

1. Introduction
When fully extended, one copy of the three billion base pair human genome reaches a length of over two meters. Yet, it must be packaged into the nucleus of a cell with an average diameter of less than 10 μm. Within this context, specific segments of the genome must be transcriptionally active or repressed in a coordinated fashion to allow a cell to react to its ever-changing environment. This is akin to arranging 30 miles of thread inside a basketball such that at moment’s notice key segments can be accessed. To establish such compaction while maintaining coordinated accessibility, organisms ranging from yeast to man organize their genomes in a polymeric complex called chromatin. The fundamental unit of the chromatin polymer is the nucleosome, which repeats every 160 to 240 bp across the genome.1 Each nucleosome contains a nucleosome core, composed of an octameric complex of the core histone proteins, which forms a spool to wrap 145 to 147 bp of DNA. The nucleosome core is connected to the adjacent nucleosome core through a segment of linker DNA, which often associates with the linker histone protein (H1 or H5). The nucleosome core with ∼165 bp of DNA together with the linker histone is called the chromatosome.2 The chromatosome and the additional linker DNA constitutes a nucleosome.2 Despite these technical definitions, the nucleosome core particle is often colloquially referred to as the nucleosome.
The nucleosome serves three primary functions. First, it brings about the first level of genomic compaction, organizing ∼200 bp of DNA. Second, the nucleosome acts as a signaling hub for chromatin-templated processes by providing a scaffold for the binding of chromatin enzymes and displaying a combinatorial array of post-translational modifications (PTMs). This array of PTMs further regulates the recruitment of chromatin enzymes3 and tunes both nucleosome stability4 and the higher-order compaction of chromatin.5−7 Third, the nucleosome can self-assemble into higher-order chromatin structures, allowing for further compaction of the genome. The first level of higher-order compaction is the 30 nm chromatin fiber for which several models have been created based on experimental data using cryogenic-electron microscopy (cryo-EM) and X-ray crystallography.8
This review focuses on recent advances in our understanding of the structure and function of the nucleosome and is divided into four parts. First, we present a primer covering the fundamentals of the nucleosome core particle structure determined at atomic scale by X-ray crystallography in 1997.9 Next we discuss recent insights into the role of DNA sequence in the structure of nucleosomal DNA based on structure–function studies of nucleosome core particles containing derivatives of the Widom 601 nucleosome positioning sequence.10 We then introduce patterns of nucleosome recognition by chromatin factors using recent crystal structures and NMR and cryo-EM models of peptide and protein macromolecular chromatin factors bound to the nucleosome core particle. Finally, we will compare a recent cryo-EM model for the 30 nm chromatin fiber11 to two previous models based on crystallographic and cryo-EM data.12,13
2. Fundamentals of the Nucleosome Core Particle Structure
While the composition of the nucleosome had long since been realized, the 1997 2.8 Å crystal structure of the nucleosome core particle (NCP) solved by Luger et al. afforded the first atomic depiction of this fundamental genomic unit.9 This structure showed 146 bp of the human alpha-satellite sequence wrapped 1.65 times around an octameric scaffold of Xenopus laevis histone proteins in a left-handed superhelix (Figure 1a). A single base pair is centered on the nucleosome dyad,14 which defines the pseudo-2-fold symmetry axis of the NCP. DNA locations are designated by superhelical locations (SHL) representing superhelical turns from the dyad (SHL 0) and ranging from SHL −7 to SHL 7. The central histone octamer contains two copies of each of the core histone proteins, H2A, H2B, H3, and H4 as established by Arents and Moudrianakis in the 1991 3.1 Å crystal structure of the histone octamer.15 The core histones are assembled into four histone-fold heterodimers (two each of H2A/H2B and H3/H4). Ten flexible tails protrude from the NCP at defined locations, one N-terminal tail from each of the eight core histone proteins and two additional C-terminal tails contributed by H2A.
Figure 1.

Nucleosome core particle structure and the histone-fold heterodimers. (a) Nucleosome core particle structure (PDB ID 1KX5). Histones and DNA are depicted in cartoon and sticks representations, respectively, and colored as indicated. (b) H3/H4 histone-fold heterodimer. (c) H2A/H2B histone-fold heterodimer. Structures (top) and schemes (bottom) with secondary structure elements indicated. All molecular graphics in this review were prepared using PyMOL software (The PyMOL Molecular Graphics System, version 1.6, Schrodinger, LLC). All structures of NCP using high-resolution structure17 (PDB ID 1KX5) unless indicated otherwise.
2.1. Histone-Fold Heterodimers
Each of the core histones contains a central α-helical region that forms a histone-fold motif, flanked by N- and C-terminal extensions. The histone-fold is constructed from three α helices connected by two intervening loops specified as α1-L1-α2-L2-α3 (Figure 1b,c).9,15,16 The two shorter α1 and α3 helices loop back to pack against the longer central α2 helix. Each histone-fold pairs with a complementary histone-fold, H3 pairs with H4 and H2A pairs with H2B, to form a histone-fold heterodimer handshake motif. The antiparallel arrangement of this heterodimer approximates the L1 loop from one histone-fold and the L2 loop of the complementary histone-fold, placing one L1L2 pair at each end of the heterodimer. The result is a crescent-shaped heterodimer with the convex surface including the L1L2 loops and the α1 helices and the concave surface including the α3 and central α2 helices. The convex surface of the H2A/H2B and H3/H4 heterodimers carries a strong positive charge and constitutes the primary DNA binding element of each histone-fold heterodimer.
2.2. Histone Octamer Architecture and DNA Binding
The histone octamer forms a spool for wrapping nucleosomal DNA. It is assembled from two H3/H4 and two H2A/H2B histone-fold heterodimers using a single structural motif, the four-helix bundle. Each four-helix bundle is formed by the α2 and α3 helices from the adjacent histone-folds. Two H3/H4 dimers interact in a head to head arrangement through an H3/H3 four helix bundle to form an (H3/H4)2 tetramer (Figure 2a). An H2A/H2B dimer binds to each half of the (H3/H4)2 tetramer using a four-helix bundle formed by the H4 and H2B histone folds (Figure 2b). Several structured N- and C-terminal extensions to the histone-fold regions also contribute to the histone octamer architecture. The αN helix between the N-terminal tail and histone-fold of H3 rests on top of the H4 histone-fold and organizes DNA at the entry/exit site of the NCP (Figure 3a). H2A and H2B also contain C-terminal extensions that contribute to the nucleosome core surface. The H2B αC helix extends from the center of the nucleosome disk to the DNA edge opposite the nucleosome dyad, packing against the underlying H2A/H2B histone-fold helices (Figure 3b). The H2A C-terminal extension includes a docking domain that interacts with the H2A/H2B histone-fold dimer after which it traverses the nucleosome surface toward the dyad resting on a platform generated by the underlying H3/H4 heterodimer from the opposite side of the octamer (Figure 3b).
Figure 2.

Histone octamer constructed with four helix bundles. (a) Nucleosome core particle structure highlighting H3–H3 four helix bundle (blue). Remainder of H3 and H4 are shown in light blue and light green, respectively. (b) Nucleosome core particle structure highlighting one H4–H2B four helix bundle (green for H4 and red for H2B). Remainder of H4 and H2B are shown in light green and pink, respectively.
Figure 3.

Histone-fold heterodimers in the nucleosome core particle structure. (a) Nucleosome core particle structure with central H3/H4 histone-fold tetramer shown in blue (H3) and green (H4). H3 and H4 extensions are shown in light blue and light green, respectively. (b) Nucleosome core particle structure with one H2A/H2B histone-fold dimer shown in yellow (H2A) and red (H2B). H2A and H2B extensions are shown in light yellow and pink, respectively.
The H2A–H2B–H4–H3–H3–H4–H2B–H2A octamer designates a nonuniform path of the nucleosomal DNA. The H2A/H2B dimers bind DNA in two planes perpendicular to the DNA superhelical axis, while the central H3/H4 tetramer forms a diagonal ramp through the nucleosomal dyad, connecting these two planes (Figure 4). Notably, the DNA gyres in neighboring planes align their major and minor grooves, respectively, as they track along the octamer surface. The histone-fold regions of the octamer bind the central ∼121 bp of nucleosomal DNA. The remaining ∼13 bp at each of the DNA ends is organized by the histone-fold extensions, especially the H3 αN helices. The histone octamer contacts the DNA superhelix at regular intervals projecting an arginine into the minor groove of the neighboring DNA segment. Histone-DNA interfaces are mediated by extensive direct and water-mediate hydrogen bonds, ionic interactions, nonpolar contacts, and the alignment of helix dipoles relative to phosphate backbone ions.9,17,18
Figure 4.

Histone-fold heterodimers form a ramp for nucleosomal DNA. (a) H2A/H2B histone-fold heterodimers interact with DNA in two different parallel planes. Structure of NCP viewed from opposite dyad, highlighting H2A and H2B in yellow and red, respectively (left) and scheme of DNA planes (right). (b) H3/H4 tetramer forms a diagonal ramp for DNA connecting two parallel planes. Structure of NCP view from dyad (black oval and orange base pair) with H3 and H4 in blue and green, respectively, (left) and scheme of diagonal DNA ramp (right). Arrows point away from central dyad base pair.
2.3. Nucleosome Topology
The ∼200 kDa disk-shaped nucleosome core particle has a diameter of approximately 100 Å and height ranging from ∼25 Å at the dyad to ∼60 Å at the H2B αC helices. It has a multifaceted, solvent accessible surface of about 74 000 Å2 (Figure 5a). The disk face furthest from the dyad is lined by three parallel ridges, formed centrally by the H2B αC helix and on the sides by the H2B α1 helix and H3 α1 helix together with the H4 N-terminal tail, respectively. These ridges create two intervening grooves, one containing the H2A/H2B acidic patch (more details below). The disk face near the dyad contains a central depression overlaying the H3–H3 interface. The complexity of the NCP surface is furthered by the histone N-terminal tails that protrude from the nucleosome surface either outside (H4 and H2A) or between (H3 and H2B) the DNA gyres (Figure 4). These tails, ranging in length from 15 to 36 amino acids, can extend great distances from the NCP, adopt flexible structures, and bind intranucleosomal DNA and/or DNA and histone surfaces in neighboring nucleosomes.19−22 The DNA phosphodiester backbone at the perimeter of the NCP presents a highly negative electrostatic surface (Figure 5b). An additional negatively charged surface is found in a groove on the H2A/H2B dimer surface that is often referred to as the nucleosome or H2A/H2B acidic patch. Eight acidic residues contribute to the acidic patch, six from H2A (E56, E61, E64, D90, E91, and E92) and two from H2B (E102 and E110). As discussed in detail below, this acidic patch may be a hot-spot for nucleosome binding by chromatin factors. In contrast to the prominent negatively charged surfaces of the NCP disk, the histone tails contain many arginine and lysine residues and carry a strong net positive charge. Overall, the topological and electrostatic complexity of the nucleosome core affords the opportunity for a diverse set of surfaces for nucleosome binding.
Figure 5.
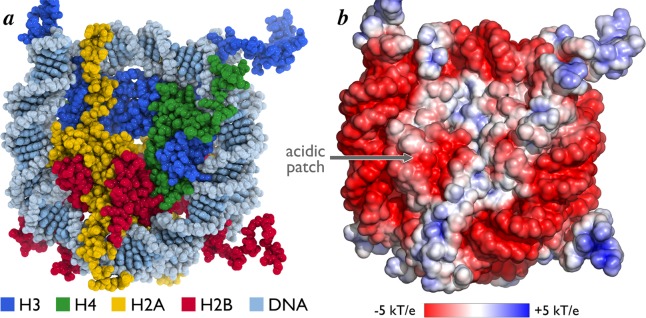
Surface topology and charge of the nucleosome core particle. (a) Surface of nucleosome core particle viewed down the DNA superhelical axis in space-filling representation. (b) Surface electrostatic potential of nucleosome core particle contoured from −5 to +5 kT/e calculated with ABPS.164 Location of acidic patch is indicated.
2.4. Variability in NCP Structure
Since the solution of the core particle containing Xenopus histones was reported, crystal structures have been solved using histones from yeast, fly, and man.23−25 While minor sequence differences result in small changes to the composition of exposed surfaces and complementary coevolution within the hydrophobic core, the architecture of the complexes remains nearly constant. A myriad of structural studies of histone variants have also revealed some variant-specific roles in the stability and exposed surfaces of the NCP.26−39 For example, H2A.Z extends the H2A/H2B acidic patch and causes a subtle destabilization of the H2A/H2B interface with H3/H4.39 Similar destabilization was observed recently with testes-specific variant H3T.30 A 2011 structure of the NCP containing the centromeric H3 variant CENP-A revealed that CENP-A nucleosomes only organize the central 121 bp of nucleosomal DNA potentially due to a shortened H3 αN helix.28 Other structural and biochemical data validate increased opening of the entry exit DNA in CENP-A nucleosomes.39−43 This trait is not specific to CENP-A as biochemical and biophysical interrogation of H2A.Bbd nucleosomes show similar opening of the DNA ends in nucleosomes containing H2A.Bbd.26,31,36,44 Much like histone variants, histone PTMs can induce structural changes in the solvent accessible nucleosome surface6 and interactions with nucleosomal DNA.45−51 Variant- and PTM-specific structural changes can contribute to the ability of nucleosomes to recruit chromatin factors as discussed below. Finally, several structures of NCPs containing different nucleosome positioning sequences have revealed that DNA sequence has effects on nucleosomal DNA structure allowing the octamer to wrap 145–147 bp of DNA.9,17,52,53 This topic is reviewed in detail in the following section.
3. Structure of Nucleosomal DNA
3.1. Nucleosome Positioning Sequences in Nucleosome Structures
Nucleosomes examined by structural studies have for the most part contained four types of DNA sequences: mixed sequence genomic DNA, the 5S RNA coding sequence, the human α-satellite repeat, and the Widom 601 nucleosome positioning sequence. Early structural studies of nucleosomes utilized nucleosomes isolated from naturally occurring sources such as beef kidney and consequently contained mixed sequence genomic DNA.54,55 Furthermore, the length of the nucleosomal DNA was variable at 147 ± 2 bp, the result of using micrococcal nuclease to digest long chromatin into nucleosome core particles.56,57 Such nucleosome core particles were used in the 7 Å low resolution crystal structure in 1984.55 To improve the internal order and diffraction limits of nucleosome core particle crystals, Richmond and colleagues prepared defined length 146 bp nucleosomal DNA fragments containing the 5S RNA coding sequence characterized by Simpson and others.58 This technical feat employing (now) classical recombinant DNA technology permitted the preparation of milligram quantities of defined sequence and defined length nucleosomal DNA in 1988, resulting in nucleosome core particle crystals which diffracted to ∼4.5 Å.57 While this marked an improvement over the 7 Å diffraction observed for mixed sequence nucleosome core particles, the diffraction was not sufficient for atomic structure determination.
The crystal structure of the nucleosome core particle determined at 2.8 Å by the Richmond group in 1997 contained instead the human alpha satellite centromeric repeat.9 Use of this nucleosome positioning sequence for reconstituting recombinant nucleosome core particles was first described by Bunick and colleagues in 1995.59 The 2.8 Å Luger et al. structure revealed important features of how the histone octamer organizes nucleosomal DNA. One such feature was the somewhat surprising finding that the dyad of the nucleosome core particle lines with a base pair and not between base pairs. This means that a nucleosome core particle with 73 bp on either side of the dyad contains 147 and not 146 bp. Previous studies had generally assumed an even number of base pairs in the nucleosome, and this was in fact the basis for using 146 bp of nucleosomal DNA in nucleosome crystallization trials. The occurrence of a base pair at the dyad axis was actually predicted earlier by single base pair resolution mapping of nucleosome positions using site-directed hydroxyl radical footprinting.14 This study examined recombinant nucleosome reconstituted with the 5S RNA sequence, providing evidence that the placement of the nucleosome dyad on the central base pair is independent of nucleosome positioning sequence. This conclusion has been borne out by all subsequent studies.
3.2. Widom 601 Nucleosome Positioning Sequence
Crystal structures of at least 20 nucleosome core particles incorporating variant histones or DNA sequence changes have been determined since the original 1997 structure. The large majority of these utilized the human α-satellite repeat DNA sequence. In contrast, the most popular DNA positioning sequence used in chromatin biochemical studies is the Widom 601 sequence. Lowary and Widom had performed an in vitro selection experiment to isolate synthetic random DNA sequences with high affinity for the histone octamer.10 The Widom 601 sequence was among the tightest binding sequences found, and its strong nucleosome positioning and high yields in nucleosome reconstitution experiments have made it a favorite among chromatin researchers. Crystal structures of nucleosome core particles containing the Widom 601 sequence were determined on their own and in complex with the RCC1 chromatin factor in 2010.52,53 Since the nucleosome core particles pack differently in the 601 nucleosome versus the RCC1/601 nucleosome crystals, the similarity of the structures indicate that the structures are not artifacts of their crystal packing. This is not an insignificant consideration given that the same crystal packing is present in all crystals of nucleosome core particles on their own to date (see below).
The reasons why the Widom 601 sequence is such a strong nucleosome positioning sequence have intrigued many since the sequence was first characterized in 1998, and we are now beginning to understand the mechanistic basis. Nucleosomal DNA must endure an aggregate bend of about 600° in approximately 150 bp, and consequently, DNA sequences that can be bent to contour the histone octamer will be favored. By sequencing chicken nucleosomal DNA, Satchwell et al. showed in 1986 that AA/TT, TA, and AT base steps were favored where the double helix minor groove faces the histone octamer and conversely that GG, GC, and CG base steps were more likely to be found 5 bp away or where the minor groove faces away from the histone.60 The particular significance of the TA base step in nucleosome positioning was suggested in several experiments, including the analysis of in vitro selected sequences (such as the 601 sequence) which highlighted a 10 bp sequence periodicity of the TA base step.10 In fact, the 601 sequence contains the TA base step at 5 of the 8 central positions where the DNA minor groove faces the histone octamer (SHL ± 0.5, ± 1.5, ± 2.5, ± 3.5) (Figure 6). Crystallographic and biochemical experiments by the Davey laboratory now provide a structural explanation for the significance of this observation. Noting that 4 of the 5 TA base steps are located on the “left” half of the 601 sequence and the remaining one on the “right” half, Davey and colleagues examined the salt stability of nucleosome core particles reconstituted with symmetrized versions of the 601 sequence.61 Nucleosomes containing the original, asymmetrical 601 sequence dissociated at a salt concentration of 1.26 M, significantly more than the 0.94 M concentration needed to dissociate nucleosome reconstituted with the human α-satellite sequence. However, nucleosome reconstituted with symmetrical DNA based on the left half of the 601 sequence (“601L”) were noticeably more stable to salt, while nucleosomes containing the right 601 sequence (“601R”) were less stable than the original 601 sequence (Figure 6). These results, as well as the fact that other high affinity in vitro selected nucleosome sequences, such as the 603 and 605 sequence also contain TA base steps in similar positions,10,62 provide evidence for an important role of the TA base step in nucleosome positioning of these high affinity in vitro selected nucleosomes.
Figure 6.
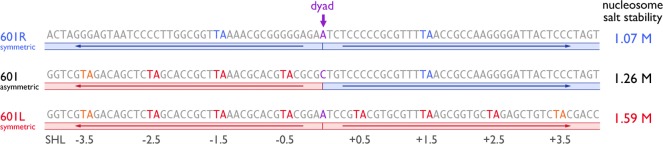
Scheme of asymmetric and symmetric 601 sequences. Sequences of 601R symmetric, (canonical) 601 asymmetric, and 601L symmetric sequences with H3/H4 TA steps highlighted in red for left half and blue for right half.61 Nucleosome salt stability values (molar monovalent salt) are listed at right and indicate stability as follows: 601L > 601 > 601R. This trend correlates with the number of H3/H4 TA steps: 601L (6), 601 (4), 601R (2). The dyad position is indicated (purple).
Crystal structures of nucleosome core particles containing the 601 and 601L sequences confirm that the TA base steps at the aforementioned central positions directly face the histone octamer.53,61 These TA base steps also exhibit significant distortions from ideality particularly in their propeller twist values. The crystal structures help explain the significance of this observation. Histones H3/H4 form the tetramer, which binds DNA around the nucleosomal dyad. The L1/L2 loops, α1/α1 N-terminal ends and L2/L1 loops of histone H3/H4 grip DNA phosphates groups where the minor groove faces the histone octamer and thus constrains the nucleosomal DNA path to these locations (termed “pressure points” by Davey and colleagues) (Figure 7). These pressure points occur at SHL ± 0.5, ± 1.5, and ± 2.5 (i.e., 5 bp from the nucleosomal dyad and then again at 10 bp intervals), precisely where TA base steps are located. Fixing these phosphate groups at these pressure points creates stress particularly in the base pairs between the phosphates. Base steps that are more flexible will more easily accommodate this stress through distortions such as propeller twisting within a base pair, rolling between base pairs or sliding one base pair with respect to the adjacent base pair.63,64 Since the TA base step is the most flexible of all base steps,65,66 it is most able to accommodate the stress created at the pressure points (Figure 7). Thus, we can understand why high affinity nucleosome positioning sequences such as the 601 sequence contain TA base steps where the minor groove faces the histone tetramer. Additional analyses of relevant DNA parameters for nucleosome positioning in the human α-satellite and 601 sequence have been described elsewhere.67,68
Figure 7.
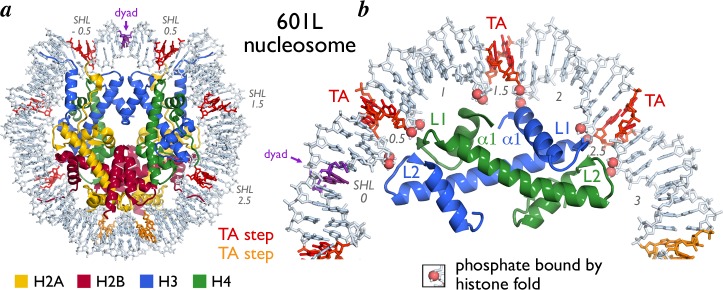
Location of TA steps in 601L nucleosome core particle structure. (a) 601L NCP structure viewed down the DNA superhelical axis with TA steps interacting with H3/H4 and H2A/H2B colored red and orange, respectively. The dyad is indicated (purple). Histones H3, H4, H2A, and H2B are shown in cartoon representation and colored blue, green, yellow, and red, respectively. Nucleosomal DNA is shown as sticks (light blue). (b) Enlarged view showing one H3/H4 heterodimer bound to DNA containing three TA steps (other histones are not shown for clarity purposes). Backbone phosphates bound to the H3/H4 histone folds are shown in space-filling representation as indicated. Secondary structure elements of dimer are shown.
Why is the TA base step so flexible? The fewer hydrogen bonds between the bases do allow for greater flexibility compared to base steps containing GC base pairs. However, this cannot be sufficient since TT, AA, and AT base steps are not as flexible despite having the same number of Watson–Crick hydrogen bonds. A simple structural explanation is that the stacking of bases is minimal in the TA base step, allowing greater flexibility for roll, propeller twisting and other distortions than the TT, AA, or AT base steps (Figure 8). The methyl group on the thymine base also plays an important role because the relatively bulky methyl group must be accommodated when a T-A base pair is distorted. In the TA base step, the minimal base stacking and the position of the methyl group allow for large roll or propeller twisting without the thymine methyl clashing with other atoms. It is for a similar reason that the eukaryotic transcriptional initiation TATA box is distorted so dramatically upon binding of the TBP TATA binding protein.69−71 In contrast, the TT, AA, and AT base steps each offer steric challenges to distortions including roll and propeller twisting. It is worth emphasizing that the geometry of the TA base steps is distinct from the AT base step despite the common alternating A/T sequence. The other base step with fewer constraints to distortions is CA = TG, and this base step has been found to be among the most flexible in protein–DNA structures.72
Figure 8.
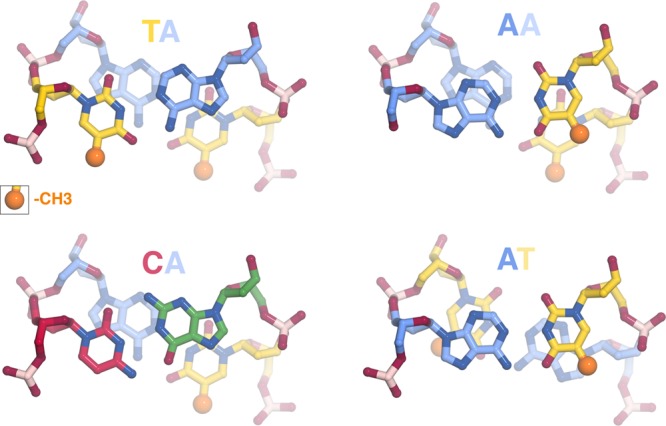
Minimal base stacking in TA and CA compared to other base pair steps. TA, CA, AA, and AT base pair steps colored as follows: T = yellow, A = blue, G = green, C = red. The thymine methyl groups are shown highlighted in space-filling representation (dark yellow), all other non-hydrogen atoms shown in sticks representation. The minimal base stacking and the absence of atoms close to the thymine methyl group permit greater flexibility of the TA and CA base pair steps.
The concept that the flexible TA base steps located between critical pressure points in nucleosomal DNA provides an explanation for surprising results of experiments studying the ability of RNA polymerases to progress through a nucleosome. Studitsky and colleagues found that transcriptional elongation by yeast and human RNA polymerase II were blocked by a nucleosome reconstituted with the 601 positioning sequence in one but not the opposite orientation73 (Figure 9). This blockage was mediated by the H3/H4 histone tetramer since the same effect was observed in the absence of the H2A/H2B dimer. Similar results were obtained for the Widom 603 and 605 nucleosome positioning sequences. These were puzzling findings because the symmetrical nature of the nucleosome structure made it difficult to imagine why a sequence block would function in one orientation but not in the opposite orientation. However, inspection of the DNA sequences with a focus on TA base steps shows a strong correlation between TA base steps at SHL +0.5, +1.5, and +2.5 and the ability to block RNA polymerase II progression. For each of the Widom 601, 603, and 605 nucleosome positioning sequences, RNA polymerase II transcriptional elongation was blocked when the TA base steps are positioned at the pressure points facing the H3/H4 tetramer downstream of the dyad (Figure 9). At first glance, the fact that the block to transcriptional elongation occurs when tight binding to the nucleosome is downstream of the dyad seems counterintuitive. If we imagine RNA polymerase II as an engine peeling off DNA from a one-dimensional histone track, we might expect that TA base steps positioned upstream of the dyad to be more efficient at blocking. The problem, of course, is that this Flatland74 analysis ignores the three-dimensionality of molecules instead of focusing on how RNA polymerase interacts with the architecture of the three-dimensional nucleosome. The finding that tight binding of nucleosomal DNA to the histone tetramer downstream of the dyad blocks RNA polymerase II indicates the ability of RNA polymerase II to unwrap DNA from the tetramer on the downstream side is a critical aspect of the mechanism of passing through a nucleosome. This insight was exploited by Studitsky and colleagues in a subsequent study where they propose a structural mechanism for this very process.62
Figure 9.
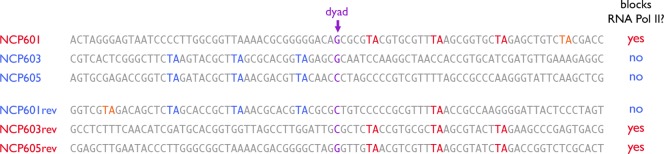
RNA polymerase II blocking by nucleosome positioning sequences. Sequences of NCP601, NCP603, and NCP605 sequences and their reversed counterparts together with ability to block RNA polymerase II.73 Multiple TA steps bound to the H3/H4 tetramer downstream (red) of the dyad (purple) blocks RNA polymerase II passage as compared with upstream (blue) of the dyad. TA steps bound to the H2A/H2B dimers are shown in orange. The sequence shown for the 601 sequence is the reverse complement of what is shown in Figure 6 to be consistent with ref (73).
3.3. DNA Stretching in the Nucleosome
The original 2.8 Å crystal structure of the nucleosome core particle contained 146 bp of symmetrized human alpha satellite DNA. Since the nucleosomal dyad lies on a base pair, there was necessarily 73 bp on one side of the dyad base pair and 72 bp on the other side. Thus, the structure was asymmetrical despite the fact that the nucleosomal DNA was symmetrical. To date, every single nucleosome core particle crystallized by itself has packed in the crystal essentially the same way: in space group P212121 with end-to-end DNA contacts between individual nucleosomes (with the possible exception of the CENP-A centromeric nucleosome where the DNA ends are not visible in the crystal structure28) (Figure 10). In order for the DNA ends to pack against each other in the crystal, the DNA on the 72 bp side had to stretch by one bp localized around SHL −2 (Figure 11a). Subsequent nucleosome core particle crystal structures containing human alpha satellite DNA of variant length or sequence show stretching of one bp around SHL ± 2 and SHL ± 5 but not at other locations. For example, the NCP146b nucleosome, which incorporates a symmetrical version of a different human α-satellite half-repeat, is stretched around SHL −5,61 while nucleosomes prepared from the original human α-satellite 146 bp DNA and recombinant human histones display stretching at SHL −2 and ±5.25 In contrast, a 147 bp pseudosymmetric human α-satellite sequence (pseudosymmetric because the symmetry of the two 73 bp halves was broken at the dyad) displayed no DNA stretching,17 and it is generally accepted that the human alpha satellite sequence forms a 147 bp nucleosome core particle in solution.
Figure 10.

DNA end-to-end packing in nucleosome core particle crystals. Three nucleosome core particles from one plane of the high resolution NCP crystal structure (PDB ID 1KX5) colored yellow, red and blue. (a) Full and (b) enlarged views of the alignment of the DNA ends from adjacent NCP in the structure. The DNA end-to-end packing exists in all crystals of the nucleosome core particle on its own.
Figure 11.

DNA stretching in nucleosome core particle structures. Cartoon representation of structure of approximately half of the nucleosomal DNA for (a) 146 bp human alpha-satellite (HAS146) (PDB ID 1AOI, blue) and (b) 145 bp 601 (PDB ID 3LZO, red) nucleosome positioning sequences relative to the HAS147 sequence (PDB ID 1KX5, yellow) (top). Stretching of 1 bp is observed at superhelical location (SHL) −2 with the HAS146 sequence and 1 bp each at SHL ± 5 with the 145 bp 601 sequence. SHLs and the dyad = SHL 0 are indicated. The length of DNA wrapped on each side of the NCP for each of the sequences is also shown (bottom).
When we crystallized the chromatin factor RCC1 in complex with the 601 nucleosome, we anticipated that the Widom 601 sequence would also likewise form a 147 bp nucleosome core particle. We therefore employed a 147 bp 601 DNA sequence in our crystallization studies. To our surprise, structure determination showed that the 601 nucleosome in the RCC1/nucleosome structure forms a 145 bp nucleosome core particle due to stretching of DNA by one bp at SHL ± 552 (Figure 11b). Two lines of evidence indicate that the DNA stretching in the 601 nucleosome did not result from crystal contacts. First, unlike crystals of nucleosome core particles on their own, the RCC1/nucleosome complex does not make DNA end to DNA end crystal contacts. A symmetry related RCC1 does make important crystal contacts with one DNA end, but the DNA end on other end of the nucleosome makes no crystal contacts.75 Second, the 601 nucleosome core particle on its own (i.e., in the absence of RCC1) crystallized in a different space group (P212121 vs P21 for RCC1/nucleosome) and via different crystal packing interactions than in the complex with RCC1.52,53 Despite the different crystal packing arrangement, the 601 nucleosome particle on its own also exhibits stretching at SHL ± 5. This stretching can explain why the 601 nucleosome core particle had evaded crystallization for such a long time: the 147 bp 601 nucleosome core particles with its extra bp extending beyond the nucleosome core particle would prevent the canonical DNA end to DNA end crystal packing found in all nucleosome only crystals. We were fortunate that our use of the 147 bp 601 nucleosome not only did not prevent the RCC1/nucleosome from crystallizing but was in fact important for the RCC1-nucleosomal DNA end crystal contact to occur.75
4. Recognition of the Nucleosome Core by Chromatin Factors
The recruitment of macromolecular chromatin factors to genomic loci is controlled at many levels. Factors can be actively sequestered in or excluded from the nucleus. The accessibility of large territories of the genome can be regulated by altering the degree of chromatin compaction. At a more local level, the binding of chromatin factors can be tuned by the positioning of nucleosomes. Some chromatin factors including many transcription factors bind to specific DNA sequences only in nucleosome free regions. On the other hand, many chromatin factors require nucleosomes for binding to chromatin. Obvious examples are histone-modifying and chromatin remodeling enzymes that by definition modify the chemical composition or architecture/location of nucleosomes, respectively. However, many other chromatin factors also bind to nucleosomal regions including high-mobility group proteins,76 heterochromatin scaffolding proteins,77,78 and even viral proteins.79 As described above, the nucleosome provides a diverse platform for binding of macromolecular chromatin factors. This platform is further diversified by replacement of canonical histones with histone variants and the chemical modification of both histone and DNA components of the nucleosome.
Chromatin factors can bind the nucleosome using one or more of the three following nucleosome surfaces: (1) the histone N- and C-terminal tails; (2) the disk faces of the histone octamer; and/or (3) the nucleosomal DNA. [Of particular note, the nucleosomal DNA affords the possibility of novel protein–DNA interactions, given its unique curvature as well as the alignment of the nucleosomal DNA gyres.] Much of the research regarding binding to histone tails is centered around histone PTMs. The molecular recognition of histone tails by numerous catalytic domains that establish and remove histone PTMs and diverse protein domains that bind tails modified at specific residues are thoroughly reviewed elsewhere.3,80−82 Due to technical challenges in the structural characterization of the nucleosome core bound to chromatin factors, much less is understood regarding the molecular recognition of the disk surfaces of the histone octamer and nucleosomal DNA. However, recent advances in the cocrystallization of macromolecular chromatin factors bound to the nucleosome core particle have permitted new atomic scale depictions of recognition of the nucleosomal disk. All crystal structures of macromolecules bound to the nucleosome solved to date share one common interaction motif, an arginine bound to the H2A/H2B acidic patch. Details of each of these structures, a discussion of this shared acidic patch arginine-anchor, and insights from further modes of nucleosomal recognition from NMR and cryo-EM studies are discussed below.
4.1. H4 N-Terminal Tail
The first instance of a protein segment binding to the nucleosomal H2A/H2B acidic patch was observed in a crystal contact of the 1997 nucleosome core particle structure.9 In these crystals, residues 16 to 25 of one H4 tail contact the acidic patch on an adjacent NCP forming a charged interaction surface (Notably, the other tail is not resolved N-terminal to residue 20 even though NMR experiments suggest the H4 tail is structured starting with residue 1683). The H4 K16 side chain projects into an acidic cavity generated by H2A acidic patch residues E61, D90, and E92 of the neighboring NCP surface. Other positively charged amino acids in the H4 tail also interact with negative side chains in the H2A/H2B acidic patch, including H4 R19-H2A E64, H4 K20–H2B E110 and H4 R23 which contacts both H2B E110 and H2A E56. Similar interactions are observed for H4 K20 and R23 in the crystal lattice of the 1.9 Å NCP structure though the other basic H4 side chains point toward intranucleosomal DNA.17 A functional role for the H4 N-terminal tail in chromatin structure has been confirmed using nucleosome arrays in solution. Truncation of the N-terminal H4 tail prior to residue 2084 or the charge neutralizing acetylation of H4 K165,85,86 leads to incomplete cation-mediated compaction of the 30 nm fiber in vitro. The case for a role in chromatin structure is furthered by disulfide formation between spatially adjacent mutant nucleosomes containing H4 V21C and H2A E64C both within a chromatin array87 and between arrays.88 It is important to note that the observed structure of the H4 N-terminal tail seen in the crystal lattice may not reflect a native conformation in chromatin fibers. Recent modeling of the H4 N-terminal tail suggests that H4 residues 15 to 20 exhibit a propensity for α helix formation. This allows H4 K16, R17, R19, and K20 to occupy a single helical face, which can be accommodated within the acidic patch groove.89 Though the evidence for the H4 tail-acidic patch in higher order chromatin structure is compelling, higher resolution structural characterization is required to accurately define the molecular details of this interaction.
4.2. Viral LANA Peptide
In 2006, Barbera et al. demonstrated that the nucleosome docking region of the Kaposi’s sarcoma-associated herpes virus (KSHV) latency-associated nuclear antigen (LANA) also interacts with the H2A/H2B acidic patch.79 The KSHV genome is contained within an episome, which is tethered to mitotic chromosomes using the basic N-terminal region of LANA by anchoring to the nucleosomal acidic patch. The 2.9 Å crystal structure of the LANA nucleosome recognition sequence bound to the nucleosome core particle was solved by soaking the peptide corresponding to LANA residues 1–23 into NCP crystals. LANA peptide forms a hairpin that fits in the acidic patch groove between the αC and α1 helices of H2B and makes multiple charged and hydrophobic interactions (Figure 12). The LANA R9 side chain inserts into the acidic patch to form ionic interactions with H2A residues E61, D90, and D92. This acidic patch “arginine-anchor” (our terminology) is shared with all crystal structures of chromatin factors bound to the NCP reported to date and overlaps the H4 K16 binding site observed in the original NCP structure crystal lattice.9 LANA R9 is critical in nucleosome binding as mutation of this residue eliminates LANA’s chromatin association.79,90 An additional LANA arginine (R7) forms an ionic interaction with H2B E110 and LANA S10 hydrogen bonds to H2A E64. LANA hydrophobic residues M6 and L8 bind a hydrophobic surface adjacent to the acidic patch including H2A residues Y50, V54, and Y57. Overall the regions of LANA observed to interact with the NCP correlate with those required for the virus to tether its episomal DNA to chromosomes.
Figure 12.
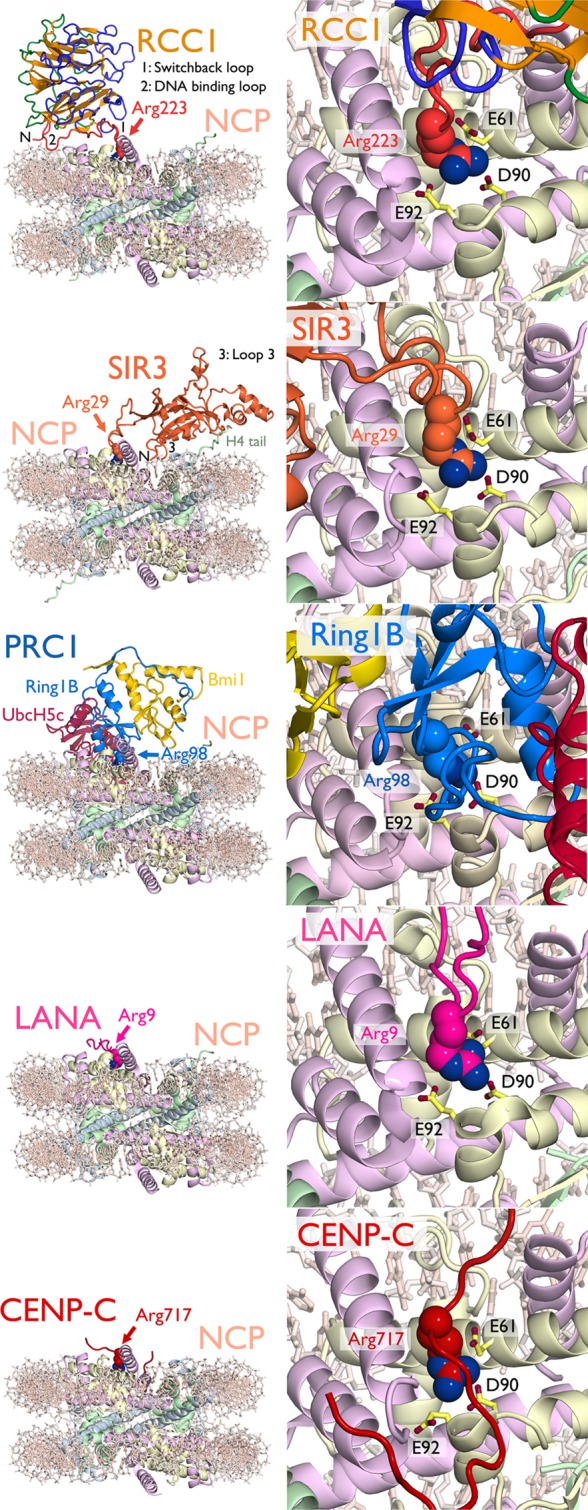
Nucleosome recognition using the acidic patch arginine-anchor. From top to bottom, structures of RCC1 (PDB ID 3MVD),52 Sir3 (PDB ID 3TU4),78 PRC1 (PDB ID 4R8P),111 LANA peptide (PDB ID 1ZLA),79 and CENP-C peptide (PDB ID 4INM)107 bound to the nucleosome core particle. Overview of structures as viewed from opposite the dyad (right) and zoomed view of acidic patch (left) with arginine-anchor in space-filling representation and key H2A residues shown as sticks. Locations of RCC1 switchback loop (1), DNA binding loop (2), and N-terminus (N) and Sir3 loop 3 (3) and N-terminus (N) are indicated. Histones H3, H4, H2A, and H2B are shown in cartoon representation and colored cornflower blue, light green, wheat, and pink, respectively. DNA (light pink) is shown as sticks.
4.3. Ran Guanine Exchange Factor RCC1
In 2010, we solved the structure of the Drosophila melanogaster β-propeller protein RCC1 (Regulator of chromatin condensation) bound to the nucleosome core particle.52 In contrast to the LANA-NCP structure, the RCC1-NCP structure was solved by cocrystallization of RCC1 on the NCP. RCC1 is a guanine exchange factor for the Ran GTPase (or RanGEF) that establishes a gradient of the GTP bound form of Ran around chromatin. This gradient plays roles in nuclear-cytoplasmic transport, mitotic spindle formation, and formation of the nuclear envelope following mitosis.91−93 RCC1 binds to the nucleosome resulting in an increase in its ability to catalyze Ran guanine exchange.94 Our 2.9 Å structure of the RCC1-NCP complex shows that RCC1 interacts with both the acidic patch and nucleosomal DNA using two β-propeller loops and its N-terminal tail52 (Figure 12). One loop, termed the switchback loop, binds to the H2A/H2B acidic patch using an intricate network of ionic and hydrogen bonds and van der Waals contacts. The switchback loop contains the RCC1 arginine-anchor residue 223 that binds H2A E61, D90, and E92 nearly identically to LANA R9. Much like LANA, RCC1 uses a second arginine 216 for additional interactions with H2A E61 and E64. Both RCC1 R223 and R216 are important for nucleosome binding in solution.95 RCC1 S217 forms hydrogen bonds with H2A V45 and E64. An additional hydrogen bond is observed between RCC1 S214 and H2A E64. These ionic and hydrogen bonding interactions are complemented by van der Waals contacts, especially with residues in the H2B αC helix that form a ridge at the edge of the acidic patch.
In addition to the acidic patch, RCC1 also binds to nucleosomal DNA using a distinct β-propeller loop and its N-terminus. The RCC1 DNA binding loop interacts with the phosphodiester backbone across a major groove near SHL ± 6 forming hydrogen bonds or charged interactions with the side chains of K241 and R239. An additional hydrogen bond is formed by the side chain of RCC1 259. The N-terminal tail of RCC1 is also implicated in nucleosome binding.94 While the N-terminal residues 2–27 are not visible in our RCC1-NCP structure, residues 28 and 29 are positioned to allow the N-terminal tail to enter the major groove of nucleosomal DNA adjacent to the DNA binding loop. Alignment of RCC1 from the RCC1-NCP and RCC1-Ran structures suggest that Ran approaches but does not contact the nucleosome surface. Therefore, either RCC1 or Ran must undergo conformational changes to allow for Ran-NCP interactions to enhance RCC1’s RanGEF activity. Further experiments are required to resolve this issue.
4.4. Silencing Protein Sir3
In 2011, Armache et al. solved the crystal structure of the BAH (bromo-associated homology) domain of the yeast silent information regulator protein Sir3 bound to the nucleosome core particle.78Saccharomyces cerevisiae uses SIR (silent information regulator) proteins Sir1, Sir2, Sir3, and Sir4 to establish a transcriptionally repressive chromatin state at telomeres, ribosomal DNA loci and silent mating-type loci.96 Silencing is thought to be accomplished in part though direct chromatin compaction by Sir3 as demonstrated in vitro.97−99 This 3.0 Å structure illustrates one of the most extensive interaction surfaces of the published high resolution chromatin factor-NCP structures, including 28 Sir3 BAH domain residues and greater than 30 histone residues (and potentially nucleosomal DNA at the BAH domain N-terminus)78 (Figure 12). The structure also suggests that weak self-interactions of the BAH domain observed in the crystal lattice and in solution may contribute to its ability to compact chromatin fibers. Analogous to LANA and RCC1, the Sir3 BAH domain binds to the H2A/H2B acidic patch, but also interacts with the nucleosome in three additional regions: the H4 N-terminal tail, surfaces of H3/H4 in the loss of rDNA silencing (LRS) domain, and the H2B C-terminal helices. Sixteen Sir3 BAH domain residues from loops 2 and 4, strand B5 and the A1 helix interact with H4 tail residues 13–23. This region of the H4 tail is often unstructured in NCP crystals in the absence of crystal lattice contacts. The Sir3 BAH domain-H4 tail interface is predominantly electrostatic in nature owing to the positively charged H4 tail and negatively charged complementary Sir3 surface. Importantly, the H4 K16 side chain forms several hydrogen bonds and ionic interactions with acidic Sir3 side chains, offering a molecular mechanism for the observed loss of Sir3 binding upon H4 K16 acetylation.98 The nucleosomal recognition surface of the Sir3 BAH domain also includes the α1 helix and L1 loop of H3, the α2 helix and L2 loop of H4 as well as the α3 and αC helices of H2B. These interactions are mediated by the Sir3 BAH loop 3 which becomes structured upon NCP binding and strands B6 and B8. This interaction surface includes the LRS domain residues 76–80 of H3 that are required for silencing in yeast.100 Much like H4 K16 acetylation, H3 K79 methylation disrupts Sir3 binding.101−103 The interactions in this region offer insight into the preference for H3 K79 in the unmodified state due to loss of potential hydrogen bonds with Sir3 BAH side chains. This is further exemplified by two recent structures of the N-α-acetylated Sir3 BAH domain bound to the NCP.104,105 The native N-terminally acetylated residue specifically interacts with regions of the Sir3 BAH domain further structuring its loop 3, enhancing contacts with the nucleosome surface in the LRS region and leading to a 30-fold increase in affinity for the NCP.105 Sir3 BAH also contacts the H2A/H2B acidic patch using the loop 1 region that is unstructured in the absence of the NCP.106 While electron density is weaker in this region of the Sir3 BAH-NCP structure, it appears that multiple arginines (R28, R29, R30, R32, and R34) line the acidic patch groove to make charged interactions with the NCP surface. Notably, Sir3 R32 occupies an identical binding site to that observed for LANA R9 and RCC1 R223 in a cavity surrounded by H2A E61, D90, and D92. Overall, the multifaceted interaction of the Sir3 BAH with the NCP explains many silencing defects observed upon mutation of both histones and Sir3 in yeast.
4.5. Centromeric Protein CENP-C
The crystal structure of the central region of rat centromere protein CENP-C bound to the nucleosome core particle illustrates the ability of a chromatin factor to recognize specific features of a variant histone in the context of the nucleosome.107 Proper segregation of chromosomes in mitosis requires the mitotic spindle to attach to the kinetochore at the centromere of each chromosome.108 Centromeric chromatin contains H3 variant CENP-A and a complex of 16 centromeric proteins including CENP-C.109,110The crystal structure of the CENP-C nucleosome binding peptide in complex with a chimeric NCP in which the C-terminal region of H3 was replaced with the LEEGLG solvent exposed sequence of CENP-A was solved by Kato et al. in 2013.107 In the structure, CENP-C forms an elongated conformation, which contacts the acidic patch and the CENP-A specific regions of the chimeric NCP (Figure 12). CENP-C binds to the acidic patch using two arginines, R717 and R719. The CENP-C R717 arginine-anchor binds in the now characteristic H2A E61, D90, E92 pocket; R719 makes additional ionic interactions with H2A E61 and E64. In the CENP-A specific region of the chimeric nucleosome, CENP-C residue Y725 binds in a hydrophobic pocket created by CENP-A residues I133 and L137. While CENP-A proteins are not highly conserved, they all contain at least one large hydrophobic residue in the CENP-C binding region that is not found in canonical H3.107 This 3.5 Å structure was validated by extensive NMR experiments using chemical shift perturbations and site-specific incorporation of paramagnetic spin labels.107
4.6. Polycomb Repressive Complex 1 (PRC1) Ubiquitylation Module
Recently, we solved the crystal structure of the PRC1 ubiquitylation module, containing the Ring1B-Bmi1 ubiquitin E3 ligase RING heterodimer together with the E2 enzyme UbcH5c, bound to its nucleosome core particle substrate at 3.3 Å resolution (Figure 12).111 PRC1 is a member of the Polycomb group family of complexes and plays a role in transcriptional repression of developmentally regulated genes at least in part through H2A K119 ubiquitylation and intrinsic chromatin compaction.112−114 PRC1 contains a RING-type ubiquitin E3 ligase composed of RING domains from Ring1B and Bmi1 that can pair with one of several ubiquitin E2 conjugating enzymes, including UbcH5c.115 Our structure reveals that all three proteins in the PRC1 ubiquitylation module bind to the nucleosome surface, together contacting all components of the nucleosome core particle. The Ring1B-Bmi1 RING heterodimer forms a saddle over the proximal end of the H2B αC helix, anchored on each side by histone interactions. The RING domain of Ring1B binds the H2A/H2B acidic patch using multiple positively charged side chains including an arginine anchor residue R98 (Figure 12). The substantial Ring1B-acidic patch interface contrasts a more modest Bmi1-nucleosome interface. The RING domain of Bmi1 interacts with a smaller acidic surface on the H3/H4 tetramer and forms a cap on the distal end of the H3 α1 helix. The structure is consistent with mutagenesis experiments implicating both the H2A/H2B acidic patch, the arginine anchor, and several other basic side chains.116,117 In addition to these E3 ligase-histone interactions, the E2 UbcH5c binds to nucleosomal DNA in two positions. Near the DNA end, the UbcH5c antiparallel β-sheet aligns several charged and polar side chains for interaction with the adjacent nucleosomal DNA backbone. UbcH5c binds nucleosomal DNA again at the dyad using basic α3 side chains. Mutagenesis at these novel E2-substrate interfaces diminishes nucleosome binding and activity by the PRC1 ubiquitylation module.111 Importantly, unlike other histone modifying enzymes structurally characterized to date, the PRC1 ubiquitylation module does not appear to directly recognize its targeted primary sequence. Rather the E3 and E2 components bind to topologically unique nucleosome surfaces distant from the site of catalysis to position the E2 active site directly over the H2A C-terminal tail near the target lysine.
4.7. NMR-Based Model of HMGN2
As a complementary approach to X-ray crystallography, Bai and colleagues have introduced NMR-based techniques to characterize chromatin factor-nucleosome interactions.107,118,119 The combination of methyl-TROSY and paramagnetic spin label NMR experiments allowed Kato et al. to map the nucleosomal surface bound by high mobility group nucleosomal protein HMGN2.103 A model for the HMGN2-NCP complex was created based on comprehensive NMR-based restraints. HMGN proteins are chromatin architectural proteins with roles in DNA damage repair, chromatin remodeling, and histone PTMs.76,120−122 They can also directly decompact chromatin and compete for chromatin binding with the linker histone H1.123 HMGNs share a common N-terminal nucleosome binding domain with the conserved basic octapeptide sequence, RRSARLSA.123 Kato et al. observed chemical shift perturbations of labeled side chain methyl groups of H2A L65 and H2B V45 and L103 upon HMGN binding.118 These residues are in proximity to the H2A/H2B acidic patch. Based on many NMR experimental restraints, a model of the HMGN2-NCP complex was proposed, suggesting arginines in the conserved HMGN nucleosome binding domain (R22, R23, and R26) interact with the H2A/H2B acidic patch. HMGN2 lysines 39, 41, and 42 were also proposed to interact with DNA near the entry/exit from the NCP. The two NCP-binding regions of HMGN2 are separated by a rigid proline-rich linker. Of note, two HMGN2 proteins bind the pseudosymmetry-related faces of the NCP with positive cooperativity (Kd of 1.5 and 0.17 μM for the first and second binding events, respectively, giving a Hill coefficient of 1.4). The authors suggest that HMGN-NCP interactions staple the DNA end to the histone octamer blocking the activity of chromatin remodelers. Moreover, the model explains the release of HMGNs from chromatin during mitosis following phosphorylation of S24 and S28.124 The negative charge carried on the phosphorylated serines would create repulsive interactions with the neighboring acidic patch.
4.8. Acidic Patch Arginine-Anchor As a Common Motif for Nucleosome Recognition
All crystal structures of chromatin factors bound to the nucleosome core particle share a common structural motif: an arginine-anchor that binds to a specific cavity generated by H2A E61, D90, and E92 side chains in the H2A/H2B acidic patch. And these are not the only examples of chromatin enzymes and factors relying on the acidic patch for nucleosome binding and/or activity. Ubiquitin E3 ligases RNF168 and BRCA1 exhibit defective ubiquitylation when the acidic patch is mutated111,125 and IL-33 binds to the acidic patch, likely in a manner similar to the LANA nucleosome targeting peptide.126 So why would such a common motif for recognition of a complex as large as the nucleosome exist? The simple answer is that the acidic patch is the most unique region of the nucleosome surface. It is topologically poised for chromatin factor interaction as a deep groove with a complex surface. The width of the groove allows the binding of multiple types of structures including loops (RCC1, Sir3), hairpins (LANA), extended conformations (HMGN2, CENP-C) but can also accommodate helical and β-strand secondary structure elements.89,127 In addition, the acidic patch carries the greatest net charge of the solvent exposed region of the histone octamer disk surface. Furthermore, the guanidinium group of the arginine-anchor is optimal for ionic interaction with all three H2A acidic side chains in the shared acidic-patch binding pocket.
Why would other distinct surfaces not be targeted to minimize interference? Luger and colleagues propose that the overlap of binding sites may serve a regulatory role in the determination of chromatin structure.127 That is, competition for the acidic patch between factors that condense chromatin (H4 tail and Sir3) with other macromolecules (HMGN2, RCC1, etc.) may tune the higher-order state of chromatin. Many questions still remain regarding this emerging paradigm for nucleosome binding. How common is it? Are we seeing it so frequently in biochemical experiments because we know to look for it? Are chromatin factors that bind to the acidic patch just easier to crystallize owing to binding affinities or resultant crystal packing opportunities? How is the binding of multiple chromatin factors to the same nucleosomal surface regulated? Are other acidic patch binders post-translationally modified to tune their binding affinities similar to HMGNs? Currently, the sample size is too small to address most of these questions. However, as described above, recent strides have been made in the structural characterization of chromatin factor-nucleosome complexes. This will provide a foundation for future work to address these unanswered questions and undoubtedly uncover new paradigms for nucleosomal recognition.
4.9. Cryo-EM Models of Chromatin Factor-Nucleosome Complexes
In addition to X-ray crystallography and NMR structures, several cryo-EM structures have enhanced our understanding of nucleosome recognition by chromatin enzymes and factors. The chromatin factors studied by cryo-EM to date fall into three functional categories: chromatin remodeling enzymes, histone modification enzyme complexes, and chromatin architectural proteins. They include large and dynamic macromolecules that present difficulties to crystallographers and NMR spectroscopists. Of note, all reported cryo-EM structures of chromatin factor-nucleosome complexes were solved at resolutions greater than ∼20 Å. This allows overall architecture to be revealed but precludes molecular description of NCP interactions. Studies using multimodality approaches, pairing cryo-EM with crystallographic characterization of subcomplexes, cross-linking mass spectrometry and/or comprehensive biochemical analysis permit nearly residue-specific understanding of interactions and thus heighten mechanistic insight as compared to cryo-EM reconstructions alone.
ATP-dependent chromatin remodeling complexes can alter the position and composition of nucleosomes by sliding them along DNA, unwrapping nucleosomal DNA, or ejecting/exchanging histone dimers or octamers.128 There are four principal families of chromatin remodelers: SWI/SNF, ISWI, Mi-2/CHD, and SWR/INO80. Nucleosome binding of representatives of all families except the Mi-2/CHD family have been characterized by cryo-EM. These three families interact with the nucleosome in distinct ways. In 2008, Chaban et al. used negative stain reconstructions of the SWI/SNF family remodeling complex RSC to show that RSC nearly engulfs the NCP,129 consistent with DNaseI protection experiments130,131 and the 2007 Leschziner et al. model docking the NCP into the central cavity of a reconstruction of RSC alone.132 Interestingly, some density was missing for both nucleosomal DNA and one H2A/H2B dimer, suggesting RSC-mediated remodeling even in the absence of ATP.
Cryo-EM reconstructions of two ISWI family remodeling complexes bound to nucleosomes show less extensive interactions. In a 2009 study, Racki et al. demonstrated that the ACF catalytic subunit, Snf2, binds with 2:1 stoichiometry to the nucleosome.133 While the ATPase domains and linker DNA are not visible in their reconstructions, biochemical data suggests that the ATPase domain binds the nucleosome at SHL ± 2 and the interaction also involves the H4 N-terminal tail. The authors propose a competition mechanism through which nucleosome spacing is accomplished by competitive sliding by two Snf2 subunits bound to opposite sides of the nucleosome. A different mechanism for nucleosome spacing was suggested for ISWI family member ISW1a by Yamada et al. based on combined cryo-EM and crystallographic data.134 Cryo-EM reconstructions of ISW1a in the absence of its ATPase domain bound to nucleosomes containing one or two DNA extensions revealed two modes of interactions with linker DNA. Together with a crystal structure of the ISW1a construct bound to free DNA, these cryo-EM reconstructions led to a model in which ISW1a acts as a ruler, using its size and shape to space adjacent nucleosomes.
Two cryo-EM studies also offered insight into the nucleosome binding of SWR/INO80 family remodelers. In 2012, Saravanan et al. generated a model for the 2:1 Arp8:NCP complex using crystal structures of the components and a 21 Å cryo-EM reconstruction.135 In this model, Arp8 interacts with the H3/H4 surface though the molecular details are unclear. More recently, Tosi et al. used cryo-EM and cross-linking mass spectrometry (XL-MS) to extensively characterize the interaction of the holo-INO80 complex with the NCP.136 The architecture of the INO80 complex alone shows four domains: the Rvb1/2 dodecamer head, the Ino80 ATPase-Ies2-Arp5-Ies6 neck, the Ino80 N-terminus-Nhp10-Ies1-Ies3-Ies5 body, and the Ino80 HAS-Act1-Arp4-Arp8-Ies4-Taf14 foot. While heterogeneity in the INO80-NCP cryo-EM images prevented proper 3D reconstruction, extensive XL-MS was observed between all four domains of INO80 and the nucleosome disk and tails. Notably, some cross-linking was observed to surfaces of the H2A/H2B dimer that are buried in the NCP structure. These contacts may facilitate opening of the NCP structure required for INO80 mediated H2A/H2B exchange. The authors created a model for the INO80-NCP complex based on the XL-MS data in which the NCP rests on a cradle surrounded by all four domains of the INO80 complex.
NCP bound cryo-EM reconstitutions have also been reported for the Saccharomyces cerevisiae Piccolo NuA4 histone acetyltransferase (HAT) complex137 and the HP1-like heterochromatin protein Swi6 from Schizosaccharomyces pombe.77 Piccolo NuA4 is an H4- and H2A-specific HAT complex that functions alone and as part of the larger NuA4 complex.138 Piccolo’s Esa1 catalytic subunit is unable to bind and acetylate nucleosomes without its accessory subunits Epl1 and Yng2.138 The 2011 Chittuluru et al. cryo-EM reconstruction shows that Piccolo binds to the NCP with 1:1 stoichiometry using two prongs that contact the NCP opposite to the dyad and over the H4 histone-fold with flexibility observed between Piccolo and the NCP.137 Subsequent cross-linking experiments place the Esa1 Tudor domain in proximity to nucleosomal DNA and the Epl1 EPcA domain near the N-terminal tail of H2A.139
Swi6 is an HP1 ortholog that binds to trimethylated H3 K9 to enable the spreading of heterochromatin. The 2013 25 Å cryo-EM reconstitution of two Swi6 dimers bound to the NCP in open/disinhibited forms reported by Canzio et al. suggests that one Swi6 chromodomain and one chromoshadow domain from each dimer contact the nucleosome near the exit of the H3 tail and at the nucleosomal DNA at SHL ± 5, respectively.77 The other chromodomain in each dimer is poised for binding H3 K9me3 in a neighboring nucleosome to facilitate spreading of Swi6 across chromatin. While the authors could thoroughly investigate the autoinhibitory function of Swi6, the low resolution of the cryo-EM structure prevented a molecular understanding of the Swi6-NCP interactions.
These cryo-EM studies offer unique views of nucleosome recognition by large and complex chromatin enzymes and factors. At this time, cryo-EM allows for the general architecture of chromatin factor-NCP complexes to be ascertained. The molecular workhorse remains other modalities, including crystallography of subcomplexes, XL-MS and comprehensive biochemistry. Yet, with technological advances in sample preparation together with higher resolution and faster detectors and more powerful image alignment algorithms, cryo-EM holds promise to complement if not rival or surpass crystallographic and NMR methods for atomic-resolution determination of chromatin factor-nucleosome complex structures.
5. Structural Studies of the Chromatosome and 30 nm Fiber
The structure of the chromatosome and the structure, and even relevance, of the 30 nm fiber are two of the most highly studied, yet controversial, topics in chromatin biology. It is clear linker histone H1 (or H5) binds to the nucleosome core particle and linker DNA and promotes compaction of chromatin arrays into 30 nm fibers.140 Linker histones contain a central globular domain and unstructured N- and C-terminal extensions. The globular domain gH1 and the C-terminal domain are primarily involved in chromatin binding and compaction. Many years of biochemical, biophysical and computational experiments have led to two distinct classes of gH1-NCP interactions: (1) symmetric in which gH1 binds at the dyad and interacts with linker DNA extending from both sides of the core particle;141−144 and (2) asymmetric in which gH1 binds near the dyad and interacts with 10–20 bp of linker DNA extending from one side of the nucleosome core.119,143,145−149 Two recent structural studies offer unique insights into H1 binding in the chromatosome and the 30 nm fiber.
5.1. NMR and Cryo-EM Models of the Chromatosome
In 2013, Zhou et al. used extensive NMR measurements to generate a unique residue-specific model for the gH1/NCP complex.119 The authors first observed chemical shift perturbation of isotopically labeled gH1 residues 37–211 and nucleosome core particles to define the regions of each involved in gH1/NCP complex formation. Then they incorporated paramagnetic spin labels to define distances between regions of gH1 and the NCP to orient the complex. Finally, they performed computational docking to generate models of the gH1/NCP complex. The favored model correlates with asymmetric binding to the nucleosome consistent with strong effects observed with spin labels attached to H3 R37 and H2A T119, as these observations are incompatible with symmetric binding models. Similar results were seen with the H1 tails that are unstructured in the chromatosome. gH1 only interacts with the 10 bp extending from the NCP given that no differences were seen with longer segments of nucleosomal DNA. In the favored computational model, gH1 uses two positively charged surfaces defined by NMR experiments (residues 119–125 and 164–174) to bridge the nucleosome core surface and 10 bp of linker DNA on one side of the NCP. However, the authors could not rule out weaker binding to the other linker DNA. No evidence was seen of gH1 binding histones within the nucleosomal disk. However, gH1 binding imparts structural organization of the H2A C-terminal tail consistent with a direct interaction. This explains the decreased binding of gH1 to H2A.Z containing nucleosomes that have been attributed to divergent C-terminal sequences.150,151 In 2014, Song et al. reported an 11 Å reconstruction of the 30 nm fiber reconstituted with histone H1. The density for the gH1 domain showed a 1:1 H1:nucleosome binding with H1 binding asymmetrically near, but off-center from, the dyad and interacting with both entry and exit linker DNAs. These studies provide exciting views of the position of H1 within the chromatosome and bolster growing evidence for the asymmetric binding model. Precise molecular details await higher-resolution structural solutions.
5.2. 11 Å Cryo-EM Structure of a Two-Start 30 nm Fiber
Traditional dogma holds that the 11 nm unfolded chromatin strand (often referred to as beads on a string) compacts into a 30 nm fiber with side-to-side packing of nucleosomes perpendicular to the fiber axis.152,153 The folding of the 30 nm fiber is encouraged by the interiorly positioned linker histones.154 The 30 nm fiber further condenses into progressively higher-order chromatin states. Most models for the 30 nm fiber fall into two categories: (1) one-start models are solenoidal with sequential nucleosomes connected by bent linker DNA segments arranged along a helical path;12,152−154 (2) two-start models separate sequential nucleosomes by straight linker DNA in a zigzag patter either longitudinally along the fiber (helical ribbon) or radially across the fiber (crossed-linker).13,154−157
Studies by two groups in the mid 2000s used reconstituted arrays with defined nucleosome positions to build opposing two- and one-start molecular models. Richmond and colleagues proposed a two-start model based on digestion patterns of short, cross-linked nucleosomal arrays.87 The two-start model was further supported by a 9 Å tetranucleosome crystal structure showing two stacks of two nucleosomes separated by a zig-zagging pattern of straight linker DNA.13 This structure was used to generate an idealized model for the two-start crossed-linker-type 30 nm fiber (Figure 13). Of note, the modeled fiber has a smaller diameter owing to the 167 nucleosome repeat length used for the tetranucleosome structure and the dependence of crossed-linker-type fiber diameters on linker length. Meanwhile, Rhodes and colleagues characterized longer H1-containing 30 nm fibers by cryo-EM.12 They observed similar fiber diameter over widely varying linker lengths characteristic of a one-start solenoid structure (Figure 13). Later modeling also suggested potential two-start solutions to the cryo-EM data in addition to the original one-start model.158
Figure 13.

Models of the 30 nm fiber. Orthogonal views perpendicular to the 30 nm fiber axis (top) and down the axis (bottom) of the Richmond two-start model (left), Rhodes one-start model (center) and Li-Zhu tetranucleosome-unit repeat two-start model (right). The sequence of nucleosomes in each model is indicated. In the Richmond model, each sequential pair of nucleosomes across the fiber is colored similarly. For the Rhodes model, all nucleosomes in the same turn of the solenoid are colored similarly. In the Li-Zhu model, each tetranucleosome repeating unit is colored similarly. Unlabeled nucleosomes in the two-start models are not shown in the bottom views for figure clarity. Linker DNA is not present in the Rhodes model but, given the nature of the solenoidal structure, must be bent. The B-form DNA double helix is shown for comparison (far right). All models shown in space-filling representation and scaled as indicated.
Despite this long-standing hierarchical paradigm for chromatin folding, recent cryo-EM and SAXS measurements with mitotic chromosomes show no evidence of the 30 nm fiber.159−162 Rather mitotic chromatin may assume a fractal-like state. Similar experiments with chicken erythrocytes did show evidence of the 30 nm fiber.162 Altogether, this implies that the 30 nm fiber does exist in certain cell-types and/or cell-cycle stages, but may not be as pervasive as once thought.163
In 2014, Song et al. solved 11 Å cryo-EM structures of 30 nm fibers reconstituted with 12 × 177 and 12 × 187 bp of the Widom 601 nucleosome positioning sequence.11 The structures clearly show a left-handed parallel double helix similar to that proposed by Richmond and colleagues (Figure 13). This structure bears some resemblance to the DNA double helix, although the DNA double helix contains right-handed antiparallel strands. The diameter of the fiber is dependent on small changes in DNA linker length, suggesting a two-start model in which straight linker DNA crossing the central channel determines the fiber diameter. This two-start crossed-linker type model is confirmed by clear density for straight segments of linker DNA crossing the center channel of the fiber. The repeating unit of the fiber is a tetranucleosome with two stacked nucleosomes on opposing sides of the superhelix. The arrangement of nucleosomes places the thinner half of the nucleosome near the dyad close to the interior of the fiber where the fiber diameter is smaller. Save the different linker length, the tetranucleosome repeating unit is very similar to the 9 Å tetranucleosome crystal structure.13 Unexpectedly, gH1 from neighboring nucleosomes in the 30 nm fiber align with alternating head-to-head arrangements within the tetranucleosomal unit and tail-to-tail arrangements between tetranucleosomal units. The tail-to-tail aligned gH1s interact with one another, imparting an additional twist to the fiber. This results in different internucleosomal contacts between adjacent stacked nucleosomes in the tetranucleosome unit and between tetranucleosome units. Notably, the H4 tail-H2A/H2B acidic patch interaction is plausible between tetranucleosome units, as in the idealized model from Richmond and colleagues. This interaction is not possible within the tetranucleosome unit due to juxtaposition of the H2B αC helix of one nucleosome with the acidic patch H2A α2 helix of the neighboring nucleosome. This phenomenon was also seen in the tetranucleosome crystal structure. While controversy remains regarding the prevalence of the 30 nm fiber in cell-cycle specific structures of chromatin in different cell types in vivo, this model provides a higher resolution view of the 30 nm fiber, which gives new insights into the orientation of H1 within each nucleosome and across the chromatin fiber.
6. Perspective
Over the past 5 years, major strides have been made in the understanding of nucleosome structure and function. Molecular details of the sequence-dependence of nucleosomal DNA structure have been elucidated. Co-crystallization and comprehensive NMR analysis of chromatin factors and the nucleosome core particle have established the acidic patch arginine-anchor as a paradigm for nucleosome recognition. Advancements in cryo-EM technology have aided in the visualization of the two-start 30 nm fiber and characterization of nucleosome binding by large and conformationally flexible chromatin enzymes. Despite this extraordinary progress, it is clear that we have only scratched the surface of the tremendously complex role of the nucleosome in coordinating chromatin-templated processes. Additional work will undoubtedly establish new paradigms for nucleosome recognition and bring about a heightened understanding of the role of the 30 nm fiber and other chromatin structures in the functional organization of the eukaryotic genome.
Acknowledgments
This work was supported by NIGMS Grants GM060489-09S1, GM088236, and GM111651 to S.T. R.K.M. is supported by a Damon Runyon Postdoctoral fellowship (DRG 2107-12).We would like to thank Tim Richmond for providing the coordinates for his two-start model of the 30 nm fiber; Phil Robinson and Daniela Rhodes for providing coordinates for their one-start model of the 30 nm fiber; and Guohong Li and Ping Zhu for providing coordinates for their two-start 30 nm fiber model.
Glossary
Abbreviations
- NCP
nucleosome core particle
- LANA
latency-associated nuclear antigen
- RCC1
regulator of chromosome condensation 1
- Sir3
silent information regulator 3
- CENP-C
centromeric protein C
- PRC1
Polycomb repressive complex 1
- cryo-EM
cryogenic electron microscopy
- SHL
superhelical location
- KSHV
Kaposi’s sarcoma-associated herpesvirus
- BAH
bromo-adjacent homology
- CENP-A
centromeric protein A
- HMGN2
high-mobility group nucleosome binding 2
- NMR
nuclear magnetic resonance
- methyl-TROSY
methyl transverse relaxation optimized spectroscopy
Biographies

Robert K. McGinty received his Ph.D. from The Rockefeller University in 2010 and M.D. from Weill Cornell Medical College as a member of the Tri-Institutional M.D.–Ph.D. program. During his graduate studies under the supervision of Prof. Tom Muir, McGinty used protein chemistry to study crosstalk between histone modifications. In 2011, he joined the laboratory of Prof. Song Tan in the Center for Eukaryotic Gene Regulation at Penn State, where he is using X-ray crystallography to study the molecular recognition of the nucleosome by histone modifying enzymes. He is currently a Damon Runyon postdoctoral fellow.

Song Tan studied physics as an undergraduate at Cornell University (1985) before pursuing his Ph.D. at the MRC Laboratory of Molecular Biology as a Marshall Scholar (1989). He continued his training as a postdoctoral fellow and project leader under Tim Richmond at the ETH-Zürich (Swiss Federal Institute of Technology) where he determined crystal structures of several transcription factor/DNA complexes. Dr. Tan joined the Center for Eukaryotic Gene Regulation and the faculty of the Department of Biochemistry and Molecular Biology in 1998. He was named a Pew Scholar in the Biomedical Sciences in 2001. Dr. Tan’s laboratory investigates how chromatin enzymes and factors interact with their nucleosome substrates through biochemical and structural approaches. His laboratory determined the first chromatin factor-nucleosome crystal structure (RCC1-nucleosome) in 2010 and, with Dr. McGinty, the first chromatin enzyme-nucleosome crystal structure (PRC1-nucleosome) in 2014.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- McGhee J. D.; Felsenfeld G. Annu. Rev. Biochem. 1980, 49, 1115. [DOI] [PubMed] [Google Scholar]
- Simpson R. T. Biochemistry 1978, 17, 5524. [DOI] [PubMed] [Google Scholar]
- Taverna S. D.; Li H.; Ruthenburg A. J.; Allis C. D.; Patel D. J. Nat. Struct. Mol. Biol. 2007, 14, 1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K.; Dechassa M. L.; Tremethick D. J. Nat. Rev. Mol. Cell Biol. 2012, 13, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogren-Knaak M.; Ishii H.; Sun J.-M.; Pazin M. J.; Davie J. R.; Peterson C. L. Science 2006, 311, 844. [DOI] [PubMed] [Google Scholar]
- Lu X.; Simon M. D.; Chodaparambil J. V.; Hansen J. C.; Shokat K. M.; Luger K. Nat. Struct. Mol. Biol. 2008, 15, 1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierz B.; Chatterjee C.; McGinty R. K.; Bar-Dagan M.; Raleigh D. P.; Muir T. W. Nat. Chem. Biol. 2011, 7, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson P. J.; Rhodes D. Curr. Opin. Struct. Biol. 2006, 16, 336. [DOI] [PubMed] [Google Scholar]
- Luger K.; Mäder A. W.; Richmond R. K.; Sargent D. F.; Richmond T. J. Nature 1997, 389, 251. [DOI] [PubMed] [Google Scholar]
- Lowary P. T.; Widom J. J. Mol. Biol. 1998, 276, 19. [DOI] [PubMed] [Google Scholar]
- Song F.; Chen P.; Sun D.; Wang M.; Dong L.; Liang D.; Xu R.-M.; Zhu P.; Li G. Science 2014, 344, 376. [DOI] [PubMed] [Google Scholar]
- Robinson P. J. J.; Fairall L.; Huynh V. A. T.; Rhodes D. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schalch T.; Duda S.; Sargent D. F.; Richmond T. J. Nature 2005, 436, 138. [DOI] [PubMed] [Google Scholar]
- Flaus A.; Luger K.; Tan S.; Richmond T. J. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arents G.; Burlingame R. W.; Wang B. C.; Love W. E.; Moudrianakis E. N. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 10148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arents G.; Moudrianakis E. N. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 11170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey C. A.; Sargent D. F.; Luger K.; Maeder A. W.; Richmond T. J. J. Mol. Biol. 2002, 319, 1097. [DOI] [PubMed] [Google Scholar]
- McGinty R. K.; Tan S. In Fundamentals of Chromatin; Springer: New York: New York, NY, 2013; pp 1–28. [Google Scholar]
- Preez du L. L.; Patterton H.-G. Subcell. Biochem. 2013, 61, 37. [DOI] [PubMed] [Google Scholar]
- Zheng C.; Hayes J. J. J. Biol. Chem. 2003, 278, 24217. [DOI] [PubMed] [Google Scholar]
- Kan P. Y.; Lu X.; Hansen J. C.; Hayes J. J. Mol. Cell. Biol. 2007, 27, 2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan P. Y.; Caterino T. L.; Hayes J. J. Mol. Cell. Biol. 2009, 29, 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White C. L.; Suto R. K.; Luger K. EMBO J. 2001, 20, 5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier C. R.; Chakravarthy S.; Petosa C.; Fernández-Tornero C.; Luger K.; Müller C. W. Proteins 2008, 71, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunaka Y.; Kajimura N.; Tate S.-I.; Morikawa K. Nucleic Acids Res. 2005, 33, 3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama M.; Arimura Y.; Shirayama K.; Fujita R.; Oba Y.; Sato N.; Inoue R.; Oda T.; Sato M.; Heenan R. K.; Kurumizaka H. Biophys. J. 2014, 106, 2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urahama T.; Horikoshi N.; Osakabe A.; Tachiwana H.; Kurumizaka H. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2014, 70, 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachiwana H.; Kagawa W.; Shiga T.; Osakabe A.; Miya Y.; Saito K.; Hayashi-Takanaka Y.; Oda T.; Sato M.; Park S.-Y.; Kimura H.; Kurumizaka H. Nature 2011, 476, 232. [DOI] [PubMed] [Google Scholar]
- Tachiwana H.; Osakabe A.; Shiga T.; Miya Y.; Kimura H.; Kagawa W.; Kurumizaka H. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 578. [DOI] [PubMed] [Google Scholar]
- Tachiwana H.; Kagawa W.; Osakabe A.; Kawaguchi K.; Shiga T.; Hayashi-Takanaka Y.; Kimura H.; Kurumizaka H. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 10454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimura Y.; Kimura H.; Oda T.; Sato K.; Osakabe A.; Tachiwana H.; Sato Y.; Kinugasa Y.; Ikura T.; Sugiyama M.; Sato M.; Kurumizaka H. Sci. Rep. 2013, 3, 3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy S.; Bao Y.; Roberts V. A.; Tremethick D.; Luger K. Cold Spring Harbor Symp. Quant. Biol. 2004, 69, 227. [DOI] [PubMed] [Google Scholar]
- Chakravarthy S.; Gundimella S. K. Y.; Caron C.; Perche P.-Y.; Pehrson J. R.; Khochbin S.; Luger K. Mol. Cell. Biol. 2005, 25, 7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy S.; Luger K. J. Biol. Chem. 2006, 281, 25522. [DOI] [PubMed] [Google Scholar]
- Abbott D. W.; Laszczak M.; Lewis J. D.; Su H.; Moore S. C.; Hills M.; Dimitrov S.; Ausio J. Biochemistry 2004, 43, 1352. [DOI] [PubMed] [Google Scholar]
- Eirín-López J. M.; Ishibashi T.; Ausio J. FASEB J. 2008, 22, 316. [DOI] [PubMed] [Google Scholar]
- Horikoshi N.; Sato K.; Shimada K.; Arimura Y.; Osakabe A.; Tachiwana H.; Hayashi-Takanaka Y.; Iwasaki W.; Kagawa W.; Harata M.; Kimura H.; Kurumizaka H. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suto R. K.; Clarkson M. J.; Tremethick D. J.; Luger K. Nat. Struct. Biol. 2000, 7, 1121. [DOI] [PubMed] [Google Scholar]
- Dechassa M. L.; Wyns K.; Li M.; Hall M. A.; Wang M. D.; Luger K. Nat. Commun. 2011, 2, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde e Silva N.; Black B. E.; Sivolob A.; Filipski J.; Cleveland D. W.; Prunell A. J. Mol. Biol. 2007, 370, 555. [DOI] [PubMed] [Google Scholar]
- Sekulic N.; Bassett E. A.; Rogers D. J.; Black B. E. Nature 2010, 467, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston I. J.; Yung J. S. Y.; Singleton M. R. J. Biol. Chem. 2011, 286, 4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchenko T.; Sorensen T. C.; Woodcock C. L.; Kan Z.-Y.; Wood S.; Resch M. G.; Luger K.; Englander S. W.; Hansen J. C.; Black B. E. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 16588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyen C.-M.; Montel F.; Gautier T.; Menoni H.; Claudet C.; Delacour-Larose M.; Angelov D.; Hamiche A.; Bednar J.; Faivre-Moskalenko C.; Bouvet P.; Dimitrov S. EMBO J. 2006, 25, 4234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove M. S.; Wolberger C. Biochem. Cell Biol. 2005, 83, 468. [DOI] [PubMed] [Google Scholar]
- Cosgrove M. S.; Boeke J. D.; Wolberger C. Nat. Struct. Mol. Biol. 2004, 11, 1037. [DOI] [PubMed] [Google Scholar]
- Mersfelder E. L.; Parthun M. R. Nucleic Acids Res. 2006, 34, 2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North J. A.; Šimon M.; Ferdinand M. B.; Shoffner M. A.; Picking J. W.; Howard C. J.; Mooney A. M.; van Noort J.; Poirier M. G.; Ottesen J. J. Nucleic Acids Res. 2014, 42, 4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North J. A.; Javaid S.; Ferdinand M. B.; Chatterjee N.; Picking J. W.; Shoffner M.; Nakkula R. J.; Bartholomew B.; Ottesen J. J.; Fishel R.; Poirier M. G. Nucleic Acids Res. 2011, 39, 6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manohar M.; Mooney A. M.; North J. A.; Nakkula R. J.; Picking J. W.; Edon A.; Fishel R.; Poirier M. G.; Ottesen J. J. J. Biol. Chem. 2009, 284, 23312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šimon M.; North J. A.; Shimko J. C.; Forties R. A.; Ferdinand M. B.; Manohar M.; Zhang M.; Fishel R.; Ottesen J. J.; Poirier M. G. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makde R. D.; England J. R.; Yennawar H. P.; Tan S. Nature 2010, 467, 562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasudevan D.; Chua E. Y. D.; Davey C. A. J. Mol. Biol. 2010, 403, 1. [DOI] [PubMed] [Google Scholar]
- Finch J. T.; Brown R. S.; Richmond T.; Rushton B.; Lutter L. C.; Klug A. J. Mol. Biol. 1981, 145, 757. [DOI] [PubMed] [Google Scholar]
- Richmond T. J.; Finch J. T.; Rushton B.; Rhodes D.; Klug A. Nature 1984, 311, 532. [DOI] [PubMed] [Google Scholar]
- Richmond T. J.; Finch J. T.; Klug A. Cold Spring Harbor Symp. Quant. Biol. 1983, 47, 493. [DOI] [PubMed] [Google Scholar]
- Richmond T. J.; Searles M. A.; Simpson R. T. J. Mol. Biol. 1988, 199, 161. [DOI] [PubMed] [Google Scholar]
- Simpson R. T.; Stafford D. W. Proc. Natl. Acad. Sci. U.S.A. 1983, 80, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harp J. M.; Palmer E. L.; York M. H.; Gewiess A.; Davis M.; Bunick G. J. Electrophoresis 1995, 16, 1861. [DOI] [PubMed] [Google Scholar]
- Satchwell S. C.; Drew H. R.; Travers A. A. J. Mol. Biol. 1986, 191, 659. [DOI] [PubMed] [Google Scholar]
- Chua E. Y. D.; Vasudevan D.; Davey G. E.; Wu B.; Davey C. A. Nucleic Acids Res. 2012, 40, 6338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulaeva O. I.; Gaykalova D. A.; Pestov N. A.; Golovastov V. V.; Vassylyev D. G.; Artsimovitch I.; Studitsky V. M. Nat. Struct. Mol. Biol. 2009, 16, 1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui F.; Zhurkin V. B. J. Biomol. Struct. Dyn. 2010, 27, 821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolstorukov M. Y.; Colasanti A. V.; McCandlish D. M.; Olson W. K.; Zhurkin V. B. J. Mol. Biol. 2007, 371, 725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers A. A. Philos. Trans. R. Soc., A 2004, 362, 1423. [DOI] [PubMed] [Google Scholar]
- Travers A. A.; Muskhelishvili G.; Thompson J. M. T. Philos. Trans. R. Soc., A 2012, 370, 2960. [DOI] [PubMed] [Google Scholar]
- Olson W. K.; Zhurkin V. B. Curr. Opin. Struct. Biol. 2011, 21, 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond T. J.; Davey C. A. Nature 2003, 423, 145. [DOI] [PubMed] [Google Scholar]
- Juo Z. S.; Chiu T. K.; Leiberman P. M.; Baikalov I.; Berk A. J.; Dickerson R. E. J. Mol. Biol. 1996, 261, 239. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Geiger J. H.; Hahn S.; Sigler P. B. Nature 1993, 365, 512. [DOI] [PubMed] [Google Scholar]
- Kim J. L.; Nikolov D. B.; Burley S. K. Nature 1993, 365, 520. [DOI] [PubMed] [Google Scholar]
- Olson W. K.; Gorin A. A.; Lu X.-J.; Hock L. M.; Zhurkin V. B. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 11163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondarenko V. A.; Steele L. M.; Ujvári A.; Gaykalova D. A.; Kulaeva O. I.; Polikanov Y. S.; Luse D. S.; Studitsky V. M. Mol. Cell 2006, 24, 469. [DOI] [PubMed] [Google Scholar]
- Abbott E. A.Flatland: A romance of many dimensions; Seely & Co.: UK, 1884. [Google Scholar]
- Makde R. D.; Tan S. Anal. Biochem. 2013, 442, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hock R.; Furusawa T.; Ueda T.; Bustin M. Trends Cell Biol. 2007, 17, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canzio D.; Liao M.; Naber N.; Pate E.; Larson A.; Wu S.; Marina D. B.; Garcia J. F.; Madhani H. D.; Cooke R.; Schuck P.; Cheng Y.; Narlikar G. J. Nature 2013, 496, 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armache K. J.; Garlick J. D.; Canzio D.; Narlikar G. J.; Kingston R. E. Science 2011, 334, 977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbera A. J.; Chodaparambil J. V.; Kelley-Clarke B.; Joukov V.; Walter J. C.; Luger K.; Kaye K. M. Science 2006, 311, 856. [DOI] [PubMed] [Google Scholar]
- Marmorstein R.; Trievel R. C. Biochim. Biophys. Acta, Gene Regul. Mech. 2009, 1789, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman C. A.; Lalonde M.-E.; Côté J.; Kutateladze T. G. Nat. Struct. Mol. Biol. 2012, 19, 1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg A. J.; Li H.; Patel D. J.; Allis C. D. Nat. Rev. Mol. Cell Biol. 2007, 8, 983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.-R.; Feng H.; Ghirlando R.; Kato H.; Gruschus J.; Bai Y. J. Mol. Biol. 2012, 421, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorigo B.; Schalch T.; Bystricky K.; Richmond T. J. J. Mol. Biol. 2003, 327, 85. [DOI] [PubMed] [Google Scholar]
- Robinson P. J. J.; An W.; Routh A.; Martino F.; Chapman L.; Roeder R. G.; Rhodes D. J. Mol. Biol. 2008, 381, 816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allahverdi A.; Yang R.; Korolev N.; Fan Y.; Davey C. A.; Liu C. F.; Nordenskiold L. Nucleic Acids Res. 2011, 39, 1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorigo B.; Schalch T.; Kulangara A.; Duda S.; Schroeder R. R.; Richmond T. J. Science 2004, 306, 1571. [DOI] [PubMed] [Google Scholar]
- Sinha D.; Shogren-Knaak M. A. J. Biol. Chem. 2010, 285, 16572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D.; Arya G. Phys. Chem. Chem. Phys. 2011, 13, 2911. [DOI] [PubMed] [Google Scholar]
- Barbera A. J.; Ballestas M. E.; Kaye K. M. J. Virol. 2004, 78, 294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke P. R.; Zhang C. Nat. Rev. Mol. Cell Biol. 2008, 9, 464. [DOI] [PubMed] [Google Scholar]
- Carazo-Salas R. E.; Guarguaglini G.; Gruss O. J.; Segref A.; Karsenti E.; Mattaj I. W. Nature 1999, 400, 178. [DOI] [PubMed] [Google Scholar]
- Kalab P.; Heald R. J. Cell Sci. 2008, 121, 1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemergut M. E.; Mizzen C. A.; Stukenberg T.; Allis C. D.; Macara I. G. Science 2001, 292, 1540. [DOI] [PubMed] [Google Scholar]
- England J. R.; Huang J.; Jennings M. J.; Makde R. D.; Tan S. J. Mol. Biol. 2010, 398, 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusche L. N.; Kirchmaier A. L.; Rine J. Annu. Rev. Biochem. 2003, 72, 481. [DOI] [PubMed] [Google Scholar]
- McBryant S. J.; Krause C.; Woodcock C. L.; Hansen J. C. Mol. Cell. Biol. 2008, 28, 3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson A.; Li G.; Sikorski T. W.; Buratowski S.; Woodcock C. L.; Moazed D. Mol. Cell 2009, 35, 769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino F.; Kueng S.; Robinson P.; Tsai-Pflugfelder M.; van Leeuwen F.; Ziegler M.; Cubizolles F.; Cockell M. M.; Rhodes D.; Gasser S. M. Mol. Cell 2009, 33, 323. [DOI] [PubMed] [Google Scholar]
- Norris A.; Bianchet M. A.; Boeke J. D. PLoS Genet. 2008, 4, e1000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer M. S.; Kahana A.; Wolf A. J.; Meisinger L. L.; Peterson S. E.; Goggin C.; Mahowald M.; Gottschling D. E. Genetics 1998, 150, 613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng H. H.; Ciccone D. N.; Morshead K. B.; Oettinger M. A.; Struhl K. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen F.; Gafken P. R.; Gottschling D. E. Cell 2002, 109, 745. [DOI] [PubMed] [Google Scholar]
- Arnaudo N.; Fernández I. S.; McLaughlin S. H.; Peak-Chew S. Y.; Rhodes D.; Martino F. Nat. Struct. Mol. Biol. 2013, 20, 1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D.; Fang Q.; Wang M.; Ren R.; Wang H.; He M.; Sun Y.; Yang N.; Xu R.-M. Nat. Struct. Mol. Biol. 2013, 20, 1116. [DOI] [PubMed] [Google Scholar]
- Connelly J. J.; Yuan P.; Hsu H.-C.; Li Z.; Xu R.-M.; Sternglanz R. Mol. Cell. Biol. 2006, 26, 3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H.; Jiang J.; Zhou B.-R.; Rozendaal M.; Feng H.; Ghirlando R.; Xiao T. S.; Straight A. F.; Bai Y. Science 2013, 340, 1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S.; Ahmad K.; Malik H. S. Science 2001, 293, 1098. [DOI] [PubMed] [Google Scholar]
- Verdaasdonk J. S.; Bloom K. Nat. Rev. Mol. Cell Biol. 2011, 12, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hori T.; Amano M.; Suzuki A.; Backer C. B.; Welburn J. P.; Dong Y.; McEwen B. F.; Shang W.-H.; Suzuki E.; Okawa K.; Cheeseman I. M.; Fukagawa T. Cell 2008, 135, 1039. [DOI] [PubMed] [Google Scholar]
- McGinty R. K.; Henrici R. C.; Tan S. Nature 2014, 514, 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao R.; Tsukada Y.-I.; Zhang Y. Mol. Cell 2005, 20, 845. [DOI] [PubMed] [Google Scholar]
- Grau D. J.; Chapman B. A.; Garlick J. D.; Borowsky M.; Francis N. J.; Kingston R. E. Genes Dev. 2011, 25, 2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon J. A.; Kingston R. E. Nat. Rev. Mol. Cell Biol. 2009, 10, 697. [DOI] [PubMed] [Google Scholar]
- Buchwald G.; van der Stoop P.; Weichenrieder O.; Perrakis A.; van Lohuizen M.; Sixma T. K. EMBO J. 2006, 25, 2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley M. L.; Corn J. E.; Dong K. C.; Phung Q.; Cheung T. K.; Cochran A. G. EMBO J. 2011, 30, 3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung J. W.; Agarwal P.; Canny M. D.; Gong F.; Robison A. D.; Finkelstein I. J.; Durocher D.; Miller K. M. PLoS Genet. 2014, 10, e1004178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H.; van Ingen H.; Zhou B.-R.; Feng H.; Bustin M.; Kay L. E.; Bai Y. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.-R.; Feng H.; Kato H.; Dai L.; Yang Y.; Zhou Y.; Bai Y. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 19390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattner B. P.; Yusufzai T.; Kadonaga J. T. Mol. Cell 2009, 34, 620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J.-H.; West K. L.; Rubinstein Y.; Bergel M.; Postnikov Y. V.; Bustin M. EMBO J. 2005, 24, 3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J.-H.; Catez F.; Birger Y.; West K. L.; Prymakowska-Bosak M.; Postnikov Y. V.; Bustin M. Mol. Cell 2004, 15, 573. [DOI] [PubMed] [Google Scholar]
- Postnikov Y.; Bustin M. Biochim. Biophys. Acta, Gene Regul. Mech. 2010, 1799, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prymakowska-Bosak M.; Misteli T.; Herrera J. E.; Shirakawa H.; Birger Y.; Garfield S.; Bustin M. Mol. Cell. Biol. 2001, 21, 5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattiroli F.; Uckelmann M.; Sahtoe D. D.; van Dijk W. J.; Sixma T. K. Nat. Commun. 2014, 5, 3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriere V.; Roussel L.; Ortega N.; Lacorre D.-A.; Americh L.; Aguilar L.; Bouche G.; Girard J.-P. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalashnikova A. A.; Porter-Goff M. E.; Muthurajan U. M.; Luger K.; Hansen J. C. J. R. Soc., Interface 2013, 10, 20121022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapier C. R.; Cairns B. R. Annu. Rev. Biochem. 2009, 78, 273. [DOI] [PubMed] [Google Scholar]
- Chaban Y.; Ezeokonkwo C.; Chung W.-H.; Zhang F.; Kornberg R. D.; Maier-Davis B.; Lorch Y.; Asturias F. J. Nat. Struct. Mol. Biol. 2008, 15, 1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asturias F. J.; Chung W.-H.; Kornberg R. D.; Lorch Y. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 13477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha A.; Wittmeyer J.; Cairns B. R. Nat. Struct. Mol. Biol. 2005, 12, 747. [DOI] [PubMed] [Google Scholar]
- Leschziner A. E.; Saha A.; Wittmeyer J.; Zhang Y.; Bustamante C.; Cairns B. R.; Nogales E. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racki L. R.; Yang J. G.; Naber N.; Partensky P. D.; Acevedo A.; Purcell T. J.; Cooke R.; Cheng Y.; Narlikar G. J. Nature 2009, 462, 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K.; Frouws T. D.; Angst B.; Fitzgerald D. J.; DeLuca C.; Schimmele K.; Sargent D. F.; Richmond T. J. Nature 2011, 472, 448. [DOI] [PubMed] [Google Scholar]
- Saravanan M.; Wuerges J.; Bose D.; McCormack E. A.; Cook N. J.; Zhang X.; Wigley D. B. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 20883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosi A.; Haas C.; Herzog F.; Gilmozzi A.; Berninghausen O.; Ungewickell C.; Gerhold C. B.; Lakomek K.; Aebersold R.; Beckmann R.; Hopfner K.-P. Cell 2013, 154, 1207. [DOI] [PubMed] [Google Scholar]
- Chittuluru J. R.; Chaban Y.; Monnet-Saksouk J.; Carrozza M. J.; Sapountzi V.; Selleck W.; Huang J.; Utley R. T.; Cramet M.; Allard S.; Cai G.; Workman J. L.; Fried M. G.; Tan S.; Côté J.; Asturias F. J. Nat. Struct. Mol. Biol. 2011, 18, 1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreault A. A.; Cronier D.; Selleck W.; Lacoste N.; Utley R. T.; Allard S.; Savard J.; Lane W. S.; Tan S.; Côté J. Genes Dev. 2003, 17, 1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Tan S. Mol. Cell. Biol. 2013, 33, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harshman S. W.; Young N. L.; Parthun M. R.; Freitas M. A. Nucleic Acids Res. 2013, 41, 9593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan J.; Hartman P. G.; Crane-Robinson C.; Aviles F. X. Nature 1980, 288, 675. [DOI] [PubMed] [Google Scholar]
- Syed S. H.; Goutte-Gattat D.; Becker N.; Meyer S.; Shukla M. S.; Hayes J. J.; Everaers R.; Angelov D.; Bednar J.; Dimitrov S. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L.; Roberts V. A. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 8384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer S.; Becker N. B.; Syed S. H.; Goutte-Gattat D.; Shukla M. S.; Hayes J. J.; Angelov D.; Bednar J.; Dimitrov S.; Everaers R. Nucleic Acids Res. 2011, 39, 9139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y. B.; Gerchman S. E.; Ramakrishnan V.; Travers A.; Muyldermans S. Nature 1998, 395, 402. [DOI] [PubMed] [Google Scholar]
- Bharath M. M. S.; Chandra N. R.; Rao M. R. S. Nucleic Acids Res. 2003, 31, 4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong J.; Li Q.; Levi B. Z.; Shi Y. B.; Wolffe A. P. EMBO J. 1997, 16, 7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachov G. V.; Gabdoulline R. R.; Wade R. C. Nucleic Acids Res. 2011, 39, 5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An W.; Leuba S. H.; van Holde K.; Zlatanova J. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogler C.; Huber C.; Waldmann T.; Ettig R.; Braun L.; Izzo A.; Daujat S.; Chassignet I.; Lopez-Contreras A. J.; Fernandez-Capetillo O.; Dundr M.; Rippe K.; Längst G.; Schneider R. PLoS Genet. 2010, 6, e1001234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakar A.; Gupta P.; Ishibashi T.; Finn R.; Silva-Moreno B.; Uchiyama S.; Fukui K.; Tomschik M.; Ausio J.; Zlatanova J. Biochemistry 2009, 48, 10852. [DOI] [PubMed] [Google Scholar]
- McGhee J. D.; Nickol J. M.; Felsenfeld G.; Rau D. C. Cell 1983, 33, 831. [DOI] [PubMed] [Google Scholar]
- Widom J.; Klug A. Cell 1985, 43, 207. [DOI] [PubMed] [Google Scholar]
- Thoma F.; Koller T.; Klug A. J. Cell Biol. 2003, 83, 403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worcel A.; Strogatz S.; Riley D. Proc. Natl. Acad. Sci. U.S.A. 1981, 78, 1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcock C. L.; Frado L. L.; Rattner J. B. J. Cell Biol. 2002, 99, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams S. P.; Athey B. D.; Muglia L. J.; Schappe R. S.; Gough A. H.; Langmore J. P. Biophys. J. 1986, 49, 233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong H.; Victor J.-M.; Mozziconacci J. PLoS One 2007, 2, e877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eltsov M.; MacLellan K. M.; Maeshima K.; Frangakis A. S.; Dubochet J. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 19732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeshima K.; Hihara S.; Eltsov M. Curr. Opin. Cell Biol. 2010, 22, 291. [DOI] [PubMed] [Google Scholar]
- Joti Y.; Hikima T.; Nishino Y.; Kamada F.; Hihara S.; Takata H.; Ishikawa T.; Maeshima K. Nucleus 2012, 3, 404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino Y.; Eltsov M.; Joti Y.; Ito K.; Takata H.; Takahashi Y.; Hihara S.; Frangakis A. S.; Imamoto N.; Ishikawa T.; Maeshima K. EMBO J. 2012, 31, 1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen J. C. EMBO J. 2012, 31, 1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker N. A.; Sept D.; Joseph S.; Holst M. J.; McCammon J. A. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 10037. [DOI] [PMC free article] [PubMed] [Google Scholar]