

Abstract
Pneumococcal infections are the leading cause of community-acquired pneumonia. Although the type 1 interferon-α (IFN-α) is a well-known antiviral cytokine, the role of IFN-α in antipneumococcal host defense and its therapeutic potential remain poorly understood. We have investigated these issues by using a murine transgene expression model. We found that in control animals, Streptococcus pneumoniae infection caused severe weight loss and excessive lung inflammation, associated with rapid bacterial outgrowth. In contrast, the animals that received a single dose of an adenoviral vector expressing IFN-α prior to pneumococcal infection demonstrated rapid and effective control of bacterial replication and lung inflammation and improved clinical outcome. Enhanced protection by IFN-α was due to increased activation of neutrophils and macrophages with increased release of reactive oxygen and nitrogen species and bacterial killing. Furthermore, we found that raised levels of IFN-α in the lung remained immune protective even when the gene transfer vector was given at a time postpneumococcal infection. Our study thus shows that the classically antiviral type 1 IFN can be exploited for enhancing immunity against pneumococcal infection via its activating effects on innate immune cells. Our findings hold implications for the therapeutic use of IFN-α gene transfer strategies to combat pneumococcal infections.
Keywords: adenoviral-mediated gene transfer, innate immune regulation, lung, pneumococcal pneumonia, type 1 interferon
Introduction
Streptococcus pneumoniae remains a major human pathogen worldwide. Pneumococcal infection is the leading cause of community-acquired pneumonia and can lead to invasive pneumococcal disease that includes bacteremia, septicemia, pneumonia, and meningitis.1,2 It is estimated that community-acquired pneumonia alone accounts for 1 million physician visits and 60,000 hospitalizations annually in Canada, costing about $100 million.3 The incidence of invasive pneumococcal disease in adults remains high, whereas the highest incidence is in the elderly and in children less than 5 years of age, particularly in aboriginal children living in northern Canada.1,4 Worldwide, S. pneumoniae continues to be the most significant bacterial pathogen in children, causing ~25% of all preventable deaths in children less than 5 years of age and 1.6 million infant deaths per year in developing countries.2,5,6 In addition, S. pneumoniae superinfection is the most common complication of influenza, which together form the leading cause of death from infectious diseases in Canada.3,7 Alarmingly, S. pneumoniae outbreaks have occurred across Canada and elsewhere in North America in the last 10 years.8 Treatment is complicated by the continuous emergence of antibiotic-resistant S. pneumoniae serotypes, the narrow serotype-specific protection provided by conjugate polysaccharide vaccines, and the ever-shifting bacterial serotypes away from the serotypes covered by the vaccine in invasive infections of vaccinated children.2,9,10 Thus, there has been a pressing need for enhanced understanding of host defense mechanisms and development of novel therapeutic agents against pneumococcal diseases, particularly those caused by antibiotic resistant strains.
The type 1 interferons (IFNs) that are mainly produced during viral and intracellular bacterial infections are IFN-α and IFN-β.11 Most research has focused on the classic antiviral role of type 1 IFNs.11,12 In this regard, a replication-deficient human type 5 adenovirus vector (Ad5) expressing the human consensus IFN-α gene, known as DEF201, has been developed to be an antiviral drug. DEF201 has been tested prophylactically and therapeutically in experimental models, with demonstrated protective efficacy for Ebola virus, yellow fever virus, arenavirus, and phlebovirus infection after a single i.n. dose.13–16 Likewise, the Ad5 vector expressing mouse IFN-α, mDEF201, has been shown to be protective from lethal severe acute respiratory syndrome (SARS) virus, Western equine encephalitis virus, Venezuelan equine encephalitis virus, and Pox Vaccinia virus infection.17–20 In spite of much established knowledge in the role of type 1 IFNs and their therapeutic effects in viral infections, it still remains poorly understood about their role and therapeutic potential in extracellular bacterial infections. Past and recent evidence has only hinted at a beneficial role of type 1 IFN in S. pneumoniae infection.12,21
In our current study, we have investigated the role of IFN-α in host defense and its therapeutic potential in S. pneumoniae infection by using a murine model of acute pneumococcal infection and an Ad5 gene transfer vector expressing IFN-α. We have provided novel evidence that transgenically raised levels of IFN-α in the lung provide enhanced immune protection against acute S. pneumoniae infection via activating effects on neutrophils and macrophages. Our findings hold implications in using type 1 IFN transgene approaches for immunotherapy against pneumococcal pneumonia.
Results
Rapid S. pneumoniae replication, pronounced immunopathology, and deleterious clinical outcomes in control animals
Before (0 hour) or upon pulmonary S. pneumoniae infection, the control mice that received an empty control Ad vector before infection (Addl group; Figure 1a) were found to have minimally detectable levels of type 1 IFN-α in the lung (Figure 1b). These mice suffered progressively deteriorating body weight losses (Figure 1c), which were associated with heightened immunopathology including intrabronchial hemorrhage in the lung (Figure 1d) and 85% death by 7 days postinfection (Figure 1c). On examination of bacterial burden, high levels of S. pneumoniae counts were found at 48 and 72 hours both in the lung and spleen (Figure 1e, black bars). These data suggest that the control animals have difficulty to effectively control S. pneumoniae infection, systemic dissemination, and associated tissue inflammation in the lung.
Figure 1.
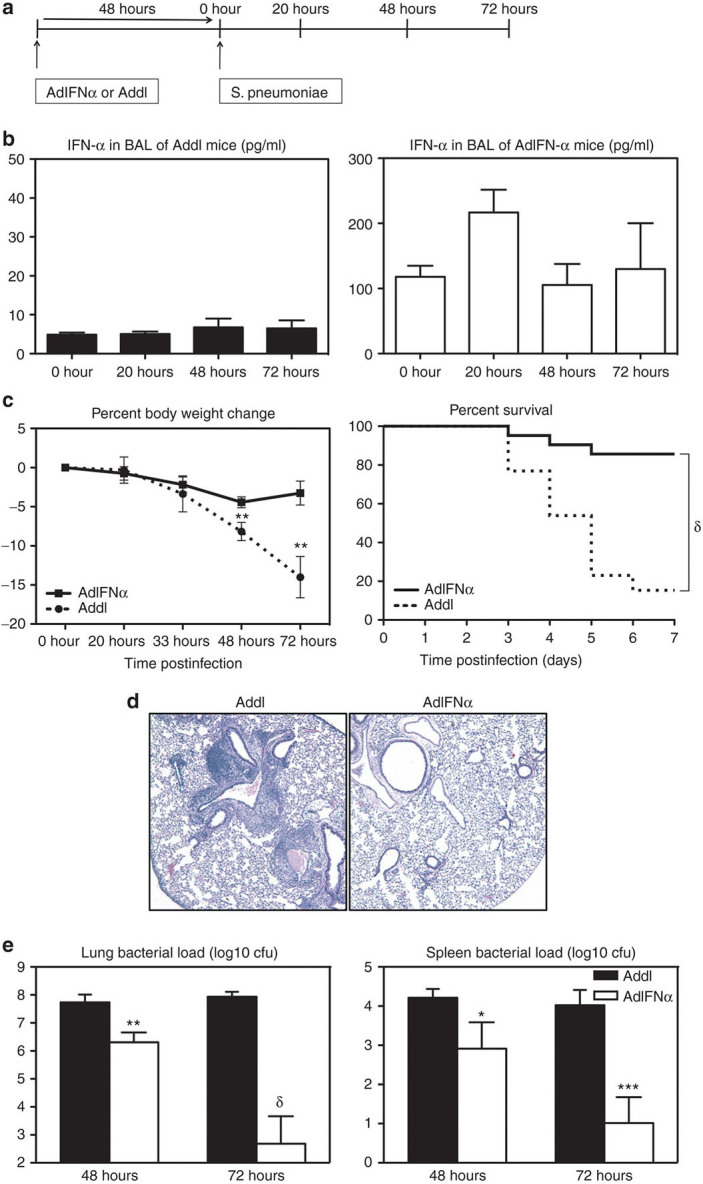
Transgenic expression of interferon (IFN)-α improves clinical outcome and bacterial control following pneumococcal infection. (a) Experimental schema. Female C57BL/6 mice were infected with 107 plaque-forming units (pfu) AdIFN-α or Addl and 48 hours later with 104 colony-forming unit (cfu) of S. pneumoniae. (b) IFN-α was measured in the bronchoalveolar lavage (BAL) 48 hours after infection with Ad (0-hour time point) and at 20-, 48-, 72-hour post-Strep by enzyme-linked immunosorbent assay. Results are from two to four independent experiments, n = 4–10/group. (c) Average percent body weight change and survival were monitored (end point was considered as 20% body weight loss; log-rank test, P < 0.0001). Results are from two to six independent experiments, n = 10–44/group/time point. (d) Histopathological changes in the lung were examined by hematoxylin and eosin staining at 72-hour post-Strep infection (magnification ×5). (e) Lung and spleen bacterial load was measured using a colony forming unit (CFU) assay at 48- and 72-hour post-Strep infection. Results are from three independent experiments, n = 5–8/group/time point. Data are expressed as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.005, δ, P < 0.0005.
IFN-α gene transfer controls S. pneumoniae replication and immunopathology and improves clinical outcomes
Given that there were only minimal levels of IFN-α detected in the lung of S. pneumoniae infected control (Addl) mice (Figure 1b), we set out to determine the role of raised lung levels of IFN-α in host defense against S. pneumoniae infection. To this end, we used a transient transgenic approach for expression of IFN-α in the lung by i.n. delivering a recombinant replication-defective Ad5 gene transfer vector encoding mIFN-α (AdIFN-α) before S. pneumoniae infection. Thus, in contrast to minimal levels of IFN-α in the lung of the mice receiving the empty vector (Addl) before and after S. pneumoniae infection (Figure 1b), the mice receiving AdIFN-α had significantly raised levels of IFN-α in the lung before S. pneumoniae infection (0 hour) (Figure 1b). There were also elevated circulating levels of IFN-α in these mice (Supplementary Figure S1). Compared with Addl controls, levels of IFN-α remained raised in the lung at 20, 48, and 72 hours after S. pneumoniae infection (Figure 1b). We found that elevated IFN-α levels significantly reduced body weight loss in these mice, which was monitored as a measure of overall illness (Figure 1c). Thus, there was only a small and transient body weight loss (4.4%) at 48 hours post-Strep infection in these mice, in sharp contrast to 8.2 and 14.0% body weight losses at 48 and 72 hours, respectively, in the control mice (Figure 1c). In addition, although 85% of control mice reached end point by 7 days postinfection, only 15% death was observed in the IFN-α–expressing group (Figure 1c).
Consistent with improved survival, immunopathology and tissue injury were much reduced in the lung of IFN-α–expressing mice (Figure 1d). Furthermore, the bacterial load both in the lungs and in the spleens of these mice was dramatically (multiple logs) lower, compared with that in the control counterparts (Figure 1e). These findings suggest that transgenically raised IFN-α levels are immune protective from acute pneumococcal pneumonia, capable of effectively controlling S. pneumoniae replication and improving lung immunopathology and clinical outcome.
IFN-α gene transfer leads to rapid induction of innate immune cellular infiltration and cytokine responses in the lung upon S. pneumoniae infection
To begin investigating the mechanisms of improved bacterial control and clinical outcome mediated by IFN-α gene transfer to the lung, we first analyzed the innate cellular responses in the lung at 20 hours post-Strep using flow cytometry. Total inflammatory infiltrate numbers were almost threefold higher in the respiratory tract (bronchoalveolar lavage (BAL)) of IFN-α–expressing mice compared with the controls (Table 1). This was accounted for primarily by markedly increased neutrophil influx in the BAL (Figure 2a,b). Higher cell numbers and neutrophil influx were also observed at 48 hours in these mice (Table 1 and data not shown). In comparison, neutrophil numbers in the lung parenchyma were similar between the two groups, as were macrophages in the BAL and lung parenchyma (Figure 2a). In addition to neutrophils, there appeared to be moderately increased natural killer (NK) cells and T cells in the lung parenchyma of IFN-α–expressing mice (Supplementary Figure S2). These data suggest that some of such altered innate immune cellular responses might be involved in enhanced immune protection by IFN-α gene transfer.10,22
Table 1. Total cell numbers in BAL following S. pneumoniae infection (×106).
|
20 hours |
48 hours |
72 hours |
|||
|---|---|---|---|---|---|
| Addl | AdIFNα | Addl | AdIFNα | Addl | AdIFNα |
| 0.35 ± 0.03 | 0.92 ± 0.26 | 0.21 ± 0.02 | 0.44 ± 0.07 | 3.2 ± 1.15 | 0.26 ± 0.02 |
Results are expressed as mean ± SEM (n = 5–8/group/time point from one to two experiments/time point).
BAL, bronchoalveolar lavage; IFN, interferon.
Figure 2.

Transgenic expression of interferon (IFN)-α leads to rapid cellular infiltration and induction of cytokine and chemokine responses in the lungs of S. pneumoniae-infected mice by 20-hour post-Strep infection. (a) Neutrophil and macrophage numbers in the bronchoalveolar lavage (BAL) and lung. Cells were assessed by flow cytometry in AdIFN-α or Addl treated mice at 20-hour post-Strep infection. (b) Dotplots show representative populations from the BAL (gates show frequency of total cells). (c) Cytokine and chemokine levels in BAL were measured in AdIFN-α or Addl treated mice at 20-hour post-Strep infection. Results are from one experiment, n = 5/group. Data are expressed as mean ± SEM. *P < 0.05; ***P < 0.005. IL, interleukin; TNF, tumor necrosis factor; IP-10, interferon γ-induced protein 10; MCP-1, monocyte chemoattractant protein-1.
Given the increased neutrophilic responses in the BAL by IFN-α gene transfer, we next assessed cytokine and chemokine levels in the BAL at 20 hours following S. pneumoniae infection. Indeed, compared with the control animals, the levels of proinflammatory cytokines tumor necrosis factor-α and interleukin-1β were increased in the IFN-α–expressing mice (Figure 2c). Associated with increased proinflammatory cytokines were higher levels of chemokines including the neutrophil chemoattractants macrophage inflammatory protein 2 (MIP-2) and keratinocyte-derived chemokine (KC) (Figure 2c). These results together show that raised IFN-α levels enhance host defense against S. pneumoniae infection via rapid induction of cytokine responses and innate cellular responses in the lung at early time points postinfection, thus leading to better control of bacterial replication and immunopathology.
Rapidly diminishing tissue inflammatory responses induced by IFN-α gene transfer in the latter stages of S. pneumoniae infection
To further investigate the potential mechanisms for improved clinical outcome by IFN-α gene transfer, we examined the innate cellular and cytokine responses at the later time point of 72 hours after S. pneumoniae infection, when the control mice were approaching end point (Figure 1c) and had uncontrolled lung inflammation (Figure 1d) and bacterial outgrowth (Figure 1e). By this time, IFN-α–expressing mice had remarkably diminished numbers of neutrophils both in the BAL and in the lung parenchyma compared with the control mice (Figure 3a). The number of macrophages also markedly decreased in the BAL of IFN-α–expressing mice (Figure 3a). The rapid clearance of cellular infiltrates from the lung of these mice was associated with a sharp decline in proinflammatory cytokines and chemokines (Figure 3b), contrary to higher levels of these cytokines at 20 hours post-Strep infection (Figure 2b). On the other hand, by 72 hours, the control mice had excessive influx of neutrophils and macrophages in the BAL and lung (Figure 3a), corresponding to 12-fold more total inflammatory cells in the airway (Table 1). Heightened inflammatory cellular responses in the control animals were associated with sustained proinflammatory cytokines, tumor necrosis factor-α and interleukin-1β, and remarkably increased levels (up to 3,900 pg/ml) of chemokines (Figure 3b). These data suggest that earlier increased innate immune responses induced by IFN-α gene transfer lead to rapid control of bacterial infection and help the lung to return to homeostasis. On the other hand, the control animals have markedly heightened inflammatory responses due to uncontrolled bacterial infection.
Figure 3.

Excessive neutrophil infiltration and dysregulated cytokine and chemokine responses in the lungs of control mice at 72-hour post-Strep infection. (a) Neutrophil numbers in the bronchoalveolar lavage (BAL) and lung were assessed by flow cytometry in AdIFN-α or Addl treated mice at 72-hour post-Strep infection. (b) Cytokine and chemokine levels in BAL were measured in AdIFN-α or Addl treated mice at 72-hour post-Strep infection. Results are from two experiments, n = 8/group. Data are expressed as mean ± SEM. *P < 0.05; **P < 0.01. IFN, interferon; IL, interleukin; TNF, tumor necrosis factor.
Enhanced neutrophil responses contribute to improved bacterial clearance by IFN-α gene transfer
To further assess the mechanisms of increased bacterial control by raised IFN-α levels, we measured reactive oxygen species (ROS) and reactive nitrogen species (RNS) (hydrogen peroxide, peroxyl radical, nitric oxide, and peroxynitrite anion) in lung homogenates collected at various time points post-Strep infection, given their critical antibacterial activities and association with activated neutrophils and macrophages. Significantly increased levels of ROS/RNS were induced by IFN-α at 8, 20, and 48 hours with equalized levels with the control mice at 72-hour postinfection (Figure 4). The largest difference in ROS/RNS between the two groups was seen at 20-hour postinfection (Figure 4), corresponding with high neutrophil influx in the lung of the mice with raised IFN-α levels at this time point (Figure 2a).
Figure 4.
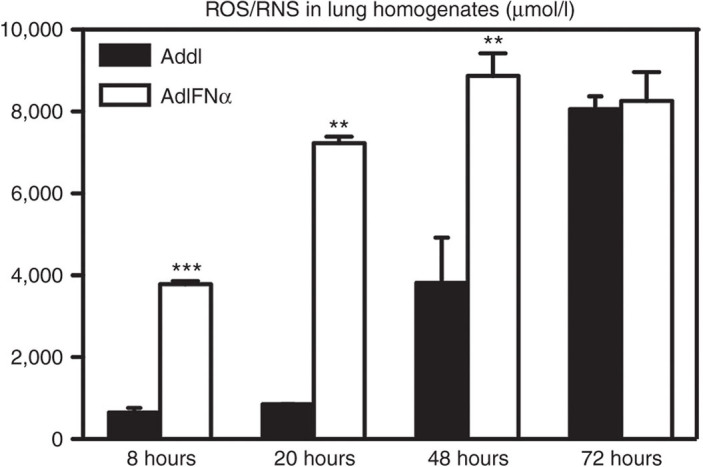
Transgenic expression of interferon (IFN)-α leads to enhanced release of reactive oxygen species (ROS) and reactive nitrogen species (RNS). Female C57BL/6 mice were infected with 107 plaque-forming units AdIFN-α or Addl and 48 hours later with 104 colony-forming unit of S. pneumoniae. Lung homogenates at various time points were measured for concentration of total ROS/RNS. Data are expressed as mean ± SEM of n = 3–4/group/time point, representative of two independent experiments. **P < 0.01; ***P < 0.005.
Based on the ROS/RNS data, we further investigated the role of increased neutrophils in improved bacterial control by IFN-α. To this end, we undertook a strategy that allowed only partial neutrophil depletion from the lungs of IFN-α–expressing mice prior to S. pneumoniae infection. Thus, the mice received the IFN-α gene transfer vector as depicted in Figure 1a and were then treated i.n. once with a neutrophil-depleting antibody (α-Ly6G) at 4 hours before S. pneumoniae infection. The control IFN-α–expressing mice received only the control immunoglobulin G. By flow cytometry, this protocol was found to render ~57% reduction in neutrophils in S. pneumoniae-infected mice by 20-hour postinfection and 30% reduction by 72 hours as neutrophil influx continued into the lung (Supplementary Figure S3). We found that such partial neutrophil depletion led to significantly greater weight loss (Figure 5a), worsened bacterial control both in the lung and in the spleen (Figure 5b), and markedly reduced measurable ROS/RNS in the lung (Figure 5c). Nevertheless, notably the extent of body weight loss and increased bacterial load in partially neutrophil-depleted IFN-α–expressing animals did not reach that previously seen in the Addl control group (Figure 1c,e). These data together suggest that enhanced neutrophil responses induced by IFN-α gene transfer contribute to improved bacterial clearance and clinical outcome.
Figure 5.

Neutrophil depletion during transgenic expression of interferon (IFN)-α partially decreases protection from pneumococcal infection. Female C57BL/6 mice were infected with 107 plaque-forming units AdIFN-α and 48 hours later with 104 colony-forming unit (cfu) of S. pneumoniae. In addition, 4 hours before Strep. infection, 50 µg of α-Ly6G depleting antibody or immunoglobulin G control was administered i.n.. (a) Average percent body weight change was monitored as an indicator of illness. (b) Lung and spleen bacterial load was measured using a CFU assay at 72-hour post-Strep infection. (c) Lung homogenates were measured for concentration of total of reactive oxygen species (ROS)/reactive nitrogen species (RNS) at 72-hour post-Strep infection. Results are from one experiment, n = 10–12/group. Data are expressed as mean ± SEM. *P < 0.05; ***P < 0.005.
Improved macrophage phagocytic killing of S. pneumoniae by IFN-α transgene expression
The observation that neutrophil depletion in IFN-α–expressing mice resulted in only a significantly reduced, but not completely eliminated, IFN-α–mediated enhancement in protection (Figure 5) suggested additional mechanisms at play. Although IFN-α expression did not increase the number of macrophages in the lung (Figure 2a), it is possible that the antibacterial property of these innate immune cells was altered by IFN-α. To examine this possibility, freshly isolated naive alveolar macrophages (AMs) and purified lung macrophages (MOs) were transduced in vitro to express IFN-α by AdIFN-α or treated with the control vector Addl, and then viable streptococci were added to these macrophages. AdIFN-α-transduced, but not the control AMs and lung MOs produced significant levels of IFN-α as measured in the culture media (Supplementary Figure S4). IFN-α–producing AMs and lung MOs also produced much higher levels of antibacterial ROS/RNS upon bacterial phagocytosis than their control counterparts (Figure 6a). Importantly, IFN-α–producing AMs were three times more effective at bacterial killing than the non–IFN-α producers (Figure 6b). In comparison, although killing efficiency was similar at 1 hour, IFN-α–producing lung MOs were significantly more effective at bacterial killing by 2 hours compared with the non–IFN-α producers, which ceased to kill any more streptococci by this time (Figure 6b). These data indicate that besides neutrophils, activation of alveolar and lung macrophages by IFN-α expression represents an additional mechanism for enhanced immune protection and improved bacterial control.
Figure 6.

AdIFN-α infection improves S. pneumoniae killing ability of lung macrophages. Alveolar macrophages and purified lung macrophages were infected with AdIFN-α or Addl for 3 hours, and the following day, S. pneumoniae was added to the cells. Phagocytosis was allowed to occur for 1 hour, and then bacterial killing was allowed for another 1–2 hours. (a) Supernatants were measured for concentration of total of reactive oxygen species (ROS)/reactive nitrogen species (RNS). (b) Cells were lysed and the live bacteria were enumerated by a CFU assay. Data are expressed as mean ± SEM of triplicate wells/group/time point, representative of three independent experiments. *P < 0.05. AMs, alveolar macrophages; IFN, interferon; MOs, macrophages.
Therapeutic IFN-α gene transfer rendered after S. pneumoniae infection controls bacterial replication and improves clinical outcomes
To determine the therapeutic potential of IFN-α for pneumococcal infections, mice were infected with S. pneumoniae, and 12 hours later, they were inoculated i.n. with AdIFN-α or Addl control vector (Figure 7a). While body weight losses were similar between the two groups for the initial course of infection, IFN-α–expressing mice experienced stalled body weight loss at 48 hours and began to recover by 68 hours, leading to 50% death in the group by 7 days postinfection (Figure 7c). This was in sharp contrast to the control mice that exhibited progressively deteriorating clinical conditions, with most approaching the end point soon after 68 hours, with 100% death in the group by 7 days postinfection (Figure 7c). Consistent with improved clinical outcomes induced by IFN-α, the lung of IFN-α–expressing mice had markedly reduced immunopathology with inflammation primarily limited to the peribronchial and perivascular regions (Figure 7b). In contrast, the control lungs had severe and diffuse tissue immunopathology and inflammation (Figure 7b). Also consistent with improved clinical outcome and immunopathology induced by therapeutic IFN-α gene transfer, these mice had significantly lower bacterial loads both in the lung and in the spleen compared with the control mice (Figure 7d). The almost one-log difference in colony-forming unit (cfu) in the lungs (Figure 7d) is correlated with significant improvement in lung pathology (Figure 7b), weight loss, and survival (Figure 7c). These findings together support the therapeutic potential of raised IFN-α in the lung by using a gene transfer strategy in effectively controlling S. pneumoniae infection and its associated detrimental clinical outcomes.
Figure 7.

Transgenic expression of interferon (IFN)-α following pneumococcal infection improves clinical outcome. (a) Experimental schema. Female C57BL/6 mice were infected with 104 colony-forming unit (cfu) of S. pneumoniae and 12 hours later with 107 plaque-forming units (pfu) AdIFN-α or Addl. (b) Histopathological changes in the lung were examined by hematoxylin and eosin staining at 68-hour post-Strep infection (magnification ×5). (c) Average percent body weight change and survival were monitored (end point was considered as 20% body weight loss; log-rank test, P < 0.01). (d) Lung and spleen bacterial load was measured using a CFU assay at the last time point. Results are from two independent experiments, n = 10/group. Data are expressed as mean ± SEM. *P < 0.05; **P < 0.01. IFN, interferon.
Discussion
Infections with S. pneumoniae continue to have significant clinical, social, and economic impacts in Canada and worldwide. As a leading cause of community-acquired pneumonia and invasive pneumococcal disease, as the most significant bacterial pathogen in children, and as the most common complication of influenza, S. pneumoniae contributes to significant morbidity and mortality worldwide.1,2,5,6 Treatment is complicated by the continuous emergence of antibiotic-resistant strains and suboptimal vaccines, making the threat of S. pneumoniae outbreaks a reality.2,9,10 The role and therapeutic potential of type 1 IFN in the host defense to extracellular bacterial infections is not well understood, as IFN-α and IFN-β have been best characterized in the host defense to viral infections. Intriguingly, it has recently been demonstrated that S. pneumoniae activates type 1 IFN signaling, and emerging evidence suggests a beneficial role of type 1 IFN in S. pneumoniae infection.12,21 However, the protective effects of IFN-α have not been fully established, and the mechanisms of protection are entirely unknown.11,12,23,24 Using a mouse model of acute respiratory S. pneumoniae infection and an Ad5-based IFN-α gene transfer approach, we have investigated these outstanding issues. We show that S. pneumoniae infection that leads to rapid and uncontrolled S. pneumoniae replication, pronounced immunopathology, and deleterious clinical outcomes in control animals is ameliorated by transgenic expression of IFN-α in the lung. Enhanced immune protection against acute S. pneumoniae infection is induced by IFN-α via rapid induction of innate immune cellular infiltration and cytokine responses in the lung and activating effects on neutrophils and macrophages. For the first time, we also provide a novel therapeutic strategy in which a single i.n. dose of AdIFN-α (mDEF201) administered after an ongoing S. pneumoniae infection controls bacterial replication and improves clinical outcomes.
In our model of pulmonary pneumococcal infection, mice were infected with S. pneumoniae 48 hours after receiving one i.n. dose of AdIFN-α or Addl control. Control mice had barely detectable levels of IFN-α in the lung. Consequently, these mice suffered rapid body weight losses and were in the process of reaching end point by 72-hour postinfection. The poor clinical outcome was associated with heightened immunopathology in the lung and high levels of bacterial burden locally in the lung and systemically in the spleen. Thus, the control animals had major difficulties in effective control of S. pneumoniae infection in our model. On the other hand, IFN-α–expressing mice had markedly raised IFN-α levels in the lung and serum at the time of infection. Measured at various time points after S. pneumoniae infection in the BAL, IFN-α expression was further induced in these mice. Thus, the adenovirus vector provided sustained and inducible expression of IFN-α. We found that raised IFN-α levels significantly reduced body weight loss in these mice, as there was only a small and transient body weight loss at 48-hour postinfection and markedly improved survival rates. Consistent with the improved clinical outcome, immunopathology and tissue injury were dramatically reduced in the lung of IFN-α–expressing mice compared with control animals, as were bacterial loads in the lung and spleen. These findings show that raised IFN-α levels are immune protective from acute pneumococcal pneumonia, controlling S. pneumoniae replication and immunopathology and improving clinical outcomes.
In our study, we next investigated the unknown mechanisms of improved bacterial control and clinical outcomes mediated by IFN-α gene transfer. We observed that cytokine and chemokine levels in the airway of IFN-α–expressing mice early after S. pneumoniae infection (20 hours) were markedly higher compared with control mice. The likely cellular sources of heightened chemokines include airway epithelial cells, neutrophils, and macrophages. Increased cytokines and chemokines correlated with rapid infiltration of immune cells into the airway and lung parenchyma including neutrophils, NK cells, and CD4+ T cells. Recently, NK cells and CD4+ T cells have been shown to have important roles in early host defense against extracellular bacteria.2,10,22,25 Importantly, higher neutrophil numbers in the airway are indicative of the improved bacterial control, as neutrophils are known to be key phagocytes involved in S. pneumoniae clearance.2 The levels of ROS and RNS were also significantly higher in the lungs of IFN-α–expressing mice throughout the infection, correlating with the improved bacterial clearance. Partial depletion of neutrophils during infection showed that neutrophil number and activity in the lungs of IFN-α+–expressing mice contributed to the observed improvement in bacterial clearance and clinical outcome. Furthermore, as macrophages are also known to be major cells involved in the first line of defense at the site of infection, and were not found to be increased in number in IFN-α–expressing mice, we investigated their functionality as a potential mechanism of improved bacterial control. Both AMs and purified lung macrophages displayed more efficient S. pneumoniae killing, increased release of ROS/RNS, and more effective control of bacterial outgrowth when previously infected with AdIFN-α as opposed to Addl. Thus, heightened macrophage and/or neutrophil activation at earlier time points in IFN-α–expressing animals likely led to much increased amounts of ROS/RNS in the lung and contributed to sustained levels of ROS/RNS at later times when bacteria were largely cleared from the lung of these animals. The similar amounts of ROS/RNS between the two groups at the later time point 72 hours most likely resulted from sustained ROS/RNS production in AdIFN-α group due to lasting macrophage activation and relatively markedly increased ROS/RNS production in the control group by this time point due to persistently high bacterial burden. Increased ROS/RNS production in the lung may represent one of the key mechanisms by which IFN-α enhances anti-pneumococcal host defense as both oxygen and nitrogen species are known to be critical to neutrophil and macrophage antibacterial activities. Our study, therefore, reveals the mechanisms mediated by IFN-α to provide improved bacterial control and clinical outcome in S. pneumoniae infection. Importantly, in the latter stages of S. pneumoniae infection (72 hours), IFN-α–expressing mice exhibited largely diminished tissue inflammatory responses. Thus IFN-α expression led to rapid control of bacterial infection and allowed the lung to return to homeostasis before the deleterious effects of lung inflammation could cause any irreparable damage. On the other hand, the control mice displayed strikingly heightened and ongoing inflammatory responses due to uncontrolled bacterial infection.
The IFN-α–expressing adenovirus (DEF201) used in our study has been proposed as a novel broad-spectrum drug, as it has proven successful in providing protection from a large variety of viruses.13–20 It has been reported that i.m. injection of mDEF201 rapidly leads to high levels of IFN-α in the serum within 3–5 hours of infection, which were undetectable by day 7.18 However, the protective effects of the IFN were sustained, as animals challenged with Western equine encephalitis virus 7 days after AdIFN-α administration were 100% protected.18 In addition, i.n. delivery of AdIFN-α was observed to be protective when given 14 and 56 days before infection with SARS and vaccinia virus, respectively.17,20 We have also previously shown that i.n. Ad5 gene transfer vector delivery leads to sustained transgene expression within the airway epithelium and macrophages for a period of 12–14 days.26 Thus, a single dose of AdIFN-α is able to induce a long-lasting antiviral state that activates protective immune mechanisms. Accordingly, we next investigated the therapeutic potential of the AdIFN-α vector for pneumococcal infections. Mice were infected with S. pneumoniae, and 12 hours later, they were inoculated i.n. with AdIFN-α or Addl control vector. Therapeutic IFN-α expression led to significantly lower bacterial replication and improved lung immunopathology and clinical outcomes. Survival was significantly improved, as 50% of mice were alive at 7 days postinfection when no control animals survived. Of note, the therapeutic application of AdIFN-α only resulted in a moderately reduced bacterial burden in the lung and yet it markedly reduced lung immunopathology and other clinical outcomes. This suggests that the moderately improved infection control by IFN-α is only part of the mechanism for improved histopathology. Recent mounting evidence indicates that lung immunopathology does not always correlate with the level of microbial burden and that the immune regulatory mechanisms not directly involved in host defense also play an important role in regulating tissue inflammation.7,27 This suggests an additional regulatory functionality that IFN-α possesses in our model. The protective effects by IFN-α transgene expression are in agreement with two previous studies that have approached this question. A murine study by Weigent et al.21 showed that either recombinant IFN-α or IFN-β markedly improved survival in streptococcal infection when administered i.p. 5 hours before infection (63–72% survival compared with 0% survival in controls). In the same study, anti-IFN-α/β serum significantly increased mortality (20% survival compared with 95% survival in controls).21 Similarly, in a colonization model study by Parker et al.24 IFN-α/β receptor null mice had significantly increased nasal colonization with S. pneumoniae compared with wild-type mice. Therefore, our findings support the therapeutic potential of transgenically expressed IFN-α in the lung for effectively controlling S. pneumoniae infection and its associated detrimental clinical outcomes.
In conclusion, our results indicate that the classically antiviral cytokine IFN-α is also protective against extracellular bacterial infection with S. pneumoniae. Furthermore, we identify for the first time that a single i.n. dose of AdIFN-α leads to rapid and effective control of bacterial replication and lung inflammation and consequently to an improvement in clinical outcome. Our findings hold important implications for transgenic expression of IFN-α in future clinical practice, in particular during outbreaks where a rapid therapy at the site of infection would be invaluable.
Materials and Methods
Mice
Female 7- to 10-week-old C57BL/6 mice were purchased from Charles River Laboratories (Wilmington, MA). Mice were housed in specific pathogen-free level facilities at the McMaster University Central Animal Facility (Hamilton, Ontario, Canada). For all experiments, mice were euthanized by exsanguination of the abdominal artery under anesthesia. All experiments were conducted in accordance with the animal research ethics board of McMaster University (Hamilton, Ontario, Canada).
Pneumococcal infection
S. pneumoniae serotype 3 (ATCC 6303) was prepared as previously described, by plating frozen stock on blood-agar plates and incubating overnight, then culturing the resultant colonies for ~5 hours at 37 °C in 5% CO2 in Todd Hewitt broth (BD Biosciences, San Jose, CA) to midlogarithmic phase.7 The bacteria were harvested and resuspended in phosphate-buffered saline (PBS). Anesthetized mice were infected i.t. with 104 cfu of S. pneumoniae in a volume of 40 µl.
Prophylactic and therapeutic IFN-α gene delivery to the lung
A replication-deficient adenoviral gene transfer vector (Ad5) expressing murine IFN-α (AdIFN-α) was obtained from Defyrus (Toronto, Ontario, Canada – construct mDEF201 prepared at the Robert Fitzhenry Vector Laboratory, McMaster University), and an empty adenoviral vector (Addl) was used as a control. Mice anesthetized by inhalation of isoflurane were infected i.n. with 107 plaque-forming units of AdIFN-α in a volume of 20 µl, 48 hours before (prophylactic) or 12 hours after (therapeutic) S. pneumoniae infection. As we have previously well documented, i.n. Ad5 gene transfer vector delivery leads to transgene expression within the airway epithelium and macrophages and raises transgene protein levels for a period of 12–14 days.26
Determination of streptococcus burden in local and systemic tissues
Levels of streptococcus in the lung and spleen were determined by homogenizing the tissues in PBS, plating serial dilutions on blood-agar plates, and incubating overnight at 37 °C, 5% CO2. Colonies were counted and calculated as cfu per whole organ.
BAL and lung mononuclear cell isolation
Airway luminal cells were collected by BAL of the lungs, and mononuclear cells were isolated from lung tissue, using our standard procedures as previously described.27 Briefly, the lungs were cut into small pieces and incubated in 10 ml of 150 U/ml collagenase type 1 (Sigma-Aldrich, St. Louis, MO) for 1 hour at 37 °C, the lung pieces were crushed through 40-µm basket filters, red blood were cells lysed with ammonium-chloride-potassium (ACK) lysis buffer, and cell pellets were resuspended in complete RPMI medium (RPMI 1640 supplemented with 10% fetal bovine serum, 1% penicillin–streptomycin, and 1% L-glutamine).
Flow cytometric analysis
Flow cytometry was performed on BAL and lung cells. Cells were incubated for 15 minutes with the CD16/CD32 Fc block antibody and then stained with extracellular antibodies (BD Biosciences unless otherwise stated) for antigen-presenting cells (APC-Cy7-anti-CD45, Alexa Fluor 700-anti-MHCII (eBioscience, San Diego, CA), Pacific Orange-anti-Gr1 (Life Technologies, Burlington, Ontario), APC-anti-CD11c, PECy7-anti-CD11b), NK cells (V450-anti-CD3, PE-anti-NK1.1), and T cells (V450-anti-CD3, APC-Cy7-anti-CD4). Stained cells were analyzed on the LSRII flow cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star, Ashland, OR). Neutrophils (CD45+CD11b+Gr1+), macrophages (CD45+Gr1-AF+CD11c+), NK cells (CD3-NK1.1+), and CD4+ T cells (CD3+CD4+) were assessed as total number of cells per tissue compartment by multiplying the “frequency of total” of the population by total cell number in the tissue.
Cytokine and chemokine quantification
Levels of cytokines and chemokines in BAL fluids were quantified using Luminex multianalyte technology (Luminex Molecular Diagnostics, Toronto, Ontario), according to the manufacturer’s protocols. Levels of IFN-α were measured in BAL, serum, and culture supernatants using the VeriKine Mouse Interferon Alpha ELISA Kit (PBL Interferon Source, Piscataway, NJ).
Measurement of ROS/RNS
Levels of total ROS/RNS (hydrogen peroxide, peroxyl radical, nitric oxide, and peroxynitrite anion) were analyzed in lung homogenates and culture supernatants using the OxiSelect In Vitro ROS/RNS Assay Kit (Cedarlane, Burlington, Ontario) according to the manufacturer’s instructions.
In vivo neutrophil depletion
Anti-Ly6G (1A8) antibody (UCSF Monoclonal Antibody Core, San Francisco, CA) or total rat immunoglobulin G (Sigma-Aldrich) as control were given to mice. A total of 50 μg of the antibodies were administered once i.n. 4 hours before bacterial infection.
Macrophage bacterial killing assay
A standard bacterial phagocytosis and killing assay was used to evaluate the effect of pretreatment of macrophages with AdIFN-α.28 Naive C57BL/6 mice were used to obtain the cells, by BAL for AMs and by mononuclear cell isolation followed by purification of CD11c+ cells and CD11b+ cells (which were then pooled) by magnetic cell sorting according to the manufacturer’s instructions (Miltenyi Biotec, Bergisch Gladbach, Germany), from lung tissue for lung MOs. In separate experiments, 5 × 105 AMs were used per well and 2 × 105 lung MOs were used per well, in 24-well plates. Cells were transduced with AdIFN-α or Addl at a multiplicity of infection of 100 infectious viral particles in serum-free cRPMI for 3 hours at 37 °C (during this time, cells adhered to the bottom of the wells), washed, resuspended in Pen/Strep-free cRPMI and left overnight at 37 °C. S. pneumoniae was freshly grown to midlogarithmic phase and then opsonized by adding 10% naive C57BL/6 serum to bacteria for 30 minutes at 37 °C. Bacteria were then washed twice with PBS to remove excess serum and added to the macrophage culture (bacteria to macrophage ratio 10:1). After incubation for 1 hour, phagocytosis was stopped by treating with Pen/Strep-containing cRPMI for 30 minutes at room temperature to kill any extracellular bacteria. Some wells were then lysed to determine cfu after phagocytosis. The other wells were washed with PBS, resuspended with Pen/Strep-free cRPMI, and returned to 37 °C for an additional 1 or 2 hours to determine cfu after bacterial killing. Specifically, to determine cfu, cells were washed with PBS, lysed with 1 ml distilled autoclaved water for 30 minutes at room temperature, diluted in PBS for twofold serial dilutions, plated in on blood-agar plates, and incubated overnight. Colonies were counted the next day and determined as intracellular cfu.
Histopathological assessment
Lungs were fixed by perfusion with 10% formalin and kept in 5 ml of 10% formalin for at least 72 hours. Tissue sections were stained with hematoxylin and eosin for histological examination.
Statistical analysis
To determine if the differences among infection groups were significant, Student’s t-test was performed for two-sample comparisons with Prism (GraphPad Software, La Jolla, CA). For comparison of body weight changes, the percentages were transformed before the t-test was performed. Survival data were compared with a log-rank (Mantel-Cox) test with prism. A P value of <0.05 was regarded as statistically significant.
Acknowledgments
The study was supported by funds from the Canadian Institutes of Health Research. The authors thank Uma Sankar and Xueya Feng for their technical assistance.
The authors declare no conflict of interest.
References
- 2005Notifiable Diseases On-Line Public Health Agency of Canada; . http://dsol-smed.phac-aspc.gc.ca/dsol-smed/ndis/index-eng.php . [Google Scholar]
- Kadioglu A, Weiser JN, Paton JC, Andrew PW. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat Rev Microbiol. 2008;6:288–301. doi: 10.1038/nrmicro1871. [DOI] [PubMed] [Google Scholar]
- 2001Respiratory Disease in Canada Canadian Institute for Health Information Canadian Lung Association, Health Canada, Statistics Canada; . https://secure.cihi.ca/free_products/RespiratoryComplete.pdf . [Google Scholar]
- 2011Pneumococcal Disease, Invasive (IPD) Alberta Health and Wellness Public Health Notifiable Disease Management Guidelines Government of Alberta; . www.health.alberta.ca/documents/Guidelines-Pneumococcal-Disease-Invasive-IPD-2011.pdf . [Google Scholar]
- Le Saux N, Robinson J. Pneumonia in healthy Canadian children and youth: Practice points for management. Paediatr Child Health. 2011;16:417–424. doi: 10.1093/pch/16.7.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien KL, Wolfson LJ, Watt JP, Henkle E, Deloria-Knoll M, McCall N, Hib and Pneumococcal Global Burden of Disease Study Team Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet. 2009;374:893–902. doi: 10.1016/S0140-6736(09)61204-6. [DOI] [PubMed] [Google Scholar]
- Damjanovic D, Lai R, Jeyanathan M, Hogaboam CM, Xing Z. Marked improvement of severe lung immunopathology by influenza-associated pneumococcal superinfection requires the control of both bacterial replication and host immune responses. Am J Pathol. 2013;183:868–880. doi: 10.1016/j.ajpath.2013.05.016. [DOI] [PubMed] [Google Scholar]
- Update on Pediatric Invasive Pneumococcal Disease and Recommended Use of Conjugate Pneumococcal Vaccines. vol. 36. National Advisory Committee on Immunization (NACI) Public Health Agency of Canada; 2010. [Google Scholar]
- Chavanet P. Pneumococcus infections: is the burden still as heavy? Med Mal Infect. 2012;42:149–153. doi: 10.1016/j.medmal.2012.02.002. [DOI] [PubMed] [Google Scholar]
- Kadioglu A, Andrew PW. The innate immune response to pneumococcal lung infection: the untold story. Trends Immunol. 2004;25:143–149. doi: 10.1016/j.it.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Decker T, Müller M, Stockinger S. The yin and yang of type I interferon activity in bacterial infection. Nat Rev Immunol. 2005;5:675–687. doi: 10.1038/nri1684. [DOI] [PubMed] [Google Scholar]
- Parker D, Prince A. Type I interferon response to extracellular bacteria in the airway epithelium. Trends Immunol. 2011;32:582–588. doi: 10.1016/j.it.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JS, Wong G, Pillet S, Schindle S, Ennis J, Turner J. Evaluation of different strategies for post-exposure treatment of ebola virus infection in rodents. J Bioterror Biodef. 2011;S1:007. doi: 10.4172/2157-2526.s1-007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julander JG, Ennis J, Turner J, Morrey JD. Treatment of yellow fever virus with an adenovirus-vectored interferon, DEF201, in a hamster model. Antimicrob Agents Chemother. 2011;55:2067–2073. doi: 10.1128/AAC.01635-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowen BB, Ennis J, Russell A, Sefing EJ, Wong MH, Turner J. Use of recombinant adenovirus vectored consensus IFN-a to avert severe arenavirus infection. PLoS ONE. 2011;6:e26072. doi: 10.1371/journal.pone.0026072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowen BB, Ennis J, Sefing EJ, Wong MH, Jung KH, Turner JD. Extended protection against phlebovirus infection conferred by recombinant adenovirus expressing consensus interferon (DEF201) Antimicrob Agents Chemother. 2012;56:4168–4174. doi: 10.1128/AAC.00376-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaki Y, Ennis J, Rahbar R, Turner JD, Wandersee MK, Smith AJ. Single-dose intranasal administration with mDEF201 (adenovirus vectored mouse interferon-alpha) confers protection from mortality in a lethal SARS-CoV BALB/c mouse model. Antiviral Res. 2011;89:75–82. doi: 10.1016/j.antiviral.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JQ, Barabé ND, Huang YM, Rayner GA, Christopher ME, Schmaltz FL. Pre- and post-exposure protection against Western equine encephalitis virus after single inoculation with adenovirus vector expressing interferon alpha. Virology. 2007;369:206–213. doi: 10.1016/j.virol.2007.07.024. [DOI] [PubMed] [Google Scholar]
- O’Brien L, Perkins S, Williams A, Eastaugh L, Phelps A, Wu J. Alpha interferon as an adenovirus-vectored vaccine adjuvant and antiviral in Venezuelan equine encephalitis virus infection. J Gen Virol. 2009;90 Pt 4:874–882. doi: 10.1099/vir.0.006833-0. [DOI] [PubMed] [Google Scholar]
- Smee DF, Wong MH, Russell A, Ennis J, Turner JD. Therapy and long-term prophylaxis of vaccinia virus respiratory infections in mice with an adenovirus-vectored interferon alpha (mDEF201) PLoS ONE. 2011;6:e26330. doi: 10.1371/journal.pone.0026330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigent DA, Huff TL, Peterson JW, Stanton GJ, Baron S. Role of interferon in streptococcal infection in the mouse. Microb Pathog. 1986;1:399–407. doi: 10.1016/0882-4010(86)90071-9. [DOI] [PubMed] [Google Scholar]
- Elhaik-Goldman S, Kafka D, Yossef R, Hadad U, Elkabets M, Vallon-Eberhard A. The natural cytotoxicity receptor 1 contribution to early clearance of Streptococcus pneumoniae and to natural killer-macrophage cross talk. PLoS ONE. 2011;6:e23472. doi: 10.1371/journal.pone.0023472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso G, Midiri A, Biondo C, Beninati C, Zummo S, Galbo R. Type I IFN signaling is crucial for host resistance against different species of pathogenic bacteria. J Immunol. 2007;178:3126–3133. doi: 10.4049/jimmunol.178.5.3126. [DOI] [PubMed] [Google Scholar]
- Parker D, Martin FJ, Soong G, Harfenist BS, Aguilar JL, Ratner AJ. Streptococcus pneumoniae DNA initiates type I interferon signaling in the respiratory tract. MBio. 2011;2:e00016–e00011. doi: 10.1128/mBio.00016-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small CL, McCormick S, Gill N, Kugathasan K, Santosuosso M, Donaldson N. NK cells play a critical protective role in host defense against acute extracellular Staphylococcus aureus bacterial infection in the lung. J Immunol. 2008;180:5558–5568. doi: 10.4049/jimmunol.180.8.5558. [DOI] [PubMed] [Google Scholar]
- Lei XF, Ohkawara Y, Stämpfli MR, Gauldie J, Croitoru K, Jordana M. Compartmentalized transgene expression of granulocyte-macrophage colony-stimulating factor (GM-CSF) in mouse lung enhances allergic airways inflammation. Clin Exp Immunol. 1998;113:157–165. doi: 10.1046/j.1365-2249.1998.00652.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damjanovic D, Divangahi M, Kugathasan K, Small CL, Zganiacz A, Brown EG. Negative regulation of lung inflammation and immunopathology by TNF-a during acute influenza infection. Am J Pathol. 2011;179:2963–2976. doi: 10.1016/j.ajpath.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small CL, Shaler CR, McCormick S, Jeyanathan M, Damjanovic D, Brown EG. Influenza infection leads to increased susceptibility to subsequent bacterial superinfection by impairing NK cell responses in the lung. J Immunol. 2010;184:2048–2056. doi: 10.4049/jimmunol.0902772. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.