CyO;dfmr150M,UAS-9557-3/TM6 progeny were raised on either vehicle or ATR food at 18°C until pupal day 4 (P4), and then shifted to 29°C for 24 h and exposed to 24 h of blue light stimulation as described above.
Immunocytochemistry
Staged brains were dissected in PBS and fixed in 4% paraformaldehyde+4% sucrose in PBS for 30 min at 25°C. Preparations were washed three times with PBS, placed in buffer (PBS, 1% BSA, 0.5% NGS, 0.2% Triton X-100) for 20 min and then incubated with primary antibodies in buffer for 12-16 h at 4°C. The following primary antibodies were used: rabbit anti-GFP (ab290; Abcam) and anti-RFP (ab62341; Abcam) at 1:2000; mouse anti-FasII (1D4, Developmental Studies Hybridoma Bank) at 1:10; mouse anti-FMRP (clone 6A15; Sigma) at 1:500. Following three 20 min washes in 0.2% PBS-Triton X-100, secondary antibodies were incubated for 2-3 h at 25°C. The following secondary antibodies were used: anti-mouse IgG conjugated to Alexa Fluor 488 or 568, anti-rabbit IgG conjugated to Alexa Fluor 488 or 568 at 1:500 and Alexa Fluor 633 conjugated to phalloidin at 1:1000 (Molecular Probes). Following three 20 min washes in 0.2% PBS-Triton X-100, one 20 min wash in PBS and one 20 min wash in distilled water, preparations were mounted in Fluoromount G (Electron Microscopy Sciences).
Imaging and quantification
Brains were imaged on a Zeiss Meta 510 confocal microscope with 40× and 100× objectives. z-stacks of 1 µm section depth were collected for the central brain region. Dendritic arbor depths were calculated by counting consecutive fluorescent marker-postive sections within the z-stack. Volume and maximum cross-sectional area calculations of dendritic arbors were determined with the Volumest plug-in in ImageJ (M. Merzin, Bachelor's thesis, University of Tartu, Estonia, 2008; Valle-Leija et al., 2012), a semi-automated program used to measure the total volume of the thresholded fluorescently labeled dendritic arbors obtained as a cumulative z-stack. Each user-defined 2D measurement generated an area value for a single section, and the plug-in then generated a 3D volume calculated from the entire stack.
Statistics
All data are presented in box and whisker plots showing minimum, 25th percentile, median, 75th percentile and maximum values for each dataset. All statistical analyses were performed using GraphPad InStat 3 (GraphPad Software). As appropriate, one-way analysis of variance (ANOVA) with Tukey–Kramer multiple comparison post-tests or unpaired, two-tailed t-tests with Welch corrections were applied, allowing for populations with variable standard deviations.
Supplementary Material
Acknowledgements
We particularly thank the Drosophila Stock Center (Bloomington, IN, USA), which provided essential genetic stocks. We are also very grateful to Leslie Griffith (Brandeis University) for the UAS-ChR2(H134R)-mCherry line and Karl Deisseroth (Stanford University) for the pAAV-CaMKIIa-eNpHR3.0-EYFP construct vector. We thank Cheryl Gatto and Neil Dani for critical input on this manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
C.A.D. performed all experiments and data analyses. K.B. and C.A.D. jointly developed concepts, designed experiments and co-wrote the manuscript.
Funding
This work was funded entirely by the National Institutes of Health [R01 MH084989 to K.B.]. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.117127/-/DC1
References
- Antar L. N., Afroz R., Dictenberg J. B., Carroll R. C. and Bassell G. J. (2004). Metabotropic glutamate receptor activation regulates fragile x mental retardation protein and FMR1 mRNA localization differentially in dendrites and at synapses. J. Neurosci. 24, 2648-2655 10.1523/JNEUROSCI.0099-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antar L. N., Dictenberg J. B., Plociniak M., Afroz R. and Bassell G. J. (2005). Localization of FMRP-associated mRNA granules and requirement of microtubules for activity-dependent trafficking in hippocampal neurons. Genes Brain Behav. 4, 350-359 10.1111/j.1601-183X.2005.00128.x [DOI] [PubMed] [Google Scholar]
- Ataman B., Ashley J., Gorczyca M., Ramachandran P., Fouquet W., Sigrist S. J. and Budnik V. (2008). Rapid activity-dependent modifications in synaptic structure and function require bidirectional Wnt signaling. Neuron 57, 705-718 10.1016/j.neuron.2008.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belmonte M. K., Cook E. H. Jr, Anderson G. M., Rubenstein J. L., Greenough W. T., Beckel-Mitchener A., Courchesne E., Boulanger L. M., Powell S. B., Levitt P. R. et al. (2004). Autism as a disorder of neural information processing: directions for research and targets for therapy. Mol. Psychiatry 9, 646-663. [DOI] [PubMed] [Google Scholar]
- Bongmba O. Y. N., Martinez L. A., Elhardt M. E., Butler K. and Tejada-Simon M. V. (2011). Modulation of dendritic spines and synaptic function by Rac1: a possible link to Fragile X syndrome pathology. Brain Res. 1399, 79-95 10.1016/j.brainres.2011.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadie K., Rushton E., Skoulakis E. M. C. and Davis R. L. (1997). Leonardo, a Drosophila 14-3-3 protein involved in learning, regulates presynaptic function. Neuron 19, 391-402 10.1016/S0896-6273(00)80948-4 [DOI] [PubMed] [Google Scholar]
- Bureau I., Shepherd G. M. G. and Svoboda K. (2008). Circuit and plasticity defects in the developing somatosensory cortex of FMR1 knock-out mice. J. Neurosci. 28, 5178-5188 10.1523/JNEUROSCI.1076-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher N. J., Friedrich A. B., Lu Z., Tanimoto H. and Meinertzhagen I. A. (2012). Different classes of input and output neurons reveal new features in microglomeruli of the adult Drosophila mushroom body calyx. J. Comp. Neurol. 520, 2185-2201 10.1002/cne.23037 [DOI] [PubMed] [Google Scholar]
- Charalambous D. C., Pasciuto E., Mercaldo V., Pilo Boyl P., Munck S., Bagni C. and Santama N. (2013). KIF1Bbeta transports dendritically localized mRNPs in neurons and is recruited to synapses in an activity-dependent manner. Cell. Mol. Life Sci. 70, 335-356 10.1007/s00018-012-1108-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L. Y., Rex C. S., Babayan A. H., Kramar E. A., Lynch G., Gall C. M. and Lauterborn J. C. (2010). Physiological activation of synaptic Rac>PAK (p-21 activated kinase) signaling is defective in a mouse model of fragile X syndrome. J. Neurosci. 30, 10977-10984 10.1523/JNEUROSCI.1077-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comery T. A., Harris J. B., Willems P. J., Oostra B. A., Irwin S. A., Weiler I. J. and Greenough W. T. (1997). Abnormal dendritic spines in fragile X knockout mice: maturation and pruning deficits. Proc. Natl. Acad. Sci. USA 94, 5401-5404 10.1073/pnas.94.10.5401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couto A., Alenius M. and Dickson B. J. (2005). Molecular, anatomical, and functional organization of the Drosophila olfactory system. Curr. Biol. 15, 1535-1547 10.1016/j.cub.2005.07.034 [DOI] [PubMed] [Google Scholar]
- Cruz-Martin A., Crespo M. and Portera-Cailliau C. (2010). Delayed stabilization of dendritic spines in fragile X mice. J. Neurosci. 30, 7793-7803 10.1523/JNEUROSCI.0577-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S., Sadanandappa M. K., Dervan A., Larkin A., Lee J. A., Sudhakaran I. P., Priya R., Heidari R., Holohan E. E., Pimentel A. et al. (2011). Plasticity of local GABAergic interneurons drives olfactory habituation. Proc. Natl. Acad. Sci. USA 108, E646-E654 10.1073/pnas.1106411108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries S. E. and Clandinin T. (2013). Optogenetic stimulation of escape behavior in Drosophila melanogaster. J. Visual Exp. 71, ppii: 50192. 10.3791/50192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dictenberg J. B., Swanger S. A., Antar L. N., Singer R. H. and Bassell G. J. (2008). A direct role for FMRP in activity-dependent dendritic mRNA transport links filopodial-spine morphogenesis to fragile X syndrome. Dev. Cell 14, 926-939 10.1016/j.devcel.2008.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolan B. M., Duron S. G., Campbell D. A., Vollrath B., Shankaranarayana Rao B. S., Ko H. Y., Lin G. G., Govindarajan A., Choi S. Y. and Tonegawa S. (2013). Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proc. Natl. Acad. Sci. USA 110, 5671-5676 10.1073/pnas.1219383110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll C. A. and Broadie K. (2014). Impaired activity-dependent neural circuit assembly and refinement in autism spectrum disorder genetic models. Front. Cell. Neurosci. 8, 30 10.3389/fncel.2014.00030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg E. H., VanHoven M. K., Bendesky A., Wang G., Fetter R. D., Shen K. and Bargmann C. I. (2008). GFP Reconstitution Across Synaptic Partners (GRASP) defines cell contacts and synapses in living nervous systems. Neuron 57, 353-363 10.1016/j.neuron.2007.11.030 [DOI] [PubMed] [Google Scholar]
- Fenno L., Yizhar O. and Deisseroth K. (2011). The development and application of optogenetics. Annu. Rev. Neurosci. 34, 389-412 10.1146/annurev-neuro-061010-113817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari F., Mercaldo V., Piccoli G., Sala C., Cannata S., Achsel T. and Bagni C. (2007). The fragile X mental retardation protein-RNP granules show an mGluR-dependent localization in the post-synaptic spines. Mol. Cell. Neurosci. 34, 343-354 10.1016/j.mcn.2006.11.015 [DOI] [PubMed] [Google Scholar]
- Gabel L. A., Won S., Kawai H., McKinney M., Tartakoff A. M. and Fallon J. R. (2004). Visual experience regulates transient expression and dendritic localization of fragile X mental retardation protein. J. Neurosci. 24, 10579-10583 10.1523/JNEUROSCI.2185-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto C. L. and Broadie K. (2008). Temporal requirements of the fragile X mental retardation protein in the regulation of synaptic structure. Development 135, 2637-2648 10.1242/dev.022244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto C. L. and Broadie K. (2009). Temporal requirements of the fragile x mental retardation protein in modulating circadian clock circuit synaptic architecture. Front. Neural Circuits 3, 8 10.3389/neuro.04.008.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto C. L. and Broadie K. (2011). Fragile X mental retardation protein is required for programmed cell death and clearance of developmentally-transient peptidergic neurons. Dev. Biol. 356, 291-307 10.1016/j.ydbio.2011.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto C. L., Pereira D. and Broadie K. (2014). GABAergic circuit dysfunction in the Drosophila Fragile X syndrome model. Neurobiol. Dis. 65, 142-159 10.1016/j.nbd.2014.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind D. H. and Levitt P. (2007). Autism spectrum disorders: developmental disconnection syndromes. Curr. Opin. Neurobiol. 17, 103-111 10.1016/j.conb.2007.01.009 [DOI] [PubMed] [Google Scholar]
- Gibson J. R., Bartley A. F., Hays S. A. and Huber K. M. (2008). Imbalance of neocortical excitation and inhibition and altered UP states reflect network hyperexcitability in the mouse model of fragile X syndrome. J. Neurophysiol. 100, 2615-2626 10.1152/jn.90752.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman R. J., Berry-Kravis E., Kaufmann W. E., Ono M. Y., Tartaglia N., Lachiewicz A., Kronk R., Delahunty C., Hessl D., Visootsak J. et al. (2009). Advances in the treatment of fragile X syndrome. Pediatrics 123, 378-390 10.1542/peds.2008-0317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris S. W., Hessl D., Goodlin-Jones B., Ferranti J., Bacalman S., Barbato I., Tassone F., Hagerman P. J., Herman H. and Hagerman R. J. (2008). Autism profiles of males with fragile X syndrome. Am. J. Ment. Retard. 113, 427-438 10.1352/2008.113:427-438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hays S. A., Huber K. M. and Gibson J. R. (2011). Altered neocortical rhythmic activity states in Fmr1 KO mice are due to enhanced mGluR5 signaling and involve changes in excitatory circuitry. J. Neurosci. 31, 14223-14234 10.1523/JNEUROSCI.3157-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry G. L., Davis F. P., Picard S. and Eddy S. R. (2012). Cell type-specific genomics of Drosophila neurons. Nucleic Acids Res. 40, 9691-9704 10.1093/nar/gks671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A. and Svoboda K. (2009). Experience-dependent structural synaptic plasticity in the mammalian brain. Nat. Rev. Neurosci. 10, 647-658 10.1038/nrn2699 [DOI] [PubMed] [Google Scholar]
- Honjo K., Hwang R. Y. and Tracey W. D. (2012). Optogenetic manipulation of neural circuits and behavior in Drosophila larvae. Nat. Protoc. 7, 1470-1478 10.1038/nprot.2012.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutsler J. J. and Zhang H. (2010). Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 1309, 83-94 10.1016/j.brainres.2009.09.120 [DOI] [PubMed] [Google Scholar]
- Inada K., Kohsaka H., Takasu E., Matsunaga T. and Nose A. (2011). Optical dissection of neural circuits responsible for Drosophila larval locomotion with halorhodopsin. PLoS ONE 6, e29019 10.1371/journal.pone.0029019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki H. K., Jung Y., Hoopfer E. D., Wong A. M., Mishra N., Lin J. Y., Tsien R. Y. and Anderson D. J. (2013). Optogenetic control of Drosophila using a red-shifted channelrhodopsin reveals experience-dependent influences on courtship. Nat. Methods 11, 325-332 10.1038/nmeth.2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin S. A., Galvez R. and Greenough W. T. (2000). Dendritic spine structural anomalies in fragile-X mental retardation syndrome. Cereb. Cortex 10, 1038-1044 10.1093/cercor/10.10.1038 [DOI] [PubMed] [Google Scholar]
- Irwin S. A., Patel B., Idupulapati M., Harris J. B., Crisostomo R. A., Larsen B. P., Kooy F., Willems P. J., Cras P., Kozlowski P. B. et al. (2001). Abnormal dendritic spine characteristics in the temporal and visual cortices of patients with fragile-X syndrome: a quantitative examination. Am. J. Med. Genet. 98, 161-167 [DOI] [PubMed] [Google Scholar]
- Ito K., Shinomiya K., Ito M., Armstrong J. D., Boyan G., Hartenstein V., Harzsch S., Heisenberg M., Homberg U., Jenett A. et al. (2014). A systematic nomenclature for the insect brain. Neuron 81, 755-765 10.1016/j.neuron.2013.12.017 [DOI] [PubMed] [Google Scholar]
- Jefferis G. S. X. E., Marin E. C., Stocker R. F. and Luo L. (2001). Target neuron prespecification in the olfactory map of Drosophila. Nature 414, 204-208 10.1038/35102574 [DOI] [PubMed] [Google Scholar]
- Jenett A., Rubin G. M., Ngo T.-T. B., Shepherd D., Murphy C., Dionne H., Pfeiffer B. D., Cavallaro A., Hall D., Jeter J. et al. (2012). A GAL4-driver line resource for Drosophila neurobiology. Cell Rep. 2, 991-1001 10.1016/j.celrep.2012.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johard H. A. D., Enell L. E., Gustafsson E., Trifilieff P., Veenstra J. A. and Nässel D. R. (2008). Intrinsic neurons of Drosophila mushroom bodies express short neuropeptide F: relations to extrinsic neurons expressing different neurotransmitters. J. Comp. Neurol. 507, 1479-1496 10.1002/cne.21636 [DOI] [PubMed] [Google Scholar]
- Jones W. D. (2009). The expanding reach of the GAL4/UAS system into the behavioral neurobiology of Drosophila. BMB Rep. 42, 705-712 10.5483/BMBRep.2009.42.11.705 [DOI] [PubMed] [Google Scholar]
- Kanai Y., Dohmae N. and Hirokawa N. (2004). Kinesin transports RNA: isolation and characterization of an RNA-transporting granule. Neuron 43, 513-525 10.1016/j.neuron.2004.07.022 [DOI] [PubMed] [Google Scholar]
- Kastanenka K. V. and Landmesser L. T. (2013). Optogenetic-mediated increases in in vivo spontaneous activity disrupt pool-specific but not dorsal-ventral motoneuron pathfinding. Proc. Natl. Acad. Sci. USA 110, 17528-17533 10.1073/pnas.1316457110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz L. C. and Shatz C. J. (1996). Synaptic activity and the construction of cortical circuits. Science 274, 1133-1138 10.1126/science.274.5290.1133 [DOI] [PubMed] [Google Scholar]
- Kelleher R. J. III and Bear M. F. (2008). The autistic neuron: troubled translation? Cell 135, 401-406 10.1016/j.cell.2008.10.017 [DOI] [PubMed] [Google Scholar]
- Kooy R. F. (2003). Of mice and the fragile X syndrome. Trends Genet. 19, 148-154 10.1016/S0168-9525(03)00017-9 [DOI] [PubMed] [Google Scholar]
- Laissue P. P., Reiter C., Hiesinger P. R., Halter S., Fischbach K. F. and Stocker R. F. (1999). Three-dimensional reconstruction of the antennal lobe in Drosophila melanogaster. J. Comp. Neurol. 405, 543-552 [DOI] [PubMed] [Google Scholar]
- Li H., Li Y., Lei Z., Wang K. and Guo A. (2013). Transformation of odor selectivity from projection neurons to single mushroom body neurons mapped with dual-color calcium imaging. Proc. Natl. Acad. Sci. USA 110, 12084-12089 10.1073/pnas.1305857110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R., Wang H., Liang Z., Ku L., O'Donnell W. T., Li W., Warren S. T. and Feng Y. (2004). The fragile X protein controls microtubule-associated protein 1B translation and microtubule stability in brain neuron development. Proc. Natl. Acad. Sci. USA 101, 15201-15206 10.1073/pnas.0404995101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuyama K., Zhang Y., Rao Y. and Wang J. W. (2012). Mapping neural circuits with activity-dependent nuclear import of a transcription factor. J. Neurogenet. 26, 89-102 10.3109/01677063.2011.642910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire S. E., Le P. T., Osborn A. J., Matsumoto K. and Davis R. L. (2003). Spatiotemporal rescue of memory dysfunction in Drosophila. Science 302, 1765-1768 10.1126/science.1089035 [DOI] [PubMed] [Google Scholar]
- Nicolai L. J. J., Ramaekers A., Raemaekers T., Drozdzecki A., Mauss A. S., Yan J., Landgraf M., Annaert W. and Hassan B. A. (2010). Genetically encoded dendritic marker sheds light on neuronal connectivity in Drosophila. Proc. Natl. Acad. Sci. USA 107, 20553-20558 10.1073/pnas.1010198107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyallon J., Apitz H., Miguel-Aliaga I., Timofeev K., Ferreira L. and Salecker I. (2012). Regulation of locomotion and motoneuron trajectory selection and targeting by the Drosophila homolog of Olig family transcription factors. Dev. Biol. 369, 261-276 10.1016/j.ydbio.2012.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer A. M., Peterka D. S., Hirtz J. J., Prakash R., Deisseroth K. and Yuste R. (2012). Two-photon optogenetics of dendritic spines and neural circuits. Nat. Methods 9, 1202-1205 10.1038/nmeth.2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan L., Zhang Y. Q., Woodruff E. and Broadie K. (2004). The Drosophila fragile X gene negatively regulates neuronal elaboration and synaptic differentiation. Curr. Biol. 14, 1863-1870 10.1016/j.cub.2004.09.085 [DOI] [PubMed] [Google Scholar]
- Pan L., Woodruff E. III, Liang P. and Broadie K. (2008). Mechanistic relationships between Drosophila fragile X mental retardation protein and metabotropic glutamate receptor A signaling. Mol. Cell. Neurosci. 37, 747-760 10.1016/j.mcn.2008.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parnas M., Lin A. C., Huetteroth W. and Miesenböck G. (2013). Odor discrimination in Drosophila: from neural population codes to behavior. Neuron 79, 932-944 10.1016/j.neuron.2013.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel A. B., Hays S. A., Bureau I., Huber K. M. and Gibson J. R. (2013). A target cell-specific role for presynaptic Fmr1 in regulating glutamate release onto neocortical fast-spiking inhibitory neurons. J. Neurosci. 33, 2593-2604 10.1523/JNEUROSCI.2447-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P., Cahill M. E., Jones K. A., VanLeeuwen J.-E. and Woolfrey K. M. (2011). Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285-293 10.1038/nn.2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perisse E., Yin Y., Lin A. C., Lin S., Huetteroth W. and Waddell S. (2013). Different kenyon cell populations drive learned approach and avoidance in Drosophila. Neuron 79, 945-956 10.1016/j.neuron.2013.07.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer B. E. and Huber K. M. (2007). Fragile X mental retardation protein induces synapse loss through acute postsynaptic translational regulation. J. Neurosci. 27, 3120-3130 10.1523/JNEUROSCI.0054-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer B. E., Zang T., Wilkerson J. R., Taniguchi M., Maksimova M. A., Smith L. N., Cowan C. W. and Huber K. M. (2010). Fragile X mental retardation protein is required for synapse elimination by the activity-dependent transcription factor MEF2. Neuron 66, 191-197 10.1016/j.neuron.2010.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portera-Cailliau C. (2012). Which comes first in fragile X syndrome, dendritic spine dysgenesis or defects in circuit plasticity? Neuroscientist 18, 28-44 10.1177/1073858410395322 [DOI] [PubMed] [Google Scholar]
- Pulver S. R., Pashkovski S. L., Hornstein N. J., Garrity P. A. and Griffith L. C. (2009). Temporal dynamics of neuronal activation by Channelrhodopsin-2 and TRPA1 determine behavioral output in Drosophila larvae. J. Neurophysiol. 101, 3075-3088 10.1152/jn.00071.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repicky S. and Broadie K. (2009). Metabotropic glutamate receptor-mediated use-dependent down-regulation of synaptic excitability involves the fragile X mental retardation protein. J. Neurophysiol. 101, 672-687 10.1152/jn.90953.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroll C., Riemensperger T., Bucher D., Ehmer J., Völler T., Erbguth K., Gerber B., Hendel T., Nagel G., Buchner E. et al. (2006). Light-induced activation of distinct modulatory neurons triggers appetitive or aversive learning in Drosophila larvae. Curr. Biol. 16, 1741-1747 10.1016/j.cub.2006.07.023 [DOI] [PubMed] [Google Scholar]
- Selby L., Zhang C. and Sun Q.-Q. (2007). Major defects in neocortical GABAergic inhibitory circuits in mice lacking the fragile X mental retardation protein. Neurosci. Lett. 412, 227-232 10.1016/j.neulet.2006.11.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinakevitch I., Grau Y., Strausfeld N. J. and Birman S. (2010). Dynamics of glutamatergic signaling in the mushroom body of young adult Drosophila. Neural Dev. 5, 10 10.1186/1749-8104-5-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K., Gaur P. and Prasad S. (2007). Fragile x mental retardation (Fmr-1) gene expression is down regulated in brain of mice during aging. Mol. Biol. Rep. 34, 173-181 10.1007/s11033-006-9032-8 [DOI] [PubMed] [Google Scholar]
- Tanaka N. K., Tanimoto H. and Ito K. (2008). Neuronal assemblies of the Drosophila mushroom body. J. Comp. Neurol. 508, 711-755 10.1002/cne.21692 [DOI] [PubMed] [Google Scholar]
- Tanaka N. K., Endo K. and Ito K. (2012). Organization of antennal lobe-associated neurons in adult Drosophila melanogaster brain. J. Comp. Neurol. 520, 4067-4130 10.1002/cne.23142 [DOI] [PubMed] [Google Scholar]
- Tessier C. R. and Broadie K. (2008). Drosophila fragile X mental retardation protein developmentally regulates activity-dependent axon pruning. Development 135, 1547-1557 10.1242/dev.015867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier C. R. and Broadie K. (2012). Molecular and genetic analysis of the Drosophila model of fragile X syndrome. Results Probl. Cell Differ. 54, 119-156 10.1007/978-3-642-21649-7_7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tutor A. S., Prieto-Sanchez S. and Ruiz-Gomez M. (2014). Src64B phosphorylates Dumbfounded and regulates slit diaphragm dynamics: Drosophila as a model to study nephropathies. Development 141, 367-376 10.1242/dev.099408 [DOI] [PubMed] [Google Scholar]
- Tye K. M. and Deisseroth K. (2012). Optogenetic investigation of neural circuits underlying brain disease in animal models. Nat. Rev. Neurosci. 13, 251-266 10.1038/nrn3171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valle-Leija P., Blanco-Hernández E., Drucker-Colín R., Gutiérrez-Ospina G. and Vidaltamayo R. (2012). Supernumerary formation of olfactory glomeruli induced by chronic odorant exposure: a constructivist expression of neural plasticity. PLoS ONE 7, e35358 10.1371/journal.pone.0035358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Wu L.-J., Zhang F. and Zhuo M. (2008). Roles of calcium-stimulated adenylyl cyclase and calmodulin-dependent protein kinase IV in the regulation of FMRP by group I metabotropic glutamate receptors. J. Neurosci. 28, 4385-4397 10.1523/JNEUROSCI.0646-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West A. E. and Greenberg M. E. (2011). Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb. Perspect. Biol. 3, pii: a005744 10.1101/cshperspect.a005744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuyama K., Meinertzhagen I. A. and Schürmann F.-W. (2002). Synaptic organization of the mushroom body calyx in Drosophila melanogaster. J. Comp. Neurol. 445, 211-226 10.1002/cne.10155 [DOI] [PubMed] [Google Scholar]
- Zalfa F., Eleuteri B., Dickson K. S., Mercaldo V., De Rubeis S., di Penta A., Tabolacci E., Chiurazzi P., Neri G., Grant S. G. N. et al. (2007). A new function for the fragile X mental retardation protein in regulation of PSD-95 mRNA stability. Nat. Neurosci. 10, 578-587 10.1038/nn1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Q., Bailey A. M., Matthies H. J. G., Renden R. B., Smith M. A., Speese S. D., Rubin G. M. and Broadie K. (2001). Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell 107, 591-603 10.1016/S0092-8674(01)00589-X [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.