

Abstract
Bone morphogenetic protein (BMP) signaling plays many roles in skull morphogenesis. We have previously reported that enhanced BMP signaling through the BMP type IA receptor (BMPR1A) in cranial neural crest cells causes craniosynostosis during postnatal development. Additionally, we observed that 55% of Bmpr1a mutant mice show neonatal lethality characterized by a distended gastrointestinal tract. Here, we show that severely affected mutants exhibit defective nasal cartilage, failure of fusion between the nasal septum and the secondary palate, and higher levels of phosphorylated SMAD1 and SMAD5 in the nasal tissue. TUNEL demonstrated an increase in apoptosis in both condensing mesenchymal tissues and cartilage of the nasal region in mutants. The levels of p53 (TRP53) tumor suppressor protein were also increased in the same tissue. Injection of pifithrin-α, a chemical inhibitor of p53, into pregnant mice prevented neonatal lethality while concomitantly reducing apoptosis in nasal cartilage primordia, suggesting that enhanced BMP signaling induces p53-mediated apoptosis in the nasal cartilage. The expression of Bax and caspase 3, downstream targets of p53, was increased in the mutants; however, the p53 expression level was unchanged. It has been reported that MDM2 interacts with p53 to promote degradation. We found that the amount of MDM2-p53 complex was decreased in all mutants, and the most severely affected mutants had the largest decrease. Our previous finding that the BMP signaling component SMAD1 prevents MDM2-mediated p53 degradation coupled with our new data indicate that augmented BMP signaling induces p53-mediated apoptosis by prevention of p53 degradation in developing nasal cartilage. Thus, an appropriate level of BMP signaling is required for proper craniofacial morphogenesis.
Keywords: BMP, SMAD, Craniofacial development, Neural crest cell, p53, MDM2, Apoptosis, Nasal septum
Summary: BMP signaling levels control p53-mediated apoptosis through SMAD1/5 phosphorylation and MDM2-p53 complex formation in cranial neural crest cells, regulating nasal cartilage morphogenesis.
INTRODUCTION
A cartilaginous nasal septum continuous with the cartilaginous nasal capsule forms the boundary of the nasal cavity (Bingham et al., 1991). This cartilage structure functions as a strut in the nasal cavity, keeping the nasal airway open to enable optimal respiratory and olfactory function (Badoux, 1966; Moss et al., 1968; Stenstrom and Thilander, 1970). Cartilaginous nasal septum resection and transplantation experiments using rabbits demonstrated that the nasal septum acts as a growth site pushing the mid-facial bones downward and forward from the cranial base (Wexler and Sarnat, 1961, 1965; Copray, 1986). Hence, nasal septal defects in rabbits affect maxillary growth, and some human cases support this notion (Moss et al., 1968; Howe et al., 2004). The importance of nasal cartilage is further illustrated by defective nasal cartilage that results in diminished airway capacity, leading to nasal obstruction (Metzinger et al., 1994).
BMP signaling molecules have been demonstrated to play crucial roles in skull morphogenesis (Dunn et al., 1997; Liu et al., 2005). Loss-of-function mutation of Acvr1 in neural crest cells results in cleft palate, reduced anterior-posterior dimension of the skull, hypotrophic mandible and failure of zygomatic bone formation (Dudas et al., 2004). Gain-of-function mutation in Msx2, a member of the homeobox gene family whose transcription is regulated by BMP and TGF-β signaling, results in mid-face clefting and mandibular hypoplasia (Winograd et al., 1997). However, the mechanism of BMP signaling in cranial cartilage development is not well understood.
We have previously reported that augmented BMP-Smad signaling through BMPR1A in cranial neural crest cells results in craniofacial deformity (Komatsu et al., 2013). Because the anterior portion of the craniofacial complex is derived from cranial neural crest cells (Noden and Trainor, 2005; Chai and Maxson, 2006; Mishina and Snider, 2014), we augmented BMP-Smad signaling in the neural crest region by crossing a transgenic mouse line carrying a conditional constitutively active form of Bmpr1a (ca-Bmpr1a) with P0-Cre mice (Yamauchi et al., 1999; Kamiya et al., 2008; Komatsu et al., 2013). This genetic manipulation allows an increase in BMP signaling specifically in neural crest-derived tissues, resulting in premature suture fusion of the anterior frontal suture, orbital hypertelorism, short snouts and thinner calvaria. Moreover, increased apoptosis was found in mutant calvarial bone (Komatsu et al., 2013). The skull abnormalities in ca-Bmpr1a:P0-Cre mice were partially rescued by the removal of one copy of endogenous Bmpr1a (ca-Bmpr1a:P0-Cre:Bmpr1a+/−, rescued mice hereafter). Rescued mice showed normalized levels of Smad-dependent signaling, suggesting that a small increase in BMP-Smad signaling is enough to cause the aforementioned craniofacial abnormalities (Komatsu et al., 2013).
BMP signaling components such as BMP4, BMP7 and MSX2 are known to be involved in apoptosis during the development of rhombomeres, limbs and teeth (Graham et al., 1994; Yokouchi et al., 1996; Zou and Niswander, 1996; Macias et al., 1997). Additionally, BMP4 is known to induce apoptosis in a p53 (TRP53)-dependent manner in cancer cells as well as oral epithelial cells (Fukuda et al., 2006; Kim et al., 2006; Haubold et al., 2010). p53 tumor suppressor is a transcription factor that induces cell growth arrest and apoptosis (Kerr et al., 1972). p53 is activated by stress signals, promoting its nuclear accumulation (Giaccia and Kastan, 1998), and it is inhibited by MDM2. Initially MDM2 was found as a p53 binding protein, and it is now known that MDM2 induces p53 ubiquitylation, prompting nuclear export and proteasomal degradation (Momand et al., 1992; Kussie et al., 1996; Lai et al., 2001). Activated SMAD1, a component of BMP signaling, was also recently found to interact with p53, preventing MDM2-mediated p53 degradation (Chau et al., 2012). However, there is no direct evidence that augmented BMP signaling leads to p53-mediated apoptosis in vivo.
In this study, we found that augmented BMP-Smad signaling through BMPR1A in neural crest cells leads to an increase in p53-mediated apoptosis in developing nasal cartilage. We documented that 55% of ca-Bmpr1a:P0-Cre mice experience neonatal lethality and have abnormal nasal cartilage structures. Here, we show that the nasal cartilage defects are caused by an increase in apoptosis. These severely affected mutants had increased levels of p53 protein without increases in p53 gene expression. Phosphorylation levels of p53 at the serine 15 residue were elevated in nasal tissue in association with increased expression of caspase 3 and Bax. MDM2 acts as an E3 ligase prompting p53 degradation by the proteasome. The formation of the MDM2-p53 complex was also decreased in the mutants. Taken together, we propose a model in which increased nuclear levels of phospho-SMAD1/5/9 prevent MDM2-p53 interaction, leading to activation of the apoptotic pathways in chondrocytes of ca-Bmpr1a:P0-Cre mice.
RESULTS
Augmented BMP signaling in neural crest cells causes neonatal lethality
ca-Bmpr1a mice were crossed with P0-Cre mice, which express Cre recombinase under the control of a neural crest-specific promoter, protein zero (Yamauchi et al., 1999). As we reported previously (Komatsu et al., 2013), mice that carried both ca-Bmpr1a and P0-Cre transgenes, ca-Bmpr1a:P0-Cre (hereafter, mutants), displayed short broad snouts and orbital hypertelorism as early as postnatal day 0 (newborn). We found that 55% of mutants died within 24 h after birth, whereas the vast majority of control mice survived (Fig. 1A). Importantly, all of the mutant mice that showed neonatal lethality (hereafter, type 2 mutants) displayed gradual abdominal distension after birth (Fig. 1B,C). By contrast, mutants that survived for >24 h (hereafter, type 1 mutants) showed no such distension. Type 1 mutants survive for >1 year and have skull malformation subsequent to premature suture fusion (Komatsu et al., 2013). Short, broad snouts and orbital hypertelorism were common craniofacial features of both type 1 and type 2 mutants (Fig. 1D); however, a crucial difference in type 2 mutants was the absence of milk in the stomach (Fig. 1C). Next, we evaluated levels of phosphorylated SMAD1, SMAD5 and SMAD9 (pSMAD1/5/9) in nasal tissues dissected from a newborn head (Fig. 1E). We observed higher levels of pSMAD1/5/9 in both types of mutants compared with controls, but type 2 mutants showed the highest pSMAD1/5/9 level. These results suggested that more BMP-Smad signaling results in a more severe phenotype.
Fig. 1.

Enhanced BMP signaling through a constitutively active form of BMPR1A causes neonatal lethality. (A) Survival curve for control (blue, n=14) and ca-Bmpr1a:P0-Cre mice (red, n=13) after cesarean section at E18.5. (B) Type 2 mutants exhibited abdominal distension (arrow). (C) Gastrointestinal tracts of type 2 mutants were filled with air bubbles, whereas controls and type 1 mutants had milk in their stomachs. (D) Newborn type 1 and type 2 mutants displayed short broad snouts and hypertelorism. (E) Western blot analysis measuring the levels of pSMAD1/5/9 in control, type 1 and type 2 mutant nasal tissues. GAPDH was used as a loading control. Results for pSMAD1/5/9 were quantified by densitometry (bar graph). All data are represented as mean±s.d. (n=4). *P<0.05, **P<0.005 (Student's t-test). Scale bars: 1 mm.
Histological analysis of the nasal cavity in ca-Bmpr1a:P0-Cre mutants
To investigate the causes of the neonatal lethality, we examined structural abnormalities unique to the type 2 mutants. Because we found air bubbles in the gastrointestinal tract (Fig. 1C), we suspected the presence of cleft palate in type 2 mutants. However, type 2 mutants had neither overt cleft palate (supplementary material Fig. S1C) nor abnormalities in other respiratory organs such as the tongue, soft palate, epiglottis and trachea (supplementary material Fig. S1). Instead, newborn type 2 mutants exhibited failure of fusion between the nasal septum and the secondary palate (Fig. 2K,L asterisks, compare with 2C,D), discontinuous cartilage in the nasal capsule (Fig. 2I, arrowheads) and reduced nasal airway volume (Fig. 2I,J, compare with 2A,B). By contrast, newborn type 1 mutants had a fused nasal septum (Fig. 2G,H) and a unilateral defect in the nasal cartilage of the nasal capsule (Fig. 2E, white arrowhead). In addition, type 1 mutants had comparable airway size to controls (Fig. 2Q,R) as well as a completely fused nasal septum (Fig. 2S,T) by postnatal day 7 (P7). These results suggest that enhanced BMP signaling in neural crest cells leads to nasal cartilage dysmorphogenesis.
Fig. 2.
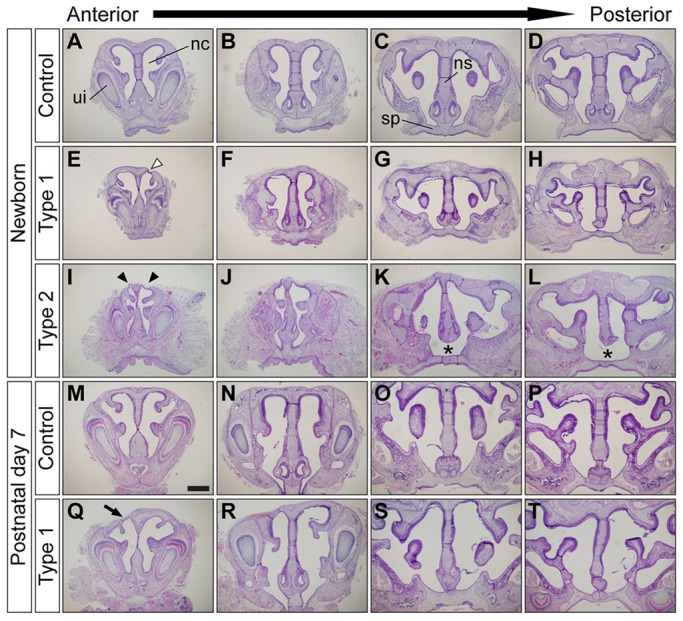
Augmentation of BMP signaling leads to nasal cartilage defects. (A-L) Frontal sections of newborn control (A-D), type 1 (E-H) and type 2 mutant (I-L) nasal cavities. Type 1 mutants had unilateral nasal cartilage defects (E, white arrowhead). Type 2 mutants had bilateral nasal cartilage defects (I, arrowheads), severe nasal cavity stenosis (I,J) and failure of fusion between the nasal septum and the secondary palate (K,L, asterisks). (M-T) Histological comparison of nasal structures between control (M-P) and type 1 surviving mutants (Q-T) at P7. Frontal sections from four different anteroposterior levels of nasal cavity were stained using Hematoxylin and Eosin (H&E). Type 1 mutants had unilateral nasal cartilage defects (Q, arrow). ui, upper incisor; nc, nasal cavity; ns, nasal septum; sp, secondary palate. Scale bar: 500 µm.
In mice, the nasal septum grows inferiorly from the nasofrontal prominence to the level of the palatal shelves (Neskey et al., 2009). The nasal septum and the secondary palate fuse between late embryonic day (E)14 and early E15; the palatal shelves fuse around the same time (Walker and Fraser, 1956). Thus, at E15.5 and E16.5, we found that ∼50% of the mutant embryos showed failure of nasal septum fusion to the secondary palate (data not shown). Next, we investigated abnormalities during nasal cartilage development at E14.5 before these fusion events. Alcian Blue staining revealed differentiated chondrocytes forming nasal capsule and nasal septum (Fig. 3A,B for the anterior portion corresponding to Fig. 2A and Fig. 3E,F for the posterior portion corresponding to Fig. 2C). In the mutants, staining intensity was reduced in nasal cartilage (Fig. 3D,H, arrows). Furthermore, histological analysis of mutant embryos showed a shorter nasal septum (Fig. 3M,O, brackets) and discontinuous nasal cartilage (Fig. 3L,P, arrowheads). During this stage, all mutants (n>20) had a very similar phenotype, thus we were unable to distinguish type 2 mutants from type 1. These findings indicate that nasal cartilage formation was markedly poor in mutants as early as E14.5.
Fig. 3.
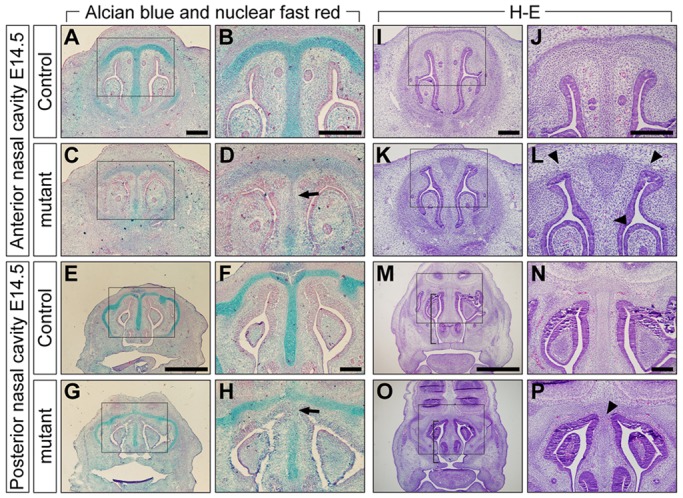
Inhibition of chondrogenesis in mutants at E14.5. (A-H) Alcian Blue and Nuclear Fast Red staining of E14.5 controls (A,B,E,F) and mutants (C,D,G,H). The level of Alcian Blue-positive mucin was reduced in mutant nasal cartilage (D,H, arrows). (I-P) H&E staining of E14.5 controls (I,J,M,N) and mutants (K,L,O,P). Mutants had a shorter nasal septum (M,O, brackets) and cartilage defects in both anterior and posterior nasal cavity levels (L,P, arrowheads). Boxed areas in A,C,E,G,I,K,M, and O are shown at higher magnification in B,D,F,H,J,L,N and P, respectively. Scale bars: 200 µm in A,B,F,I,J,N; 1 mm in E,M.
Expression level of FGF signaling components in the nasal cavity
Many studies have demonstrated that fibroblast growth factor (FGF) signaling components act as negative regulators of bone and cartilage growth (Coffin et al., 1995; Colvin et al., 1996; Sahni et al., 2001; Murakami et al., 2004; Raucci et al., 2004). We previously reported that FGF signaling components were upregulated in the anterior frontal suture of ca-Bmpr1a:P0-Cre mutants at E17.5 (Komatsu et al., 2013). These results raise the possibility that enhanced FGF signaling leads to nasal cartilage dysmorphogenesis in the mutants. To address this possibility, we measured the level of phosphorylated ERK1 and ERK2 (MAPK3 and MAPK1 – Mouse Genome Informatics) (hereafter referred to as pERK1/2) as an indicator of FGF signaling in the nasal portion of newborn control and mutant mice (Fig. 4A). As expected, we confirmed enhanced FGFR1 and FGFR2 production in the anterior frontal suture of both types of mutants (supplementary material Fig. S2); however, pERK1/2 level in the nasal region was comparable between all mice (Fig. 4A). We also examined levels of pERK1/2 and FGF pathway components in the nasal cartilage at E14.5 by immunohistochemistry. FGF2, FGFR1 and FGFR2 were abundant in both mucosal epithelium and nasal septal chondrocytes of embryonic controls and mutants (Fig. 4F-Q). FGFR3 and pERK1/2 signals were weak in all mice at E14.5 (Fig. 4B-E,R-U). These results indicate that altered FGF signaling is not likely to be the etiology for the mutant nasal cartilage phenotype.
Fig. 4.
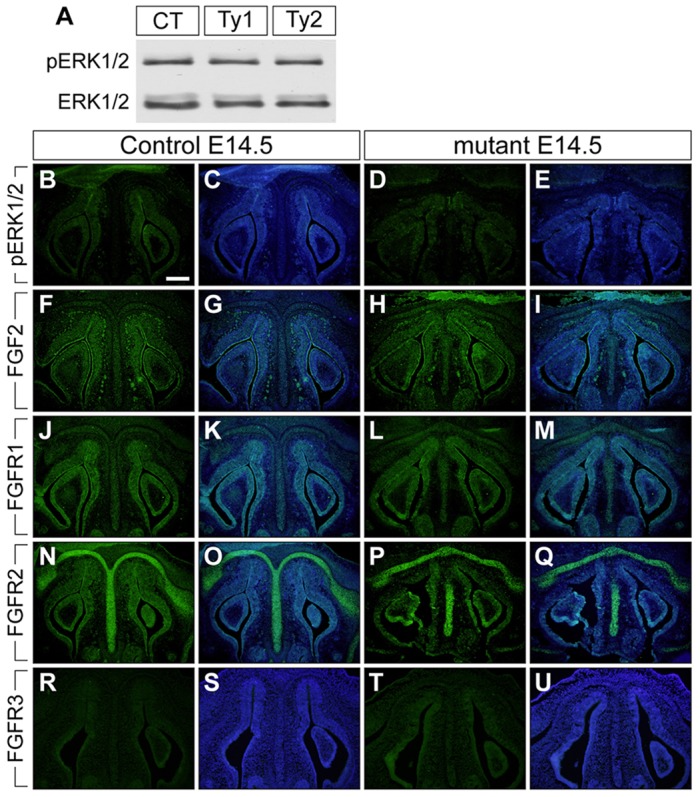
Augmentation of BMP signaling did not change FGF signaling components in the nasal portion of mutant at E14.5. (A) The levels of pERK1/2 were examined using protein lysate from control (CT), type 1 (Ty1), and type 2 mutant (Ty2) nasal tissues. Total ERK1/2 was used as a loading control. (B-U) Immunohistochemistry was performed using antibodies against pERK1/2, FGF2, FGFR1, FGFR2 and FGFR3 (green). The levels of FGF2, FGFR1 and FGFR2 were upregulated in both control and mutant nasal cartilage and mucosal epithelium at E14.5 (F-Q). C,E,G,I,K,M,O,Q,S and U are merged images, with DAPI staining shown in blue. Scale bar: 200 µm.
Cell survival in nasal mesenchymal tissues
In our previous study of ca-Bmpr1a:P0-Cre mice, increased cell death was observed in neural crest-derived skull tissues such as the anterior frontal suture (Komatsu et al., 2013). Therefore, we focused on cell death to investigate the mechanisms responsible for morphological changes in the nasal cavity. First, we examined the distribution of neural crest-derived cells in E12.5 nasal tissues by crossing P0-Cre mice with ROSA26-tdTomato Cre reporter mice (Fig. 5A). The cartilage primordia in these mice were positive for tdTomato, which is consistent with previous studies in avians indicating that chondrocytes in nasal cartilage derive from cranial neural crest cells (Le Lievre, 1978; Le Douarin et al., 2004). Next, we performed terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay to examine apoptosis in frontal sections of E12.5 controls and mutants. By E12.5, mutants already exhibited nasal cavity abnormalities such as reduced nasal septal size (Fig. 5B,C). In cartilage primordia of control mice, TUNEL-positive cells were sparse (Fig. 5E; supplementary material Fig. S3N,P,R,T for higher magnification). By contrast, apoptotic cells were substantially increased in mutant nasal septa (Fig. 5F; supplementary material Fig. S3O,Q,S,U for higher magnification). As we previously reported, reducing Bmpr1a dosage by superimposing Bmpr1a heterozygosity (Bmpr1a+/−) (Mishina et al., 1995) onto the mutant genome rescues the craniosynostosis phenotype (Komatsu et al., 2013). To understand how this genetic alteration contributes to the nasal phenotype, we examined both morphology and cell death in ca-Bmpr1a:P0-Cre mice carrying one copy of endogenous Bmpr1a, ca-Bmpr1a:P0-Cre:Bmpr1a+/− (rescued mice). Genetically rescued mice at E12.5 had comparable nasal septal length to controls with almost no increase in either the number of apoptotic cells or p53 production in their nasal cartilage primordia (Fig. 5D,G,J).
Fig. 5.
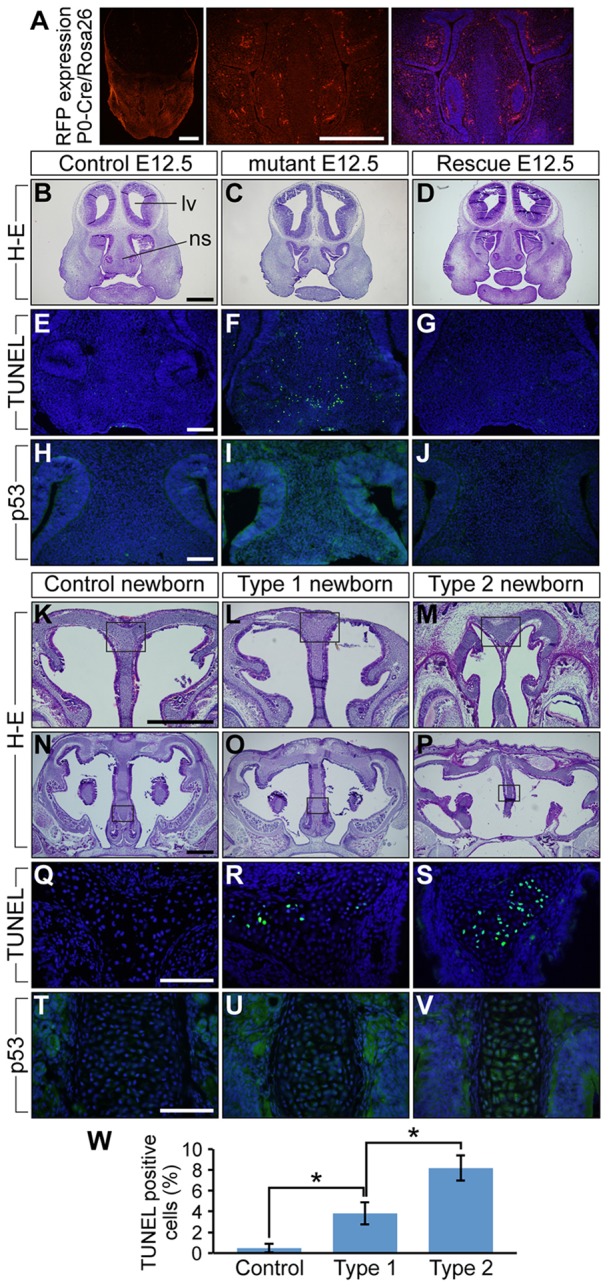
Augmentation of BMP signaling leads to increased levels of cell death and p53 protein in developing nasal cartilage of E12.5 mice and newborns. (A) tdTomato distribution in E12.5 P0-Cre transgenic mice crossed with ROSA-tdTomato Cre reporter mice. The right panel is the tdTomato image merged with DAPI staining, which is shown in blue. (B-D) Frontal sections of E12.5 control, mutant and ca-Bmpr1a:P0-Cre:Bmpr1a+/− (rescue) mice were stained using H&E. Control and rescue mice had comparable nasal septum morphology; however, mutants had shorter nasal septa. lv, lateral ventricle; ns, nasal septum. (E-G) TUNEL assay was performed to detect apoptosis. E12.5 mutants exhibited increased numbers of apoptotic cells shown in green (F). Blue indicates DAPI staining. (H-J) Immunohistochemistry using p53 antibody revealed increased p53 levels in mutant nasal septa (I). (K-P) Frontal sections of newborn controls, type 1 and type 2 mutants were stained using H&E. Boxed areas in K-P are shown at high magnification in Q-V, respectively. (Q-S) TUNEL assay was performed to detect apoptosis. Both type 1 and type 2 newborn mutants exhibited more apoptotic cells, which are shown in green (R,S). (T-V) Immunohistochemistry using p53 antibody revealed increased p53 levels in both mutant nasal septa (U,V). (W) Quantification of TUNEL assay results in newborns. All data are represented as mean±s.d. (n=3); *P<0.01 (Student's t-test). Scale bars: 500 µm in A,B,K,N; 100 µm in E,H,Q,T.
Next, we examined the extent to which cell death is involved in morphological differences between type 1 and type 2 mutants by comparing the number of TUNEL-positive cells and p53 levels in newborn pups. The number of apoptotic cells was significantly higher in the nasal cartilage of type 2 mutants (Fig. 5R,S,W). These results suggest a link between BMP signaling levels and the amount of cell death and subsequent severity of nasal cartilage defects. Additionally, we examined mesenchymal cell proliferation at E12.5 by Ki67 immunohistochemical staining. Large numbers of Ki67-positive cells were identified in nasal mesenchymal tissues (supplementary material Fig. S3C-L); however, there was no significant difference between controls and mutants (supplementary material Fig. S3M). We also investigated chondrocyte differentiation in condensing mesenchymal nasal tissues at E12.5. SOX9, required for mesenchymal cell condensation and differentiation, was highly expressed in condensed mesenchymal cells as expected; however, no significant differences were found between controls and mutants (supplementary material Fig. S3A,B). In the context of augmented BMP signaling, our results lend credence to the idea that increasingly severe nasal defects trend with increased cell death rather than reduced proliferation through FGF signaling.
Enhanced p53-mediated apoptosis is responsible for nasal cartilage abnormalities
There is mounting evidence showing that BMP4 treatment can induce p53-mediated apoptosis (Fukuda et al., 2006; Kim et al., 2006; Haubold et al., 2010). However, the molecular mechanisms connecting these two pathways are poorly understood. Because we demonstrated that pSMAD1 physically interacts with p53, reducing its degradation in cultured cells (Chau et al., 2012), we tested the possibility that enhanced BMP-Smad signaling prevents degradation of p53 in ca-Bmpr1a:P0-Cre mutants. First, we compared p53 production in nasal septa harvested at E12.5 and from newborn mice from each group. Notably, p53 was highly abundant in mutants (Fig. 5I,U,V). To evaluate the involvement of p53 in nasal cartilage dysmorphogenesis, we intraperitoneally administered pifithrin-α (PFT), a p53-specific inhibitor, to pregnant females from E8.5 to E18.5. PFT is known to reduce apoptosis through inhibition of transcription events dependent upon p53 (Komarov et al., 1999), and it also rescues the craniofacial defects found in Tcof1+/− mutants by reducing apoptosis in the neural tube (Jones et al., 2008). Excitingly, PFT-treated ca-Bmpr1a:P0-Cre mutants sacrificed shortly after birth exhibited a nasal septum that was normally fused with the secondary palate as well as nasal capsule cartilage that was structurally similar to that of newborn controls (Fig. 6J,N). Skeletal abnormalities of the mutants were partially rescued by PFT treatment. Treated mutants showed improvement with regards to both hypertelorism (Fig. 6E-H, brackets, 6.39 mm±0.19 mm for PFT-treated mutants versus 7.02 mm±0.17 mm for vehicle-treated mutants, n=5, P=0.00062, ±s.d.) and nasal bone length (supplementary material Fig. S4D,H,I, 1.72 mm±0.07 mm for PFT-treated mutants versus 0.89 mm±0.11 mm for vehicle-treated mutants, n=5, P=0.00001, ±s.d.). Neonatal lethality in mutants was also ameliorated [PFT treatment from E8.5 to E18.5 resulted in death of 2 out of 19 mutants (9.0%), vehicle injection resulted in death of 10 out of 19 mutants (52.5%), P=0.00087]. Furthermore, treated mutants showed significantly reduced levels of apoptosis in condensed mesenchymal cells at E12.5 when treated with PFT from E8.5 to 11.5 (Fig. 6R,U). By contrast, vehicle-treated mutants showed no improvement with regards to apoptosis (Fig. 6T,U). We next evaluated the effect of PFT treatment on BMP signaling activity by western blotting utilizing the anti-pSMAD1/5/9 antibody. The BMP signaling levels of PFT-treated mutants were significantly higher than those of controls but comparable to untreated type 1 or type 2 mutants (Fig. 6V,W). Taken together, these results strongly support the notion that p53-mediated apoptosis is the downstream event resulting from augmented BMP-Smad signaling activity and is one of the causes of the nasal cartilage defects.
Fig. 6.

p53-specific chemical inhibition by pifithrin-α rescues nasal tissue defects in vivo. (A-H) Newborn skulls from PFT-treated controls (A,E), PFT-treated mutants (B,F), vehicle-treated controls (C,G) and vehicle-treated mutants (D,H). PFT was intraperitoneally injected to pregnant mice from E8.5 to E18.5. Nasal bone defects found in vehicle-treated mutants were ameliorated by PFT treatment (F,H, white arrows). Hypertelorism found in vehicle-treated mutants was also partially rescued by PFT treatment (F,H, brackets). (I-P) Histological analysis of PFT- and vehicle-treated mice. PFT-treated mutants (J,N) had structurally comparable nasal cartilage to both PFT- and vehicle-treated controls (I,K,M,O), whereas vehicle-treated mutants had nasal cartilage defects (L,P). (Q-T) TUNEL assay in E12.5 PFT- and vehicle-treated mice. PFT was intraperitoneally injected from E8.5 to E11.5. Green indicates TUNEL-positive nuclei and blue indicates DAPI. PFT treatment reduced the number of TUNEL-positive cells. Scale bars: 1 mm in A,E; 500 μm in L,M; 100 μm in Q. (U) Quantification of TUNEL assay. Vehicle-treated mutants showed a significantly higher number of TUNEL-positive cells (n=5). *P<0.005 (Student's t-test). CT, control; mut, mutant. (V) The levels of pSMAD1/5/9 were evaluated by western blotting. Protein lysates were collected from vehicle-treated controls, PFT-treated controls, PFT-treated mutants, vehicle-treated type 1 mutants and vehicle-treated type 2 mutants. GAPDH was used as a loading control. MT, mutant. (W) Results of western blotting were quantified. pSMAD1/5/9 levels of PFT-treated mutants were significantly higher than those of PFT- and vehicle-treated controls (n=3-9 per group). Ty1, type 1 mutant; Ty2, type 2 mutant. *P<0.005 (Student's t-test). All data are represented as mean±s.d.
Augmented BMP signaling prompts the p53-mediated apoptotic pathway by preventing interaction between p53 and MDM2
To dissect the molecular mechanism governing how augmented BMP signaling results in p53-mediated apoptosis, we examined the mRNA expression levels of p53 and its regulatory network by real-time reverse transcription PCR (RT-PCR). Bax expression was increased in both type 1 and type 2 mutants. Bax is one of the downstream targets of p53 and is an apoptosis-promoting gene (Oltvai et al., 1993). The levels of caspase 3, which is involved in the execution phase of apoptosis, were increased in type 2 mutants (Fig. 7A). These findings are consistent with increased levels of p53 phosphorylated at serine 15 (p-p53-Ser15) in both mutants, as revealed by western blot analysis (Fig. 7B). p-p53-Ser15 is known to activate the apoptotic cascade (Shieh et al., 1997; Siliciano et al., 1997; Sluss et al., 2004). It is noteworthy that type 2 mutants showed much higher levels of p-p53-Ser15 than type 1 mutants, despite no significant difference in total p53 levels between control and either of the mutants (Fig. 7B; see Fig. 8 for more details). Interestingly, there were no differences in expression levels of Cdkn1a (which encodes p21) or Gadd45a, both of which are involved in the p53-mediated cell cycle arrest pathway (Fig. 7A). The p53 expression levels were unchanged in both mutants (Fig. 7A). DNA damage response kinases such as ataxia telangiectasia mutated kinase (ATM) and ATM-Rad3-related protein (ATR), which are known to activate p53 by phosphorylation (Canman et al., 1998; Tibbetts et al., 1999), were also unchanged (Fig. 7A). These results suggest that activation of p53-mediated apoptosis is likely caused by post-translational modification of p53 rather than by DNA damage or higher expression of p53. This prompted us to evaluate expression levels of Mdm2 and Pten. It is reported that MDM2 ubiquitylates p53 to prompt its degradation (Momand et al., 1992; Kussie et al., 1996; Lai et al., 2001). PTEN negatively regulates phosphoinositide 3-kinase (PI3K)-initiated activation of MDM2 (Stambolic et al., 1998; Mutter, 2001) and its expression is positively regulated by BMP signaling (Waite and Eng, 2003a,b). RT-PCR demonstrated there was no significant difference in Mdm2 and Pten expression levels between control and mutant samples (Fig. 7A). We also evaluated MDM2 production level in cell lysates from nasal tissue. Western blot analysis using an anti-MDM2 antibody showed no difference between control and mutant samples (Fig. 7B).
Fig. 7.

Augmentation of BMP signaling leads to p53-mediated apoptosis by preventing MDM2-mediated p53 degradation. (A) Expression levels of p53, Bax, caspase 3, p21, Gadd45a, Mdm2, Pten, Atm and Atr were measured by RT-PCR. Total RNA was isolated from the nasal tissue of newborn controls, type 1 and type 2 mutants (n=4-6 per group). Expression levels of these mRNAs were normalized to Gapdh mRNA. *P<0.01, **P<0.005 (Student's t-test). (B) p-p53-Ser15, total p53 and MDM2 levels were examined by western blotting. GAPDH was used as a loading control. Protein lysate was isolated from nasal tissues of controls (CT), type 1 (Ty1) and type 2 (Ty2) mutants. The levels of p-p53-Ser15 (left), total p53 (center) and MDM2 (right) were normalized to GAPDH and compared between control (CT), type 1 and type 2 mutants (n=4 per group). *P<0.05 (Student's t-test). (C) MDM2-p53 and pSMAD1/5/9-p53 complex formation levels were examined by immunoprecipitation (IP) and immunoblotting (IB). Immunoprecipitation was performed using p53 antibody. MDM2 and pSMAD1/5/9 levels were examined using cell lysates after immunoprecipitation. Input indicates levels of MDM2 or pSMAD1/5/9 in 10% protein lysates before immunoprecipitation. MDM2 (left graph) and pSMAD1/5/9 (right graph) interaction levels were quantified and compared between control, type 1 and type 2 mutants (n=3 per group). *P<0.05, **P<0.005 (Student's t-test). All data are represented as mean±s.d.
Fig. 8.
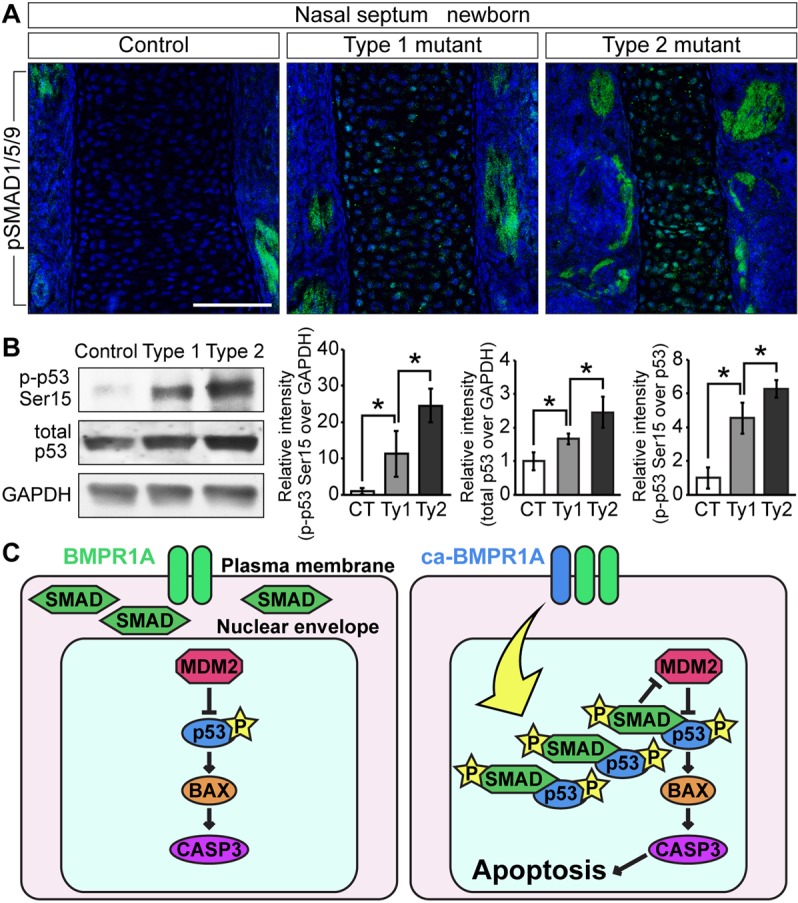
Nuclear accumulation of pSMAD leads to activation of p53 by inhibiting MDM2-mediated p53 degradation in the nasal cartilage. (A) Immunohistochemistry was performed in newborn controls, type 1 and type 2 mutants using pSMAD1/5/9 antibody (green). pSMAD1/5/9 was accumulated in the nuclei of mutant nasal septal chondrocytes. These images were taken by confocal laser-scanning microscopy. Blue indicates DAPI staining. Scale bar: 100 µm. (B) p-p53-Ser15, total p53 and GAPDH levels were quantified by western blotting. Protein lysate was isolated from dissected nasal cartilage of controls (CT), type 1 (Ty1) and type 2 (Ty2) mutants. The levels of p-p53-Ser15 were normalized to GAPDH (left) or total p53 (right). The levels of p53 were normalized to GAPDH (center). These protein levels were compared between controls, type 1 and type 2 mutants. The data are represented as mean ± s.d. (n=3 per group). *P<0.05 (Student's t-test). (C) Cartoon depicting BMP signaling activating p53-mediated apoptosis. Nuclear accumulation of pSMAD facilitates its interaction with p53. This p53 stabilization results in inhibition of MDM2-mediated p53 degradation. This event activates the p53-mediated apoptosis pathway in mutant nasal cartilage.
In the previous study, we demonstrated that SMAD1 physically interacts with p53, thus preventing MDM2-mediated p53 degradation in cultured cells (Chau et al., 2012). Considering the fact that increased p53 protein levels are found in the mutants whereas p53 transcription levels remain constant (Fig. 5I,U,V; Fig. 7A), we hypothesized that augmented BMP signaling increases the nuclear localization of pSMAD1/5/9 to prevent interaction between p53 and MDM2, thus reducing MDM2-mediated p53 degradation. We isolated cells from newborn nasal tissues to prepare lysate. Immunoprecipitation using an antibody against p53 followed by western blot analysis revealed that the amount of complex between MDM2 and p53 was lower in type 1 mutants than in controls and was barely detectable in type 2 mutants (Fig. 7C). Next, we evaluated the amount of pSMAD1/5/9 interacting with p53 in controls and mutants. Western blot analysis utilizing anti-pSMAD1/5/9 antibody revealed that mutants had higher levels of pSMAD1/5/9 bound to p53. In particular, type 2 mutants showed more pSMAD1/5/9 bound to p53 than type 1 (Fig. 7C).
Although BMPR1A is expressed broadly in nasal tissue (supplementary material Fig. S5A), p53 (Fig. 5U,V) and apoptotic cells (Fig. 5R,S) were readily observed in mutant nasal cartilage. To understand why the nasal cartilage is the most affected nasal tissue, we evaluated the patterns of Cre excision in P0-Cre/ROSA26RlacZ mice (supplementary material Fig. S5B), as well as pSMAD1/5/9 and cell death between controls and type 2 mutants (supplementary material Fig. S5C,D). Concordant with the lacZ staining patterns, we observed increases in pSMAD1/5/9 in both cartilage and bones. It is, however, noteworthy that the nasal cartilage in the mutants shows a significantly higher number of TUNEL-positive cells when compared with either mutant nasal bones or corresponding tissues in controls. Confocal laser-scanning microscopy also confirmed that pSMAD1/5/9 accumulated in the nuclei of mutant cartilaginous nasal septa to a higher degree in type 2 than type1 mutants (Fig. 8A). Thus, we repeated the immunoblot analyses using the protein lysates extracted from dissected nasal cartilage. Total p53 levels in the lysates from nasal cartilage increased 1.7-fold in the type 1 mutants and 2.5-fold in the type 2 mutants, the ratio between p-p53-Ser15 and total p53 increased 4.5-fold in the type 1 mutants and 6.3-fold in the type 2 mutants (Fig. 8B). Because the levels of p53 were locally higher in mutant nasal cartilage (Fig. 5T-V), it is likely that increases in total p53 were masked when proteins were prepared from the entire nasal portion. These results strongly suggest that increased nuclear levels of pSMAD1/5/9 prevent the MDM2-p53 interaction, consequently inhibiting p53 degradation, which results in activation of apoptotic pathways as seen in nasal cartilage of ca-Bmpr1a:P0-Cre mice.
DISCUSSION
Our present results provide a detailed picture of how augmented BMP signaling through BMPR1A in neural crest cells induces p53-mediated apoptosis in developing cartilage. In ca-Bmpr1a:P0-Cre mice, increased BMP type 1 receptor kinase activity results in increased nuclear accumulation of pSMAD1/5/9. pSMAD1/5/9 then interacts with p53, inhibiting the association between p53 and MDM2. This decreases p53 degradation. Stabilization of p53 leads to transcriptional activation of its downstream targets such as Bax and caspase 3 in the nasal cartilage primordia. Such alterations induce caspase 3-mediated apoptosis and cause abnormal nasal structures such as short snouts and nasal cartilage defects. Furthermore, we demonstrated that inhibition of p53 activity reduced apoptosis in developing nasal cartilage and partially rescued the mutant phenotype. Based on these findings, we propose a molecular model that attempts to account for the phenotype (Fig. 8C).
In both mouse and human embryos, a cartilaginous nasal capsule surrounds the nasal cavity and is continuous with the cartilage of the nasal septum (Neskey et al., 2009). This cartilaginous capsule acts as a strut providing structural support to the nasal cavity. Therefore, reduced nasal airway volume due to choanal atresia, nasal septal defects or deviation leads to difficulties in nasal respiration (Hall, 1979; Tardy and Garner, 1990; Yilmaz and Altuntas, 2005). In severe cases, the nasal obstruction leads to asphyxia or sudden death in human infants (Shaw, 1968; Hall, 1979). Congenital septal defects (CSDs) are also documented in humans. Failure of nasal septum fusion to the secondary palate was discovered in these cases, and these patients also exhibited signs of nasal obstruction (Dogru et al., 2004; Yilmaz and Altuntas, 2005). We found abdominal distension and air bubbles in the gastrointestinal tract of type 2 mutants. Similar findings are reported in neonatal mice with cleft palate and retrognathia (Winograd et al., 1997; Dudas et al., 2004; Morita et al., 2004). These abnormal oral structures have been thought to prevent normal nasal respiration and lead to neonatal lethality in mice; however, we did not observe cleft palate in our mutant mice. The only overt morphological changes we observed were nasal cavity defects. We are not certain whether these defects are the direct cause of gradual abdominal distension and neonatal lethality observed in type 2 mutants.
Mutations in BMP inhibitors such as Noggin (Nog) and Chordin (Chrd) are also reported to induce craniofacial abnormalities. Nog−/− mutants show a severe skull phenotype that includes exencephaly (McMahon et al., 1998). Nog+/−;Chrd−/− mutants exhibit phenotypes involving medial forebrain tissue, such as cyclopia as well as a lack of jaw elements derived from cranial neural crest cells (Bachiller et al., 2000; Anderson et al., 2002). Consistent with our findings in these mutants, programmed cell death in cranial tissues is dramatically increased (Anderson et al., 2002). The origin of abnormalities in these mice could possibly be traced back to patterning defects developed at the gastrulation stage, because these are global knockouts in which BMP signaling is augmented in several tissues. Unfortunately, neural crest-specific mutation of these genes has not been reported yet. In our ca-Bmpr1a:P0-Cre mice, BMP signaling is augmented only in neural crest-derived cells after their emergence, which provides a plausible explanation for deviations from phenotypes reported for those mice mutant for BMP antagonists. In chondrogenesis, condensed mesenchymal cells give rise to chondrocytes, and SOX9 is required for this fate (Lefebvre et al., 1997). Mounting evidence also suggests that FGF signaling negatively regulates chondrogenesis (Coffin et al., 1995; Colvin et al., 1996; Sahni et al., 2001; Murakami et al., 2004; Raucci et al., 2004). FGF ligand, FGF receptors and downstream FGF components were enhanced in the anterior frontal suture of ca-Bmpr1a:P0-Cre mice (Komatsu et al., 2013). These results prompted us to evaluate the levels of SOX9 and FGF signaling components. In our immunohistochemistry experiments, levels of SOX9 and FGF signaling components in mutants were comparable to those of control mice, whereas the number of apoptotic cells was significantly increased in mutant condensing mesenchymal cells and chondrocytes. These results indicate that cell death in mesenchymal cells and chondrocytes is likely to be the cause of the nasal phenotype in our mutants.
In our study, PFT treatment successfully reduced apoptosis in developing nasal septal primordia and rescued the nasal cartilage phenotype. Furthermore, PFT-treated mutants exhibited comparable pSMAD1/5/9 levels to vehicle-treated mutants. These results indicate that p53-mediated cell death is crucial to the etiopathophysiology underlying nasal cartilage dysmorphogenesis. TUNEL assay revealed that cell death was increased in the chondrocytes of newborns as well as in mesenchymal cells at E12.5. It remains unclear, however, which of these cells is responsible for the nasal cartilage phenotype. There is mounting evidence that p53 induces chondrocyte apoptosis both in developing cartilage and in cell culture. It is known that p53 induces cell death in articular as well as Meckel's cartilage (Trichilis and Wroblewski, 1997; Okazaki et al., 2003). In human chondrocyte culture, PFT treatment suppresses p-p53-Ser15 as well as chondrocyte apoptosis induced by mechanical stress (Hashimoto et al., 2009). Additional studies with PFT are necessary to identify the cell type that is responsible for the nasal phenotype.
In ca-Bmpr1a:P0-Cre mutants, p-p53-Ser15 level and downstream targets of p53 such as Bax and caspase 3 were upregulated. By contrast, Atm and Atr, DNA damage signaling kinases, and p53 mRNA expression levels were unchanged. A possible explanation of this phenomenon is post-translational regulation of p53 by ubiquitin ligase. It is reported that p53 is ubiquitylated by ubiquitin E3 ligases (Brooks and Gu, 2006). Among several E3 ligases, MDM2 is identified as a key E3 ligase in the p53 regulatory network (Montes de Oca Luna et al., 1995). In this study, we quantified levels of MDM2-p53 complex formation and found a reduction in the mutants. Moreover, we previously found that SMAD1 decreases the ubiquitylation level of p53 in cultured cell lines (Chau et al., 2012). These results suggest that ubiquitylation levels of p53 might be reduced in ca-Bmpr1a:P0-Cre mutants. Thus, it is an interesting future endeavor to determine p53 ubiquitylation levels in our mutants.
We also found that levels of p21 and Gadd45a, and proliferating cell number were unchanged in ca-Bmpr1a:P0-Cre mutants. The mechanism by which p53 mediates either apoptosis or growth arrest is poorly understood. However, it is reported that certain viral or mammalian proteins can direct p53 transcriptional specificity towards apoptosis or growth arrest. For instance, BRCA1 as well as the virus protein NS5A, both of which physically associate with p53, have been shown to stabilize p53, thus activating p53 transcriptional specificity towards genes involved in cell cycle arrest but not the apoptotic pathway (Majumder et al., 2001; MacLachlan et al., 2002). Additionally, the bacterial redox protein azurin is known to induce only p53-mediated apoptosis (Yamada et al., 2004). Interestingly, mutations in azurin (M44KM64E) shift p53 specificity towards growth arrest (Yamada et al., 2004). Although we do not have experimental results at this moment, we speculate that complex formation between pSMAD and p53 might direct p53 transcriptional specificity towards apoptosis.
One of the outstanding questions is whether the same mechanisms are applicable for normal developmental processes. There is mounting evidence that BMP signaling components, such as BMP2, BMP4, BMP7 and MSX2, are involved in apoptosis during skin and tooth development (Vaahtokari et al., 1996a,b; Sharov et al., 2003). In cancer cells, BMP4 can induce apoptosis through a p53-dependent mechanism (Fukuda et al., 2006; Kim et al., 2006; Haubold et al., 2010). We previously found that the BMP-Smad1 pathway participates in the DNA damage response by interacting with p53 (Chau et al., 2012). Thus, it is possible to speculate that some BMP-induced apoptosis during embryogenesis is p53 dependent. However, p53-null mice are viable and develop normally (Donehower et al., 1992). By contrast, mice lacking the Tp63 gene (encoding p63), which is closely related to p53, show severe developmental defects in structures requiring epithelial-mesenchymal interactions, such as hair follicles, limbs, teeth and mammary glands (Mills et al., 1999). Notably, p63 was found to be a direct transcriptional target of SMAD4 and SMAD5 in zebrafish (Bakkers et al., 2002). These findings suggest the possibility that BMP signaling regulates both p53 and p63 to induce apoptosis during embryogenesis. In the nasal cartilage of ca-Bmpr1a:P0-Cre mutants, we found nuclear accumulation of pSMAD1/5/9 as well as increased expression of the proapoptotic genes Bax and caspase 3. We also found increased levels of p-p53-Ser15 and total p53, and increased physical interaction between p53 and pSMAD1/5/9. In conjunction with the finding that MDM2-p53 complex formation was reduced in mutants, we propose a model where augmented BMP signaling through BMPR1A enhances p53 activity by inhibiting MDM2-mediated p53 degradation (Fig. 8C).
MATERIALS AND METHODS
Animals
Generation of the mouse line expressing the constitutively active form of Bmpr1a (ca-Bmpr1a) used in this study has been described previously (Kamiya et al., 2008; Komatsu et al., 2013). The P0-Cre mouse line, C57BL/6J-Tg(P0-Cre)94Imeg (ID 148), was provided by CARD, Kumamoto University, Japan (Yamauchi et al., 1999). They were maintained in a mixed background of 129S6 and C57BL/6J. Embryos were collected from timed pregnant mice, and embryonic ages were determined by the day when the vaginal plug was discovered, which was designated as E0.5. Embryonic tails were used for PCR-based genotyping. All mouse experiments were performed in accordance with University of Michigan guidelines covering the humane care and use of animals in research.
Histology, skeletal staining and immunohistochemistry
E12.5, E14.5, newborn and P7 heads were fixed in 4% paraformaldehyde (PFA) at 4°C overnight. P7 heads were decalcified in 10% EDTA for 10 days. They were embedded in paraffin and sectioned at 7 µm for Hematoxylin and Eosin staining or Alcian Blue and Nuclear Fast Red staining. For immunohistochemistry, these embryos were fixed in 4% PFA at 4°C overnight, incubated in 20% sucrose/PBS at 4°C overnight, embedded in O.C.T. compound (Sakura Finetek, Tokyo, Japan) and serially sectioned at 10 µm. The samples were incubated with rabbit anti-FGF2 (1:100, AB1458, Chemicon), rabbit anti-FGFR1 (1:100, sc-121, Santa Cruz Biotechnology), rabbit anti-FGFR2 (1:100, sc-122, Santa Cruz Biotechnology), rabbit anti-FGFR3 (1:100, sc-123, Santa Cruz Biotechnology), rat anti-Ki67 (1:50, M7249, Dako), rabbit anti-SOX9 (1:100, sc-20095, Santa Cruz Biotechnology), rabbit anti-p53 (1:50, sc-6243, Santa Cruz Biotechnology), rabbit anti-BMPR1A (1:100, AP2004B, Abgent) and rabbit anti-pSMAD1/5/9 (1:100, 13820, Cell Signaling Technology) at 4°C overnight. Alexa Fluor 488-conjugated donkey anti-rabbit IgG (1:100, A21206, Invitrogen) or Alexa Fluor 488-conjugated goat anti-mouse IgG1 (1:100, A21121, Invitrogen) were used as secondary antibodies. Sections were mounted with ProLong Gold antifade reagent with DAPI (P36935, Invitrogen).
TUNEL assays
E12.5 and newborn heads were fixed in 4% PFA at 4°C overnight, incubated in 20% sucrose in PBS at 4°C overnight, embedded in O.C.T. compound and sectioned at 10 µm. Levels of cell death were measured by employing the TdT-mediated dUTP nick-end labeling (TUNEL) method using the In Situ Cell Death Detection Kit (Roche). DNA strand breaks were labeled with fluorescein and visualized with FITC.
Treatment of animals with the p53 inhibitor pifithrin-α
For treatment of animals, a 1.68 mg/ml stock solution of pifithrin-α (Sigma) was made with 50% DMSO in PBS. This stock solution was diluted ten times with PBS prior to injection and injected intraperitoneally (2.2 mg/kg) into pregnant mice on days E8 to E18. Injection of the vehicle alone (5% DMSO in PBS) was performed for vehicle-treated groups.
Immunoblot analysis
Nasal portions of each embryo were lysed in RIPA buffer (20 mM Tris-HCl, 0.1% SDS, 1% Triton X-100, 1% sodium deoxycholate) using a Bio-Gen Pro200 homogenizer (Pro Scientific). The resulting lysates were run on 10% Mini-Protean TGX gels (Bio-Rad) and transferred to Amersham Hybond-P membrane (GE Healthcare). The following antibodies were used for immunodetection: rabbit anti-pSMAD1/5/9 (1:1000, 9511, Cell Signaling Technology), rabbit anti-pSMAD1/5/9 (1:1000, 13820, Cell Signaling Technology), rabbit anti-p44/42 MAPK (ERK1/2) (1:1000, 4695, Cell Signaling Technology), rabbit anti-phospho-p44/42 MAPK (pERK1/2) (1:1000, 4376, Cell Signaling Technology), mouse anti-MDM2 (1:500, sc-965, Santa Cruz Biotechnology), rabbit anti-phospho-p53 (p-p53) (Ser15) (1:500, ab1431, Abcam), rabbit anti-p53 (1:2000, sc-6243, Santa Cruz Biotechnology) and rabbit anti-GAPDH antibody (1:2000, 2118, Cell Signaling Technology). Super Signal West Pico Chemiluminescent Substrate (Pierce Biotechnology) was used to detect bound antibody against ERK1/2, pERK1/2, p-p53 (Ser37) and GAPDH. SuperSignal West Femto Maximum Sensitivity Substrate (Pierce Biotechnology) was used to detect bound antibodies against pSMAD1/5/9, p-p53 (Ser15) and MDM2.
Immunoprecipitation of p53-associated protein
Primary cells from nasal tissue were lysed in co-immunoprecipitation buffer (20 mM Tris pH 7.5, 100 mM NaCl, 0.5% NP-40, 0.5 mM EDTA) with protease inhibitors. Immunoprecipitation was performed with anti-p53 antibody (sc-6243, Santa Cruz Biotechnology) and Dynabeads Protein G (Life Technologies) for 12 h at 4°C. The immunoprecipitates were washed four times in lysis buffer and analyzed by immunoblot analysis.
RNA isolation and quantitative real-time RT-PCR
Total RNA was isolated from the nasal portion of the skulls using RNeasy (Qiagen), according to the manufacturer's protocol. cDNA was synthesized using Superscript First-Strand Synthesis System for RT-PCR (Invitrogen). TaqMan probes were purchased and real-time RT-PCR was performed by using the ABI PRISM 7500 (Applied Biosystems). Data were normalized to GAPDH mRNA levels using the 2−ΔΔCt method.
Supplementary Material
Acknowledgements
We thank Dr Kenichi Yamamura for providing P0-Cre mice; Dr Honghao Zhang for excellent technical advice; and Drs Vesa Kaartinen, Yoshiyuki Mochida, Noriaki Ono, Taylor Nicholas Snider, Greg Scott and Honghao Zhang for critical reading and editing of the manuscript.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
S.H. and Y.M. designed the study. S.H., Y.K., H.P. and Y.M. generated materials. S.H. conducted the study and collected and analyzed data. S.H. and Y.M. interpreted data. S.H. and Y.M. wrote the manuscript. S.H., Y.K., H.P. and Y.M. approved the final version of the manuscript. S.H. and Y.M. take responsibility for the integrity of the data analysis.
Funding
This study is supported by the National Institutes of Health [grant number R01DE020843 to Y.M.]. The molecular biology core at the School of Dentistry is funded by University of Michigan. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.118802/-/DC1
References
- Anderson R. M., Lawrence A. R., Stottmann R. W., Bachiller D. and Klingensmith J. (2002). Chordin and noggin promote organizing centers of forebrain development in the mouse. Development 129, 4975-4987. [DOI] [PubMed] [Google Scholar]
- Bachiller D., Klingensmith J., Kemp C., Belo J. A., Anderson R. M., May S. R., McMahon J. A., McMahon A. P., Harland R. M., Rossant J. et al. (2000). The organizer factors Chordin and Noggin are required for mouse forebrain development. Nature 403, 658-661 10.1038/35001072 [DOI] [PubMed] [Google Scholar]
- Badoux D. M. (1966). Framed structures in the mammalian skull. Acta Morphol. Neerlando Scandinavica 6, 239-250. [PubMed] [Google Scholar]
- Bakkers J., Hild M., Kramer C., Furutani-Seiki M. and Hammerschmidt M. (2002). Zebrafish DeltaNp63 is a direct target of Bmp signaling and encodes a transcriptional repressor blocking neural specification in the ventral ectoderm. Dev. Cell 2, 617-627 10.1016/S1534-5807(02)00163-6 [DOI] [PubMed] [Google Scholar]
- Bingham B., Wang R.-G., Hawke M. and Kwok P. (1991). The embryonic development of the lateral nasal wall from 8 to 24 weeks. Laryngoscope 101, 992-997 10.1288/00005537-199109000-00011 [DOI] [PubMed] [Google Scholar]
- Brooks C. L. and Gu W. (2006). p53 ubiquitination: Mdm2 and beyond. Mol. Cell 21, 307-315 10.1016/j.molcel.2006.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canman C. E., Lim D.-S., Cimprich K. A., Taya Y., Tamai K., Sakaguchi K., Appella E., Kastan M. B. and Siliciano J. D. (1998). Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 281, 1677-1679 10.1126/science.281.5383.1677 [DOI] [PubMed] [Google Scholar]
- Chai Y. and Maxson R. E. Jr (2006). Recent advances in craniofacial morphogenesis. Dev. Dyn. 235, 2353-2375 10.1002/dvdy.20833 [DOI] [PubMed] [Google Scholar]
- Chau J. F. L., Jia D., Wang Z., Liu Z., Hu Y., Zhang X., Jia H., Lai K. P., Leong W. F., Au B. J. et al. (2012). A crucial role for bone morphogenetic protein-Smad1 signalling in the DNA damage response. Nat. Commun. 3, 836 10.1038/ncomms1832 [DOI] [PubMed] [Google Scholar]
- Coffin J. D., Florkiewicz R. Z., Neumann J., Mort-Hopkins T., Dorn G. W. II, Lightfoot P., German R., Howles P. N., Kier A. and O'Toole B. A. (1995). Abnormal bone growth and selective translational regulation in basic fibroblast growth factor (FGF-2) transgenic mice. Mol. Biol. Cell 6, 1861-1873 10.1091/mbc.6.12.1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colvin J. S., Bohne B. A., Harding G. W., McEwen D. G. and Ornitz D. M. (1996). Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat. Genet. 12, 390-397 10.1038/ng0496-390 [DOI] [PubMed] [Google Scholar]
- Copray J. C. (1986). Growth of the nasal septal cartilage of the rat in vitro. J. Anat. 144, 99-111. [PMC free article] [PubMed] [Google Scholar]
- Dogru H., Yasan H. and Tuz M. (2004). Congenital vomeral bone defect in two thalassemia trait cases. Eur. Arch. Otorhinolaryngol. 261, 136-138 10.1007/s00405-003-0619-6 [DOI] [PubMed] [Google Scholar]
- Donehower L. A., Harvey M., Slagle B. L., McArthur M. J., Montgomery C. A. Jr, Butel J. S. and Bradley A. (1992). Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356, 215-221 10.1038/356215a0 [DOI] [PubMed] [Google Scholar]
- Dudas M., Sridurongrit S., Nagy A., Okazaki K. and Kaartinen V. (2004). Craniofacial defects in mice lacking BMP type I receptor Alk2 in neural crest cells. Mech. Dev. 121, 173-182 10.1016/j.mod.2003.12.003 [DOI] [PubMed] [Google Scholar]
- Dunn N. R., Winnier G. E., Hargett L. K., Schrick J. J., Fogo A. B. and Hogan B. L. M. (1997). Haploinsufficient phenotypes in Bmp4 heterozygous null mice and modification by mutations in Gli3 and Alx4. Dev. Biol. 188, 235-247 10.1006/dbio.1997.8664 [DOI] [PubMed] [Google Scholar]
- Fukuda N., Saitoh M., Kobayashi N. and Miyazono K. (2006). Execution of BMP-4-induced apoptosis by p53-dependent ER dysfunction in myeloma and B-cell hybridoma cells. Oncogene 25, 3509-3517 10.1038/sj.onc.1209393 [DOI] [PubMed] [Google Scholar]
- Giaccia A. J. and Kastan M. B. (1998). The complexity of p53 modulation: emerging patterns from divergent signals. Genes Dev. 12, 2973-2983 10.1101/gad.12.19.2973 [DOI] [PubMed] [Google Scholar]
- Graham A., Francis-West P., Brickell P. and Lumsden A. (1994). The signalling molecule BMP4 mediates apoptosis in the rhombencephalic neural crest. Nature 372, 684-686 10.1038/372684a0 [DOI] [PubMed] [Google Scholar]
- Hall B. D. (1979). Choanal atresia and associated multiple anomalies. J. Pediatr. 95, 395-398 10.1016/S0022-3476(79)80513-2 [DOI] [PubMed] [Google Scholar]
- Hashimoto S., Nishiyama T., Hayashi S., Fujishiro T., Takebe K., Kanzaki N., Kuroda R. and Kurosaka M. (2009). Role of p53 in human chondrocyte apoptosis in response to shear strain. Arthritis Rheum. 60, 2340-2349 10.1002/art.24706 [DOI] [PubMed] [Google Scholar]
- Haubold M., Weise A., Stephan H. and Dünker N. (2010). Bone morphogenetic protein 4 (BMP4) signaling in retinoblastoma cells. Int. J. Biol. Sci. 6, 700-715 10.7150/ijbs.6.700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe A. M., Hawkins J. K. and Webster W. S. (2004). The growth of the nasal septum in the 6–9 week period of foetal development--Warfarin embryopathy offers a new insight into prenatal facial development. Aust. Dent. J. 49, 171-176 10.1111/j.1834-7819.2004.tb00069.x [DOI] [PubMed] [Google Scholar]
- Jones N. C., Lynn M. L., Gaudenz K., Sakai D., Aoto K., Rey J.-P., Glynn E. F., Ellington L., Du C., Dixon J. et al. (2008). Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat. Med. 14, 125-133 10.1038/nm1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya N., Ye L., Kobayashi T., Mochida Y., Yamauchi M., Kronenberg H. M., Feng J. Q. and Mishina Y. (2008). BMP signaling negatively regulates bone mass through sclerostin by inhibiting the canonical Wnt pathway. Development 135, 3801-3811 10.1242/dev.025825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr J. F. R., Wyllie A. H. and Currie A. R. (1972). Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 26, 239-257 10.1038/bjc.1972.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.-G., Chae C.-H., Cho B.-O., Kim H.-N., Kim H.-J., Kim I.-S. and Choi J.-Y. (2006). Apoptosis of oral epithelial cells in oral lichen planus caused by upregulation of BMP-4. J. Oral Pathol. Med. 35, 37-45 10.1111/j.1600-0714.2005.00373.x [DOI] [PubMed] [Google Scholar]
- Komarov P. G., Komarova E. A., Kondratov R. V., Christov-Tselkov K., Coon J. S., Chernov M. V. and Gudkov A. V. (1999). A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 285, 1733-1737 10.1126/science.285.5434.1733 [DOI] [PubMed] [Google Scholar]
- Komatsu Y., Yu P. B., Kamiya N., Pan H., Fukuda T., Scott G. J., Ray M. K., Yamamura K.-i. and Mishina Y. (2013). Augmentation of Smad-dependent BMP signaling in neural crest cells causes craniosynostosis in mice. J. Bone Miner. Res. 28, 1422-1433 10.1002/jbmr.1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussie P. H., Gorina S., Marechal V., Elenbaas B., Moreau J., Levine A. J. and Pavletich N. P. (1996). Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274, 948-953 10.1126/science.274.5289.948 [DOI] [PubMed] [Google Scholar]
- Lai Z., Ferry K. V., Diamond M. A., Wee K. E., Kim Y. B., Ma J., Yang T., Benfield P. A., Copeland R. A. and Auger K. R. (2001). Human mdm2 mediates multiple mono-ubiquitination of p53 by a mechanism requiring enzyme isomerization. J. Biol. Chem. 276, 31357-31367 10.1074/jbc.M011517200 [DOI] [PubMed] [Google Scholar]
- Le Douarin N. M., Creuzet S., Couly G. and Dupin E. (2004). Neural crest cell plasticity and its limits. Development 131, 4637-4650 10.1242/dev.01350 [DOI] [PubMed] [Google Scholar]
- Le Lievre C. S. (1978). Participation of neural crest-derived cells in the genesis of the skull in birds. J. Embryol. Exp. Morphol. 47, 17-37. [PubMed] [Google Scholar]
- Lefebvre V., Huang W., Harley V. R., Goodfellow P. N. and de Crombrugghe B. (1997). SOX9 is a potent activator of the chondrocyte-specific enhancer of the pro alpha1(II) collagen gene. Mol. Cell. Biol. 17, 2336-2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Sun X., Braut A., Mishina Y., Behringer R. R., Mina M. and Martin J. F. (2005). Distinct functions for Bmp signaling in lip and palate fusion in mice. Development 132, 1453-1461 10.1242/dev.01676 [DOI] [PubMed] [Google Scholar]
- Macias D., Ganan Y., Sampath T. K., Piedra M. E., Ros M. A. and Hurle J. M. (1997). Role of BMP-2 and OP-1 (BMP-7) in programmed cell death and skeletogenesis during chick limb development. Development 124, 1109-1117. [DOI] [PubMed] [Google Scholar]
- MacLachlan T. K., Takimoto R. and El-Deiry W. S. (2002). BRCA1 directs a selective p53-dependent transcriptional response towards growth arrest and DNA repair targets. Mol. Cell. Biol. 22, 4280-4292 10.1128/MCB.22.12.4280-4292.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder M., Ghosh A. K., Steele R., Ray R. and Ray R. B. (2001). Hepatitis C virus NS5A physically associates with p53 and regulates p21/waf1 gene expression in a p53-dependent manner. J. Virol. 75, 1401-1407 10.1128/JVI.75.3.1401-1407.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon J. A., Takada S., Zimmerman L. B., Fan C.-M., Harland R. M. and McMahon A. P. (1998). Noggin-mediated antagonism of BMP signaling is required for growth and patterning of the neural tube and somite. Genes Dev. 12, 1438-1452 10.1101/gad.12.10.1438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzinger S. E., Boyce R. G., Rigby P. L., Joseph J. J. and Anderson J. R. (1994). Ethmoid bone sandwich grafting for caudal septal defects. Arch. Otolaryngol. Head Neck Surg. 120, 1121-1125 10.1001/archotol.1994.01880340061010 [DOI] [PubMed] [Google Scholar]
- Mills A. A., Zheng B., Wang X.-J., Vogel H., Roop D. R. and Bradley A. (1999). p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature 398, 708-713 10.1038/19531 [DOI] [PubMed] [Google Scholar]
- Mishina Y. and Snider T. N. (2014). Neural crest cell signaling pathways critical to cranial bone development and pathology. Exp. Cell Res. 325, 138-147 10.1016/j.yexcr.2014.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishina Y., Suzuki A., Ueno N. and Behringer R. R. (1995). Bmpr encodes a type I bone morphogenetic protein receptor that is essential for gastrulation during mouse embryogenesis. Genes Dev. 9, 3027-3037 10.1101/gad.9.24.3027 [DOI] [PubMed] [Google Scholar]
- Momand J., Zambetti G. P., Olson D. C., George D. and Levine A. J. (1992). The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 69, 1237-1245 10.1016/0092-8674(92)90644-R [DOI] [PubMed] [Google Scholar]
- Montes de Oca Luna R., Wagner D. S. and Lozano G. (1995). Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378, 203-206 10.1038/378203a0 [DOI] [PubMed] [Google Scholar]
- Morita H., Mazerbourg S., Bouley D. M., Luo C.-W., Kawamura K., Kuwabara Y., Baribault H., Tian H. and Hsueh A. J. W. (2004). Neonatal lethality of LGR5 null mice is associated with ankyloglossia and gastrointestinal distension. Mol. Cell. Biol. 24, 9736-9743 10.1128/MCB.24.22.9736-9743.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss M. L., Bromberg B. E., Song I. G. and Eisenman G. (1968). The passive role of nasal septal cartilage in mid-facial growth. Plast. Reconstr. Surg. 41, 536-542 10.1097/00006534-196806000-00004 [DOI] [PubMed] [Google Scholar]
- Murakami S., Balmes G., McKinney S., Zhang Z., Givol D. and de Crombrugghe B. (2004). Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes Dev. 18, 290-305 10.1101/gad.1179104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutter G. L. (2001). Pten, a protean tumor suppressor. Am. J. Pathol. 158, 1895-1898 10.1016/S0002-9440(10)64656-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neskey D., Eloy J. A. and Casiano R. R. (2009). Nasal, septal, and turbinate anatomy and embryology. Otolaryngol. Clin. North Am. 42, 193–205 10.1016/j.otc.2009.01.008 [DOI] [PubMed] [Google Scholar]
- Noden D. M. and Trainor P. A. (2005). Relations and interactions between cranial mesoderm and neural crest populations. J. Anat. 207, 575-601 10.1111/j.1469-7580.2005.00473.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki R., Sakai A., Ootsuyama A., Sakata T., Nakamura T. and Norimura T. (2003). Apoptosis and p53 expression in chondrocytes relate to degeneration in articular cartilage of immobilized knee joints. J. Rheumatol. 30, 559-566. [PubMed] [Google Scholar]
- Oltvai Z. N., Milliman C. L. and Korsmeyer S. J. (1993). Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programed cell death. Cell 74, 609-619 10.1016/0092-8674(93)90509-O [DOI] [PubMed] [Google Scholar]
- Raucci A., Laplantine E., Mansukhani A. and Basilico C. (2004). Activation of the ERK1/2 and p38 mitogen-activated protein kinase pathways mediates fibroblast growth factor-induced growth arrest of chondrocytes. J. Biol. Chem. 279, 1747-1756 10.1074/jbc.M310384200 [DOI] [PubMed] [Google Scholar]
- Sahni M., Raz R., Coffin J. D., Levy D. and Basilico C. (2001). STAT1 mediates the increased apoptosis and reduced chondrocyte proliferation in mice overexpressing FGF2. Development 128, 2119-2129. [DOI] [PubMed] [Google Scholar]
- Sharov A. A., Weiner L., Sharova T. Y., Siebenhaar F., Atoyan R., Reginato A. M., McNamara C. A., Funa K., Gilchrest B. A., Brissette J. L. et al. (2003). Noggin overexpression inhibits eyelid opening by altering epidermal apoptosis and differentiation. EMBO J. 22, 2992-3003 10.1093/emboj/cdg291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw E. B. (1968). Sudden unexpected death in infancy syndrome. Am. J. Dis. Child. 116, 115-119. [DOI] [PubMed] [Google Scholar]
- Shieh S.-Y., Ikeda M., Taya Y. and Prives C. (1997). DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91, 325-334 10.1016/S0092-8674(00)80416-X [DOI] [PubMed] [Google Scholar]
- Siliciano J. D., Canman C. E., Taya Y., Sakaguchi K., Appella E. and Kastan M. B. (1997). DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 11, 3471-3481 10.1101/gad.11.24.3471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluss H. K., Armata H., Gallant J. and Jones S. N. (2004). Phosphorylation of serine 18 regulates distinct p53 functions in mice. Mol. Cell. Biol. 24, 976-984 10.1128/MCB.24.3.976-984.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic V., Suzuki A., de la Pompa J. L., Brothers G. M., Mirtsos C., Sasaki T., Ruland J., Penninger J. M., Siderovski D. P. and Mak T. W. (1998). Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95, 29-39 10.1016/S0092-8674(00)81780-8 [DOI] [PubMed] [Google Scholar]
- Stenstrom S. J. and Thilander B. L. (1970). Effects of nasal septal cartilage resections on young guinea pigs. Plast. Reconstr. Surg. 45, 160-170 10.1097/00006534-197002000-00010 [DOI] [PubMed] [Google Scholar]
- Tardy M. E. and Garner E. T. (1990). Inspiratory nasal obstruction secondary to alar and nasal valve collapse: technique for repair using autogenous cartilage. Techniq. Otolaryngol. Head Neck Surg. 1, 215-217 10.1016/S1043-1810(10)80028-4 [DOI] [Google Scholar]
- Tibbetts R. S., Brumbaugh K. M., Williams J. M., Sarkaria J. N., Cliby W. A., Shieh S.-Y., Taya Y., Prives C. and Abraham R. T. (1999). A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 13, 152-157 10.1101/gad.13.2.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trichilis A. and Wroblewski J. (1997). Expression of p53 and hsp70 in relation to apoptosis during Meckel's cartilage development in the mouse. Anat. Embryol. 196, 107-113 10.1007/s004290050083 [DOI] [PubMed] [Google Scholar]
- Vaahtokari A., Aberg T. and Thesleff I. (1996a). Apoptosis in the developing tooth: association with an embryonic signaling center and suppression by EGF and FGF-4. Development 122, 121-129. [DOI] [PubMed] [Google Scholar]
- Vaahtokari A., Åberg T., Jernvall J., Keränen S. and Thesleff I. (1996b). The enamel knot as a signaling center in the developing mouse tooth. Mech. Dev. 54, 39-43 10.1016/0925-4773(95)00459-9 [DOI] [PubMed] [Google Scholar]
- Waite K. A. and Eng C. (2003a). BMP2 exposure results in decreased PTEN protein degradation and increased PTEN levels. Hum. Mol. Genet. 12, 679-684 10.1093/hmg/ddg069 [DOI] [PubMed] [Google Scholar]
- Waite K. A. and Eng C. (2003b). From developmental disorder to heritable cancer: it's all in the BMP/TGF-beta family. Nat. Rev. Genet. 4, 763-773 10.1038/nrg1178 [DOI] [PubMed] [Google Scholar]
- Walker B. E. and Fraser F. C. (1956). Closure of the secondary palate in 3 strains of mice. J. Embryol. Exp. Morphol. 4, 176-189. [Google Scholar]
- Wexler M. R. and Sarnat B. G. (1961). Rabbit snout growth. Effect of injury to septovomeral region. Arch. Otolaryngol. 74, 305-313 10.1001/archotol.1961.00740030312013 [DOI] [PubMed] [Google Scholar]
- Wexler M. R. and Sarnat B. G. (1965). Rabbit snout growth after dislocation of nasal septum. Arch. Otolaryngol. 81, 68-71 10.1001/archotol.1965.00750050073015 [DOI] [PubMed] [Google Scholar]
- Winograd J., Reilly M. P., Roe R., Lutz J., Laughner E., Xu X., Hu L., Asakura T., vander Kolk C., Strandberg J. D. et al. (1997). Perinatal lethality and multiple craniofacial malformations in MSX2 transgenic mice. Hum. Mol. Genet. 6, 369-379 10.1093/hmg/6.3.369 [DOI] [PubMed] [Google Scholar]
- Yamada T., Hiraoka Y., Ikehata M., Kimbara K., Avner B. S., Das Gupta T. K. and Chakrabarty A. M. (2004). Apoptosis or growth arrest: modulation of tumor suppressor p53's specificity by bacterial redox protein azurin. Proc. Natl. Acad. Sci. USA 101, 4770-4775 10.1073/pnas.0400899101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi Y., Abe K., Mantani A., Hitoshi Y., Suzuki M., Osuzu F., Kuratani S. and Yamamura K.-i. (1999). A novel transgenic technique that allows specific marking of the neural crest cell lineage in mice. Dev. Biol. 212, 191-203 10.1006/dbio.1999.9323 [DOI] [PubMed] [Google Scholar]
- Yilmaz M. D. and Altuntas A. (2005). Congenital vomeral bone defect. Am. J. Otolaryngol. 26, 64-66 10.1016/j.amjoto.2004.06.016 [DOI] [PubMed] [Google Scholar]
- Yokouchi Y., Sakiyama J., Kameda T., Iba H., Suzuki A., Ueno N. and Kuroiwa A. (1996). BMP-2/-4 mediate programmed cell death in chicken limb buds. Development 122, 3725-3734. [DOI] [PubMed] [Google Scholar]
- Zou H. and Niswander L. (1996). Requirement for BMP signaling in interdigital apoptosis and scale formation. Science 272, 738-741 10.1126/science.272.5262.738 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.