

Significance
Despite dramatically improved therapeutic properties of single enantiomer drugs over the racemic mixtures, numerous drugs and drug candidates are still being developed and sold as racemates, including the class of immunomodulatory drugs derived from thalidomide. The chiral center of these thalidomide analogs is chemically unstable, resulting in interconversion of the enantiomers both in vitro and in vivo. Through stabilization of the chiral center with deuterium, we show for the first time, to our knowledge, that the in vitro antiinflammatory and in vivo antitumorigenic activities of a thalidomide analog currently in clinical development (CC-122) are caused exclusively by one enantiomer. Our findings enable the development of improved thalidomide analogs as therapeutics following stated regulatory guidance for the development of single enantiomers.
Keywords: thalidomide, enantiomer, deuterium, CC-122, cancer
Abstract
Therapeutics developed and sold as racemates can exhibit a limited therapeutic index because of side effects resulting from the undesired enantiomer (distomer) and/or its metabolites, which at times, forces researchers to abandon valuable scaffolds. Therefore, most chiral drugs are developed as single enantiomers. Unfortunately, the development of some chirally pure drug molecules is hampered by rapid in vivo racemization. The class of compounds known as immunomodulatory drugs derived from thalidomide is developed and sold as racemates because of racemization at the chiral center of the 3-aminoglutarimide moiety. Herein, we show that replacement of the exchangeable hydrogen at the chiral center with deuterium allows the stabilization and testing of individual enantiomers for two thalidomide analogs, including CC-122, a compound currently in human clinical trials for hematological cancers and solid tumors. Using “deuterium-enabled chiral switching” (DECS), in vitro antiinflammatory differences of up to 20-fold are observed between the deuterium-stabilized enantiomers. In vivo, the exposure is dramatically increased for each enantiomer while they retain similar pharmacokinetics. Furthermore, the single deuterated enantiomers related to CC-122 exhibit profoundly different in vivo responses in an NCI-H929 myeloma xenograft model. The (−)-deuterated enantiomer is antitumorigenic, whereas the (+)-deuterated enantiomer has little to no effect on tumor growth. The ability to stabilize and differentiate enantiomers by DECS opens up a vast window of opportunity to characterize the class effects of thalidomide analogs and improve on the therapeutic promise of other racemic compounds, including the development of safer therapeutics and the discovery of new mechanisms and clinical applications for existing therapeutics.
Chirality plays an important role in a variety of disciplines, including pharmaceuticals, foods and flavorings, materials science, and agricultural chemicals. In pharmaceuticals, changing just one chiral center can affect critical compound properties, including potency, off-target side effects, and pharmacokinetics (1–5), thus impacting efficacy and therapeutic index. Since the 1990s, drug molecules originally developed as racemates (a racemate is a 1:1 mixture of two mirror-image compounds or enantiomers) have been separated and developed as single preferred enantiomers (eutomers) because of improved synthesis, purification, and analytical methods. This approach, known as chiral switching, resulted in several new therapeutics based on existing drugs, including esomeprazole (Nexium), escitalopram (Lexapro), levalbuterol (Xopenex), eszopiclone (Lunesta), and levomilnacipran (Fetzima). It also led to new Food and Drug Administration guidance for the characterization and development of stereoisomers (6).
There are numerous racemic compounds where chiral switching is impossible, because the chiral center has an exchangeable hydrogen that interconverts on a timescale that is incompatible with storage or dosing of a single pure enantiomer. Some examples of drugs that are still marketed as a mixture of two enantiomers include thalidomide, pioglitazone, bupropion, prasugrel, donepezil, and lorazepam. For each of these molecules, the rate of interconversion of enantiomers under physiological conditions is fast compared with the elimination rate of each molecule.
Immunomodulatory drugs derived from thalidomide are an important class of antiinflammatory and antitumorigenic drugs, of which thalidomide is the prototype (7, 8). These compounds (Fig. 1) are all characterized by a nitrogen-substituted 3-aminoglutarimide moiety essential for their therapeutic activity with an exchangeable hydrogen at the chiral center. In addition to thalidomide, a number of analogs, including lenalidomide (Revlimid), pomalidomide (Pomalyst and Imnovid), CC-11006, CC-122, and CC-220, have been or are being developed for the treatment of blood cancers and hematological conditions (e.g., multiple myeloma, myelodysplastic syndrome, lymphoma, and chronic lymphocytic leukemia), solid tumors, and inflammatory diseases (e.g., sarcoidosis, systemic sclerosis, and systemic lupus erythematosus). [We tentatively assign N-{[2-(2,6-dioxopiperidin-3-yl)-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl]methyl}cyclopropanecarboxamide (compound 1) as CC-11006, a former development compound for hematological cancers. The chemical structure is disclosed in patents and patent applications related explicitly to this 3-aminoglutarimide (9–11). Experimental data are explicitly disclosed for CC-11006 in the pharmacology review sections for new drug applications (NDAs) 021880 and 204026 corresponding to Revlimid and Pomalyst, respectively. The chemical structure of CC-122 is disclosed in figure 5b in International Patent Application No. WO 2012/125459 (12). The experimental data disclosed in this patent and other patent applications related to the explicit chemical structure (12–16) are also identical to the experimental data in published poster abstracts related to CC-122 (17, 18).]
Fig. 1.

Structures of thalidomide, pomalidomide, lenalidomide, and protonated and deuterated enantiomers of compound 1 [CC-11006; i.e., (S)-H-1, (R)-H-1, (S)-D-1, and (R)-D-1] as well as protonated and deuterated enantiomers of compound 2 [CC-122; (S)-H-2, (R)-H-2, (S)-D-2, and (R)-D-2 (absolute configuration was not established for compound 2; enantiomers are referred to by the sign of their optical rotation; i.e., (+) or (−); details in the text)].
The challenges of dosing and maintaining levels of a single preferred enantiomer of thalidomide analogs are apparent comparing the generally short racemization half-life (rac t1/2) in human plasma or blood with the longer elimination half-life (elim t1/2) of thalidomide (19–24) (rac t1/2 = 2–6 h; elim t1/2 = 3–8 h), lenalidomide (25–27) (rac t1/2 < 3 h; elim t1/2 = 3–8 h), and pomalidomide (28–30) (rac t1/2 < 1 h; elim t1/2 = 7.5–9.5 h). The propensity to racemize prior to elimination makes it extremely difficult to assess the properties of the individual enantiomers and impedes mechanistic studies. In fact, the broad spectrum of activity for thalidomide analogs has been extensively studied and suggests multiple target sites of action (7, 31–35). Recently, CC-122 has been defined as a pleiotropic pathway modulator (Celgene Corp) because of its broad range of activity (17, 18). In actuality, each enantiomer may have unique pharmacological and safety profiles. Although there is limited data available and the enantiomers interconvert during the time course of the studies, a few groups have shown that the teratogenicity, in vitro antiinflammatory activity, and in vivo efficacy of protonated thalidomide analogs are caused, in large part, by the (S)-enantiomer (22, 36–38). (S)-pomalidomide was originally advanced into clinical trials as ENMD 0995 (39) but soon abandoned because of the rapid racemization of the exchangeable chiral center (28, 29). Finally, it has recently been shown by X-ray crystallography that the (S)-enantiomers of thalidomide, lenalidomide, and pomalidomide preferentially bind a newly identified target, cereblon (CRBN), believed to be responsible for their efficacy and teratogenicity in a cocrystal with the DDB1–CRBN complex, where DDB1 stands for DNA damage-binding protein 1 (40).
Attempts to stabilize the (S)-enantiomer of thalidomide analogs have included replacement of the exchangeable hydrogen with methyl (20, 37, 41, 42) or fluorine groups (43, 44). If and when the stable enantiomer of these analogs was studied, none were superior to the racemic, protonated thalidomide analog. The effects observed included similar or decreased potency, increased degradation, increased toxicity, and/or increased teratogenicity. In vitro stabilization of enantiomers has also been achieved by replacement of the carbonyl group adjacent to the chiral center with an oxetane (45). The impact of this change on in vivo dosing or efficacy has not been reported.
Recently, deuterium has been explored to stabilize interconverting enantiomers. Deuterium is a stable isotope of hydrogen with a natural abundance of 0.015%, and it is known for its potential to stabilize chemical bonds. Therefore, deuterium is predicted to not affect the pharmacological properties of a compound, contrary to what can be anticipated with methyl, fluoro, or oxetane functional groups. Furthermore, given the natural abundance of deuterium and its ubiquitous use in past human pharmacokinetic studies, the use of deuterium in therapeutics does not present a safety concern.
The use of deuterium to stabilize drugs against undesirable metabolism, known as metabolic switching, began in the 1960s (46–51) and is the predominant approach to deuterated drugs today. Metabolic switching can be a challenging strategy, because it is often difficult to translate from in vitro to in vivo (52), is limited to defined metabolic pathways, and requires the synthesis and testing of numerous analogs.
The approach that we describe in this paper, deuterium-enabled chiral switching (DECS), is uniquely differentiated from metabolic switching, in that it is based on chemical stability, is generally independent of metabolism, thus resulting in little or no change in pharmacokinetics, translates from in vitro to in vivo, and requires the synthesis and testing of just two analogs.
We are the only group, to our knowledge, to previously and herein report the stabilization and differentiated in vitro and in vivo properties of monodeuterated enantiomers of several thalidomide analogs, including reduced degradation, improved pharmacokinetics, and separation of in vitro antiinflammatory effects (53). Yamamoto et al. (54) previously reported the stabilization of monodeuterated thalidomide enantiomers in aqueous solution but did not differentiate their biochemical properties or their pharmacokinetic and pharmacodynamic properties. Another group has shown improved stability, pharmacokinetics, and separation of in vitro pharmacological effects with pentadeuterated lenalidomide enantiomers (55, 56). However, the latter publications do not differentiate the properties in vivo or allow discrimination of the effects of the additional four deuteria on the 3-aminoglutarimide ring and their pharmacodynamic contributions (55, 56).
Herein, we report the synthesis, in vitro characterization, and differentiation of stabilized enantiomers of two unique thalidomide analogs, compounds 1 (57) and 2 (CC-122) (58), and for the first time to our knowledge, we differentiate stabilized enantiomers of thalidomide analogs in in vitro and in vivo efficacy models.
Results
Synthesis of Deuterated (R)- and (S)-N-{[2-((3-2H)-2,6-Dioxopiperidin-3-Yl)-1,3-Dioxo-2,3-Dihydro-1H-Isoindol-4-Yl]Methyl}Cyclopropanecarboxamide and Deuterated rac-3-(5-Amino-2-Methyl-4-Oxoquinazolin-3(4H)-Yl)-(3-2H)-Piperidine-2,6-Dione.
Compounds were synthesized as shown in Figs. 2 and 3 and are described in more detail in Materials and Methods. Enantiomers (R)-N-{[2-((3-2H)-2,6-dioxopiperidin-3-yl)-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl]methyl}cyclopropanecarboxamide [(R)-D-1] and (S)-N-{[2-((3-2H)-2,6-dioxopiperidin-3-yl)-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl]methyl}cyclopropanecarboxamide [(S)-D-1] were prepared using commercially available deuterated chiral starting materials (R)-(3-2H)-3-amino-2,6-dioxopiperidine and (S)-(3-2H)-3-amino-2,6-dioxopiperidine and isolated with deuterium contents at the chiral center of 92% and 80%, purities > 99% and 98.5%, and enantiomeric excesses (%ee; %ee = percentage of enantiomer 1 − percentage of enantiomer 2) of 92%ee and 86%ee for (R)-D-1 and (S)-D-1, respectively. Enantiomers (+)-3-(5-amino-2-methyl-4-oxoquinazolin-3(4H)-yl)-(3-2H)-piperidine-2,6-dione [(+)-D-2] and (−)-3-(5-amino-2-methyl-4-oxoquinazolin-3(4H)-yl)-(3-2H)-piperidine-2,6-dione [(−)-D-2] were obtained as a mixture in a deuterium incorporation and quinazoline formation step. They were separated by chiral chromatography, and both were isolated with deuterium content at the chiral center of 87%, purity > 99%, and >98%ee. Isotopomeric enantiomers (+)-H-2 and (−)-H-2 were synthesized similarly and isolated with similar purities and %ee.
Fig. 2.
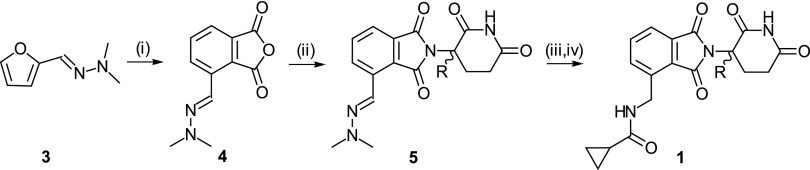
Synthesis of rac-H-1 and isotopomeric enantiomers (R)-D-1 and (S)-D-1: (i) maleic anhydride, trifluoroacetic acid, 50 °C; (ii) rac-3-amino-2,3-dioxopiperidine, imidazole, acetic acid, 77 °C (for rac-H-1); (R)- or (S)-(3-2H)-3-amino-2,3-dioxopiperidine, diisopropylethylamine, 70 °C [for (R)-D-1 or (S)-D-1, respectively]; (iii) H2, palladium on carbon (Pd/C); and (iv) cyclopropanecarbonyl chloride, diisopropylethylamine.
Fig. 3.

Synthesis of rac-D-2: (i) acetic anhydride, 120 °C; (ii) water, reflux; (iii) rac-3-amino-2,6-piperidinedione hydrochloride, diisopropylethylamine, hydroxybenzotriazole, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide, N,N-dimethylformamide; (iv) chlorotrimethylsilane, triethylamine, acetonitrile, 75 °C and then deuterium oxide, room temperature; and (v) H2, Pd(OH)2, N,N-dimethylformamide.
Deuteration at the Chiral Center Reduces Interconversion of Enantiomers in Plasma.
The enantiomers of deuterated rac-3-(5-amino-2-methyl-4-oxoquinazolin-3(4H)-yl)-(3-2H)-piperidine-2,6-dione (rac-D-1) and rac-D-2 in plasma can degrade or through slow deuterium/hydrogen (D/H) exchange, form the isotopomeric enantiomers, which can themselves enantiomerize and degrade (SI Appendix, Fig. S1). These reactions were observed in studies performed in human plasma for compound 1 (SI Appendix, Fig. S2) and mouse and human plasma for compound 2 (SI Appendix, Figs. S3 and S4). The experimental data were analyzed as described in Materials and Methods using the appropriate kinetic equations to generate rate constants for the individual reactions.
For rac-D-1 (SI Appendix, Table S1), degradation of the deuterated enantiomers in human plasma was found to be about 1.5-fold slower than D/H exchange. Additionally (S)-D-1 was shown to be twofold more stable than (R)-D-1 to both D/H exchange and degradation. Data obtained from incubation of protonated (S)-enantiomer, (S)-H-1, show that enantiomerization is stereoselective, because (S)-H-1 enantiomerizes at least 15-fold faster than (R)-H-1. Therefore, the enantiomerization rate of (R)-H-1 and the degradation rate of (S)-H-1 could not be calculated accurately, because (S)-H-1 rapidly enantiomerized to (R)-H-1, which then degraded quickly. Degradation of protonated (R)-H-1 was at least threefold faster than that of (R)-D-1.
In the case of compound 2 (SI Appendix, Table S2), all reaction rates (degradation, D/H exchange, and enantiomerization) were about twofold faster in human plasma than mouse plasma. In both species, the degradation rate was two- to threefold slower than the enantiomerization rate and similar to D/H exchange. Meanwhile, D/H exchange was 1.6-fold faster for (−)-D-2 than (+)-D-2 in human plasma, whereas the difference was not as pronounced in mouse plasma. D/H exchange was also stereoselective in mouse plasma and stereospecific in human plasma, where (+)-D-2 formed only (+)-H-2 and (−)-D-2 formed only (−)-H-2. D/H exchange was also significantly slower than enantiomerization in both species (1.5- to 3-fold), further showing stabilization of the deuterated enantiomers.
Oral Administration of a Single Deuterated Enantiomer of 2 to SCID Mice Results Predominantly in Exposure to That Enantiomer.
Quantitative pharmacokinetics are presented in Fig. 4 and SI Appendix, Fig. S5. Oral gavage of female CB.17.SCID mice with rac-3-(5-amino-2-methyl-4-oxoquinazolin-3(4H)-yl)-piperidine-2,6-dione (rac-H-2; 30 mg/kg) resulted in exposure (area under the curve [AUC]) to both (+)-H-2 and (−)-H-2 in a 3:2 ratio; (+)-D-2 or (−)-D-2 was administered at one-half of the dose of rac-H-2 (i.e., 15 mg/kg), because the dose of each enantiomer in the racemate is 1:1, corresponding to one-half the quantity within the mixture. Tmax (time to maximum plasma concentration) was not affected by deuteration (0.25–1 h). Deuteration also did not change the fast elimination, with half-lives (t1/2) of 1–2 h for both protonated and deuterated enantiomers (Table 1 and SI Appendix, Table S3). Almost exclusive exposure to the corresponding single enantiomer was observed, with 96% and 98% of the total AUC for mice dosed with (−)-D-2 and (+)-D-2, respectively (Table 1). Little to no D/H exchange was observed during the study. This observation is supported by comparing the percentage of total AUC corresponding to the deuterated enantiomers in the pharmacokinetic study [86% and 90% for (+)-D-2 or (−)-D-2, respectively] with the percentage of deuterium in the dosed compounds (87% for both enantiomers by NMR). Unexpectedly, double the exposure (Table 1) and approximately threefold larger Cmax (maximal plasma concentration) (SI Appendix, Table S3) were observed in animals administered the pure deuterated enantiomers at one-half of the dose of rac-H-2. Also, the 3:2 exposure ratio of (+)-2 to (−)-2 [e.g., AUC((+)-2) = AUC((+)-H-2) + AUC((+)-D-2)] observed in animals dosed with rac-H-2 was the same comparing exposures to (+)-2 with exposures to (−)-2 in animals dosed with (+)-D-2 and (−)-D-2, respectively.
Fig. 4.

Semilogarithmic plot of the pharmacokinetics of (−)-2 (blue circles) and (+)-2 (red diamonds; sum of isotopomers) in female CB.17 SCID mice dosed orally with (A) protonated racemate rac-H-2 (30 mg/kg), (B) (−)-D-2 (15 mg/kg), or (C) (+)-D-2 (15 mg/kg).
Table 1.
Elimination half-life (t1/2) and plasma exposure (AUC0−t) for (−)-H-2, (+)-H-2, (−)-D-2, and (+)-D-2 (h−, h+, d−, and d+) in female CB.17 SCID mice orally administered protonated racemate, rac-H-2 (30 mg/kg), or deuterated enantiomers (−)-D-2 and (+)-D-2 (15 mg/kg each)
| Compound dosed | Enantiomer observed | |||||||
| (−)-H-2 | (+)-H-2 | (−)-D-2 | (+)-D-2 | |||||
| t1/2 (h) | AUC (ng⋅h/mL) | t1/2 (h) | AUC (ng⋅h/mL) | t1/2 (h) | AUC (ng⋅h/mL) | t1/2 (h) | AUC (ng⋅h/mL) | |
| rac-H-2 (30 mg/kg) | 2.1 | 22,500 | 2.0 | 35,900 | — | — | — | — |
| (−)-D-2 (15 mg/kg) | 1.2 | 6,160 | N/A | 1,180 | 1.2 | 44,100 | N/A | 898 |
| (+)-D-2 (15 mg/kg) | N/A | 993 | 1.0 | 6,120 | N/A | 661 | 1.1 | 63,600 |
N/A, not applicable.
TNF-α Production by LPS-Stimulated Peripheral Blood Mononuclear Cells Is Inhibited by Deuterated Enantiomers of Compounds 1 and 2 with Significantly Different Potencies.
When treated with LPS, human peripheral blood mononuclear cells (PBMCs) release cytokines, including TNF-α (59). The inhibitory effects of the deuterated enantiomers of phthalimide derivative 1 and quinazolinone derivative 2 against TNF-α production were evaluated in vitro using human PBMCs. As shown in Table 2, (S)-D-1 was about 10-fold more potent than (R)-D-1, whereas the deuterated quinazolinone derivative, (−)-D-2, was about 20-fold more potent than (+)-D-2.
Table 2.
Inhibition of TNF-α production by LPS-stimulated human PBMCs
| Compound | IC50 (nM) |
| (S)-D-1 | 13.4 |
| (R)-D-1 | 123 |
| (−)-D-2 | 48.5 |
| (+)-D-2 | 945 |
(−)-D-2 Is More Antitumorigenic than CC-122 (rac-H-2) in an H929 Myeloma Xenograft Model, and (+)-D-2 Has Limited Efficacy in the Same Xenograft Model.
The stabilized enantiomers of 2 were compared with the protonated racemate, rac-H-2, in an s.c. NCI-H929 (H929) myeloma CB.17 SCID mouse xenograft model. All treatments were well-tolerated, with no differences in body weight measurements between dosing regimens (SI Appendix, Fig. S6) and no abnormal cage-side observations. A marked difference in efficacy was observed between the enantiomers (Fig. 5 A and B); (−)-D-2 significantly inhibited tumor growth compared with vehicle [tumor growth inhibition (TGI) of 58% and 71% at 1.5 and 15 mg/kg, respectively], whereas the (+)-D-2 enantiomer significantly accelerated tumor growth at 1.5 mg/kg (TGI = −43%) and nonsignificantly slowed tumor growth at 15 mg/kg (TGI = 11%). The parent racemic compound (rac-H-2) statistically significantly reduced tumor growth at 3 mg/kg (TGI = 52%) but not as efficiently as (−)-D-2 at 1.5 or 15 mg/kg. In a repeat study, the same rank order was observed, with (−)-D-2 being the most efficacious deuterated enantiomer (Fig. 5C and SI Appendix, Figs. S7 and S8), although the compounds seemed more potent and the tumor growth in vehicle-dosed animals was faster. Average tumor volumes on the last day of dosing for the two studies combined (Fig. 5C) show that (−)-D-2 (TGI of 84% and 89% at low and high dose, respectively) has significantly higher antitumorigenic potential than rac-H-2 or (+)-D-2. At low dose, (+)-D-2 (TGI = 26%) seems similar to vehicle, whereas at high dose, its efficacy is approaching that of rac-H-2 [TGI of 68% and 80% for (+)-D-2 and rac-H-2, respectively]. This efficacy is likely caused by ongoing racemization of ∼13% of the nondeuterated (+)-H-2 contaminant in (+)-D-2. Based on this contamination with the rapidly racemizing protonated enantiomer, at the high 15-mg/kg dose of (+)-D-2, the exposure to the active (−)-H-2 is predicted to become almost equal to the exposure to (−)-H-2 when rac-H-2 is dosed at 3 mg/kg.
Fig. 5.
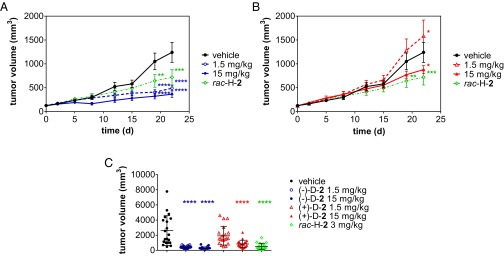
H929 xenograft model. Average tumor volume (± SEM) as function of time in female CB.17 SCID mice (n = 10 per group) treated daily by oral gavage with vehicle (black circles), rac-H-2 (3 mg/kg; green diamonds), and (A) (−)-D-2 [1.5 (open blue squares) or 15 mg/kg (filled blue squares)] or (B) (+)-D-2 [1.5 (open red triangles) or 15 mg/kg (filled red triangles)]. Two-way ANOVA of tumor volume vs. time with Bonferroni’s multiple comparisons posttest against vehicle group. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (C) Average tumor volume (± SD) on the last day of dosing for the two repeats (n = 20 per group). One-way ANOVA of tumor volume with Tukey posttest against vehicle group. ****P < 0.0001.
Discussion
To evaluate the potential differences in antiinflammatory and antitumorigenic properties of their enantiomers, we synthesized recently disclosed thalidomide analogs rac-N-{[2-(2,6-dioxopiperidin-3-yl)-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl]methyl}cyclopropanecarboxamide (rac-H-1) and rac-H-2 (CC-122) (Fig. 1) and separated their enantiomers. Similar to other compounds within the class of thalidomide analogs, they are characterized by a glutarimide moiety bearing an amino substituent on the carbon-α to a carbonyl functional group, rendering it chiral. Because of the acidic nature of the proton at the chiral center, thalidomide, lenalidomide, pomalidomide, and related analogs have been developed as racemic mixtures, because the enantiomers interconvert rapidly under physiological conditions. Enantiomers of 1 and 2 also rapidly racemize, despite bearing different substituents (isoindolinone and quinazolinone, respectively) on the nitrogen of the shared 3-aminoglutarimide moiety.
We, therefore, used DECS (the substitution of the proton at the chiral center by a deuterium to facilitate chiral switching) as a method to slow down interconversion between enantiomers and characterize the potential therapeutic differences between the enantiomers. Replacing the hydrogen at the chiral center with a deuterium may impart improved stability against racemization caused by the deuterium kinetic isotope effect (2H-KIE = kH/kD, where kH and kD are the rate constants for the reaction involving hydrogen- and deuterium-containing isotopomers, respectively). The enantiomerization reaction mechanism of compounds with an acid-labile hydrogen at the chiral center involves the formation of a planar enol (or enolate) intermediate resulting from the breaking of the chiral carbon–hydrogen bond. If formation of the intermediate is the rate-determining step in the racemization reaction, a primary 2H-KIE should be expected. The theoretical maximal primary 2H-KIE based on zero-point energy difference is 7–10 (60), although significantly larger effects, as high as 70 (61), have been reported and can be explained with tunneling mechanisms. In other cases, no 2H-KIE or even an inverse effect (62, 63) may be observed. It is, therefore, difficult to predict the magnitude of a 2H-KIE.
The stabilization effect of deuterium against enantiomerization was shown ex vivo for both thalidomide analogs, 1 and 2, by incubation in physiologically relevant media (plasma) at physiological temperature (37 °C). The increase in stability varied in magnitude as a function of the pendant chemical scaffold (phthalimide or quinazolinone) and the individual enantiomer.
The enantiomerization reactions seemed to be stereoselective, particularly for (S)-H-1 and (R)-H-1, where there is at least a 15-fold difference in enantiomerization rate constants. Stereoselective enantiomer interconversion reactions in the presence of albumin, a major plasma protein, have been reported previously (64). This stereoselectivity is likely caused by catalysis of the reaction by stereoselective plasma protein interactions. However, because of the unique property of rapid enantiomerization of the compounds studied herein, we are unable to show that the stereoselectivity is caused by plasma protein binding, because the experiments require incubation for several hours, during which enantiomerization would occur. Our D/H exchange studies of compound 1 monitored the racemic mixture; therefore, we cannot assess whether deuterium at the chiral center slowed the formation of the enolate to the same extent for both enantiomers.
The (+)-enantiomer of compound 2 was found to be slightly more stable than the (−)-enantiomer. This effect was identical for both the protonated and deuterated enantiomers, with a 1.36-fold difference between the (+)- and (−)-enantiomerization rate constants in human plasma. The species (mouse vs. human) was also shown to play an important role in enantiomerization reactions of quinazolinone derivative 2, where all reactions were found to be faster in human than mouse plasma. Similar to what has been observed for thalidomide (65, 66), both compounds were subject to in vitro degradation, likely by chemical hydrolysis of the glutarimide and/or phthalimide moiety.
Deuteration at the chiral center of phthalimide derivative 1 slowed down degradation by a factor of 3, similar to what we have described for the isoindolinone derivative, lenalidomide (53). Interestingly, no effect of deuterium on the in vitro degradation rate of 2 was observed. Based on these results and the development status of the racemic parent compounds, in vivo studies were only performed with quinazolinone 2.
There are numerous biological effects of thalidomide analogs (7). To show the difference in antiinflammatory activity of the enantiomers, we measured their ability to inhibit TNF-α production by LPS-stimulated PBMCs (Table 2). The inhibition of TNF-α production is a measure of their immunomodulatory action, essential for their efficacious use in autoimmune diseases, such as lupus erythematosus or Behçet’s disease, inflammatory diseases, such as Crohn’s disease or erythema nodosum leprosum, and also, cancers, such as multiple myeloma and selected solid tumors, where TNF-α production has been shown to play an important tumor-promoting role (31, 67, 68). Despite the potential for enantiomerization during the long incubation times (cells are incubated with compound overnight at 37 °C in the presence of 10% plasma), (S)-D-1 was shown to be about 10-fold more potent than (R)-D-1 at inhibiting TNF-α production. Similarly, (−)-D-2 was about 20-fold more potent at inhibiting TNF-α production than (+)-D-2.
The pharmacokinetics of rac-H-2, (−)-D-2, and (+)-D-2 were evaluated in CB.17 SCID mice, the strain used for the xenograft model. In animals dosed with the protonated racemate, stereoselective exposure to (+)-H-2 was observed. Stereoselective exposure was also observed comparing exposure to the enantiomers (+)-2 and (−)-2 (sum of exposure to protonated and deuterated isotopomers) between groups of animals dosed with the individual deuterated enantiomers (+)-D-2 and (−)-D-2. This stereoselectivity, which is independent of the isotopomer, may be a consequence of stereoselective absorption, metabolism, elimination, and/or degradation. The deuterated enantiomers seemed very stable in vivo and showed almost no D/H exchange, leading to an almost stereospecific exposure to one enantiomer with 96% and 98% of the total exposure to the single (−)- or (+)-enantiomer, respectively (sum of corresponding protonated and deuterated isotopomeric enantiomers). This stereospecific exposure corresponds to enantiomeric excesses of 92% and 96% for (−)-2 and (+)-2, respectively, slightly lower than the %ee of the deuterated enantiomers used (%ee > 99%). This difference is likely caused by some small amount of D/H exchange and enantiomerization of the protonated enantiomer present in the administered deuterated enantiomer [∼13% of (+)-D-2 or (−)-D-2 is (+)-H-2 or (−)-H-2, respectively]. This effect was too limited to be analyzed in more detail. Studies in larger species (rat or nonhuman primate) could be used to further evaluate this observation, because the elimination rate will likely be slower. It was also observed that, although administered at one-half of the dose of the protonated racemate (same dose of enantiomer, because the racemate is composed of a 1:1 mixture of both enantiomers), each deuterated enantiomer showed an unexpected twofold increase in exposure to the corresponding enantiomer in the racemate. Because this twofold increase in exposure is accompanied by a threefold increase in Cmax without a change in elimination half-life, the higher exposure is likely because of better absorption of the individual enantiomers than the racemate. Exposure to (−)-2 in animals administered (+)-D-2 was about 7% of the exposure to (−)-H-2 in mice dosed with rac-H-2. This exposure to the (−)-enantiomer is close to the exposure to (−)-H-2 if the protonated racemate had been administered at one-tenth the dose (i.e., 3 mg/kg). The same was observed for exposure to the (+)-2 enantiomer in animals dosed orally with (−)-D-2.
An in vivo xenograft model of myeloma (NCI-H929) showed that the pharmacodynamic effects of the deuterated enantiomers of 2 were clearly differentiated. At 1.5 mg/kg, only (−)-D-2 showed profound efficacy, significantly reducing tumor growth. This activity was better than the protonated racemate when dosed at two times the dose (3.0 mg/kg), showing that the antitumorigenic activity of compound 2 is almost exclusively caused by the (−)-enantiomer. A 15-mg/kg dose of the same compound was moderately more potent than the 1.5-mg/kg dose, and we conclude that we have reached the top of the dose–response curve in this model. In contrast, at 1.5 mg/kg, enantiomer (+)-D-2 exhibited increased tumor volume in this experiment compared with vehicle. The tumor volume increase reached statistical significance on the last day of the study. At 15 mg/kg, the (+)-D-2–dosed enantiomer trended to slightly decrease tumor volume. However, the antitumorigenic efficacy observed after a high dose of (+)-D-2 was still lower than that of the rac-H-2 at only 3 mg/kg. This efficacy, observed at the highest dose of (+)-D-2 only, is likely because of the predicted enantiomerization of the (+)-H-2 contaminant and the subsequent accumulation of the efficacious (−)-H-2 enantiomer. This accumulation is predicted to lead to exposure to the antitumorigenic enantiomer at a concentration that is close to the exposure obtained when administering rac-H-2 at 3 mg/kg, which was observed in our pharmacokinetic study discussed above.
In our repeat of the in vivo xenograft model of myeloma (NCI-H929), the (−)-enantiomer is still profoundly more potent than the (+)-enantiomer in curtailing tumor growth, but the low dose of the (+)-enantiomer did not promote tumor growth. In addition, in this second study, (−)-D-2 is more potent than in the first study, and therefore, the slow accumulation of the strongly antitumorigenic (−)-enantiomer after dosing of (+)-D-2 may mask any tumor growth-promoting effects. Synthetic method development aimed at further reducing the amount of the protonated enantiomer in the deuterated formulation is required to further delineate the effects of the individual deuterated enantiomers.
These experiments show that DECS can stabilize chiral compounds against epimerization and clearly differentiate the beneficial and potential detrimental effects of enantiomers. In the exemplary case of thalidomide analogs 1 and 2, preparation, separation, and characterization of the individual deuterated enantiomers allowed us to show for the first time, to our knowledge, that efficacy is caused almost exclusively by a single enantiomer. The determination of absolute configuration of the enantiomers of deuterated compound 2 was not possible, because there is no prior data available on the enantiomers, and we did not obtain crystals of the enantiomers. However, based on available data for other thalidomide analogs (22, 37, 38) and the TNF-α inhibition data that we obtained for the enantiomers of deuterated compound 2, it is likely that the most efficacious enantiomer, (−)-D-2, is (S)-D-2. The same tentative stereochemistry assignment can be made from the reported crystal structure of thalidomide, lenalidomide, and pomalidomide with target protein complex DDB1-CRBN, where the (S)-enantiomer was systematically shown to be preferentially bound through the constant and required 3-aminopiperidine-2,6-dione moiety (40). It is, therefore, likely that, similar to other thalidomide analogs, (−)-D-2 exerts its therapeutic effects through binding to CRBN and modulation of its activity.
Today, most chiral drugs entering the market are single stereoisomers (69, 70). However, 50% and 20% of the chiral new molecular entities approved by the Food and Drug Administration in 2009 and 2011, respectively, were racemates. Several of these racemates cannot be stored or dosed as a single stereoisomer, because they are prone to racemization. Using our approach of chiral switching with deuterium, described herein for thalidomide analogs, we have stabilized and begun to differentiate the enantiomers of several other racemic drugs and drug candidates (71–74), enabling the continued improvement of therapeutics for patients.
Materials and Methods
All reagents were purchased from commercial sources and used as received. (R)- and (S)-3-amino-(3-2H)-piperidine-2,6-dione hydrochloride were obtained from C/D/N Isotopes Inc. (Pointe-Claire). Solvents used for analytical purposes were of HPLC grade and used as purchased. All compounds were characterized by liquid chromatography (LC)/MS and NMR. Purity of the final products was also evaluated by HPLC/UV and chiral HPLC/UV. Synthetic details are presented in SI Appendix, Supplementary Material. Animal studies were performed in accordance with the recommendations of the Guide for Care and Use of Laboratory Animals (75) with respect to restrain, husbandry, surgical procedures, feed and fluid regulation, and veterinary care. All animal studies were approved by Charles River Laboratories International, Inc. Institutional Animal Care and Use Committee.
Synthesis.
The synthesis of deuterated analogs (S)-D-1, (R)-D-1, (+)-D-2, and (−)-D-2 was achieved using different approaches. Use of the advanced synthon, deuterated (S)- or (R)-3-aminoglutarimide, was successful only in the preparation of the deuterated enantiomers of phthalimide derivative 1. Preparation of deuterated 2 required formation of the quinazoline ring from open-chain amide intermediate 9, where the amino group on the quinazolinone moiety is protected as a nitro group. Although we did not optimize the cyclization reaction, equivalents of chlorotrimethylsilane and triethylamine as well as time and temperature during the deuterium oxide quench were found to be critical variables in obtaining pure 10 from 9 without contamination by bis- and tris-deuterated analogs containing deuterium atoms at carbon 5 of the glutarimide ring.
As shown in Fig. 2, the deuterated enantiomers of compound 1 were synthesized stereospecifically using commercially available deuterated chiral starting materials. The hydrochloride salts of (R)- and (S)-(3-2H)-3-amino-2,6-dioxopiperidine were used to prepare the 1,3-dioxo-2,3-dihydro-1H-insoindole bicyclic ring system by reaction with phthalic anhydride 4 bearing a protected aminomethyl substituent. The reaction proceeded without significant loss of deuterium or chiral purity. The cyclopropylcarboxamide moiety was introduced last by reaction of the aminomethyl pendant group with cyclopropanecarbonyl chloride after reductive cleavage of the hydrazone moiety by hydrogenation over palladium on carbon (Pd/C). Enantiomers (R)-D-1 and (S)-D-1 were characterized by chiral supercritical fluid chromatography (SFC) with purities > 99% and 98.5% and enantiomeric excesses 92%ee and 86%ee for (R)-D-1 and (S)-D-1, respectively. Deuterium contents of 92% and 80% at the chiral center were confirmed by 1H NMR and MS. The protonated racemate, rac-H-1, was prepared in a similar way, and enantiomers (R)-H-1 and (S)-H-1 were separated by chiral SFC with purity > 99% and %ee > 99% for both enantiomers.
Deuteration of rac-H-2 at position 3 of the piperidine-2,6-dione moiety, shown in Fig. 3, required development of a quinazoline ring formation step compatible with introduction of deuterium. Thus, 2-acetamido-6-nitrobenzoic acid 8 was prepared by hydrolysis of benzoxazinone 7 synthesized by heating 2-amino-6-nitrobenzoic acid 6 with neat acetic anhydride. 2-Acetamido-6-nitrobenzoic acid 8 was then coupled with rac-3-aminoglutarimide to give rac-2-acetamido-N-(2,6-dioxopiperidin-3-yl)-6-nitrobenzamide 9. Ring closure was effected by heating 9 in acetonitrile in the presence of trimethylsilyl chloride and triethylamine followed by quenching in deuterium oxide to give the deuterated intermediate 5-nitroquinazolin-4-one 10, which was reduced to rac-D-2 by hydrogenation in the presence of palladium(II) hydroxide as catalyst. Proton NMR analysis showed regioselective deuteration at position 3 vs. position 5 of the piperidine-2,6-dione. The cyclization reaction was further optimized to result in the single 3-deuterated regioisomer by adjusting molar ratios of trimethylsilyl chloride and triethylamine. The enantiomers of deuterated rac-D-2 and isotopomeric rac-H-2, obtained by quenching in water, were separated by chiral SFC and isolated with purity > 99%, %ee > 98%, and deuterium content of 87% at the chiral center as confirmed by NMR. Chirality was determined by measuring optical rotation; thus, enantiomers of 2 are labeled (+)-2 and (−)-2.
Plasma Stability.
For a, (S)-H-1, (S)-D-1, and (R)-D-1 were incubated in human plasma at 37 °C in duplicates. Aliquots were removed at preset time points. Plasma proteins were precipitated by addition of acetonitrile containing internal standard tolbutamide (ISTD). The supernatants were analyzed by reverse-phase LC/MS-MS (0.1% formic acid in water:methanol, 40:60 vol/vol; Phenomenex Synergi Polar-RP) for the D/H exchange studies of (S)-D-1 and (R)-D-1 and chiral LC/MS-MS (water:acetonitrile, 20:80 vol/vol; Daicel ChiralPak IE-3) for the enantiomerization of protonated enantiomer (S)-H-1. Peak areas were normalized to the ISTD, and areas for the deuterated molecule were corrected for the isotopic peak of the protonated isotopomer if present. Corrected data were plotted using Microsoft Excel 2013 (Microsoft Corp.) and analyzed using the Excel Solver Generalized Reduced Gradient Nonlinear method with central derivatives to minimize the sum of sums of weighted Δ2 (square of difference between ISTD-normalized experimental data and calculated value divided by the experimental data). The data from the deuterium isotopomer stability study were analyzed using analytical Eqs. 1 and 2, whereas the enantiomerization data were fitted using numerical approximation of differential Eqs. 3 and 4 using the Euler method (Eq. 5). The step between calculated time points was minimized to minimize the local error (proportional to the square of the step size) and the global error (proportional to the step size):
| [1] |
| [2] |
| [3] |
| [4] |
where [D] and [H] are the concentrations of the deuterated and protonated isotopomers, respectively (D-1 and H-1; sum of deuterated and protonated enantiomers, respectively), [R] and [S] are the concentrations of (R)-H-1 and (S)-H-1, respectively, and ki indicates the rate constants for D/H exchange (i = DH), enantiomerization (i = SR from S to R, and i = RS from R to S), and degradation [i = Dd, Hd, Rd, and Sd for degradation of D-1, H-1, (R)-H-1, and (S)-H-1, respectively].
| [5] |
where [X]ti is the concentration of compound X at time ti, t1 is a time at which [X] is known, t2 is a time at which [X] is calculated, and [d[X]]t1 is the calculated value of the differential equation (Eqs. 1, 2, 3, and 4) at time t1.
For b, (+)-H-2, (−)-H-2, and (rac)-D-2 [50:50 racemic mixture of (+)-D-2 and (−)-D-2] were incubated in CD-1 mouse plasma (K3EDTA as an anticoagulant) or human plasma (K3EDTA as an anticoagulant) at 37 °C in duplicates. Studies and analyses were performed as described above. Samples were analyzed by chiral LC/MS-MS (0.1% acetic acid in water:acetonitrile, 20:80 vol/vol; Daicel ChiralPak IE-3). Peak areas for (+)-D-2 and (−)-D-2 were normalized to the ISTD and corrected for the isotopic peak of the corresponding protonated enantiomer [(+)-H-2 and (−)-H-2, respectively] if present. Corrected data were plotted using Microsoft Excel 2013 (Microsoft Corp.) and analyzed within Excel using the Excel Solver Generalized Reduced Gradient Nonlinear method with central derivatives to minimize the sum of sums of weighted Δ2 (square of difference between ISTD-normalized experimental data and calculated value divided by the experimental data). Calculated values were obtained through numerical approximation of differential Eqs. 6 and 7 for the stability studies of (+)-H-2 and (−)-H-2 and Eqs. 8, 9, 10, and 11 for the stability study of deuterated racemate rac-D-2 by the Euler method (Eq. 5). Independent analyses of the data for (+)-H-2 and (−)-H-2 were performed first. The average rate constants for the enantiomerization reactions, k+− and k−+, from these two analyses were used as constants in the fitting of the stability data of rac-D-2. Furthermore, to limit the complexity of calculations, the assumption was made that degradation was not affected by the isotopic substitution or the chirality; hence, kh+d = kh−d = hd+d = kd−d = kd. The average degradation rate constant obtained by fitting the data for (+)-H-2 and (−)-H-2 was used in the fit of the stability of rac-D-2:
| [6] |
| [7] |
| [8] |
| [9] |
| [10] |
| [11] |
where [h+], [h−], [d+], and [d−] are the concentrations of (+)-H-2, (−)-H-2, (+)-D-2, and (−)-D-2, respectively; k+− and k−+ are the rate constants for the enantiomerization reactions (+)-H-2 to (−)-H-2 and (−)-H-2 to (+)-H-2, respectively; kD++, kD+−, kD−+, and kD−− are the rate constants for the D/H exchange reactions (+)-D-2 or (−)-D-2 to (+)-H-2 or (−)-H-2, and kd is the rate constant for the degradation of all four compounds.
TNF-α inhibition in LPS-stimulated human PBMCs.
Human PBMCs were isolated from whole blood using a standard density separation protocol. Cell viability was assessed by Trypan Blue. Cells were seeded in 96-well plates at 50,000 cells per well in RPMI media with 10% (vol/vol) serum. Cells were allowed to equilibrate for 1 h before compound stimulation. Compounds (S)-D-1, (R)-D-1, (+)-D-2, and (−)-D-2 were dissolved in DMSO and then, cell culture media, and they were diluted to final concentrations in half-log intervals (final concentration of DMSO < 0.1%). Compound solution or vehicle was added to the cells, and plates were incubated for 1 h before addition of LPS (1,000 ng/mL final) or an equivalent volume of media. After 18 h of incubation, plates were centrifuged, and the supernatant was removed for TNF-α quantitation by Luminex [(S)-D-1 and (R)-D-1] or ELISA [(+)-D-2 and (−)-D-2]. Percentage inhibition (vs. vehicle-treated PBMCs) was plotted against concentration, and the IC50 was calculated by fitting the log(concentration) vs. response to the logistic Hill equation with variable slope (GraphPad Prism 6.0; GraphPad Software). For studies of (+)-D-2 and (−)-D-2, cytotoxicity was evaluated by incubation for another 2 h in the presence of WST-1 (Sigma) and measured for absorbance at 450 nm. The compounds were found to be noncytotoxic.
Pharmacokinetics of rac-H-2, (+)-D-2, and (−)-D-2.
Female C.B-17/Icr-Prkdcscid SCID mice (8–12 wk of age; Charles River Labs) were administered a single dose of rac-H-2 (30 mg/kg), (+)-D-2 (15 mg/kg), or (−)-D-2 (15 mg/kg) by oral gavage. Blood was collected into potassium EDTA containers by terminal cardiac puncture under carbon dioxide anesthesia from n = 3 animals per group per time point at 0.25, 0.5, 1, 2, 4, 8, and 24 h postdose. Plasma samples were prepared for quantitative analysis by liquid–liquid extraction in methyl-t-butylether, with ondansetron as ISTD. Methyl-t-butylether was evaporated under nitrogen flow, and samples were reconstituted in 1% acetic acid in water:acetonitrile (20:80 vol/vol). Analysis was performed by chiral LC/MS-MS on a Daicel ChiralPak IE-3 Column using an isocratic LC method of 0.1% acetic acid in water and acetonitrile (20:80 vol/vol). ISTD-normalized peak areas for (+)-D-2 and (−)-D-2 were corrected from interference from naturally occurring isotopic peaks of the corresponding protonated analytes if present. Data were plotted in Excel 2013 (Microsoft Corp.) and analyzed within Excel using the PKSolver add-in (version 2.0) (76) to determine pharmacokinetic parameters.
H929 xenograft model of rac-H-2, (+)-D-2, and (−)-D-2.
Female C.B-17/Icr-Prkdcscid SCID mice (8–12 wk of age; n = 60) were administered 1 × 107 H929 tumor cells in 50% (vol/vol) Matrigel s.c. in their flanks (0.2 mL per mouse). Animals were returned to their cages, and tumor growth was monitored. Tumor size was reported as tumor volume (Vol) calculated as Vol = 1/2 (L × W2), where L and W are the length and width of the tumor as measured by caliper (L > W), respectively. A pair match was performed when tumors reached an average size of 100–150 mm3, and the animals were divided into six treatment groups of n = 10 animals each. Animals were then treated daily by oral gavage (10 mL/kg) with vehicle (0.5% wt/vol hydroxypropyl methylcellulose in water), rac-H-2 (3 mg/kg), (+)-D-2 (1.5 or 15 mg/kg), or (−)-D-2 (d+; 1.5 or 15 mg/kg). Body weight measurements were taken daily for the first week and then, biweekly. Tumor sizes were measured biweekly. The study was terminated after 23 d of dosing, and all animals were euthanized.
Supplementary Material
Acknowledgments
The authors thank Robert Gadwood at Kalexsyn and Laszlo Varady at Rilas Technologies for their assistance in the preparation of compounds 1 and 2.
Footnotes
Conflict of interest statement: V.J. is a shareholder and employee of DeuteRx LLC. A.W.C. is a director and shareholder of DeuteRx LLC. T.M.J. has no conflict of interest. L.H.T.V.d.P. is a shareholder and advisor of DeuteRx LLC. S.H.D. is a director, shareholder, and employee of DeuteRx LLC.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1417832112/-/DCSupplemental.
References
- 1.Smith SW. Chiral toxicology: It’s the same thing...only different. Toxicol Sci. 2009;110(1):4–30. doi: 10.1093/toxsci/kfp097. [DOI] [PubMed] [Google Scholar]
- 2.Csuk R. In: Biocatalysis in the Pharma and Biotech Industries. Patel RN, editor. CRC; Boca Raton, FL: 2007. pp. 699–716. [Google Scholar]
- 3.Hutt AJ, Valentova J. The chiral switch: The development of single enantiomer drugs from racemates. Acta Fac Pharm Univ Comen. 2003;50:7–23. [Google Scholar]
- 4.Ali I. Homochiral drug design and development by racemization. Comb Chem High Throughput Screen. 2007;10(5):326–335. doi: 10.2174/138620707781662835. [DOI] [PubMed] [Google Scholar]
- 5.Somogyi A. Inside the isomers: The tale of chiral switches. Aust Prescr. 2004;27(2):47–49. [Google Scholar]
- 6.Department of Health and Human Services, Food and Drug Administration 1992. FDA's Policy Statement for the Development of New Stereoisomeric Drugs. Available at www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm122883.htm. Accessed September 9, 2014.
- 7.Quach H, et al. Mechanism of action of immunomodulatory drugs (IMiDS) in multiple myeloma. Leukemia. 2010;24(1):22–32. doi: 10.1038/leu.2009.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vallet S, Witzens-Harig M, Jaeger D, Podar K. Update on immunomodulatory drugs (IMiDs) in hematologic and solid malignancies. Expert Opin Pharmacother. 2012;13(4):473–494. doi: 10.1517/14656566.2012.656091. [DOI] [PubMed] [Google Scholar]
- 9.Zeldis JB, Rohane PEW, Schafer PH. 2006. World Patent Appl WO 2006/127938.
- 10.Robarge MJ, Chen RS-C, Muller GW, Man H. 2002. World Patent Appl WO 02/059106.
- 11.Muller GW, et al. 2007. World Patent Appl WO 2007/136640.
- 12.Muller GW, et al. 2012. World Patent Appl WO 2012/125459.
- 13.Gandhi A, Schafer PH. 2012. World Patent Appl WO 2012/125475.
- 14.Muller GW, et al. 2012. World Patent Appl WO 2012/125438.
- 15.Muller GW, Man H. 2008. World Patent Appl WO 2008/039489.
- 16.Schafer PH, Lopez-Girona A, Daniel TO, Gandhi A, Mendy D. 2012. World Patent Appl WO 2012/149299.
- 17.Gandhi AK, et al. 2013. A first in human dose escalation study of CC-122, a first-in-class pleiotropic pathway modulator (PPM) compound in subjects with relapsed or refractory solid tumors, multiple myeloma and non-Hodgkin’s lymphoma. Proceedings of the 55th ASH Annual Meeting and Exposition. Blood (ASH Annu Meet Abstr) 122(21):2905 (abstr)
- 18.Gandhi AK, et al. 2012. CC-122 is a novel pleiotropic pathway modifier with potent in vitro immunomodulatory and anti-angiogenic properties and in vivo anti-tumor activity in hematological cancers. Proceedings of the 54th ASH Annual Meeting and Exposition. Blood (ASH Annu Meet Abstr) 120(21):2963 (abstr)
- 19.Knoche B, Blaschke G. Investigations on the in vitro racemization of thalidomide by high-performance liquid chromatography. J Chromatogr A. 1994;666(1-2):235–240. [Google Scholar]
- 20.Nishimura K, Hashimoto Y, Iwasaki S. (S)-form of alpha-methyl-N(alpha)-phthalimidoglutarimide, but not its (R)-form, enhanced phorbol ester-induced tumor necrosis factor-alpha production by human leukemia cell HL-60: Implication of optical resolution of thalidomidal effects. Chem Pharm Bull (Tokyo) 1994;42(5):1157–1159. doi: 10.1248/cpb.42.1157. [DOI] [PubMed] [Google Scholar]
- 21.Eriksson T, Björkman S, Roth B, Fyge A, Höglund P. Stereospecific determination, chiral inversion in vitro and pharmacokinetics in humans of the enantiomers of thalidomide. Chirality. 1995;7(1):44–52. doi: 10.1002/chir.530070109. [DOI] [PubMed] [Google Scholar]
- 22.Wnendt S, et al. Enantioselective inhibition of TNF-alpha release by thalidomide and thalidomide-analogues. Chirality. 1996;8(5):390–396. doi: 10.1002/(SICI)1520-636X(1996)8:5<390::AID-CHIR6>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 23.Teo SK, et al. Clinical pharmacokinetics of thalidomide. Clin Pharmacokinet. 2004;43(5):311–327. doi: 10.2165/00003088-200443050-00004. [DOI] [PubMed] [Google Scholar]
- 24. Celgene Corp (2014) Thalomid Prescribing Information. Available at www.thalomid.com/pdf/Thalomid_PI.pdf. Accessed September 8, 2014.
- 25.Chen N, Wen L, Lau H, Surapaneni S, Kumar G. Pharmacokinetics, metabolism and excretion of [(14)C]-lenalidomide following oral administration in healthy male subjects. Cancer Chemother Pharmacol. 2012;69(3):789–797. doi: 10.1007/s00280-011-1760-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Celgene Corp (2014) Revlimid Prescribing Information. Available at www.revlimid.com/wp-content/uploads/2013/11/PI.pdf. Accessed September 8, 2014.
- 27. Committee for Medicinal Products for Human Use (CHMP) (2007) Revlimid European Public Assessment Report (EPAR) Scientific Discussion, June 26, 2007. Available at www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Scientific_Discussion/human/000717/WC500056022.pdf. Accessed September 8, 2014.
- 28.Teo SK, et al. Chiral inversion of the second generation IMiD CC-4047 (ACTIMID) in human plasma and phosphate-buffered saline. Chirality. 2003;15(4):348–351. doi: 10.1002/chir.10221. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Zhou S, Hoffmann M, Kumar G, Palmisano M. Modeling and simulation to probe the pharmacokinetic disposition of pomalidomide R- and S-enantiomers. J Pharmacol Exp Ther. 2014;350(2):265–272. doi: 10.1124/jpet.114.215251. [DOI] [PubMed] [Google Scholar]
- 30. Celgene Corp (2014) Pomalyst Prescribing Information. Available at www.pomalyst.com/wp-content/uploads/2013/08/prescribing_information.pdf. Accessed September 8, 2014.
- 31.Melchert M, List A. The thalidomide saga. Int J Biochem Cell Biol. 2007;39(7-8):1489–1499. doi: 10.1016/j.biocel.2007.01.022. [DOI] [PubMed] [Google Scholar]
- 32.Kotla V, et al. Mechanism of action of lenalidomide in hematological malignancies. J Hematol Oncol. 2009;2:36. doi: 10.1186/1756-8722-2-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davies F, Baz R. Lenalidomide mode of action: Linking bench and clinical findings. Blood Rev. 2010;24(Suppl 1):S13–S19. doi: 10.1016/S0268-960X(10)70004-7. [DOI] [PubMed] [Google Scholar]
- 34.Krönke J, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343(6168):301–305. doi: 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shortt J, Hsu AK, Johnstone RW. Thalidomide-analogue biology: Immunological, molecular and epigenetic targets in cancer therapy. Oncogene. 2013;32(36):4191–4202. doi: 10.1038/onc.2012.599. [DOI] [PubMed] [Google Scholar]
- 36.Blaschke G, Kraft HP, Fickentscher K, Köhler F. [Chromatographic separation of racemic thalidomide and teratogenic activity of its enantiomers (author’s transl)] Arzneimittelforschung. 1979;29(10):1640–1642. [PubMed] [Google Scholar]
- 37.Muller GW, et al. Amino-substituted thalidomide analogs: Potent inhibitors of TNF-alpha production. Bioorg Med Chem Lett. 1999;9(11):1625–1630. doi: 10.1016/s0960-894x(99)00250-4. [DOI] [PubMed] [Google Scholar]
- 38.Lentzsch S, et al. S-3-Amino-phthalimido-glutarimide inhibits angiogenesis and growth of B-cell neoplasias in mice. Cancer Res. 2002;62(8):2300–2305. [PubMed] [Google Scholar]
- 39.Aragon-Ching JB, Li H, Gardner ER, Figg WD. Thalidomide analogues as anticancer drugs. Recent Patents Anticancer Drug Discov. 2007;2(2):167–174. doi: 10.2174/157489207780832478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fischer ES, et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature. 2014;512(7512):49–53. doi: 10.1038/nature13527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knabe J, Omlor G. Synthesis of racemates and enantiomers of 3-alkylthalidomide analogs and determination of their absolute configuration. Arch Pharm (Weinheim) 1989;322(8):499–505. doi: 10.1002/ardp.19893220809. [DOI] [PubMed] [Google Scholar]
- 42.Chung F, et al. Effect of 3-fluorothalidomide and 3-methylthalidomide enantiomers on tumor necrosis factor production and antitumor responses to the antivascular agent 5,6-dimethylxanthenone-4-acetic acid (DMXAA) Oncol Res. 2003;14(2):75–82. doi: 10.3727/000000003108748621. [DOI] [PubMed] [Google Scholar]
- 43.Takeuchi Y, Shiragami T, Kimura K, Suzuki E, Shibata N. (R)- and (S)-3-Fluorothalidomides: Isosteric analogues of thalidomide. Org Lett. 1999;1(10):1571–1573. [Google Scholar]
- 44.Lee CJJ, Shibata N, Wiley MJ, Wells PG. Fluorothalidomide: A characterization of maternal and developmental toxicity in rabbits and mice. Toxicol Sci. 2011;122(1):157–169. doi: 10.1093/toxsci/kfr086. [DOI] [PubMed] [Google Scholar]
- 45.Burkhard JA, Wuitschik G, Plancher J-M, Rogers-Evans M, Carreira EM. Synthesis and stability of oxetane analogs of thalidomide and lenalidomide. Org Lett. 2013;15(17):4312–4315. doi: 10.1021/ol401705a. [DOI] [PubMed] [Google Scholar]
- 46.Foster AB. Deuterium isotope effects in the metabolism of drugs and xenobiotics: Implications for drug design. Adv Drug Res. 1985;14(14):1–40. [Google Scholar]
- 47.Shao L, Hewitt MC. The kinetic isotope effect in the search for deuterated drugs. Drug News Perspect. 2010;23(6):398–404. doi: 10.1358/dnp.2010.23.6.1426638. [DOI] [PubMed] [Google Scholar]
- 48.Yarnell AT. Heavy-hydrogen drugs turn heads, again. Chem Eng News. 2009;87(25):36–39. [Google Scholar]
- 49.Nelson SD, Trager WF. The use of deuterium isotope effects to probe the active site properties, mechanism of cytochrome P450-catalyzed reactions, and mechanisms of metabolically dependent toxicity. Drug Metab Dispos. 2003;31(12):1481–1498. doi: 10.1124/dmd.31.12.1481. [DOI] [PubMed] [Google Scholar]
- 50.O’Driscoll C. Heavyweight drugs. Chem Ind. 2009;2009(5):24–26. [Google Scholar]
- 51.Tung R. 2010. The Development of Deuterium-Containing Drugs. Available at www.iptonline.com/pdf_viewarticle.asp?cat=2&article=622. Accessed September 9, 2014.
- 52.Gant TG. Using deuterium in drug discovery: Leaving the label in the drug. J Med Chem. 2014;57(9):3595–3611. doi: 10.1021/jm4007998. [DOI] [PubMed] [Google Scholar]
- 53. DeWitt SH (2012) Affidavit Filed on June 6, 2012 in U.S. Patent No. 8,288,414, Which Is Publicly Available at the United States Patent & Trademark Office. Available at portal.uspto.gov/pair/PublicPair. Accessed September 9, 2014.
- 54.Yamamoto T, Tokunaga E, Nakamura S, Shibata N, Toru T. Synthesis and configurational stability of (S)- and (R)-deuteriothalidomides. Chem Pharm Bull (Tokyo) 2010;58(1):110–112. doi: 10.1248/cpb.58.110. [DOI] [PubMed] [Google Scholar]
- 55.Wu L, Aslanian AM, Liu JF, Hogan K, Tung R. 2012. CTP-221, a deuterated S-enantiomer of lenalidomide, possesses significantly enhanced immunomodulatory and anti-proliferative properties relative to the R-enantiomer and to racemic lenalidomide. Proceedings of the 54th ASH Annual Meeting and Exposition. Blood (ASH Annu Meet Abstr) 120(21):2463 (abstr)
- 56.Uttamsingh V, et al. 2013. CTP-221, a deuterated S-enantiomer of lenalidomide, is greatly stabilized to epimerization and results in a more desirable pharmacokinetic profile than racemic lenalidomide. Proceedings of the 104th Annual Meeting of the American Association for Cancer Research. Cancer Res 73(Suppl 8):3357 (abstr)
- 57.DeWitt S. 2012. World Patent Appl WO 2012/135299.
- 58.DeWitt S. 2014. World Patent Appl WO 2014/110558.
- 59.Greig NH, et al. Thalidomide-based TNF-alpha inhibitors for neurodegenerative diseases. Acta Neurobiol Exp (Warsz) 2004;64(1):1–9. doi: 10.55782/ane-2004-1486. [DOI] [PubMed] [Google Scholar]
- 60.More O’Ferrall RA. A pictorial representation of zero-point energy and tunnelling contributions to primary hydrogen isotope effects. J Phys Org Chem. 2010;23(7):572–579. [Google Scholar]
- 61.Slaughter LM, Wolczanski PT, Klinckman TR, Cundari TR. Inter- and intramolecular experimental and calculated equilibrium isotope effects for (silox)2(tBu3SiND)TiR + RH (silox = tBu3SiO): Inferred kinetic isotope effects for RH/D addition to transient (silox)2TiNSitBu3. J Am Chem Soc. 2000;122(33):7953–7975. [Google Scholar]
- 62.Parker VD, et al. Radical cation-nucleophile combination reactions. The effect of structure of nitrogen-centered nucleophiles on reaction rates. Acta Chem Scand. 1997;51(10):1035–1040. [Google Scholar]
- 63.Slebocka-Tilk H, et al. Electrophilic bromination of 7-norbornylidene-7’-norbornane. The observation of an unusually large inverse deuterium kinetic isotope effect. J Am Chem Soc. 1995;117(34):8769–8776. [Google Scholar]
- 64.Aso Y, Yoshioka S, Takeda Y. Epimerization and racemization of some chiral drugs in the presence of human serum albumin. Chem Pharm Bull (Tokyo) 1990;38(1):180–184. doi: 10.1248/cpb.38.180. [DOI] [PubMed] [Google Scholar]
- 65.Schumacher H, Smith RL, Williams RT. The metabolism of thalidomide: The spontaneous hydrolysis of thalidomide in solution. Br Pharmacol Chemother. 1965;25(2):324–337. doi: 10.1111/j.1476-5381.1965.tb02053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schumacher H, Smith RL, Williams RT. The metabolism of thalidomide: The fate of thalidomide and some of its hydrolysis products in various species. Br Pharmacol Chemother. 1965;25(2):338–351. doi: 10.1111/j.1476-5381.1965.tb02054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Filella X, et al. Cytokines (IL-6, TNF-alpha, IL-1alpha) and soluble interleukin-2 receptor as serum tumor markers in multiple myeloma. Cancer Detect Prev. 1996;20(1):52–56. [PubMed] [Google Scholar]
- 68.Balkwill FR, Mantovani A. Cancer-related inflammation: Common themes and therapeutic opportunities. Semin Cancer Biol. 2012;22(1):33–40. doi: 10.1016/j.semcancer.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 69.Lin GQ, Zhang JG, Cheng JF. In: Chiral Drugs: Chemistry and Biological Action. Lin GQ, You QD, Cheng JF, editors. Wiley; Hoboken, NJ: 2011. pp. 3–28. [Google Scholar]
- 70.Agranat I, Wainschtein SR, Zusman EZ. The predicated demise of racemic new molecular entities is an exaggeration. Nat Rev Drug Discov. 2012;11(12):972–973. doi: 10.1038/nrd3657-c1. [DOI] [PubMed] [Google Scholar]
- 71. DeWitt SH (2012) Affidavit Filed on June 6, 2012 in U.S. Patent No. 8,304,435, Which Is Publicly Available at the United States Patent & Trademark Office. Available at portal.uspto.gov/pair/PublicPair. Accessed September 9, 2014.
- 72. Jacques V (2013) Affidavit Filed on May 20, 2013 in U.S. Patent No. 8,669,268, Which Is Publicly Available at the United States Patent & Trademark Office. Available at portal.uspto.gov/pair/PublicPair. Accessed September 9, 2014.
- 73. Jacques V (2013) Affidavit Filed on April 22, 2013 in U.S. Patent No. 8,524,780, Which Is Publicly Available at the United States Patent & Trademark Office. Available at portal.uspto.gov/pair/PublicPair. Accessed September 9, 2014.
- 74. DeWitt SH (2012) Affidavit Filed on May 14, 2012 in U.S. Patent No. 8,722,710, Which Is Publicly Available at the United States Patent & Trademark Office. Available at portal.uspto.gov/pair/PublicPair. Accessed September 9, 2014.
- 75.Committee for the Update of the Guide for the Care and Use of Laboratory Animals . Guide for the Care and Use of Laboratory Animals. 8th Ed National Academy Press; Washington, DC: 2011. [Google Scholar]
- 76.Zhang Y, Huo M, Zhou J, Xie S. PKSolver: An add-in program for pharmacokinetic and pharmacodynamic data analysis in Microsoft Excel. Comput Methods Programs Biomed. 2010;99(3):306–314. doi: 10.1016/j.cmpb.2010.01.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.