

1. Introduction
In eukaryotic cells, inheritable information is stored in a nucleoprotein complex referred to as chromatin.1 This genome architecture serves two key purposes. On the one hand, wrapping DNA (approximately 145–147 basepairs) twice around a spool composed of two copies each of the highly basic core histones H2A, H2B, H3, and H4 leads to compaction of DNA strands (Figure 1a,b). These assemblies are called nucleosomes. Contacts between individual nucleosomes are often mediated by cationic tails at the N- and C-termini of all histone proteins that protrude from the core and further tighten the chromatin fiber (Figure 1c). Additional packing is achieved through attachment of histone H1 to the DNA that links neighboring nucleosomes or by nonhistone proteins that are able to bridge units within or between chromatin fibers.2 The second pivotal function of storing genetic information as a DNA–protein complex is the additional layer of regulation that this feature provides.3−5 For instance, the very presence of histones on DNA sequences can occlude access to these sites by transcription factors and other DNA binding proteins.6 Thus, nucleosome positioning, shaped in part by DNA sequence preferences and shifted by ATP-powered molecular motors (referred to as chromatin remodelers), directly affects chromatin transactions.7 Beyond their location, the biochemical makeup of nucleosomes provides further opportunity for regulation. Canonical histones can be replaced with closely resembling variants, and all histones are dynamically decorated with post-translational modifications (PTMs). These biochemical marks can be as small as just a few atoms, such as methyl (Lys, Arg, Gln), acetyl (Lys), or phosphoryl groups (Ser, Thr), or as large as an entire protein in the case of ubiquitin or SUMO. Upon attachment by dedicated transferase enzymes, PTMs can directly alter the biophysical properties of the target protein, provide a docking site for specific interaction partners, interfere with binding events of other factors, or act through a combination of these mechanisms. In this way, signaling through histone PTMs serves to orchestrate chromatin-templated processes, including fine-tuning transcriptional outputs. Remarkably, transcriptional states can be inherited through cell division cycles, thus providing a mode of epigenetic memory.8,9 Not surprisingly, misregulation of the inputs and outputs of chromatin signaling occurs in many diseases, especially cancer.10−13
Figure 1.
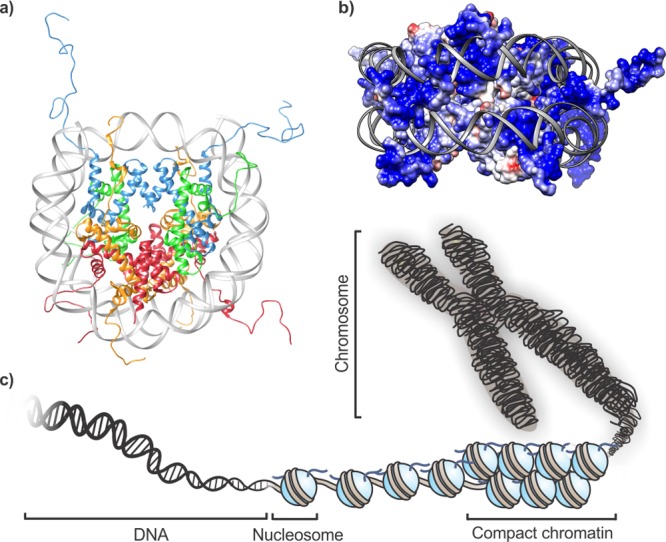
Chromatin architecture in eukaryotic cells. (a) Structure of a mononucleosome. DNA (gray) is wrapped around two copies each of H2A (orange), H2B (red), H3 (blue), and H4 (green); pdb code: 1kx5. (b) Electrostatic surface rendering of a histone octamer. Highly cationic patches (blue) guide the trajectory of DNA wrapping. (c) Schematic representation of genome architecture.
Lysine acetylation, serine/threonine phosphorylation, and lysine ubiquitylation have a strong propensity to directly influence the structure of chromatin. Both acetylation and phosphorylation reduce the net positive charge of histones, and thereby weaken electrostatic interactions with the negatively charged DNA. In particular, acetylation at multiple lysine residues is associated with decompaction of chromatin, providing space for the transcription machinery to engage with acetylated chromatin domains. These transcriptionally active, open regions are referred to as euchromatin. Attaching an entire protein such as ubiquitin (8.5 kDa) to histones (10–15 kDa) can also preclude tight packing of nucleosomes. Consequently, histone ubiquitylation is associated with active transcription (specifically, ubiquitylation of histone H2B at lysine 120; abbreviated as H2B-K120ub) and DNA damage repair (H2A-K119ub).14 In contrast, lysine and arginine methylation events only slightly change the biophysical properties of nucleosomes. These modifications are often targeted by protein factors present in the nucleus that discern the methylation states and the surrounding sequences, and thereby act as signaling hubs. A paradigm for this mechanism is the binding of heterochromatin protein 1 (HP1) to histone H3 carrying a trimethylation mark at lysine 9 (H3K9me3).15 Through oligomerization, HP1 can noncovalently link multiple nucleosomes to create a compact architecture that impedes transcription, and simultaneously provides a docking platform for a cohort of associated proteins.16 Such inactive chromatin domains are commonly referred to as heterochromatin.
Several factors drive the interactions between nuclear proteins and histones. Remarkably, all histones display a strong compositional bias in their amino acid content. They are highly enriched in basic residues, whereas acidic and aromatic residues as well as cysteines are strongly underrepresented (Figure 2a). In addition, the protruding tail regions of all four of the core histones, as well as the linker histone H1, contain strikingly few hydrophobic amino acids. Notably, high charge density and low hydrophobicity are defining features of intrinsically disordered proteins.17 Thus, histone binding frequently relies on electrostatic contributions and hydrogen bonding rather than complementary hydrophobic surfaces that typically drive protein–protein interactions. Perhaps as a consequence of the low building block diversity found in histone tails, recurring sequence motifs can be discerned (Figure 2b). Most prominently, the ARKS tetrapeptide occurs twice in the H3 tail, encompassing Lys9 and Lys27, both associated with heterochromatin-specific methylation. A permutated variation, ARTK, is located at the very N-terminus of H3. Similarly, the H4 tail contains three instances of a GKG tripeptide. Many histone binders engage these short linear motifs,18 often in a PTM-dependent fashion, allowing for tightly controlled interactions between chromatin and designated binding proteins.4,19 Many endogenous proteins contain histone-like sequences, suggesting that such motifs play an important role in cellular physiology.20,22 Interestingly, an influenza protein mimics the ARTK sequence of the H3 tail (ARSK) to highjack the host cell’s transcription machinery.21
Figure 2.
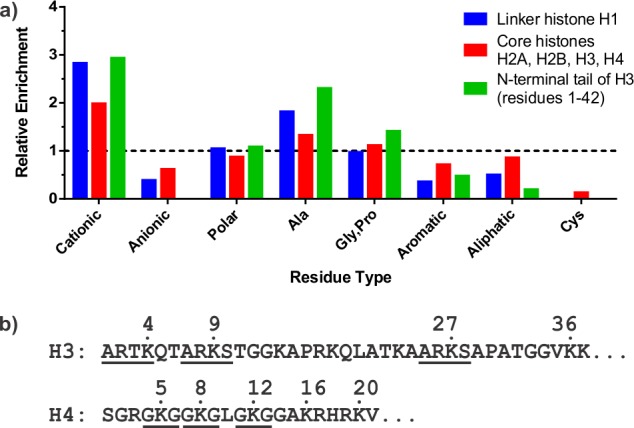
Histone sequence features. (a) Histones contain a skewed amino acid composition. Amino acid frequencies are normalized to the average occurrence found in all proteins contained in the uniprot database (www.uniprot.org). Cationic residues, Arg, Lys; anionic, Asp, Glu; polar, Asn, Gln, Ser, Thr; aromatic, His, Phe, Trp, Tyr; aliphatic, Ile, Leu, Met, Val; Ala and Cys are plotted individually; the secondary structure breaking residues Gly and Pro are binned together. (b) Recurring sequence motifs in histone tails surrounding modified lysine residues.
Given that many chromatin-related processes involve interactions with the unstructured histone tails that protrude from nucleosomes and are subject to a plethora of PTMs, peptide chemistry has aided tremendously in assigning functional roles to these modifications. The small size of these tails makes them ideal targets for peptide synthesis, and their lack of a defined 3D-structure obviates the need for refolding synthetic material. In particular, peptide models have contributed to the characterization of enzymes that attach and remove histone PTMs, and proteins that interact with specific marks. These proteins are often anthropomorphically called histone mark writers, erasers, and readers, respectively.
With increasing sophistication of proposed mechanisms for the regulation of chromatin structure and function by signaling cascades, there is a growing need for chemically defined model systems with which to directly address these emerging hypotheses. Contemporary protein chemistry and chromatin assembly strategies can fulfill this requirement to a large degree. In this Review, we first summarize the contributions of peptide chemistry over the last 40+ years in an eclectic journey that aims to provide a glimpse into the variety of histone PTMs that modulate chromatin. We focus on the synthesis of modified histone peptides and their contribution to deciphering the supramolecular chemistry that controls the function of histone PTMs. We then discuss modern approaches to generate chemically defined chromatin templates, involving innovative uses of protein chemistry and synthetic biology, and how “designer” chromatin has furthered our understanding of key molecular recognition events that govern nuclear biochemistry.
2. Historic Perspective
Since the early days of chromatin biology, protein chemistry has played a pivotal role in exploring the mechanisms of chromatin transactions. Following the discoveries that histones inhibit RNA synthesis in nuclear extracts,23−25 and that histones are also heavily acetylated,26 Allfrey and co-workers surmised that these modifications are installed post-translationally and serve to regulate transcription.27 Indeed, limited chemical acetylation of isolated histones using acetic anhydride diminished their ability to inhibit transcription. In the following years, it became evident that histone acetylation occurs largely on lysine side-chains28 and is biochemically reversible.29 As determined by then emerging protein sequencing technologies, the main sites of acetylation on histone H4 correspond to Lys16, and to a lesser degree Lys5, 8, and 12.30,31
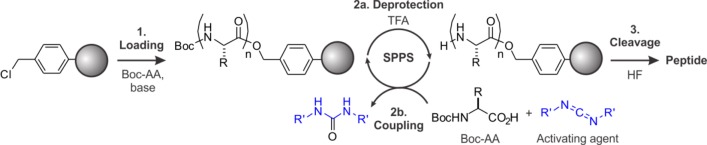
These early biochemical investigations coincided with the development of solid-phase peptide synthesis (SPPS) by Merrifield (Scheme 1),32 a process that led to a tremendous surge in efficiency of oligopeptide preparation, as exemplified by the total synthesis of Ribonuclease S, a 124 residue protein.33 Because Allfrey and Merrifield were colleagues at the Rockefeller University, the stage was set for the first targeted studies on the biochemistry of specific histone post-translational modifications employing synthetic peptides.
Scheme 1. Solid-Phase Peptide Synthesis (SPPS) Using the N-α-Boc-Protection Strategy.
3. Histone Peptide Chemistry
Chromatin biochemistry is fertile ground for peptide chemists. Chromatin-associated proteins perform many molecular transactions with the flexible histone tails that protrude from the compact nucleosome core. Thus, assays based on synthetic peptides can recapitulate certain key aspects of the interplay between chromatin and the nuclear proteome. In this section, we will highlight the contributions of peptide chemistry to solving some of the mysteries that chromatin biology harbors. We will discuss histone PTMs, as well as the utility of cross-linking and combinatorial methods to investigate their functions, with an emphasis on synthesis and molecular recognition.
3.1. Lysine Acetylation
3.1.1. Pioneering Studies with Acetylated Histone Peptides
Early studies on the biochemistry of specific histone PTMs focused on delineating the substrate scope of deacetylases (HDACs).34,35 To narrow the substrate specificity of calf thymus histone deacetylase, Merrifield, Allfrey, and co-workers prepared histone peptides by limited proteolysis of H4, purified from calf thymus nuclei that were previously incubated with radioactive acetate.35 Digestion with CNBr and chymotrypsin yielded two fragments, H4(1–84) and H4(1–37), respectively (Figure 3a). In addition, a small peptide spanning residues 15–21 containing radiolabeled Ac-Lys at position 16 was prepared by SPPS using standard N-α-tert-butyloxycarbonyl (Boc) protected building blocks, as well as N-α-Boc-N-ε-[14C]acetyllysine (Figure 3b). Cleavage from the resin was achieved with hydrofluoric acid (HF), and the resulting peptide was purified by ion exchange chromatography. HDAC activity, monitored by release of radiolabeled acetate, was detected only when using long peptide constructs; the synthetic peptide was not a substrate. Because H4K16 is a prominent histone acetylation site, these results suggested that long N-terminal peptides were required for substrate recognition. As SPPS became more routine, and the first automated peptide synthesizers were built,36−38 peptides of such length became accessible. Thus, a doubly modified peptide encompassing H4(1–37) was prepared with a 3H-labeled and a 14C-labeled acetyl group at positions 16 and 12, respectively (Figure 3c). This setup allows for a straightforward distinction between the acetyl groups at each position. HDAC-catalyzed release of 3H and 14C was equal, demonstrating that this enzyme is able to remove both marks efficiently.
Figure 3.
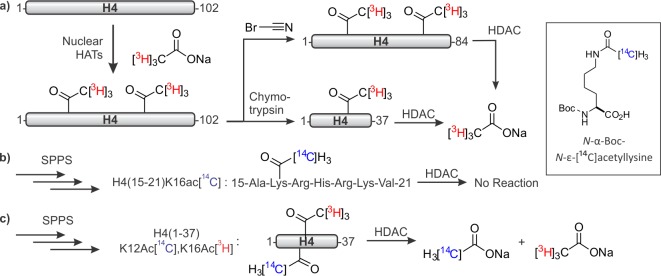
Synthesis of acetylated H4 peptides to define the substrate specificity of an HDAC. (a) Limited proteolysis of acetylated H4 yields long peptidic HDAC substrates. HATs = histone acetyltransferases. (b) Solid-phase peptide synthesis (SPPS) using a radiolabeled acetyllysine building block (inset) yields a hepta-peptide that is not an HDAC substrate. (c) A long synthetic peptide bearing two distinctly radio-labeled acetyl groups illustrates promiscuity in HDAC activity.
3.1.2. Molecular Recognition of Acetyllysine in Histones
Electrostatic interactions contribute strongly to nucleosome formation. Lysine and arginine residues, present at the lateral surface of histone octamers and on the flexible tails, direct DNA wrapping and mediate internucleosomal contacts to establish higher order chromatin structure, respectively.39,40 Accordingly, lysine acetylation is expected to affect DNA binding because this modification decreases the basicity of histones. Consistent with this hypothesis, several studies employing either full-length histone H4, or peptide fragments thereof, indicate that acetylation weakens histone DNA interactions.27,41−43
Besides a direct biophysical effect on chromatin structure, histone acetylation also serves a biochemical function by recruiting specialized reader domains. Characterization of these interactions was made possible by the ease of access to site-specifically acetylated histone peptides granted by SPPS. Currently, the most well-studied acetyllysine reader module is the Bromodomain (BD), a small protein domain encompassing approximately 110 amino acids that is often found in transcriptional coactivators.44−46 A structural investigation by NMR of the BD from one such coactivator, the acetyltransferase P/CAF, revealed a 4-helix-bundle fold with a prominent hydrophobic pocket.47 The small molecule acetyllysine analogue, N-acetyl-histamine, was able to bind to this site, with the acetamide moiety facing toward the protein interior.47 Local chemical shift perturbations upon titration of the BD with a synthetic H4 peptide containing a single acetyl mark, K8ac, revealed an interaction with a high micromolar Kd. A cocrystal structure of the BD from another acetyltransferase, GCN5, with an H4 peptide acetylated at Lys16 provides further insight into the binding interface (Figure 4a).48 The nature of the interaction is predominantly hydrophobic with a tight fit of the lysine side-chain methylene groups into an apolar cleft and a somewhat more loose fit of the terminal methyl group within the pocket. Specific hydrogen bonds with a conserved Asn residue in the BD, as well as several ordered water molecules in the partially solvent-accessible binding crevice, orient the acetamide modification. Additional interactions with residues surrounding the acetylated lysine originate from shape complementarity to the BD surface, a limited number of backbone–backbone hydrogen bonds, and, in the case of the GCN5 BD, an ion pair at the i+3 position (to Arg19 of H4).48,49 Consequently, associations of BDs with acetylated peptides are often weak and rather unspecific.50 Moreover, thermodynamic analyses performed on the binding of a typical BD to H3K9ac confirm that such interactions are primarily driven by the hydrophobic effect.51
Figure 4.
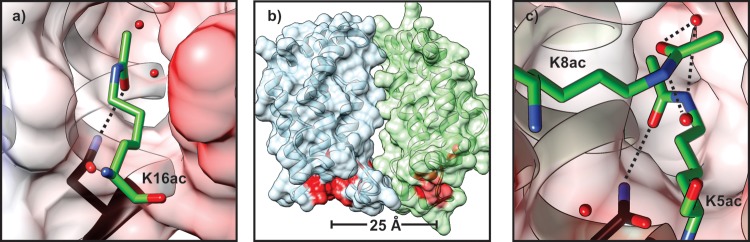
Recognition of acetyllysine residues by bromodomains. (a) Binding pocket of the GCN5 BD in complex with an H4 peptide acetylated at Lys16 (green). A hydrogen bond between Asn407 of the BD (black) and the acetyl group is indicated with a dotted line; pdb code: 1E6I. (b) Architecture of the double-BD module of TAFII250. The acetyllysine binding pocket of each lobe is indicated in red; pdb code: 1EQF. (c) Simultaneous binding of two acetyllysine residues by BD1 of Brdt. A synthetic H4 peptide bearing K5ac and K8ac is depicted in green with hydrogen-bonding networks indicated by dotted lines; pdb code: 2WP2. Surfaces in subfigures (a) and (c) are shown in electrostatic rendering (blue, positive; white, neutral; red, negative). Ordered water molecules are shown as red spheres.
The frequent occurrence of multiple acetyl marks on a single histone tail52 raises the question as to how two or more acetyllysine residues are recognized. Crystal structures and binding studies of the double-BD containing proteins TAFII250 (a general transcription factor)53 and Brdt (a testis-specific genome organizing factor)54 shed light on this question. In TAFII250, the two BDs are oriented by protein–protein interactions to enable simultaneous binding of i, i+7, or i+8 acetyl marks with low micromolar affinity (Figure 4b).53 In contrast, the first BD of Brdt preferentially binds multiple acetyl marks in a single pocket.54 This feature is accomplished due to the open BD cleft, where one residue (K5ac) is bound in an orientation typical for BD–acetyllysine interactions, including a hydrogen bond to Asn108 (Figure 4c). The side-chain of K8ac reaches into the open pocket, forming a hydrogen-bond network from its amide oxygen through an ordered water molecule to the amide nitrogen of K5ac. The orientation of K8ac is reinforced by hydrophobic interactions with the methylene groups as well as the terminal methyl group of the acetamide moiety.
3.1.3. Generation of Histone-PTM-Specific Antibodies
Modern chromatin biology relies heavily on antibodies that recognize distinct histone PTMs. Modification-specific antibodies serve to detect the presence of their cognate mark within a biological sample, for instance, in a Western blot format. Moreover, they represent indispensable affinity reagents for chromatin immunoprecipitation (ChIP),55 enabling the isolation of mono- or oligonucleosomes bearing a designated histone PTM (Figure 5a). Subsequent analysis of isolated chromatin segments by proteomic (e.g., mass spectrometry) or genomic (e.g., DNA sequencing) methods provides detailed information about the biochemistry of the targeted histone PTM, and its genomic distribution.
Figure 5.

PTM-selective antibodies as tools in chromatin biochemistry. (a) General outline of the chromatin immunoprecipitation (ChIP) workflow. (b) Production of site-specific acetyllysine antibodies using synthetic peptides. Specifically acetylated peptides are used to immunize rabbits to elicit a collection of antibodies that recognize defined acetylation marks. In this example, antibody selectivity was probed using the synthetic peptide substrates (right). Plus symbols denote a strong recognition, (+) stands for weak binding, whereas minus signs indicate no cross-reactivity. Data taken from ref (60).
Access to site-specificically modified histone peptides through SPPS represents the basis for generating these invaluable tools. Initial efforts to elicit antibodies that recognize acetylated H4 focused on purified acetylated forms of the protein,56 chemically acetylated full length protein (ref (57)), or H4 N-terminal peptides.58 The resulting antisera were capable of distinguishing acetylated from nonacetylated H4, but lacked the ability to distinguish individual acetylation sites. To address this limitation, Turner et al. synthesized a series of acetylated H4 peptides and used these as epitopes for antibody generation (Figure 5b).59 These same peptides were then used to probe antibody specificity, enabling estimates of acetylation site usage during cell division59 and in human cells.60
This seminal series of studies served as a template for many future endeavors. Indeed, a cohort of poly- and monoclonal antibodies that recognize site-specificic acetylation marks with improved selectivity have been raised, and many are commercially available.61 Similarly, antibodies against essentially all known histone PTMs, elicited using synthetic peptides featuring the modification in question, have been added to the toolkit of chromatin biochemists. This list is continuously growing, and newly discovered histone PTMs are immediately incorporated into peptide epitopes for antibody generation (see also examples in section 3.8).
3.1.4. Mechanism of Histone Deacetylases
Chemical synthesis permits the installation of non-natural analogues of acetyllysine. To scrutinize the mechanism of substrate recognition and turnover by class III histone deacetylases (these enzymes consume NAD+ during deacetylation, yielding O-acetyl-ADP-ribose and nicotinamide as byproducts), Smith and Denu prepared versions of the H3 tail containing acetyllysine mimics at position 14 (Figure 6).62,63 Hydrophobicity was found to correlate with binding strength,63 and nucleophilicity of the amide oxygen with catalysis.62 These results suggest a concerted SN2-like mechanism for NAD+ cleavage, and highlight that some HDACs tolerate bulkier substrates such as propionyllysine (see also section 3.8).
Figure 6.

Mechanism of class III HDACs (a) probed with histone peptides carrying analogues of acetyllysine (b). n.d. stands for not determined. Data taken from refs (62) and (63).
3.2. Lysine Methylation
The protein sequencing efforts performed in the 1960s revealed not only that some histone lysine residues are acetylated, but also the presence of methyllysine isoforms.31,64,65 However, biochemical investigations of histone lysine methylation lagged behind the more conveniently assayed histone acetylation.66 A further complication is that lysine side-chains are mono-, di-, and trimethylated, and each methylation state may confer a distinct biological impact.67−69 Nevertheless, a tremendous body of research has been amassed on the biochemistry of histone lysine methylation, sparked by the discoveries of S-adenosylmethionine (SAM)-dependent, lysine-specific histone methyltransferases,70 protein domains that specifically interact with lysines in different methylation states,15,71 and the importance of lysine methylation in the regulation of gene expression72,73 and the DNA damage response.74−76 While initially thought of as irreversible marks, lysine methyl groups can be removed through the action of site-specific histone lysine demethylases.77,78 As for acetyllysine, synthetic peptides bearing homogeneously modified methyllysine residues were instrumental in this endeavor.
3.2.1. Synthesis of Methyllysine-Containing Peptides
Since the turn of the millennium, when histone lysine methylation became a prolific area of study, routine SPPS of methyllysine-containing peptides has been performed using the N-α-Fmoc protecting group scheme.79 In this strategy, base-labile main chain protection is combined with side-chain protecting groups and resin linkages sensitive to TFA treatment (Scheme 2), thereby bypassing the hazardous HF cleavage step commonly employed in Boc-SPPS. In addition, SPPS benefited from improved coupling chemistries based on novel uronium80−82 and phosphonium83 reagents, as well as auxiliary nucleophiles such as oximes84 (Figure 7a). Building blocks for the incorporation of all lysine methylation states are readily available synthetically, or can be obtained commercially (Figure 7b). N-α-Fmoc-protected di- and trimethyllysine are prepared by reductive alkylation with formaldehyde and electrophilic alkylation with iodomethane, respectively.85 The monomethylated isoform is typically employed in the N-ε-Boc protected form, accessible through reductive alkylation of an N-ε-benzyl-protected intermediate.86 Peptides synthesized with these building blocks are at the routine disposal of chromatin biochemists, and have been harnessed to obtain a palette of PTM-specific antibodies87 and have found use in countless biochemical and biophysical studies.
Scheme 2. Solid-Phase Peptide Synthesis (SPPS) Using the N-α-Fmoc-Protection Strategy.

Figure 7.

Synthesis of methyllysine-containing peptides. (a) Commonly used activating agents and additives. (b) Standard methyllysine building blocks used for Fmoc-based SPPS.
3.2.2. Molecular Recognition of Methyllysine-Containing Histone Peptides
As lysine methylation does not change the side-chain charge, this class of modification exerts its biochemical effects predominantly by serving as a docking platform for protein–protein interactions.88 Trimethylation at H4K20 represents a prominent exception to this rule, and will be treated in section 4.1.4.89 Although many modules capable of interpreting lysine methyl marks do exist,69,88 we will focus here on chromodomains (CDs) to discuss the energetics of methyllysine binding and how specificity between methylation states is achieved. For a more comprehensive survey of the range of protein modules that specifically interact with histone PTMs, including methyllysine, the reader is directed to the recent review by Patel and colleagues.90
CDs are small protein modules (approximately 50 residues in size) initially identified in heterochromatin protein 1 (HP1) and polycomb protein (Pc), key organizers of heterochromatin.91−94 The CD of HP1 specifically recognizes H3K9me2/3,15 and its structure was solved in complex with a series of short peptides containing either H3K9me3, H3K9me2, or H3K9me2 in combination with H3K4me2.95,96 The methylated ammonium side-chain is enveloped in an aromatic cage, formed by an induced fit mechanism upon peptide binding (Figure 8a).95,97 The structures of the CD bound to di- and trimethyllysine are highly similar, as are their binding affinities, both in the low micromolar range.95 Imperfect size selection in dimethyllysine binding is compensated for by a water-mediated hydrogen bond between the lysine ε-amine and a glutamate side-chain (Figure 8a). The HP1 chromodomain discriminates strongly against the lower methylation states of Lys9: its affinity for monomethyllysine and unmodified lysine is reduced by 1.3 and >2.7 kcal/mol, respectively.98 An analogue of the dimethyllysine side-chain, 3-dimethylamino-1-propanol, does not bind appreciably.96 Instead, additional residues on the substrate peptide, bound as an extended strand, contribute to histone recognition based on size and charge, and concomitantly confer specificity for designated methyllysine sites.95,96 In agreement with this mechanism, the K4/K9 doubly modified peptide binds HP1 exclusively through K9me2.96
Figure 8.

Recognition of methyllysine. (a) Structures of the HP1 chromodomain in complex with methyllysine residues (pdb codes for K9me3, 1kne; K9me2, 1kna; K9me1, 1q3l) or in its apo form (right, pdb code: 1ap0). The CD is depicted in green, the ligand in yellow. Note that the apo-structure was solved with murine HP1 while the liganded structures were obtained from drosophila HP1, which contains a Tyr residue in place of Phe45. (b) Selective recognition of lower methylation states by the chromodomain of MSL3 (pdb code: 3m9p). The CD is depicted in cyan, the ligand in pink. For comparison, the corresponding residues in the HP1 CD are indicated in pale rendering. (c) Structure of tert-butylnorleucine (1), a trimethyllysine isostere. (d) Structure of a calix[4]arene receptor (2) for methyllysine-containing peptides.
Specificity for lower methylation states is exemplified by the interaction of the CD of MSL3 (a transcriptional regulator) with mono- and dimethyllysine.99 As compared to the CD from HP1, the MSL3 CD contains an additional Trp residue that serves as a tight lid for the aromatic cage to favor binding of secondary and tertiary ammonium ions over the quaternary trimethyllysine (Figure 8b).99,100 Additional strategies to favor lower methylation states, discussed in detail in ref (90), include steric restriction as well as ionic hydrogen bonds to the ε N–H group.101,102
The driving force for methyllysine binding is the cation−π interaction, a common motif for recognition of cations in biology.103−105 As is typical for this type of interactions,104 complex formation between HP1 and K9me3-modified peptides is mediated by a strong favorable enthalpy, with a slightly unfavorable entropic contribution.106 To gain more insight into the forces governing CD binding, Waters and co-workers prepared an H3 peptide containing tert-butylnorleucine (1) at position 9 (Figure 8c).98 This residue is isosteric to trimethyllysine but lacks the charge, and therefore precludes electrostatic interactions with the aromatic cage of HP1. CD binding of the H3 peptide was reduced by approximately 2 kcal/mol upon replacing K9me3 with its neutral isostere,98 in agreement with typical values for cation−π interactions (on the order of 0.4–2.4 kcal/mol).104
Synthetic receptors have been generated that mimic the biological mode of binding methyllysine residues.107,108 For example, sulfonated calix[4]arene-based hosts (2, Figure 8d) can engulf methyllysine residues by harnessing cation−π and electrostatic interactions.108,109 By matching the dimensions of the aromatic cage to the size of methylated lysine, specificity for methylation states can be achieved. Such supramolecular receptors are able to compete for the binding of H3K9me3 with its natural readers, and, as a consequence, perturb chromatin structure in cells.109
3.2.3. Identification of New Methyllysine Binders
To identify proteins that specifically bind a given histone PTM, Wysocka et al. performed pull-down experiments with synthetic peptides and cell lysates.110,111 To this end, H3 peptides, carrying the K4me3 mark and a biotin tag, were immobilized on avidin beads (Figure 9a). Incubation with nuclear extracts, followed by SDS page analysis of bound proteins, yielded a band at molecular weight >300 kDa.111 Mass spectrometry identified this H3K4me3 binder as BPTF, the largest subunit of the chromatin remodeling complex NURF.112 BPTF contains two zinc finger motifs termed plant homeodomains (PHDs) and a bromodomain. Repeating the peptide pull-down assays with purified truncated BPTF constructs demonstrated that the second PHD was necessary and sufficient for H3K4me3 binding. Subsequent structural characterization indicated that the H3K4me3 mark is bound in an aromatic cage, and sequence specificity is granted by additional cation−π interactions and an ionic H-bond to Arg2 in the peptide (Figure 9b).113 Notably, this peptide pull-down workflow has been applied to numerous histone PTMs and has provided a vast body of knowledge on stable interactions between nuclear proteins and specific histone marks.114
Figure 9.

Identification of new histone PTM binders. (a) Schematic of the workflow for peptide pull-downs of nuclear proteins. Modified peptides are immobilized on avidin beads and used to fish out specific binders such as the H3K4me3 binder BPTF. (b) Structure of the BPTF PHD finger (mauve) in complex with H3K4me3 (yellow, pdb code: 2f6j). An ion pair between Arg2 of histone H3 and an Asp residue of the PHD finger contributes to selectivity. (c) SILAC-based identification of methyllysine binders. Modified and control histone peptides are immobilized and incubated with isotopically labeled nuclear extracts. A hypothetical mass spectrum illustrating different selectivities of detected proteins is depicted on the right.
The use of stable isotope labeling by amino acids in cell culture (SILAC)115 greatly increases the sensitivity and throughput of this pull-down approach.116 Vermeulen et al. generated a map of the human histone-methyllysine interactome using histone peptides containing one of the key trimethyl marks: H3K4me3, H3K9me3, H3K27me3, H3K36me3, or H4K20me3. Methylated peptides were used to pull down nuclear factors from HeLa cells grown in normal media. In parallel, unmodified peptide controls were used to enrich binding proteins from cells grown in the presence of 13C- and 15N-labeled Arg and Lys (“heavy” medium). The “light” proteins isolated with a specific methyllysine peptide are combined with “heavy” proteins from unmodified peptide pull-downs, and the mixture analyzed by mass spectrometry (Figure 9c). For each protein identified, the ratio of “light” versus “heavy” signal (L/H) obtained by MS reveals its binding preference: L/H > 1 indicates a Kme3-dependent interaction, while analytes with L/H < 1 favor unmethylated lysine residues. Proteins with L/H ≈ 1 are nonspecific interactors, and are typically ignored in further analyses. This approach yielded between 10 and 60 specific binding protein candidates for each mark, thus significantly expanding the catalog of potential trimethyllysine reader proteins.
Reactivity-based probes have been developed to enable specific isolation of histone demethylases from nuclear lysates. To achieve this, Cole and co-workers installed a propargyllysine residue in place of Lys4 of an H3 peptide.117 Peptides armed with this warhead were recognized by the H3K4-specific demethylase LSD1, triggering their oxidation with FAD (Figure 10). This reaction yields a potent electrophile that covalently links the reduced flavin cofactor to the probe. Thus, propargyllysine peptides represent potent mechanism-based inhibitors of FAD-dependent lysine demethylases. Immobilized versions of these probes successfully pulled down LSD1 and its binding partner, the corepressor CoREST, from nuclear lysates. A panel of related probes has since been devised by the same group.118 In conjunction with SAM cofactor analogues developed by the Luo group,119 these reagents facilitate chemical proteomic approaches to delineate histone lysine methylation and demethylation pathways.
Figure 10.

Mechanism-based histone demethylase inhibitors. Propargyllysine is oxidized by LSD1 via its FAD cofactor. The resulting Michael acceptor forms a covalent adduct with the reduced cofactor.
3.3. Arginine Methylation
Histone arginine methylation occurs in three flavors: monomethylarginine (Rme) as well as the asymmetric (Rme2a) and symmetric (Rme2s) isoforms of dimethylarginine (Figure 11a). Methylarginine marks are installed by a panel of protein arginine methyltransferases (PRMTs), which are specific in regard to the Rme2 isomer they produce, but rather promiscuous in terms of site.120,121 Chemically, the synthesis of methylarginine-containing peptides using Fmoc-SPPS is straightforward. Methylarginine isoforms that contain a free N-ω atom (Rme and Rme2a) are commonly sold in Pbf-protected forms, while Rme2s is available with di-Boc protection (Figure 11b). Given the importance of arginine residues in mediating both histone–DNA and histone–protein interactions, it is not surprising that its methylation has a range of critical functions in chromatin biology, including transcription regulation.122−124 However, the majority of PRMT substrates are nonhistone proteins, often involved in RNA biochemistry, which complicates the assignment of cellular roles for histone arginine methylation.124
Figure 11.

Methylarginine structure and recognition. (a) Isoforms of methylarginine residues. (b) Standard methylarginine building blocks for Fmoc-based SPPS. (c) Structure of the aromatic cage of the TDRD3 tudor domain (pdb code: 2lto). The Rme2a residue is colored in yellow, the specificity-determining tyrosine in pale green. (d) Structure of a synthetic Rme2a receptor isolated from a dynamic combinatorial library.
3.3.1. Arginine Methylation and Protein–Histone Interactions
Arginine methylation often exerts its biological effect by interfering with the biochemistry of other histone PTMs, in particular with methyllysine.125 Many key sites of lysine methylation contain an arginine residue at the −1 (H3K9, H3K27, H4K20, all typically considered repressive marks) or the −2 position (H3K4, an activating mark). Inspired by the negative correlation between the presence of H3K4me3 and H3R2me2a,126 Guccione et al. tested if peptides containing preinstalled methyl marks at K4 or R2 were substrates of PRMT6 and ASH2, the corresponding arginine and lysine methyltransferases, respectively.127 In agreement with their hypothesis, H3K4me3-peptides were poor substrates for PRMT6, and, reciprocally, peptides containing H3R2me2a were not methylated by ASH2. Furthermore, the presence of H3R2me2a impeded the interaction of K4me3 with many of its known readers.127,128 In contrast, some effectors of K4me2/3, such as the recombinase RAG2, benefit slightly from an additional R2me2s mark.129
The search for histone methylarginine readers gained a boost with the discovery that certain tudor domain proteins, some of which were previously known to be methyllysine binders, can specifically recognize Rme2s residues.130 In 2009, the DNA methyltransferase DNMT3A was shown to bind the H4 tail in an R3me2s specific manner.131 To find additional site-specific readers of histone me2a marks, Bedford and co-workers employed a microarray featuring more than 100 chromatin associated domains132 including bromo, chromo, and tudor domains, among others.133 To generate the array, individual domains were produced and purified as fusions with the enzyme glutathione S-transferase (GST) and spotted on a glass slide precoated with nitrocellulose polymer and immobilized by drying.134 When the array was probed with H3 peptides containing R17me2a and a Cy3 label, a single protein domain, the tudor domain of TDRD3, displayed a fluorescent spot.133 This interaction was confirmed using peptide pull-down experiments, and promiscuous binding between TDRD3 and several histone-derived Rme2a marks was observed. TDRD3 functions as a transcriptional coactivator; thus, another link between histone arginine methylation and transcription regulation was found.133
The structure of the TDRD3 tudor domain has been solved by crystallography in its apo form135 and by NMR in complex with an RNA polymerase-derived peptide containing the Rme2a mark.136 The domain features a spacious aromatic cage, ideally suited to accommodate methyl arginine residues (Figure 11c).137 Selectivity for the asymmetric isomer is controlled, at least in part, by a tyrosine residue that stacks with the guanidinium group.136 However, the molecular mechanisms that underlie the discrimination for histone sites remain unclear.
Synthetic receptors for Rme2 have been generated using dynamic combinatorial libraries.138 Several aromatic dithiol building blocks were incubated in the presence of a short Rme2a-containing peptide (Figure 11d). Upon prolonged incubation, a three-membered, disulfide-bonded host molecule had formed that recognized histone peptides featuring an Rme2a mark. The same peptides containing Rme2s or Rme were bound less tightly by approximately 1 kcal/mol, although the host displayed no selectivity against trimethyllysine.138 Conceivably, such receptors may find application as affinity reagents for enriching methylated histones and other proteins for proteomics studies.
3.3.2. Histone Citrullination
Whether histone arginine methylation marks can be removed is contentious.120,139 One possibility under active research is the potential for methyl-deimination of methylarginine into citrulline by peptidyl arginine deiminases such as PAD4 (Figure 12).140−143 Interestingly, histone citrullination steers diverse biochemical functions independent of arginine methylation. Examples include the regulation of transcription140,144 and linker histone binding.145 It is currently unknown whether histone citrullination is reversible. However, given that biological mechanisms to convert free citrulline into arginine exist,146 it is tempting to speculate that related enzymes might also operate on proteins containing this residue.
Figure 12.

PAD4-catalyzed deimination and possibly demethylimination to citrulline. Whether mechanisms exist to convert citrulline back to arginine in the context of histones is unknown.
3.4. Histone Phosphorylation
Protein phosphorylation plays a central role in signaling, and histone substrates are no exception. Regulation of chromatin structure by histone phosphorylation is particularly important during cell cycle progression. As is common for protein phosphorylation in eukaryotes in general, serine and threonine phosphorylation have been studied in most detail, although histone tyrosine phosphorylation is also known to control chromatin structure and function.147−152 In addition, phosphoarginine153,154 and phosphohistidine154−156 residues have been detected in histones, but their biochemistry is much less well studied due to the chemical instability of these marks.
3.4.1. Synthesis of Histone Phosphopeptides
Incorporation of residues with O-linked phosphoryl groups by Fmoc-based solid-phase synthesis is in most cases routine nowadays. Typically, monobenzyl groups are used to protect the phosphoryl group during synthesis (Figure 13a). The presence of a negative charge on the monoprotected phosphoryl group during Fmoc deprotection with piperidine drastically reduces beta elimination for phosphoserine and phosphothreonine as compared to when dialkylated phosphoamino acids are used.157 However, during the coupling of monoprotected phosphorylated amino acids, additional base is required for efficient coupling, and reversible acylation of the phosphoryl group can occur.157,158
Figure 13.

Building blocks for the synthesis of O-linked (a) and N-linked (b) phosphopeptides and their analogues.
The synthesis of peptides containing acid-labile N-linked phosphoryl groups is much more challenging.159,160 Nevertheless, recent developments have enabled the incorporation of phosphoarginine residues through the use of trichloroethyl (Tc) protecting groups that are selectively removed by hydrogenolysis after global deprotection using a TFA/scavenger cocktail (Figure 13b).161 Furthermore, stable analogues for both isomers of phosphohistidine, where the phosphoryl group is attached to either N-τ (3-pHis, analogues 3 and 4) or N-π (1-pHis, analogue 5), have been synthesized, using a click reaction, for SPPS using Boc and Fmoc strategies (Figure 13b).162,163 These analogues permitted the generation of pan-antiphosphohistidine antibodies164 as well as variants that selectively recognize phosphohistidine in histone peptides.162 Thus, chemical and biochemical tools to study histone phosphorylation at basic residues are coming of age, enabling long-awaited investigations into the biochemistry of these intriguing PTMs.
3.4.2. Effects of Ser/Thr Phosphorylation in Protein–Protein Interactions
The close proximity of Ser/Thr residues to the major sites of histone lysine methylation on histone H3 (Thr3,Lys4; Lys9,Ser10; Lys27,Ser28) led Fischle, Wang, and Allis to propose that phosphorylation can switch the function of adjacent methylation marks.165 Experiments using site-specifically phosphorylated histone peptides were able to directly confirm this hypothesis. The binding of the CD of HP1 to H3K9me3 is abolished in the presence of a phosphorylation mark on the neighboring H3S10 (Figure 14a).166,167 Phosphorylation of H3S10 occurs during mitosis, and serves to evict HP1. In addition, H3S10ph precludes methylation of H3K9 by the heterochromatin-specific methyltransferase Suv39h1.70
Figure 14.
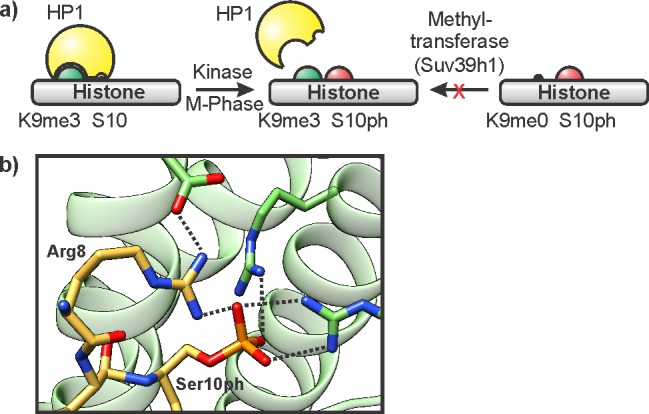
Biochemical readout of histone phosphorylation. (a) Illustration of a meLys/pSer switch. Phosphorylation at H3S10 ejects the K9me3 binding protein HP1, and prevents K9 methylation by the methyltransferase Suv39h1. (b) Structure of 14-3-3γ (green, pdb code: 2c1j) in complex with an H3 peptide containing S10ph and K9ac (yellow). Hydrogen bonds are indicated by dotted lines.
Histone serine phosphorylation can also be recognized by dedicated reader modules. To isolate binders of pSer in the context of the N-terminal tail of histone H3, Mahadevan and co-workers affinity purified cell lysate using an immobilized synthetic peptide containing H3S10ph and acetyl marks at Lys9 and Lys14.168 Using mass spectrometry, they identified a member of the 14-3-3 family,169 a helical pSer/pThr binding motif.168 Structural studies revealed that the phosphoryl group was accommodated in a cationic binding pocket featuring two arginine residues from 14-3-3 that form salt bridges with the ligand (Figure 14b). In addition, Arg8 on the histone peptide was sandwiched between pSer and a glutamate residue, thereby contributing to substrate specificity. Consistent with this binding mode, 14-3-3 also binds to H3S28ph with Arg26 at the −2 position.
Synthetic phosphopeptides also aided in illuminating the biochemistry of histone tyrosine phosphorylation. For example, the Drosophila transcription regulator Eyes Absent (EYA) was determined to be a histone tyrosine phosphatase that was able to dephosphorylate peptides of the histone variant H2A.X containing pTyr142 but not pSer139.150,151 Currently, no specific binding module for histone tyrosine phosphorylation is known, and the positions of Tyr residues in histones (only two of 15 histone Tyr residues are surface-exposed)152,170 suggest that pTyr may be able to exert its functions by directly modulating nucleosome structure and DNA access.
3.5. Glycosylation
Glycosylation has important implications for protein structure and function.171 Among the myriad of biologically pivotal carbohydrate modifications, attachment of β-N-acetylglucosamine (GlcNac) to Ser and Thr residues is most germane to histone biochemistry.172 Using lectins173 (carbohydrate-binding proteins), GlcNac-specific antibodies,173 or metabolic labeling with azide-modified GlcNac172 (and subsequent derivatization with a biotinylated alkyne moiety) to enrich GlcNac-ylated proteins, all core histones have been shown by mass spectrometry to carry this PTM. Biochemically, GlcNac-ylation of histone H2B at Ser112 promotes the ubiquitination of the proximal Lys120, and is associated with transcription activation.174 To test the effect of the GlcNAc modification in vitro, the ubiquitylation of nucleosomes by the E3 ligase BRE1A and its associated complex members was studied in the presence of H2B peptides. GlcNAc modified H2B peptides inhibited the reaction, while unmodified congeners or free GlcNAc-ylated serine did not. These results suggest that the ligase binds strongly to site-specifically glycosylated H2B.
The study of histone glycosylation is still in its infancy, but advanced methods to study protein glycosylation may be borrowed from other fields of research,175−177 and highly complex glycopeptides and glycoproteins can be synthesized.178 These tools might provide a means to answer the remaining biochemical questions about how glycosylation intersects with chromatin biology.
3.6. ADP-Ribosylation
Histones are subject to mono- and poly-ADP-ribosylation (MAR and PAR, respectively) involving many different side-chains, including lysine, arginine, asparagine, and glutamate.179,180 These modifications are associated with a plethora of important biological functions,181,182 yet the mechanistic contributions of individual ADP-ribosylation marks are difficult to dissect due to a dearth of (bio)chemical tools to study this diverse class of PTMs.183 Nevertheless, recent progress has provided strategies to incorporate ADP-ribosylated building blocks and analogues into peptides. Orthogonally protected ribose conjugates to Asn and Gln have been synthesized that allow selective phosphorylation of the 5′-OH group, followed by coupling with an activated AMP building block during Fmoc-SPPS (Figure 15a).184 In this way, a heptapeptide corresponding to the N-terminus of H2B containing an analogue of mono-ADP-ribosylated Glu has been created.
Figure 15.

Synthesis of mono-ADP-ribosylated peptides. (a) On-resin phosphorylation and AMP conjugation of an orthogonally protected ribosyl moiety. (b) Chemoselective ADP-ribose (inset) ligation to aminoxy-functionalized peptides. (c) ADP-ribose conjugates of N-methyl aminoxy-functionalized peptides retain the ribo-furanosyl-form. AMP = adenosine monophosphate, ADP = adenosine diphosphate.
Stable analogues of mono-ADP-ribosylated Glu residues can be generated using a chemoselective ligation approach.185 Peptides encompassing residues 1–19 of histone H2B, functionalized with a nucleophilic aminoxy group at position 2, form oximes with ADP-ribose at pH 4.5 (Figure 15b). This reaction is selective because lysine and arginine residues are protonated under these conditions. While the use of a secondary alkoxyamine was beneficial for retaining the ADP-ribose conjugate in the furanose form as opposed to an open configuration (Figure 15c), the yield of the ligation was poor.185 ADP-ribosylated proteins specifically interact with macrodomain-containing proteins.183 The histone variant macroH2A is the founding member of this family.186 Indeed, chemically ADP-ribosylated H2B(1–19) interacted with macroH2A,185 suggesting a role for this modification in regulating chromatin structure.181 Conceivably, the synthetic advances discussed above will enable the generation of antibodies recognizing mono-ADP-ribosylated proteins, and will thus provide a much needed tool to study ADP-ribosylation.183
3.7. Ubiquitylation
Histone ubiquitylation represents a particularly intriguing PTM given that the size of the modification (76 amino acids) rivals the size of the histone substrate. In contrast to polyubiquitylation, which commonly serves to flag proteins for degradation, the monoubiquitylation signals observed on H2A and H2B are associated with regulation of gene expression.14 Detailed evaluation of the genomic distribution of H2B modified with ubiquitin at Lys120 (H2B-K120ub) was enabled by an antibody that specifically recognizes this species.187 As described by Minsky et al.,187 a branched peptide encompassing residues 116–124 of H2B and the C-terminal four residues of ubiquitin, conjugated to H2B-K120 via an isopeptide bond, served as a surrogate for H2B-ubiquitin in the immunization process. The authors subsequently performed ChIP assays on human cell lines with this antibody and found that H2B ubiquitination occurs in the transcribed regions of highly expressed genes. Beyond antibody preparation, fully understanding the diversity of direct biochemical and biophysical consequences of attaching ubiquitin and related proteins to histones required the development of synthetic strategies for site-specific attachment of the complete ubiquitin protein to histones. A key step toward this goal involved the synthesis of peptide-ubiquitin conjugates.188 As this process hinges upon protein ligation techniques, we defer its detailed description to section 4.3.5.
3.8. A Growing List of Histone PTMs
Novel histone PTMs continue to be discovered. Highly sensitive mass spectrometry has revealed, for instance, that lysine residues can be modified with a diverse set of acyl groups.189 The prime example in this category is lysine crotonylation (Figure 16a), a mark widely distributed through active chromatin regions.189 The presence of this PTM was authenticated by synthesis; the chromatographic and mass spectrometric properties of cell-derived and synthetic histone peptides were identical. Antibodies that recognize this mark are already commercially available. Still, little is known about nuclear factors that attach, remove, or specifically bind this modification, although HDAC3, as well as members of the sirtuin family, have been found to possess measurable but small decrotonylase activity.189−191
Figure 16.
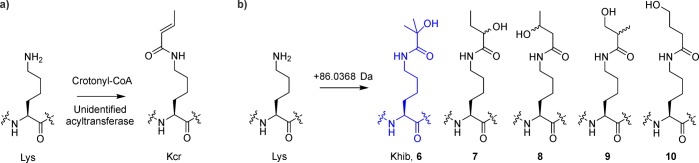
Newly discovered lysine acylation marks. (a) Lysine crotonylation. (b) Lysine hydroxyisobutyrylation (6), and control isomers (7–10).
The latest addition to the histone lysine acylation roster is 2-hydroxyisobutyrylation (Khib, 6).192 Zhao and co-workers detected a mass shift of +86.0354 in tryptic digests of histones from mouse testis cells, corresponding to the addition of a C4H7O2 fragment. Several isoforms of this composition are plausible (Figure 16b, 6–10). Therefore, five peptides encompassing residues 68–78 of H4 were synthesized with different lysine modifications. Among these, the variant where lysine 77 was acylated with 2-hydroxyisobutyric acid was indistinguishable from the biological sample in LC/MS/MS assays, thus confirming the identity of the novel PTM. The genomic localization of H4K8hib was found to differ from the distribution of H4K8ac, suggesting a distinct biochemical function for these two marks.
Recently, Tessarz et al. described that the amide side-chain of Gln104 in human histone H2A can also be selectively methylated in vivo.193 This modification abrogates binding of H2A to the histone chaperone FACT (facilitates chromatin transcription), as evidenced by a peptide-based pull-down in vitro. Glutamine methylation was only detected in the nucleolus, where it regulates the expression of the 35S rDNA gene, and hence represents the first histone mark that is associated with only one specific polymerase.
With the ever increasing sensitivity of mass spectrometers, as well as advances in sample workup, more histone PTMs will likely appear in the near future.194 These new marks contribute to the immense complexity of biological signaling, and challenge the analytical creativity of protein biochemists, not to mention the synthetic skills of peptide chemists for subsequent mechanistic investigations.
3.9. Proline Isomerization
Amino acids in proteins can occur in the cis and the trans conformation with respect to the backbone amide bond. While for most residues the equilibrium lies far on the side of the trans isomer, Pro residues populate a significant extent of the cis conformer (Figure 17a).195 The position of the equilibrium can be fine-tuned by tertiary interactions that stabilize either state. Interconversion between the distinct forms occurs spontaneously, albeit slowly on the time scale of minutes. Dedicated proline isomerases such as the yeast enzyme Fpr4 catalyze this process, and several Pro residues on histone tails have been identified as substrates.196 Kouzarides and co-workers proposed that the H3K36-specific methyltransferase Set2 is only active when the neighboring Pro38 is in the trans conformation, and that the H3K36-specific demethylase, JMJD2A, prefers the cis isomer.197 Reciprocally, K36 trimethylation inhibits the activity of Fpr4, leading to a model where genes can be activated quickly through the combined action of Fpr4 and Set2 (Figure 17b).197 The slow isomerization of the methylated trans conformer to the cis state followed by JMJD2A-mediated demethylation could act to set a timer for the duration of the active state.
Figure 17.
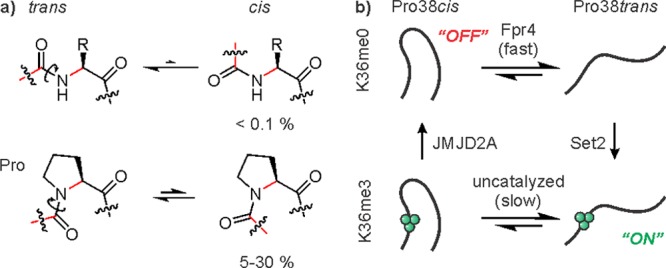
Proline isomerization. (a) Amino acid cis/trans equilibria. (b) Proposed switches through coupled Pro isomerization and Lys methylation to activate associated genes. Lys36 methyl marks are indicated as green spheres.
Chemical tools for the synthesis of peptides and proteins containing proline analogues with distinct conformational preferences have found application in the study of ion channels198 and protein aggregation,199 among others.200 They might also lend themselves to directly probe the structural and functional consequences of this noncovalent histone PTM.
3.10. Probing the Function of Cancer-Derived Histone Mutations
Genomic sequencing efforts have revealed that several cancers are associated with histone mutations.201−204 In particular, the mutation Lys27Met on histone H3 isoforms occurs in the majority of cases of a subtype of pediatric brain tumor (diffuse intrinsic pontine glioblastomas, DIPG). Lys 27 is the target site of the multisubunit methyltransferase, Polycomb Repressor Complex 2 (PRC2, Figure 18a). This molecular machine and its associated histone PTM, H3K27me3, play a central role in gene silencing, and thus are essential for cell differentiation and development of multicellular organisms.205 In mammalian cells, histone proteins are encoded on many synonymous genes. Consequently, it came as a surprise that cells carrying the K27M mutation on only one H3 gene, corresponding to a total of 3–18% of the total histone H3 protein pool,206 display strongly reduced H3K27me3 on all wild-type histones (Figure 18b).206−209 Similarly, the presence of the H3K27M mutant dramatically lowered Lys27 methylation in cell lines206,208,209 and in Drosophila.210
Figure 18.
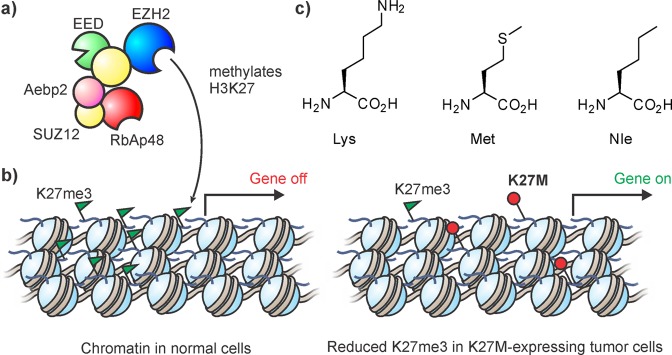
Cancer-derived H3K27M mutants inhibit PRC2 activity. (a) Molecular architecture of PRC2 according to Ciferri et al.218 (b) PRC2 inhibition by K27M causes aberrant gene expression. PRC2 serves to silence certain genes through its HMT activity (left). In K27M tumor cells (right), trimethylation at Lys27 is dramatically reduced, preventing gene repression. K27me3 marks are shown as green flags, K27M mutant as a red circle. (c) Structure of Lys, Met, and Nle.
Recent collaborative efforts from the Allis and Muir groups provided unequivocal proof that these mutant histones directly inhibit PRC2.206,211 In vitro histone methyltransferase activity on recombinant unmodified nucleosome substrates was strongly reduced by a synthetic peptide bearing the K27M mutation.206 Substituting the thioether moiety in methionine with a methylene group in norleucine (Nle, Figure 18c) resulted in even more potent inhibition of PRC2. By contrast, peptides with polar and branched residues at position 27 were poor inhibitors.211 Peptide-based inhibitor studies revealed extensive contacts between the entire H3 tail and EZH2, the catalytic subunit of the complex (see also section 3.11.1). Intriguingly, many naturally occurring PTMs of the H3 tail drastically reduced inhibitor potency of K-to-M mutant peptides, illustrating that chromatin context influences the downstream effect of “oncohistones”.211 Lewis et al. also demonstrated that Lys to Met mutations inhibit many different HMTs (all sharing a common catalytic SET domain) in vitro and in vivo.206 Specific peptide inhibitors, derived from these initial observations, would be tremendously useful to understand the biochemistry of histone methyltransferases, and might find use in combatting diseases associated with hyperactive HMTs.
The recent discovery that Lys to Met histone mutations at H3K36 are also associated with pathologies212 underscores the importance of investigating the interactions of histone methyltransferases with their substrates and inhibitors. Simultaneously, these findings provide an enormous challenge to medicinal chemists and (chemical) biologists alike to devise novel strategies to inhibit the inhibition of pivotal nuclear factors, such as PRC2, by pathological histone mutants.
3.11. Cross-linkers
Synthetic peptides can be furnished with a broad range of invaluable probes, including cross-linkers. These are stable molecules that, upon activation with a chemical or physical stimulus, become extremely reactive and covalently attach themselves to diverse functional groups in spatial proximity.213,214 Cross-linking can be harnessed to capture ephemeral interactions, and thus it lends itself to the study of transient protein–protein contacts and detection of binding partners and surfaces in complex mixtures.
3.11.1. Analysis of PRC2 Regulation
PRC2 (see section 3.10) interacts with nucleosomes through several of its subunits, and many of these binding events are regulated by chromatin state.205 Cross-linking strategies have been exploited to aid in disentangling the PRC2 regulatory network, specifically by identifying to which subunit cancer-derived histone mutants bind, and how PRC2 detects the nucleosome density of genomic targets.
The methionine analogue photomethionine215 (Figure 19a) can be incorporated into peptides by Fmoc-based SPPS, and represents an excellent tool to study the binding site of H3K27M mutants. Upon irradiation with UV light, the diazirine moiety decomposes into N2 and a highly reactive carbene, immediately inserting into nearby bonds, including C–H bonds.213,214 An H3 peptide (residues 23–34) containing K27photoMet and a biotin tag cross-linked efficiently to EZH2, the catalytic subunit of PRC2, suggesting that histone mutants act as orthosteric active site-directed inhibitors (Figure 19b).206
Figure 19.
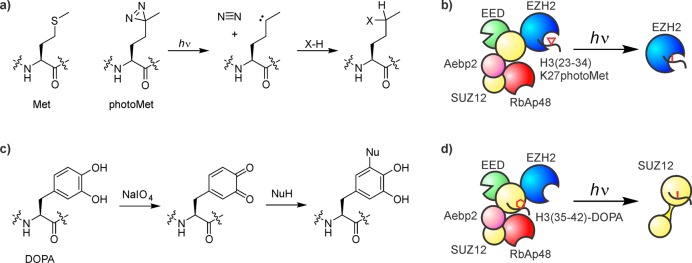
Cross-linking strategies to study PRC2 regulation. (a) Structure and photo-cross-linking mechanism of photomethionine. (b) H3(23–34)K27photoMet cross-links to the catalytic subunit EZH2. The diazirine cross-linker is shown as a red triangle, the covalent adduct as a red line. (c) Structure and oxidative cross-linking mechanism of DOPA. (d) H3(35–42) cross-links to SUZ12. The DOPA cross-linker is shown as a red hexagon, the covalent adduct as a red line.
Dense chromatin is methylated more efficiently by PRC2 than dispersed arrays.216 This stimulation is mediated by an octapeptide corresponding to residues 35–42 of H3. To determine which component of PRC2 senses local chromatin density, this peptide was synthesized with a DOPA217 residue and a biotin tag (Figure 19c). Treatment of the H3(35–42)-DOPA peptide, bound to PRC2, with periodate led to covalent cross-linking to SUZ12, the central scaffolding subunit218 of the complex (Figure 19d).216
3.11.2. Capture of Transient Interactions
Cross-linking turns weak or transient interactions into covalent ones. This feature is particularly useful to isolate binding partners from complex mixtures such as cell lysates. For example, ADP-ribosylated peptides (see section 3.6) were not able to pull down macroH2A doped into nuclear extracts, but furnishing the peptide with a benzoyl-phenylalanine (BPA) residue219 (Figure 20a) and performing the purification step after UV irradiation enabled trapping of this weak interaction.185
Figure 20.
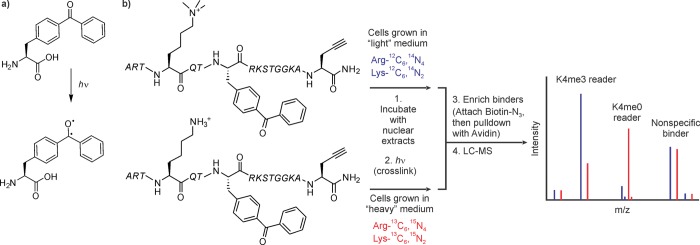
Photo-cross-linking strategies. (a) Structure and photoexcitation of p-benzoyl-phenylalanine. (b) Cross-linking-based workflow to identify proteins that are sensitive to the methylation state of H3K4.
Kapoor and co-workers used photo-cross-linking to identify PTM-specific histone binding proteins in an unbiased fashion. Initially, H3K4me3 peptides furnished with a BPA residue and an alkyne group were used to evaluate the cross-linking reaction in vitro and in vivo.220 As expected, the probe modified the PHD finger protein ING2, a known binding module for this PTM, but not HP1, which binds H3K9me3 (section 3.2.2). In follow-up studies, mass spectrometric analysis of cross-linked samples enabled proteome-wide analysis of PTM binders.221 For this work, the group synthesized a second version of their probe without the K4me3 mark and performed a SILAC experiment (Figure 20b, see also section 3.2.3). Cells grown in “light” media were lysed, incubated with the K4me3 probe, and subjected to UV irradiation. In parallel, the K4me0 probe was cross-linked to extracts from cells grown in “heavy” media. Subsequently, the two experiments were combined and reacted with biotin-N3 allowing for enrichment of cross-linked species using streptavidin beads. MS analysis provided a list of known K4me3 binders, along with a set of potentially novel readers of this mark. Similarly, the panel of proteins that prefer K4me0 included familiar and candidate interactors. Several of the newly discovered interactions were verified using ITC, demonstrating the validity of the approach.221 Extension of this methodology to H3 tails modified with K9me3, H3T3ph, and the doubly modified T3ph/K4me3 has been reported since.222
3.12. Combinatorial Approaches To Study Histone Biochemistry
Over 100 distinct histone PTMs are currently known.189 These marks seldom occur in isolation. Instead, many histone PTMs coassociate into so-called chromatin states,223−225 characterizing the biochemical environment of genomic loci. For instance, the activating signature H3K4me2/3 often manifests in combination with H3K9ac and H2B-K120ub.225 Thus, it is not surprising that many histone PTMs exert their full effect only in conjunction with other marks. Individual reader domains are sensitive to the presence of histone modifications close to their main target residue, and most chromatin associated proteins contain several histone binding domains. It is therefore important to interrogate the molecular consequences of histone modification in a combinatorial fashion.
Peptide libraries provide an ideal means to screen interactions between histone PTMs with nuclear proteins, in particular when synergism and antagonism of local PTM combinations on the same peptide ought to be explored.226 In this section, we will discuss various strategies to assemble histone peptide libraries, and highlight their key applications.
3.12.1. Histone Peptide Microarrays
Individually synthesized and purified peptides, each bearing a biotin handle and a unique PTM signature, can be printed onto avidin-coated glass slides to yield a densely covered microarray (Figure 21a). While the synthesis of such a collection is time and labor intensive, a typical synthesis scale provides enough material for hundreds of chips.227 The synthetic effort is rewarded by a dramatically increased throughput based on the simultaneous analysis of pairwise interactions between effectors and each member of the peptide library. Readout is most easily achieved by fluorescently labeled antibodies, and the use of epitope tagged proteins of interest facilitates this process (Figure 21a). Upon hybridization, bright spots are simply matched with the peptide identity through their position on the microchip. Peptide arrays containing tens to hundreds of peptides displaying varying histone PTMs at distinct residues have been utilized to screen the binding specificity of known and novel chromatin interacting domains.228−236
Figure 21.

Histone peptide microarrays. (a) Preparation of microarrays and protein binding assay. POI stands for protein of interest, AB for antibody. (b) Structure of the coupled TTD (light blue) and PHD (pale green) of UHRF1 (pdb code: 3ask). The H3 peptide trimethylated at residue 9 is depicted in yellow, the linker between the two modules in black. (c) HDAC assay using SAMDI. Xaa and Yaa denote any amino acid.
A case in point is the study by Matthews et al. on how RAG2, a protein essential to V(D)J recombination during immune cell maturation, engages chromatin.229 A 45-membered histone peptide array featuring different methyllysine, methylarginine, acetyllysine, and phosphothreonine marks identified the PHD finger of RAG2 as a K4me3-binding module. This interaction and its specificity were verified by classical pull-down approaches. Notably, mutations that cripple the aromatic cage of the RAG2 PHD finger caused a reduction in V(D)J recombination, and similar mutations occur in patients suffering from immunodeficiency.237
The same approach led to the characterization of ORC1, a component of the origin of replication complex (ORC).232 This protein contains a BAH domain (bromo-adjacent homology),238 which mediates selective binding to H4K20me2, as determined with a 82-peptide microarray. Again, the results of the screen were verified in vitro, in this case by ITC, and in vivo. Indeed, H4K20me2 binding by ORC1 is important for recruitment of ORC to designated genomic loci, and the loss of this interaction is linked to a growth retardation syndrome.239,240
Strahl and co-workers profiled several methyllysine binding domains with a peptide microarray containing 130 peptides with up to six simultaneous PTMs including lysine and arginine methylation, serine and threonine phosphorylation, and lysine acetylation.233 In most cases, the presence of a phosphoryl group proximal to the target methyllysine residue abolished binding. In contrast, the tandem tudor domain (TTD) of the E3 ubiquitin ligase UHRF1 tolerated a peptide epitope containing both H3K9me3 and S10ph. This feature enables UHRF1 to remain bound to H3K9me3 during mitosis when Aurora B-mediated S10 phosphorylation ejects many known K9me3 binders.166
A rescreen of the binding preference of the UHRF1 TTD coupled to its neighboring PHD finger suggested that the PHD, which recognized the unmodified N-terminus of H3,241 dominates the association with histone peptides.234 Variants with a mutated PHD finger unable to bind the H3 tail did not interact significantly with any peptide probe on the chip. A crystal structure of the coupled TTD-PHD domains demonstrates that the two modules associate and compactly bind to an H3 tail containing K9me3 (Figure 21b).242 Interestingly, the lipid phosphatidylinositol 5-phosphate can allosterically activate the TTD of UHRF for H3K9me3 binding, thus providing a link between lipid metabolism and chromatin architecture.243
Microarrays consisting of 250 biotinylated peptides encompassing all monoacetyllysine marks on the H3 and H4 tails, as well as di- and poly acetylated versions, have been used to profile commercial, site-specific acetyllysine antibodies.244 Surprisingly, all antibodies tested preferentially bound to polyacetylated peptides, suggesting that there is a need for improved acetyllysine detection reagents.
When peptides are immobilized on gold plates covered with a self-assembled monolayer, the resulting arrays can be used in laser desorption ionization mass spectrometry (SAMDI-MS).245,246 Gold surfaces are covered with alkane-thiolates, and subsequently functionalized with maleimide groups. Hexapeptides, centered around an acetylated lysine, were attached to the surface via C-terminal cysteine residues. Subsequent treatment with various HDACs, followed by SAMDI-MS, enabled the substrate scope of these eraser enzymes to be profiled (Figure 21c).246
3.12.2. SPOT Synthesis of Peptide Arrays
Direct synthesis of peptides on cellulose paper (so-called SPOT synthesis) provides a convenient route to spatially addressable microarrays.247 This strategy parallelizes the library synthesis and bypasses labor intensive purification steps associated with the immobilization strategies discussed above, but, as a consequence, limits the length (6–18 residues are common)248 and complexity of peptide targets.247 To commence peptide synthesis, spots on cellulose membranes are first esterified with Fmoc-β-Ala-OH or a similar protected amine. The membrane is then capped with acetic anhydride. Subsequent Fmoc deprotection is followed by iterative, parallelized peptide synthesis, where each spot is reacted with a desired Fmoc-protected amino acid separately by dispensing only enough reagents to cover the spot (Figure 22). Because cellulose membranes are resilient to short exposures in TFA, side-chain deprotection can be achieved while retaining peptide attachment and the integrity of the support. Alternatively, peptides can be cleaved from the membrane by base treatment for analytical purposes, or if soluble peptides are required. Binding assays are performed in analogy to dot-blot detection. SPOT arrays are incubated with epitope tagged proteins of interest, which are subsequently detected with primary and, if required, secondary antibodies conjugated to horseradish peroxidase or alkaline phosphatase. Peptides, identified by their position on the membrane, targeted by the protein of interest are visualized using bioluminescent or chromogenic substrates. In this way, SPOT arrays containing hundreds of modified peptides have been used to profile the specificity of a range of sequence specific methyllysine reader domains, including the CDs of HP1β249 and HP1γ,250 the PHD finger of the chromatin remodeler ATRX,251 and the PWWP domain of a DNA methyltransferase,252 among others.249,250 In addition, the diversity of sequences that can be synthesized on spot arrays is ideally suited to assess the promiscuous binding of readers, as exemplified by the interaction of MBT repeats of L3MBTL1 with dimethyllysine residues.250
Figure 22.
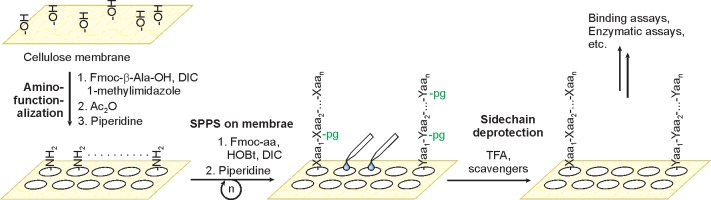
SPOT synthesis of histone peptide arrays on cellulose membranes. Xaa and Yaa denote any amino acid, pg stands for side chain protecting group.
Of particular interest is a recent comprehensive analysis of human bromodomains.253 SPOT arrays containing all possible acetylation sites on human histones were used to profile 33 individual BD family members, together spanning thousands of pairwise interactions. In general, BD binding to acetylated peptides was weak. Some binding modules displayed remarkable specificity (e.g., the BDs of TRIM28 and MLL), while others bound most acetylated peptides (e.g., the BDs of SP140 and PCAF). SPOT arrays with numerous combinations of acetyl marks were synthesized to evaluate cooperative binding. Several BDs, including those of the transcriptional coactivator BRD4, were shown to strongly prefer multiply acetylated histone peptides. A fraction of the hits were assayed by ITC using soluble peptides, and almost 30 crystal structures of BDs were determined in this landmark study.
SPOT arrays are compatible with a range of different detection strategies, and are well-suited for enzymatic assays. The substrate specificity of the histone methyltransferase G9a was evaluated using a SPOT membrane encompassing residues 1–20 of H3 with numerous mutations and PTMs.254 G9a activity was determined by fluorography upon incubation with 3H-S-adenosylmethionine. A minimal recognition motif includes an unmethylated Arg in the −1 position, with moderate selectivity at the −2,+1,+2 positions, indicating that G9a is quite promiscuous. Indeed, several nonhistone targets were found to be methylated in vitro, and the products recognized by HP1β. These results suggest that G9a exerts its effects through a combination of histone and nonhistone pathways. Similar analyses were carried out for the methyltransferases Dim-5,255 NSD1,256 and SET7/9257 to determine the substrate specificities of these important enzymes.
A 384-membered SPOT library of 19-mers was used to probe a variety of different PTM-recognizing antibodies.258 Overall, many antibodies displayed the desired specificity, but noncognate binding to the same PTM at different sites was certainly an issue. False negatives due to epitope occlusion by additional modifications surrounding the targeted residues were also frequently observed.258,259 Thus, the thorough profiling of antibody specificity using a range of different peptide approaches has provided valuable insight into the applicability of some of the most used reagents in chromatin biochemistry. While many antibodies display the proclaimed specificity, some suffer from severe cross-reactivity, and most exhibit additional preferences for the modification state of adjacent residues.
Using a particularly comprehensive array (746 peptides), Denu, Garcia, and co-workers evaluated histone-PTM reader domains as specific reagents to isolate nucleosomes from particular chromatin states.260 Consistent with previous observations, the authors found that the ADD domain (a type of zinc finger) of ATRX binds with high specificity to H3K9me3 in the context of unmodified H3K4. In contrast, antibodies raised against H3K9me3 displayed poor selectivity for their cognate marks. Chromatin affinity purifications with the ADD domain led to the enrichment of histones that were hypermethylated at H3K9 and H4K20, and hypomethylated at H3K4 and H3K79, as judged by mass spectrometry. These results demonstrate that reader domains can serve as valuable alternatives to antibodies to interrogate the composition and distribution of chromatin states.
3.12.3. One Bead-One Compound Peptide Libraries
Libraries containing thousands of peptides are produced most easily by split-pool synthesis.261,262 In this approach, peptides are synthesized using Fmoc chemistry on beads that are resilient to TFA cleavage. Additionally, for every coupling step, resin beads are split into different vials, each containing a unique activated amino acid. Upon completion of the reaction, beads are pooled again, and randomly redistributed for subsequent couplings. Finally, peptides are deprotected with TFA containing scavengers. In this way, each bead will carry only one peptide sequence, although several beads may contain the same peptide. Identification of peptides upon isolation of individual beads is achieved by microsequencing or by mass spectrometry, facilitated by performing partial capping steps at strategic sites, thus generating a mass ladder.263 Cyanogen bromide can be used to cleave peptides from the resin prior to MS analysis when a C-terminal methionine residue is included in the sequence.264
One bead-one compound libraries are particularly useful when a large number of closely related peptides are desirable, as is the case when synergies between PTMs on a histone tail are queried. Denu and co-workers have prepared peptide collections encompassing 800 and 5000 members with combinations of known histone PTMs on the H4 (ref (265)) and H3 (refs (264,266)) tail, respectively (Figure 23). A colorimetric on-bead western assay was used to evaluate the binding profile of a range of GST-tagged domains to the H3 library (residues 1–10) in an unbiased manner. The expected preferences for methylation states at Lys4 of the interrogated PHD domains were observed along with various degrees of sensitivity to proximal PTMs.264 Switch-like behavior occurred in the case of phosphorylation at Thr3 in that this modification abrogated binding to surrounding residues by all proteins tested. Regulation of binding by arginine methylation followed a rheostat model in some cases. For example, ING2 binding was gradually decreased by each additional methyl group at Arg2. Some domains (the PHD fingers of RAG2, BHC80, AIRE) were ejected by Thr6ph, while the double tudor domain (DTD) of the demethylase JMJD2A was insensitive to this mark. The potential for reader-specific responses to Thr6ph prompted a search for this modification in vivo. Indeed, MS analysis detected this mark upon phosphopeptide enrichment by affinity chromatography.264
Figure 23.

One bead-one compound libraries of modified H3 and H4 tails.
3.12.4. Toward Nucleic Acid Encoded Histone Peptide Libraries
Suga and co-workers performed in vitro translation of RNA sequences coding for histone peptides with an expanded genetic code.267 Redundant codons were reassigned to be interpreted by tRNA molecules acylated with modified lysine building blocks (Figure 24a). Ironically, this strategy entails the incorporation of desired post-translational modifications prior to ribosomal translation on the residue level. Peptides containing Kme1, Kme2, Kme3, and Kac residues at positions 4, 9, 27, and 36 on the H3 tail were synthesized, although the yield for monomethylated products was poor. Up to four PTMs were incorporated simultaneously, allowing synergies between different marks to be explored. As expected, HP1 bound specifically to peptides containing H3K9me3, with a slight increase in affinity when K27 is methylated as well.
Figure 24.
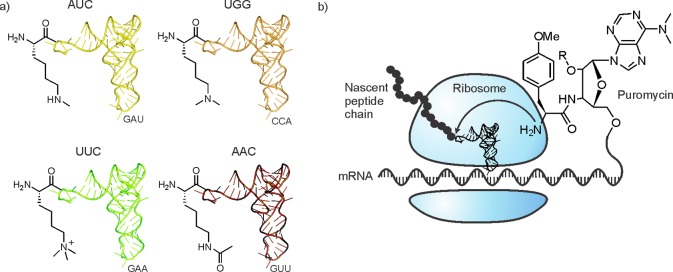
In vitro translation of histone peptides. (a) Reassigned codons with corresponding amino-acyl-tRNAs. (b) Schematic representation of mRNA display with the puromycin-mediated attachment of the mRNA to the growing peptide chain.
Notably, peptides translated in vitro can be tethered to their coding mRNA sequence, for example, through the use of puromycin-tagged mRNAs (Figure 24b).268 The link between the translated peptide or protein to its mRNA enables decoding of molecules that exhibit a given phenotype, such as binding of the peptide to a receptor, simply by sequencing the RNA portion. This strategy, combined with an expanded genetic code, has been applied by the Suga laboratory to select macrocyclic peptide inhibitors for diverse targets.269 Thus, mRNA display and related technologies270 harbor great potential for the synthesis of large encoded histone peptide libraries.
3.13. Beyond Peptides
Chemical synthesis enables routine preparation of histone peptides carrying most known PTMs and, if desired, a wealth of probes, including cross-linkers, affinity reagents, and spectroscopic handles. Such peptides have proven to be indispensable in chromatin research because many central molecular transactions in chromatin biochemistry occur at the unstructured histone tails. Specifically, the biochemical rules and the physical chemical driving forces for how histone PTMs mediate interactions with nuclear proteins have been elucidated using the peptide chemistry toolbox. Peptides are thus a first resort utensil to validate and characterize biological discoveries. Despite their utility, peptides are, however, insufficient for certain protein interaction studies and functional assays. For example, nucleosomes are required to investigate processes that involve multivalency through different histones or depend on the presence of DNA. Nucleosomal DNA can provide electrostatic interactions to strengthen otherwise weak binding events, or even serve as the substrate per se, as is the case in transcription or remodeling assays, among others. Access to modified nucleosomes requires corresponding access to modified histones, all of which are over 100 amino acids in length. Because routine SPPS approaches are limited to approximately 50 residues, and thus fall short of attaining entire histone proteins, continuative and complementary technologies are required to increase the level of complexity of chromatin-related phenomena that can be scrutinized in vitro.
4. Chemical Approaches To Manufacture Histones and Chromatin
In this section, we review modern approaches to synthesize site-specifically modified histones and chemically defined “designer” chromatin templates, and their application in investigating chromatin biochemistry. Robust protocols for the assembly of nucleosomes and chromatin templates have been developed for structural, biochemical, and biophysical studies.271 Besides isolation from eukaryotes, histones can be produced recombinantly in E. coli as inclusion bodies.272,273 Recombinant proteins are devoid of PTMs, and thus represent clean slates for in vitro studies. Their small size and positive charge permit facile purification of recombinant histones through size exclusion, ion exchange, and reverse phase chromatography. Stoichiometric amounts of each of the core histones are then refolded into octamers and supplied with DNA sequences with a high propensity to bend, that is, wrap around histone octamers. Nucleosomes assembled in this way have been crystallized, and their structure solved to <2 Å by the Richmond group.274,275 The histone octamer forms a disk with a cationic lateral surface, which is enveloped by DNA (Figure 25a). The histone tails protrude from this compact structure, with residues 16–23 of H4 docking into an acidic patch on the H2A/H2B interface of an adjacent particle in the crystal lattice (Figure 25b).274 These contacts, initially observed in crystal packing, are believed to play a central role in the folding of the chromatin fiber.276,277 Homogeneous nucleosome arrays can be assembled from repeats of a strong nucleosome positioning sequence such as the “Widom 601” (ref (278)) or the 5S rDNA sequence.279−281 Structural studies with tetranucleosome arrays by X-ray crystallography282 and dodecanucleosome arrays by cryo-EM283 reveal details on the packing interactions that govern chromatin folding. In both structures, chromatin adopts a two-start helix with close interactions between i, i+2 nucleosomes (Figure 25c and d). However, alternative packing models for the chromatin fiber have been supported experimentally,284,285 and the existence of highly ordered fibers in vivo is still contentious.286 Several recent accounts cover this controversy in detail.287−289
Figure 25.

Nucleosome and chromatin architecture. (a) Electrostatic surface rendering of the mononucleosome (pdb code: 1kx5). Cationic areas are colored in blue, anionic patches in red, the DNA backbone is drawn in gray. (b) Interaction of the acidic patch on H2A/H2B (red surface) with the H4 tail of a neighboring particle (yellow). (c) Crystal structure of a tetranucleosome array (pdb code: 1zbb). (d) Dodecanucleosome arrays fold into a two-start helix as suggested by a cryo-EM structural model (EMD-2600).
To understand how specific histone PTMs alter chromatin behavior, access to homogeneously modified chromatin templates is crucial. Initial studies relied on enzymatic preparation of modified histones and, by extension, chromatin carrying the desired marks. This approach provided invaluable insight into chromatin signaling, but suffers from lack of specificity. Many histone-modifying enzymes target multiple sites on histones as well as other nuclear proteins, thereby rendering it difficult to produce unique PTMs without unintentionally affecting alternate aspects of the biochemical pathway in question. In addition, the low activity of many isolated histone mark writers precludes complete modification. Fortunately, chemical biology has provided chromatin biochemists with a rich toolbox geared toward the assembly of site-specifically modified “designer” chromatin. Below we discuss the contributions of protein chemistry to our understanding of chromatin structure and function, and how PTMs modulate these properties from a biophysical and biochemical point of view.
4.1. Site-Specific Modifications of Histones and Chromatin
Most histone proteins are devoid of cysteine residues. Only H3 contains one completely conserved cysteine residue, which has been shown to be inessential in yeast,290 and is frequently mutated to alanine in biochemical and biophysical studies. This feature greatly facilitates site-directed modification of histones and nucleosomes upon genetically incorporating Cys residues at desired locations due to the unique reactivity of the cysteine sulfhydryl group toward a diverse repertoire of electrophilic probes.291
4.1.1. Site-Specific Protein Cross-linking of Chromatin
Dorigo et al. exploited non-native cysteine residues to investigate predicted contacts between the H4 tail and the acidic patch in chromatin fibers.292 Upon compaction with MgCl2, 12-mer nucleosome arrays containing H2A-E64C and H4 V21C were cross-linked by treatment with a mixture of oxidized and reduced glutathione (Figure 26a). The resulting disulfide bond between H2A and H4 stabilized the compact state of the array, and additionally revealed the fold of the chromatin fiber. Limited digestion with a nonspecific nuclease, followed by native gel electrophoresis, yielded cross-linked arrays containing maximally six nucleosomes. This result confirms a two-start helix, reinforced by contacts between i, i+2 nucleosomes, but lacking i, i+1 interactions (Figure 25c and d). Similar interactions also occur to stabilize interstrand association, as seen when arrays containing exclusively H2A-E64C are mixed with arrays containing only H4 V21C.293
Figure 26.
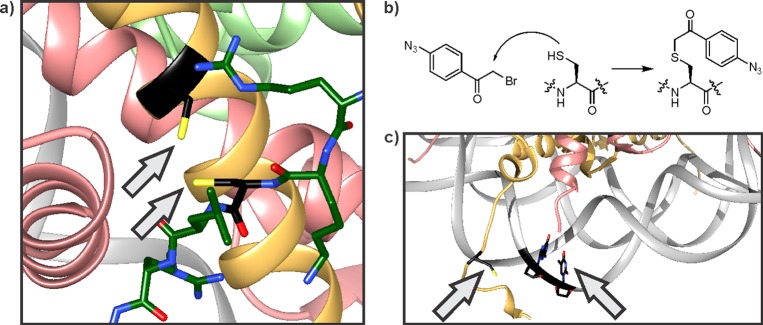
Nucleosome cross-linking. (a) Disulfide cross-linking from the H4 tail (green) to the acidic patch of H2A/H2B with engineered cysteines (black). (b) Structure of 4-azidophenacyl bromide (APB) and its reaction with cysteine. (c) Photo-cross-linking reveals the position of the H2A N-terminal tail. APB is attached to an engineered cysteine within the H2A tail (black), the cross-linking site on DNA is shown in black.
Chemical cross-linkers installed at specific histone sites complemented earlier approaches294,295 that relied on nonspecific protein–DNA cross-linking to study nucleosome and chromatin architecture.296 Cysteine residues, introduced, for example, at positions 2 or 12 of the H2A tail, can be conveniently alkylated with 4-azidophenacyl bromide (APB, Figure 26b).297 Upon UV irradiation, the APB moiety decomposes to yield a nitrene that covalently inserts itself at two specific DNA sites, hence suggesting a defined arrangement for the H2A N-terminal tail with respect to nucleosomal DNA (Figure 26c). A similar strategy has been used to map intra- and internucleosomal contacts of other histone tails,298,299 the position of linker histone H1 on a nucleosome,300 as well as the mechanism of chromatin remodelers.301
4.1.2. Footprinting Analysis of Nucleosome Positioning
Information about the register in which nucleosomal DNA wraps around the histone octamer can be obtained from site-directed footprinting studies. By tethering an Fe(II)–EDTA complex (via disulfide 7) to a cysteine residue mutated into position 47 of H4, Richmond and co-workers were able to target hydroxyl radicals generated by the Fenton reaction302,303 to specific DNA sites close to the dyad axis (Figure 27a and b).304 The resulting strand cleavage was then used to map H3/H4 tetramer and nucleosome position at basepair resolution.
Figure 27.
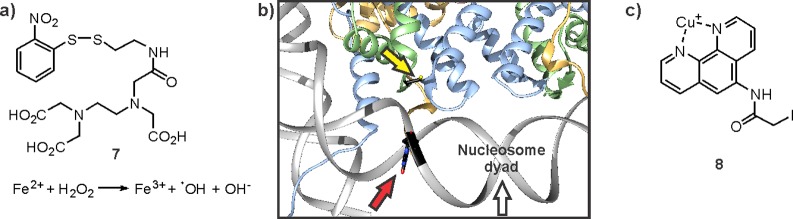
Site-directed footprinting to map nucleosome positioning. (a) Activated disulfide reagent (7) to attach an EDTA derivative to cysteine residues (top) and hydroxyl radical generation by the Fenton reaction employing Fe(II) (bottom). (b) Model of the preferred cleavage site (red) upon introducing a sensitizer at H4S47C (yellow). The nucleosome dyad is indicated with a white arrow. (c) Structure of N-(1,10-phenanthroline-5-yl)iodoacetamide (8) in complex with Cu(I).
Widom and co-workers have used a related approach to accurately map nucleosome positioning in yeast.305 A copper chelator, N-(1,10-phenanthroline-5-yl)iodoacetamide (8), was used to modify the histone mutant H4S47C (Figure 27c), enabling Cu(I)- and H2O2-dependent strand cleavage in permeabilized cells. Next generation sequencing of the resulting fragments yielded a portrait of nucleosome architecture in cells at unprecedented resolution. This map provided detailed insight into DNA sequence patterns that govern histone positioning rules, and how distinct regions within nucleosomes impact the transactions of DNA with nuclear factors such as RNA polymerase.
4.1.3. Cysteine Labeling with Biophysical Probes
Spectroscopic probes have played a central role in characterizing protein structure and function, and the ability to introduce labels into DNA or by cysteine conjugation into histones makes nucleosomes attractive targets for diverse types of spectroscopy. Biophysical studies on nucleosomes have been comprehensively reviewed recently,276,306 and we will here only provide a glimpse into the types of probes that are commonly attached to histones.
Fluorophores, available in all forms and colors as cysteine reactive dyes, provide a handle to study nucleosome stability and dynamics. FRET measurements were performed with donor-labeled DNA and acceptor-labeled histone octamers, for example, on H2AK119C or H3 V35C, both close to the DNA entry/exit site (Figure 28a and b).307 A decrease in FRET efficiency, corresponding to an unwrapping of DNA, was found under physiological ionic strength, suggesting that nucleosomes breathe to facilitate access of trans acting factors. Luger and co-workers used FRET pairs strategically installed at H2B-T112C and on the histone chaperone, NAP1, to dissect the interaction of this factor with its substrates.308,309 In addition, this assay could be harnessed to measure nucleosome stability through a coupled equilibrium cycle.310 FRET-based assays of nucleosome properties have been adapted to the single-molecule level, reviewed in refs (311,312).
Figure 28.

Spectroscopic characterization of nucleosomes and histones. (a) FRET assay to investigate DNA breathing. DNA (gray) is labeled with Cy3 (green star), H3 V35C (black), with Cy5 (red star). DNA unwrapping increases the distance between the fluorophores, leading to a loss in FRET signal. (b) Structure of cysteine-reactive Cy5-maleimide. (c) Structure of the MTSL spin label. (d) Distance measurement with PELDOR between spin labels (arrow) installed at H3Q125C. H3 is drawn in blue, H4 in green. (e) Asymmetric positioning of H1 (blue) on the nucleosome core particle. A spin label (arrow) placed at H3K37C perturbs NMR signals in its vicinity (dashed sphere).
Zhang et al. probed the interactions of the histone chaperones RbAp48 and ASF1 with H3–H4 complexes by EPR spectroscopy.313 MTSL ((1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl)-methanethiosulfonate, 9) spin labels were installed at various positions within the histones to monitor structural changes in the H3/H4 tetramer through pulsed electron–electron double-resonance (PELDOR)314 spectroscopy (Figure 28c and d).313,315 This technique can measure distances ranging from 20 to 80 Å,316 and is therefore ideally suited to probe histone assemblies. Association with RbAp48 disrupted the H3–H3 interaction (probed through labeling at H3Q125C), and changed H3–H4 distance distributions, demonstrating that this chaperone binds an H3–H4 dimer rather than a tetramer, the prominent oligomerization state in solution, and causes major structural rearrangements of the H3–H4 folds.313
Introduction of paramagnetic probes into proteins also facilitates characterization of protein–protein interactions by NMR spectroscopy.317 To investigate how the globular domain of linker histone H1 engages the nucleosome core, Bai and co-workers conjugated MTSL or Mn2+–EDTA complexes to cysteine residues at the periphery of the H1 globular domain,318 or close to the dyad axis on the nucleosome (H2A-T119C and H3K37C).319 By identifying NMR signals that are perturbed by the spin labels, the authors were able to triangulate the position of H1 on the nucleosome, and found that the complex is asymmetric; that is, H1 binds DNA on only one of its exit sites (Figure 28e).
Thus, site-specific conjugation of probes to cysteine residues strategically engineered into histones has enabled characterization of the structure, stability, and dynamics of nucleosomes. Ever more sophisticated spectroscopic methods facilitate analysis of increasingly large chromatin templates, interactions with bigger complexes, and more detailed aspects of the properties of chromatin, even at the single molecule level.
4.1.4. Installation of PTM Mimics
Inspired by the ease with which diverse probes can be conjugated to sulfhydryl groups,291,320 Simon et al. used a cysteine alkylation strategy to prepare analogues of methyllysine residues.321N-Methylated 2-haloethylamines represent convenient reagents for such transformations (Figure 29a). Alkylation of cysteine with the mono- and dimethyl species to yield the thialysine products, KCme1 and KCme2, respectively, proceeds through an aziridine intermediate that is readily formed from the corresponding chloride. In contrast, the electrophile needed to produce the trimethyl analogue, KCme3, lacks a lone pair on nitrogen and consequently the ability to form an aziridine, and so requires the stronger bromide leaving group. Methyllysine analogues installed in this way at positions H3K4, H3K9, H3K36, H3K79, and H4K20 were recognized by cognate antisera. In addition, K9Cme2 peptides bound to HP1α, and were further methylated by the HMT Suv39h1.321 Later studies confirmed that methyllysine-analogue containing nucleosomes could serve as substrates for other histone methyltransferases322 and demethylases.323 In general, nucleosomes constructed from alkylated histones behaved exactly like unmodified counterparts, auguring well for the use of these versatile reagents in chromatin biochemistry and biophysics,321 or even as quantification standards in ChIP experiments.324
Figure 29.

Structure and applications of methyllysine analogues. (a) Synthesis of methyllysine analogues by cysteine alkylation. (b) Comparison between methyllysine and a thioether analogue. (c and d) Subtle local changes in nucleosome structure upon histone lysine methylation. Modified nucleosomes are depicted in light colors (H3K79Cme2 in light blue, pdb code 3c1c in (c); H4K20Cme3 in pale green, pdb code 3c1b in (d)). Unmodified versions are shown in corresponding dark tones (pdb code: 1kx5). Yellow arrows indicate modified residues. (e) Model of the structure of a nucleosome containing H3K36me3 (yellow arrow) in complex with the PSIP1 PWWP domain (the backbones of neutral, basic, and acidic side chains are shown in white, blue, and red, respectively; pdb code: 3ZH1).
A methylene-to-sulfide substitution causes an increase in length, flexibility, and acidity of the side-chain (Figure 29b).321,325,326 To assess how these structural differences translate into energetic penalties for methyllysine analogue binding by reader modules, Fischle and co-workers investigated computational and experimental models.327 Experimentally determined ΔΔG values for association of Kme versus KCme substrates to binder modules were highly context dependent, and ranged between −0.2 kcal/mol (the PHD finger of ING1 preferentially binds to H3K4Cme3 over H3K4m3) to +1.2 kcal/mol (L3MBT preferentially binds to H4K20me1 over the analogue H4K20Cme1). In the majority of cases, methyllysine analogues recapitulate the function of the native PTM, although there exist isolated examples where this approach falls short.
The ease of obtaining large amounts of homogeneously modified histones through cysteine alkylation permitted the determination of crystal structures of mononucleosomes containing either H3K79Cme2 or H4K20Cme3 (Figure 29c and d).89 Both PTMs have known genomic associations, H3K79me2 is found in actively transcribed genes328 and H4K20me3 is enriched in heterochromatin,329 but little is known about how and if they modulate chromatin structure and function. Overall, the structures of the modified histones are superimposable with previously solved X-ray structures, but both modifications cause subtle local differences in conformation. Remarkably, nucleosome arrays prepared with H4K20Cme3 or H4K20Cme2 compacted much more readily than arrays containing unmodified H4 or a control protein with H4K20Cme0. This study demonstrates that methyllysine marks can directly affect the biophysical properties of chromatin and need not necessarily rely on protein effectors.
The interaction between HP1 and nucleosomes marked with H3K9me3 is central to the formation of large repressive chromatin domains.330 HP1 self-assembles into dimers and higher order oligomers through its C-terminal chromo-shadow domain (CSD), providing a polyvalent scaffold. Designer chromatin featuring H3 with the trimethyllysine analogue KCme3 at position 9 has enabled detailed studies of how HP1 recognizes its targets. For instance, biophysical studies based on NMR spectroscopy by Munari et al. revealed that only the CD (note, this module is distinct from the CSD) of HP1 stably contacts mononucleosomes bearing the H3K9Cme3 mark (Figure 30).331 Additionally, the highly charged hinge region that bridges the N-terminal CD and dimerization domain interacts nonspecifically with DNA and nucleosomes. Overall, the complex of HP1 with an H3K9Cme3-mononucleosome displays considerable flexibility, granted by the mobility of the H3 tail and the HP1 hinge.331,332 This plasticity presumably enables selection of diverse substrate arrangements. Notably, HP1 binding to heterochromatin domains is highly mobile in vivo, as evidenced by fluorescence recovery after photobleaching (FRAP) on the time scale of seconds.333 Accordingly, the binding of methylated mononucleosomes by HP1 is weak, and, in the case of the yeast homologue Swi6, rather unselective.334 Instead, Canzio et al. demonstrated that specificity for heterochromatin domains is imparted by cooperative engagement of HP1 oligomers with multiple H3KC9me3-modified nucleosomes in arrays.334 Intriguingly, polyvalent binding based on multiple weak interactions is a typical feature of systems that undergo phase transition to provide compartments with liquid-like properties,335 and might govern the formation of spatially distinct chromatin domains.
Figure 30.
Model for HP1 binding to heterochromatin domains. The chromodomain (CD, blue) binds to H3K9me3 (red flag), although this interaction is weak and dynamic. Stable dimerization through the chromoshadow domain (CSD, light blue and pale green) provides a polyvalent scaffold for chromatin binding. Plasticity is granted by the flexibility of the H3 tail and the linker between the HP1 domains (red arrows).
Site-specifically modified histones are crucial reagents for structural analysis of PTM binding modules that are sensitive to the nucleosomal context. The molecular recognition of methylated H3K36 is a case in point, due to its proximity to nucleosomal DNA. Peptides carrying K36me3 bind poorly to the PWWP domain (a methyllysine binding domain characterized by a Pro-Trp-Trp-Pro sequence) of the coactivator PSIP1.336 Binding studies with nucleosomes modified with K36Cme3 revealed that a basic surface on the PWWP domain reinforces K36me3 engagement.337 NMR analysis of K36Cme3-containing nucleosomes and the PSIP1 PWWP domain, enabled by strategic isotopic labeling of the methyl groups of isoleucine, leucine, and valine (methyl-TROSY),338 led to the construction of a model of this multivalent interaction (Figure 29e).337 Binding of H3K36me3 by the aromatic cage of the PWWP domain positions the basic surface of PSIP1 proximal to the nucleosomal DNA, thereby reinforcing binding approximately 10 000 fold.
Methyllysine analogues have also aided in characterizing histone modifying enzymes that require or prefer nucleosome substrates. For instance, the methylation state specificity of the methyltransferases NSD2 and SET2 was elucidated using mononucleosomes containing methyllysine analogues in place of Lys36.339 Histone methyltransferase assays demonstrated that NSD2 mediates mono- and dimethylation, whereas SET2 is capable of trimethylating H3K36. Similarly, the interplay between activating trimethyl marks and the repressive PRC2-dependent methylation at Lys27 was studied using site-specifically modified mononucleosomes.340 Templates carrying the methyllysine surrogates H3KC4me3 or H3KC36me3 were less efficiently methylated by PRC2, further illustrating the diversity of histone PTMs that this complex can sense.
Roeder and co-workers have harnessed modified chromatin templates to characterize the biochemistry of transcription. To this end, histone octamers can be loaded onto a supercoiled plasmid backbone with the help of the chaperone NAP1 and the remodeler ACF in the presence of ATP.281,341 When octamers carrying H3K4Cme3 are used, the impact of this mark, commonly associated with active genes, on transcription can be studied. H3K4Cme3 facilitated the recruitment of the preinitiator complex (an assembly of several general transcription factors that guide the positioning of RNA polymerase II to transcription start sites) to promoters,342,343 thereby increasing transcription.342 In addition, this analogue enhanced transcription activation by the coactivators p53 and p300.344
Strategies to prepare analogues of several other histone PTMs have also been reported. For example, cysteine can be modified with N-vinyl-acetamide in a radical promoted thiol–ene reaction (Figure 31a).345 This transformation results in a thia-analogue of Kac, KCac. Histones bearing H4KC16ac were recognized by an H4K16ac-specific antibody, and the PTM-analogue caused chromatin decompaction, which is a hallmark of this modification. An acetyllysine analogue featuring a methylthiocarbamoyl group, installed through alkylation of cysteine with an aziridine moiety (Figure 31b), was found to be resilient toward the deacetylases HDAC8 and Sir2.346 When placed at position 5 or 8 of H4 tail peptides, this mimic is recognized by the bromodomain of Brdt, albeit with slightly lower affinity than the corresponding acetylated peptides. H3 variants functionalized with methylthiocarbamoyl-thialysine cross-reacted with designated antibodies, providing further evidence that this acetyllysine analogue is suitable for biochemical studies, in particular when HDAC resistance is desirable.
Figure 31.

Synthesis of (a,b) acetyllysine and (c) methylarginine analogues from cysteine-containing histones.
Methylarginine analogues with different geometries were similarly prepared by a conjugate addition of a Michael acceptor to cysteine-containing histones (Figure 31c).347 Besides the methylene-to-sulfur substitutions, the resulting residues contain an amidine functional group rather than the native guanidinium group. Nevertheless, full-length histones and peptides displaying methylarginine analogues were recognized by cognate antibodies and the H4R3me2a-binder TDRD3.
The chemoselectivity of cysteine functionalization with versatile probes has been a workhorse in protein biochemistry and biophysics. Serendipitously, this reaction can be considered bio-orthogonal in chromatin research, because there is only one conserved yet nonessential cysteine residue in histones. Spectroscopic probes and cross-linkers, attached to engineered cysteines, have informed on the structure and dynamics of nucleosomes as well as the chromatin fiber. In addition, PTM analogues generated from cysteine mutants have contributed valuable data on the structural effects and molecular recognition of histone modifications, especially in the case of lysine methylation.
4.2. Synthetic Biology Meets Chromatin Research
The genetic code specifies 20 standard amino acids. In addition, natural mechanisms exist to expand the scope of genetically encoded building blocks to include selenocysteine and pyrrolysine. The same strategies have been exploited to incorporate unnatural monomers into proteins in cells (Figure 32).348 This methodology relies on the ability to suppress a stop-codon with a tRNA containing a complementary anticodon. The tRNA must be charged with the desired nonstandard amino acid in vivo using an engineered aminoacyl-tRNA synthetase (aaRS). Importantly, this system needs to be orthogonal to the cell’s endogenous apparatus in two ways: (a) the exogenous tRNA and amino acid must be recognized only by the exogenous aaRS, but not by any endogenous aaRSs, and (b) the exogenous aaRS must utilize only the exogenous building blocks, but none of the cell’s natural raw materials. This feature is usually achieved through elaborate directed evolution schemes, starting with a tRNA-aaRS pair from a different host organism such as the tRNACUA (complementary to the “amber” UAG stop codon) and the Pyrrolysyl-tRNA synthetase (PylRS) from Methanosarcinae. A comprehensive review by Lang and Chin on strategies to incorporate unnatural amino acids into proteins has recently been published in this journal.349
Figure 32.
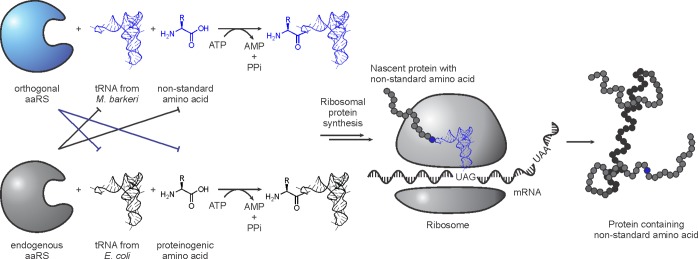
Incorporation of nonstandard amino acids (blue) into proteins in E. coli. An engineered orthogonal aaRS (top) charges a cognate tRNA with a designated amino acid, but does not interact with natural amino acids or E. coli tRNAs (bottom). Similarly, neither the exogenously introduced tRNA (here from M. barkeri) nor the nonstandard amino acid is recognized by any aaRS from E. coli (gray). Translation of the unnatural amino acid occurs opposite an amber stop codon (UAG).
4.2.1. Genetic Incorporation of Acetyllysine Residues
In the context of histones, the ability to genetically encode residues containing PTMs has been tremendously useful. In particular, acetyllysine can be integrated into ribosomal protein synthesis using an engineered aaRS originally dedicated to pyrrolysine in Methanosarcina barkeri.350 Neumann et al. harnessed the amber suppression strategy to define the biophysical and biochemical effects of H3K56 acetylation.351 By replacing the codon that specifies lysine 56 with an amber stop codon, and supplementing the growth medium with acetyllysine, the authors were able to produce H3 homogeneously modified with K56ac in E. coli carrying the orthogonal tRNACUA and the evolved aaRS. This protein was subsequently incorporated into nucleosomes and nucleosome arrays using standard techniques. Surprisingly, K56ac did not alter chromatin compaction, and only had minor effects on chromatin remodeling by bromodomain-containing motor proteins. In contrast, single-molecule FRET measurements revealed that DNA “breathing” was enhanced in K56ac-containing mononucleosomes as compared to unmodified versions, consistent with the position of K56 close to the DNA entry/exit site (Figure 33).351 K56ac marks also facilitated binding of the pluripotency factor Oct4 to nucleosomes in vitro,352 yet inhibited interactions with the components of the yeast silencing apparatus Sir2–4.353
Figure 33.
K56ac (black) increases breathing of nucleosomal DNA.
The modularity of the genetic acetyllysine incorporation strategy enabled Schneider and co-workers to insert this modified residue at several H3 sites and study the impact of site-specific histone acetylation on transcription. H3K64ac354 and H3K122ac,355 both present at the lateral surface of the histone octamer,356 are found at the transcription start site (TSS) of actively transcribed genes. In vitro assays demonstrated that these PTMs, presumably installed by the histone acetyltransferase p300, stimulate transcription and facilitate histone eviction by NAP1.354,355 In addition, K64ac directly destabilizes nucleosomes, as evidenced by FRET measurements.354
The Schultz and Carell laboratories have independently reported that amber suppression methods can also be harnessed to produce histones with crotonyllysine residues in E. coli.357,358 Schultz and co-workers evolved a PylRS to recognize this residue, enabling the biosynthesis of H2B with a crotonyl modification on Lys11.357 Carell and co-workers speculated that wild-type PylRS is able to accommodate modified lysines as well.358 Indeed, this enzyme could be used to prepare H3 crotonylated at Lys9. In addition, PylRS tolerated propionyl- and butyryllysine, providing access to H3 with the corresponding residues at position 9.
4.2.2. Genetic Incorporation of Protected Species
Direct incorporation of methyllysine residues into proteins through reengineered aaRSs has so far been unsuccessful.359,360 In contrast, the spacious binding pocket of Pyl-tRNA synthetase is ideally suited to accommodate protected lysine species. This feature has been harnessed to achieve the incorporation of N-ε-Boc- or N-ε-Alloc-protected methyllysine residues into histones (Figure 34a).360,361 Post-translational deprotection using dilute TFA or a ruthenium complex for Boc and Alloc protected building blocks, respectively, afforded monomethylated histones. H3K9me1 produced in this way interacted specifically with cognate antibodies and HP1, thus demonstrating the authenticity of the epitope.360
Figure 34.

Strategies to genetically encode methyllysine residues. (a) Incorporation of protected Kme1-species. (b) Protection-modification scheme to access Kme2-containing proteins. Boc-protected lysine is incorporated into histones through amber suppression. Orthogonal protection of other lysine residues, followed by removal of the Boc group and reductive alkylation enables site-specific modification. Global deprotection then provides the desired histone. TFMSA = trifluoromethylsulfonic acid, TFA = trifluoroacetic acid, DMS = dimethylsulfide.
Dimethyllysine can be incorporated into histones using a molecular detour involving a more elaborate protecting group scheme (Figure 34b).359 First, N-ε-Boc-Lysine was site-specifically incorporated at H3K9. Global protection of all other amine groups using Cbz-OSu ensued, followed by chemoselective Boc deprotection in TFA/H2O. Reductive methylation with formaldehyde and a borane reagent preceded global deprotection with a mixture of trifluoromethanesulfonic acid, TFA, and dimethylsulfide. The resulting protein, modified with H3K9me2 to >90%, was recognized by HP1, as expected.
A range of additional PTM analogues have been incorporated into histones upon addition of phenylselenocysteine, a caged electrophile, to the genetic code.362,363 Being prone to oxidation, this residue readily undergoes selenoxide pyrolysis in the presence of H2O2 to yield dehydroalanine, a Michael acceptor (Figure 35).364 Subsequent functionalization with N-acetylcysteamine or N-methylcysteamines provides acetyllysine and methyllysine analogues.363 H3 variants containing H3K9Cac or a thialysine control residue prepared in this way confirmed that the Ser10-targeting kinase AuroraB is sensitive to acetylation at the neighboring Lys9.363,365 The yield of dehydroalanine-containing histones can be improved through the use of selenocysteine derivatives that are incorporated into proteins more readily.366 Overall, however, this strategy is limited by undesired oxidation of methionine residues,362 as well as a lack of stereochemical control; that is, both diastereoisomers are generated.367
Figure 35.

Biosynthetic incorporation of PTM analogues through dehydroalanine intermediates. Dehydroalanine can be generated through selenoxide pyrolysis (left) or cysteine-specific reagents (right). Michael addition of thiols to dehydroalanine generates PTM analogues, albeit with loss of stereochemical information.
Chalker et al. devised a mild approach to generate dehydroalanine sites from cysteine residues, thereby bypassing the need for unnatural amino acid incorporation.368 2,5-Dibromohexanediamide selectively bis-alkylates cysteine at sulfur and causes elimination (Figure 35). Subsequent derivatization of dehydroalanine with sulfur-containing nucleophiles provided analogues for acetyllysine, methyllysine, phosphoserine (i.e., phosphocysteine), and GlcNAc-serine.369 Phosphocysteine at residue 10 of H3 was detected by an anti-S10ph antibody, H3K9Cac was deacetylated by HDAC1 and HDAC2, and even doubly modified histones carrying two copies of either dimethyllysine or acetyllysine analogues at positions 4 and 79 could be prepared, demonstrating the versatility of the approach. While this mild method steers clear of problems with methionine oxidation, PTMs are still incorporated as stereochemical mixtures, which may be of concern in certain instances.
While amber suppression is typically limited to one residue per protein, more sophisticated approaches to increase the efficiency of incorporating specific370 or multiple371−376 unnatural amino acids have been developed. For example, by using in vitro translation systems with cell extracts derived from E. coli strains with deleted release factors, Mukai et al. were able to produce H4 acetylated simultaneously at K5, K8, K12, and K16.375 In addition, the design of innovative systems to add complex building blocks, including phosphoserine377 or photocaged amino acids,378,379 to the genetic code further strengthens the potential of synthetic biology to contribute to chromatin biochemistry.
4.2.3. A Synthetic Biology Strategy To Probe Chromatin Structure in Vivo
The key advantage of the synthetic biology approach is that, in principle, histones can be generated with site-specific modifications in vivo and studied in situ, as demonstrated by Neumann and co-workers in an impressive study on mitotic chromatin compaction.380 Use of amber suppression allowed the introduction of a BPA photo-cross-linker at position 58 of H2A to report on chromatin condensation in live yeast (Figure 36). Upon UV irradiation, a cross-link to H4 formed, presumably mediated by the H4 tail engaging the acidic patch (see also section 4.1.1). Indeed, deletion of H4 tail fragments or mutagenesis of H4K16 to alanine reduced the magnitude of cross-linking. Intriguingly, the H2A–H4 interaction was highly dependent on cell cycle stage, peaking during M phase in synchrony with aurora B-dependent phosphorylation of H3S10, a classical mitotic marker.381,382 In contrast, the K16ac mark is anticorrelated with cross-linking, that is, at its lowest during M phase, suggesting that this PTM prevents chromatin condensation in vivo.380 In agreement with this conjecture, cross-linking in yeast carrying an H4K16R mutant was not cell-cycle dependent. Dissection of the signaling pathway led to a model where H3S10ph recruits the deacetylase Hst2p, and this interaction was required for H2A–H4 cross-link formation and hence chromatin compaction. This study traced the signaling pathway that governs chromatin condensation during cell division, and simultaneously demonstrated that synthetic biology strategies are well suited to investigate chromatin biology in live cells.
Figure 36.
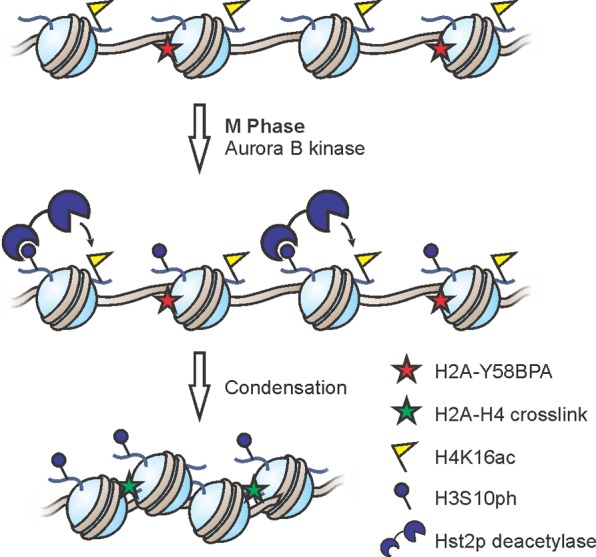
Schematic of the signaling cascade that controls chromatin condensation during mitosis. Decompacted chromatin, partially labeled with a photo-cross-linker (BPA, red star), is acetylated at H4K16 (yellow flag). Upon entry into M phase, aurora B kinase phosphorylates H3S10 (blue lollipop), which recruits the HDAC Hst2p (blue). Once H4K16 is deacetylated, chromatin compacts, observed by an H2A–H4 cross-link (green star).
4.3. Chemical (Semi)-Synthesis of Histones
Solid-phase synthesis has provided access to peptides containing essentially any histone PTM in the context of histone tails. The virtues of chemical synthesis can be extended to the manufacture of site-specifically modified histones and chromatin templates through convergent assembly strategies. Rather than synthesizing histones as a single linear chain, two or more fragments are prepared individually by SPPS and ligated upon purification. The archetypical methodology to achieve this feature is native chemical ligation (NCL), that is, the condensation of a synthetic peptide α-thioester with a second peptide carrying an N-terminal cysteine (Figure 37).383,384 The reaction is initiated by trans-thioesterification to join the two peptide fragments, arranging the thioester intermediate for an intramolecular S-to-N acyl shift to yield a native amide bond. Importantly, thioesters, often used in nature for acyl group transfers (including in nonribosomal peptide synthesis),385 are soft electrophiles. They are therefore uniquely activated toward thiol nucleophiles at neutral pH and react much more sluggishly with the harder O- and N-nucleophiles also present in proteins.386,387 In addition, although trans-thioesterification can proceed with internal cysteine residues, this side reaction is reversible because the absence of a proximal amino group precludes stable amide bond formation. Because of its exquisite chemoselectivity, NCL can be performed with unprotected peptide segments in water. In this section, we will discuss various convergent synthetic and semisynthetic strategies to produce designer histones, as well as applications of these powerful reagents in chromatin biochemistry.
Figure 37.

Mechanism of native chemical ligation (NCL).
4.3.1. Semisynthesis of N-Terminally Modified Histones
Because of the frequency of PTMs on the N-terminal histone tails, semisynthetic approaches are ideally suited for the preparation of designer histones. Semisynthesis integrates the expanded scope of peptide chemistry with the ease of generating biopolymers recombinantly (Figure 38).388 Specifically, peptides encompassing the PTM site are chemically synthesized as α-thioesters and reacted with histone fragments carrying an N-terminal cysteine. Peptide α-thioesters can be directly synthesized on solid phase using Boc chemistry (Figure 39a), but special measures are required to access α-thioesters via Fmoc-SPPS due to the copious use of base necessary for Fmoc deprotection.389 Most simply, fully protected peptides, synthesized on very acid-labile chlorotrityl resins, can be converted to α-thioesters in solution using HBTU in the presence of thiols (Figure 39b). Epimerization at the C-terminus is very common in this procedure, and much care is needed to avoid this unwanted side reaction.390,391 Preferably, peptides can be synthesized on 2-hydroxy-3-mercaptopropionic acid,392 diaminobenzoyl,393 or hydrazine394 linkers, allowing activation of the C-terminal residue via O-to-S acyl shift, as an acylurea or as an acylazide moiety, respectively (Figure 39c–e). Recombinantly produced histone fragments bearing an N-terminal cysteine are typically generated from a fusion protein using site-specific proteolysis. Factor Xa,395 TEV protease,396 Thrombin,397 and SUMO398 protease are most frequently used in this context.399,400 Alternatively, in some cases precise removal of the N-terminal methionine residue in E. coli can provide protein fragments featuring a newly exposed N-terminal cysteine.401 Given the scarcity of cysteine residues in histones, an engineered cysteine has to be placed strategically at a desired ligation junction.
Figure 38.

Protein semisynthesis by native chemical ligation.
Figure 39.

Comparison of peptide α-thioester synthesis by Boc- (a) and Fmoc-SPPS (b–e). (a) Synthesis of peptide α-thioesters on a mercaptopropionic acid linker by Boc-SPPS. (b) Direct conversion of a protected peptide acid into an α-thioester. (c) Latent thioester synthesis on a 2-hydroxy-3-mercaptopropionic acid linker. (d) α-Thioester synthesis through an acylthiourea intermediate. (e) Acyl-hydrazide method for α-thioester synthesis. Pg = protecting group.
In 2003, Shogren-Knaak et al. reported the first preparation and application of a semisynthetic histone, H3 containing S10ph (Figure 40).402 A synthetic phosphopeptide encompassing residues 1–31 was synthesized by Fmoc-based SPPS and converted into an α-thioester following cleavage of the protected peptide from the resin. In parallel, an H3 fragment consisting of residues 33–135 furnished with an N-terminal cysteine in place of Thr32 was produced recombinantly. The fragments were joined by NCL, and purified by ion exchange chromatography. The resulting H3 variant as well as a control protein synthesized with unmodified Ser10 and also containing the T32C mutation were subsequently incorporated into nucleosome arrays. In agreement with previous reports based on peptide substrates,403,404 semisynthetic chromatin modified with S10ph was acetylated more readily by GCN5 than the control array.402 Surprisingly, however, in the context of chromatin, S10ph did not stimulate acetylation by the SAGA complex (which contains GCN5), suggesting that other subunits override the expected preference.
Figure 40.
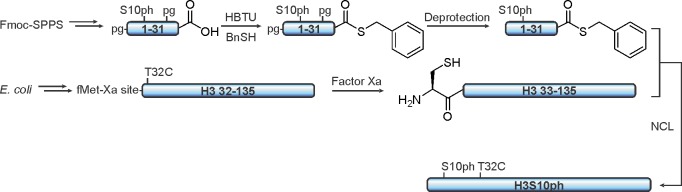
Semisynthesis of H3S10ph. A synthetic peptide is converted into an α-thioester in solution with HBTU and benzyl mercaptan (top). Simultaneously, a recombinant fragment with an N-terminal cysteine residue (in place of Thr32) is prepared by site-specific proteolysis using Factor Xa (middle). Joining of the two fragments by NCL yields full-length H3 site-specifically phosphorylated at Ser10. A T32C mutation remains at the ligation junction (below). pg = protecting group.
Shortly thereafter, He et al. described the traceless semisynthesis of several methylated and acetylated histones.401 Peptide α-thioesters corresponding to residues 1–24 of H3 or 1–14 of H4 were synthesized via Boc-SPPS on a mercaptopropionic acid linker or by Fmoc SPPS employing a postcleavage thioesterification.405 NCL with appropriate recombinant fragments (H3 residues 26–135 and H4 residues 16–102, both with an additional N-terminal cysteine) yielded H3 variants containing K9me3 or acetyl marks at K4, K9, K14, K18, and K23, as well as H4 acetylated at K5 and K8 or K5, K8, and K12 (Figure 41a).401 Notably, the ligation junctions were judiciously chosen to entail Ala-to-Cys substitutions. These mutations were reverted in the full length proteins through hydrogenolytic desulfurization of cysteine to alanine in the presence of Raney nickel.406 The resulting modified histones and unmodified controls were successfully assembled into chromatin, attesting to their integrity.401
Figure 41.

Strategies to fix ligation scars in histone semisyntheses. (a) Conversion of Cys to Ala by desulfurization with Raney nickel in the semisynthesis of H3K9me3. (b) Alkylation of cysteine with bromoethylamine to produce thialysine in the semisynthesis of H4K16ac. (c) Radical-based desulfurization in the semisynthesis of H2B-S14ph.
To scrutinize the effect of histone acetylation on chromatin structure and remodeling, several research groups have produced site-specifically acetylated nucleosomes and arrays. In a landmark study, Peterson and co-workers harnessed histone semisynthesis to investigate the biophysical and biochemical consequences of acetylating H4K16,407 a key modulator of chromatin structure and function in development and disease.408 Because of the lack of alanine residues in proximity to K16, the authors chose to use an R23C mutation to mediate native chemical ligation. Nucleosome arrays carrying K16ac and R23C, but not control variants with only the R23C mutation, displayed drastically reduced propensity to compact and self-assemble.407 In addition, H4K16ac stimulated acetylation on H3 by the SAGA complex in a bromodomain-dependent manner,409 and slightly inhibited chromatin remodeling by the ACF complex.407
In another example, Ferreira et al. studied Snf2-dependent remodeling of nucleosomes acetylated at H3 (at lysines 9, 14, 18, and 23) or H4 (at lysines 5, 8, 12, and 16).410 To enable NCL, cysteine residues were engineered in place of Ser28 and Val21, respectively. Native gel electrophoresis was used to monitor the position of a nucleosome (which affects its electrophoretic mobility) within a short DNA fragment. Remodeling assays revealed that acetylation at H3, especially at Lys14, drastically increased recruitment of the enzyme complex to modified nucleosomes. This effect was later shown to be mediated by the bromodomain of SWI/SNF.411 By contrast, acetylation of H4 inhibited remodeling of mononucleosomes by Chd1 and Isw2 in vitro.410 No such inhibition could be detected in nucleosome arrays containing semisynthetic H4K16ac, however.412
Nordenskiöld, Liu, and co-workers dissected the contribution of H4 acetylation marks to chromatin compaction and self-assembly.413 H4 peptide α-thioesters, monoacetylated at K16, triacetylated at K5, K8, and K12, or tetra-acetylated at K5, K8, K12, and K16, were ligated to truncated H4 using a K20C mutation (Figure 41b). The ligation “scar” was neatly covered up by alkylation of the cysteine with bromoethylamine. Nucleosome arrays containing differentially acetylated H4 tails were subjected to compaction and self-assembly assays. Acetylation at K16 was specifically found to disrupt intra-array folding413 (which is consistent with previous work) and mononucleosome aggregation.414 In contrast, nucleosome array aggregation was dependent on the number of acetylation marks rather than their position.413 These results suggest that intra-array folding and interarray assembly are governed by specific interactions and coarse electrostatic effects, respectively.
Semisynthesis of site-specifically acetylated and phosphorylated H2B informed on the ability of Mst1 kinase and a commercial antibody to recognize modified targets.415 H2B tails corresponding to residues 1–16 were synthesized as latent thioesters containing S14ph, four acetyl marks, or a combination of these PTMs (Figure 41c). The truncated histone (residues 18–125) was produced in E. coli with a Met-Cys dipeptide leader. The initiator Met was spontaneously removed in vivo, and treatment of the protein with methoxylamine freed the N-terminal cysteine from thiazolidine adducts. Upon ligation, the full-length histones were desulfurized to restore Ala17, rendering the procedure traceless. In this process, the radical desulfurization approach developed by Danishefsky and co-workers416 was found to be superior to Raney-nickel treatment.415 Mst1 was able to phosphorylate unmodified and polyacetylated H2B in vitro. Immunoblots of semisynthetic H2B variants with an antibody directed against S14ph, however, clearly demonstrated that H2B acetylation masks the epitope of this antibody. This finding agrees with the general conclusions from peptide array-based mapping of antibody specificity (discussed in section 3.12), jointly raising the issue that PTM detection needs to be considered in the context of neighboring marks, that is, epitope occlusion.
The examples described above illustrate the diversity of histones with N-terminal modifications that can be generated using protein semisynthesis. Recent developments in the synthesis of peptide α-thioesters using Fmoc chemistry and commercially available modified α-thioester peptides have made protein semisynthesis an amenable approach for molecular biology laboratories. When ligation junctions are chosen appropriately, chemical traces of semisynthesis can be removed through desulfurization strategies or covered up with the installation of amino acid analogues.
4.3.2. Semisynthesis of C-Terminally Modified Histones
To access modifications at the C-terminal tails of histones, a short synthetic peptide bearing the PTM and an N-terminal cysteine is condensed with an α-thioester encompassing the majority of the histone sequence. Protein α-thioesters can be obtained recombinantly with the help of inteins,417−419 protein domains that effect their own excision from a protein precursor.388,420 The splicing reaction is initiated by an N-to-S acyl shift at a cysteine residue at the N-terminus of the intein (Figure 42a). Subsequently, the linear thioester is converted to a branched thioester by an acyl transfer to a cysteine residue adjacent to the intein domain. Amide bond cleavage through succinimide formation liberates an amine, onto which the acyl chain collapses to yield a new, native peptide bond joining the protein sequences that previously flanked the intein. When succinimide formation is precluded by an Asn-to-Ala mutation, the internal thioester intermediates can be captured by exogenously added thiols (Figure 42b).421 The resulting protein α-thioester can be purified and used for NCL, a process usually referred to as expressed protein ligation (EPL).388,420
Figure 42.

Intein-mediated protein splicing. (a) Mechanism of intein autoprocessing. (b) Recombinant preparation of a protein α-thioester using a mutated intein. Thiolysis is mediated by a large excess of soluble thiol, such as sodium 2-mercaptoethanesulfonate (MesNa).
Ottesen, Poirier, and co-workers have taken advantage of EPL to synthesize H3 and H4 variants with PTMs close to the C-termini.422−425 Lys115, Lys 122, and Thr118 of H3 are all positioned in proximity to the nucleosome dyad axis where they form contacts with DNA, and Lys77 and Lys79 of H4 interact with DNA on the lateral surface (Figure 43a). Mutation of these residues affects DNA-templated processes in yeast,426 and acetylation of the lysine residues or phosphorylation of Thr118 might similarly control nucleosomal functions. This conjecture was directly addressed by protein semisynthesis. An H3 fragment (residues 1–109) was fused to an intein to generate an α-thioester, which was subsequently ligated to a synthetic peptide carrying the K115ac, K122ac marks, taking advantage of the only conserved naturally occurring cysteine in histones (Cys110, Figure 43b).425 Formation of nucleosomes with the modified H3 was possible, although the acetylation marks decreased the affinity of the histone octamer for DNA. Note that nucleosomes containing the corresponding K-to-Q mutations did not affect DNA binding, thereby illustrating the need for native PTMs rather than amino acid surrogates. Once formed, the acetylated nucleosomes were shown to slide more easily than unmodified counterparts without affecting DNA breathing. Nucleosomes phosphorylated at H3T118, prepared in the same fashion, displayed a similar phenotype.424 DNA at the nucleosome dyad is substantially more accessible, and unusual nucleosome architectures were observed in the presence of H3T118ph.423,424 These topologies might include structures where the DNA loops around two octamers to minimize crossing over the phosphorylated dyad axis.427 The H4 variant was synthesized from a recombinant α-thioester (residues 1–75) and a synthetic peptide acetylated at K77 and K79, using Ala76Cys for the ligation (Figure 43c).422 Upon ligation and desulfurization, the modified histone was incorporated into nucleosomes. FRET and DNA accessibility measurements demonstrated that acetylation of lysine residues at the lateral surface increases DNA breathing, as hypothesized.
Figure 43.

Preparation of modified histones by EPL. (a) Location of selected residues at the DNA binding surface. Residues on H3 and H4 are indicated with blue and green arrows, respectively. For clarity, labels are only placed on one copy of each histone. (b) Semisynthesis of H3 with acetyl marks close to the C-terminus using the native C110 for NCL. (c) EPL strategy to synthesize H4K77,79ac via an Ala76Cys mutation.
Recent discoveries of ultrafast inteins that are naturally split, that is, they perform protein splicing from two separate polypeptides, have improved the production of recombinant protein α-thioesters.428,429 Because the individual intein segments associate with high affinity, their assembly can be co-opted for purification.430 In this streamlined EPL, the C-terminal intein fragment is immobilized on a solid support, and incubated with crude lysate from bacteria producing the protein of interest fused to the N-terminal intein portion (Figure 44). The intein segments assemble into a tight complex, allowing the removal of contaminating proteins by washing. Elution of the protein α-thioester is achieved by incubation with excess exogenous thiol. This expedited procedure was applied to access an H2B α-thioester (residues 1–116), which was subsequently ligated with a synthetic peptide acetylated at K120.
Figure 44.
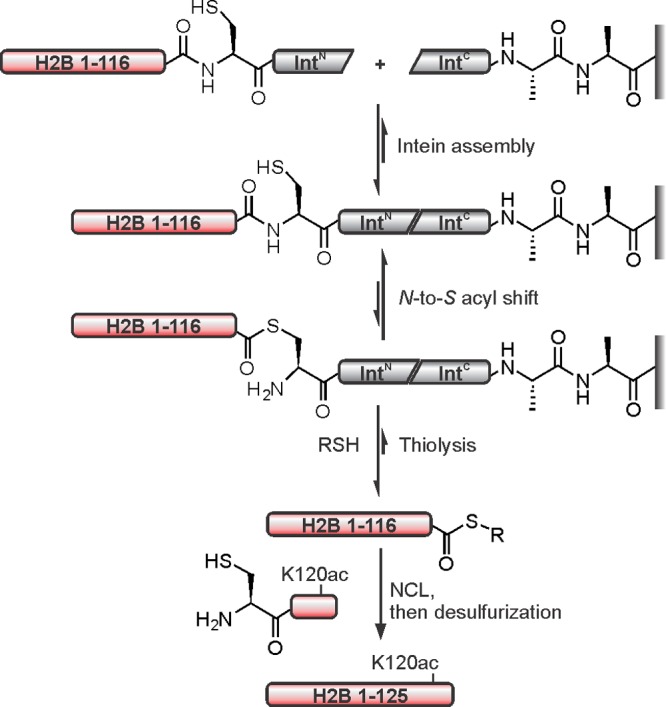
Streamlined expressed protein ligation to synthesize H2B-K120ac. Intein self-assembly is harnessed for affinity purification in a column-format. α-Thioester intermediates are subsequently captured by washing the column with excess thiols. The isolated H2B α-thioester is condensed with a synthetic peptide containing an N-terminal cysteine and the K120ac modification. The ligation product is subsequently desulfurized to render the process traceless.
Development of strategies to extend NCL and EPL to involve noncysteine residues can alleviate some constraints on choosing ligation junctions.431 Thiolated derivatives of lysine (e.g., 10)432 and arginine (e.g., 11)433 are particularly useful in the context of histones, and the ability to selectively desulfurize thiolated aspartate analogues (e.g., 12) in the presence of cysteine434 may find use in the synthesis of native H3 (Figure 45). Amino acids related to 10–12 are compatible with genetic code expansion, suggesting that biosynthetic strategies can complement chemical methods to provide raw materials for NCL reactions at noncysteine sites.435,436
Figure 45.

Thiolated amino acid derivatives used for NCL.
A variation on the NCL theme is the use of α-thioacid capture to join histone fragments.437 In this approach, an intein-derived α-thioester is converted into an α-thioacid with H2S. This nucleophile reacts rapidly with activated disulfides to form an acyldisulfide, which rearranges by an S-to-N acyl shift (Figure 46). Upon reduction, a cysteine residue with a native amide bond is obtained. The same strategy can be extended to the ligation of a synthetic peptide α-thioacid with a recombinant protein carrying an N-terminal cysteine, previously activated as an asymmetric disulfide with 2,2̀-dithiobis(5-nitropyridine). H3 variants with K4me2 and without modification have been synthesized using this strategy. In certain cases, the fast reaction rate (on the order of minutes) of the α-thioacid capture and the ensuing acyl transfer steps could outweigh the extra steps needed for this mode of protein semisynthesis.
Figure 46.
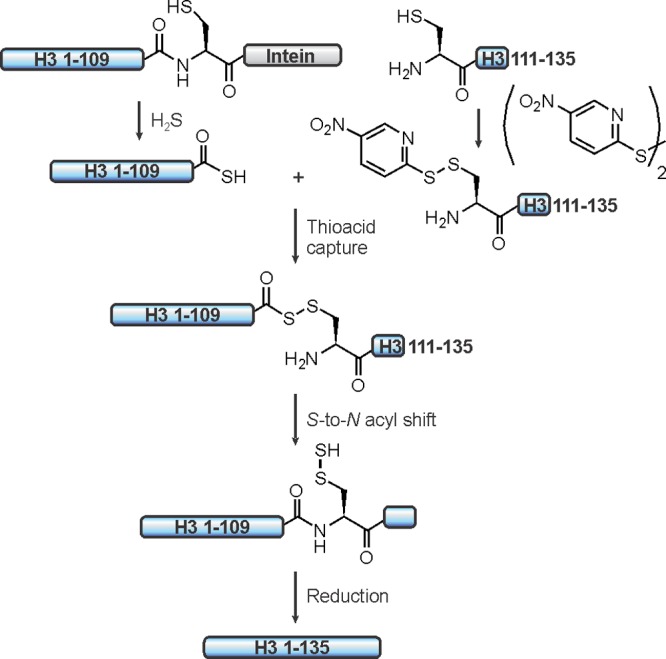
Histone semisynthesis using a thioacid capture strategy. A truncated histone-intein conjugate is converted to a thioacid with H2S (left). This fragment is coupled with a C-terminal peptide, activated as an asymmetric disulfide (right). Disulfide exchange is followed by an intramolecular acyl shift and reduction to a native cysteine residue.
By combining intein technology with peptide chemistry, histones bearing PTMs at their C-terminal tails are readily accessible. Extensions and variations of these methodologies further broaden the scope of potential histone targets, thereby contributing to elucidating the molecular function of the flexible histone C-termini.
4.3.3. Enzyme-Assisted Semisynthesis of Modified Histones
Enzyme-catalyzed ligation reactions harbor great potential for the semisynthesis of proteins. Sortases, bacterial transpeptidases that cross-link proteins of cell walls, are particularly promising.438 Sortase A recognizes a pentapeptide stretch (LPxTG) at the C-terminus of its substrate. The enzyme cleaves the terminal glycine residue and concomitantly forms a thioester using an active site cysteine. The acyl group is thereafter transferred to a glycine-rich sequence of the peptidoglycan. By reengineering the substrate specificity of Sortase A to recognize an H3 peptide sequence (APATG, residues 29–33), Piotukh et al. achieved a traceless synthesis of full length H3 (Figure 47).439 In this process, neither the synthetic peptide (residues 1–33) nor the truncated H3 variant with a native Gly-Gly N-terminus (residues 33–135) required preactivation. Further engineering to include different recognition motifs and to boost the rate and yield (currently below 50% in case of the engineered Sortase variant) of the transpeptidase reaction will boost the application of this intriguing biosynthetic tool to the generation of modified histones.
Figure 47.
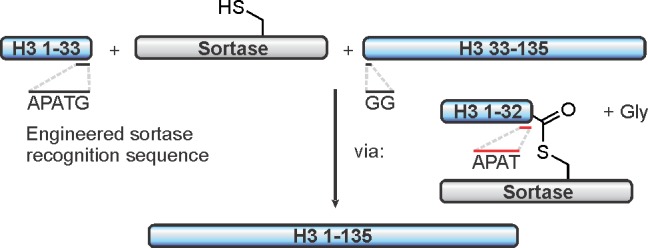
Histone semisynthesis using an engineered Sortase variant.
4.3.4. Multistep Synthesis of Histones
With the discovery of increasing numbers of PTMs that are positioned within the globular domains of histones,189 chemical methods to access these site-specifically modified histones require refinement. Synthetic peptides with core modifications approach lengths prohibitive for routine SPPS if a two-piece ligation is attempted. Instead, convergent strategies to access full length histones from three or four fragments have been developed.
In a synthetic tour de force, Ottesen and co-workers assembled histone H3 with K56ac using a three-piece ligation strategy (Figure 48a).440 The ligation sites were chosen to exclusively involve Ala residues (A47, A91) because preliminary results indicated that the introduction of non-native cysteine residues in a test protein impacted nucleosome structure. All peptides were synthesized with N-α-Boc protected building blocks and, where appropriate, as α-thioesters on a mercaptopropionamide linker. The central segment (residues 47–90) with K56ac, and A47C protected as a thiazolidine species, was first ligated to the C-terminal piece (residues 91–135, A91C). Subsequently, C47 was unmasked with methoxylamine, and the N-terminal fragment (residues 1–46) was added. Because of the presence of Val46, the second ligation reaction proceeded sluggishly, requiring 4–6 days for completion. Finally, desulfurization provided access to fully synthetic H3 bearing K56ac and the popular C110A mutation in an overall 7% yield. FRET measurements with labeled nucleosomes containing K56ac confirmed that this PTM promotes DNA breathing.
Figure 48.

Multistep histone synthesis. (a) Total synthesis of H3K56ac using a three-step NCL procedure. (b) Total synthesis of H3K9me3 using Cys-Pro ester fragments, joined by NCL and direct aminolysis in the presence of Ag+ ions. (c) Three-piece semisynthesis to generate H3R42me2a. Initially, two synthetic peptides are joined by NCL. Subsequent activation of a C-terminal acyl hydrazide by oxidation enables a second NCL step to attach a recombinant fragment. pg denotes protecting groups.
Aimoto and co-workers reported a three-piece total synthesis of H3 trimethylated at K9.441 The authors prepared an N-terminal segment (residues 1–43) and a central segment (residues 44–95) as latent thioesters using the cysteine-proline ester autoactivation motif. This unit promotes transpeptidation by forming an α-thioester upon condensing into a diketopiperazide moiety (Figure 48b).442 NCL of the middle segment, still N-α-Fmoc protected, with the unprotected C-terminal portion took advantage of Cys96, which naturally occurs in some isoforms of H3.441 To prepare for the second ligation step, cysteine residues were protected as disulfides, lysine side-chains masked with Boc-OSu, and the N-terminal glycine residue exposed with piperidine treatment. This fragment was then joined to the protected N-terminal α-thioester fragment (containing a C-terminal proline residue, Pro43) by Ag+-promoted aminolysis. DTT and TFA were sequentially used to deprotect the cysteine and lysine residues, respectively, to yield H3K9me3 in an overall yield of approximately 6%. Although shown for the synthesis of H3K9me3, this multistep procedure is equally suitable to generate H3 variants with modifications in the central domain.
Arginine 42 of H3 is situated close to the nucleosomal DNA entry site. This residue was recently identified by mass spectrometry to be dimethylated.443 Given that the methyltransferases that install this PTM catalyze asymmetric dimethylation reactions at other sites,124 R42 is most likely converted to the me2a form as well. To directly probe the biochemical function of R42 methylation, H3R42me2a was assembled in a three-step semisynthesis.443 The N-terminal fragment (residues 1–28) was prepared by Boc chemistry as an α-thioester, while the central segment containing R42me2a (residues 29–46, A29C) was assembled by Fmoc chemistry as an acylhydrazide (Figure 48c). The C-terminal portion (residues 47–135, A47C) was produced recombinantly as a SUMO fusion. Cleavage by Ulp1 liberated the histone fragment bearing an N-terminal cysteine. The fragments were joined by NCL in an N-to-C direction. Importantly, the acylhydrazide functionality is inert under the conditions of the first ligation, but is activated by oxidation during the second step,444 granting full regioselectivity to the sequential ligation process. Radical-based desulfurization concluded the semisynthesis of the homogeneously modified histone. Chromatin assembled with H3R42me2a was found to be more permissive to transcription as compared to unmodified congeners, supporting a direct role for arginine methylation in the control of DNA-templated processes, congruent with the position of this residue close to the DNA entry/exit site.
The examples discussed above demonstrate the feasibility of chemically synthesizing entire histones. A selection of different ligation strategies exists to place modified residues at diverse positions within the histone core. While these approaches are technically much more challenging than the more prevalent two-component semisyntheses, they significantly broaden the scope of chemically accessible modified histones. Total synthesis or multistep semisynthesis represents the method of choice for preparing histones where centrally located modifications are inaccessible by state of the art biosynthetic means, and analogues are not available or are inadequate. In addition, and very importantly, sequential ligations allow multiple different modifications to be placed at distinct sites along the polypeptide chain.
4.3.5. Synthesis of Ubiquitylated Histones
Investigating the functions of histone ubiquitylation poses unique chemical challenges. Given its size, the ubiquitin PTM cannot be installed as a single building block at the peptide synthesis stage. Instead, a convergent strategy drawing from expressed protein ligation can be harnessed to obtain site-selectively ubiquitylated peptides, a first step in the challenging paths toward H2A-K119ub and H2B-K120ub.188 Chatterjee et al. produced ubiquitin recombinantly as an intein fusion to yield an α-thioester lacking the C-terminal residue, Gly76. A surrogate for this residue, bromoacetic acid, is attached to the ε-amine of an orthogonally protected lysine (corresponding to K120 of H2B) via an isopeptide bond on a synthetic histone peptide. The branch is reacted with an 1,2-amino-thiol auxiliary that provides the nitrogen atom of Gly76 and enables ligation of the ubiquitin α-thioester (Figure 49a). Upon ubiquitylation, photolysis cleaves the ligation handle, thereby restoring a native ubiquitin C-terminus, site-specifically attached to a single lysine residue.
Figure 49.

Auxiliary-based semisynthesis of uH2B. (a) Site-specific ubiquitylation of histone peptides. An amino-thiol ligation auxiliary permits ligation of a ubiquitin α-thioester to a glycine residue attached to the side-chain of Lys120. (b) Semisynthesis of native, full-length uH2B via a two-step ligation. MesNa = sodium 2-mercaptoethanesulfonate, TCEP = tris(2-carboxyethyl)phosphine.
This procedure can be conveniently extended with a second ligation step to produce full-length H2B-K120ub.445 An Ala117Cys mutation in H2B permits this extension, and protecting this residue with the photolabile o-nitrobenzyl group prevents double-ubiquitylation during the first ligation step (Figure 49b). Upon completion of the synthesis, desulfurization provided native H2B-K120ub in an overall yield of 20%. This valuable reagent enabled McGinty et al. to explore the functional consequences of histone ubiquitylation.445 In vivo, H2B-K120ub is associated with increased HMT activity of the Set1 complex and Dot1 toward H3K4 and H3K79, respectively.446−448 Methyltransferase assays with semisynthetic H2B-K120ub-containing nucleosomes confirmed that this mark directly stimulates the catalytic domains of Set1449,450 and Dot1.445 Nevertheless, despite its association with actively transcribed genes, H2B-K120ub did not enhance transcription in vitro.449 Also, nucleosomes containing semisynthetic H2B-K120ub or enzymatically generated H2A-K119ub were used to define the substrate specificity of the deubiquitinase Calypso.451 This polycomb group protein efficiently removed the ubiquitin mark from H2A-K119ub but not H2B-K120ub.
McGinty et al. further subjected the stimulation of Dot1 by H2B-K120ub to detailed structure–activity relationship studies. To facilitate this process, an innocuous Gly76Ala mutation was introduced at the C-terminus of ubiquitin to streamline the synthesis of an H2B-K120ub analogue (Figure 50a).452 In this strategy, residue 76 is attached to Lys120 of H2B as a cysteine, enabling direct ligation of the ubiquitin α-thioester at this site; the auxiliary is no longer necessary. Subsequent completion of the H2B sequence by a second NCL step is followed by desulfurization, providing tens of milligrams of H2B-K120ub with one additional methyl group. When incorporated into nucleosomes, this protein was found to be biochemically indistinguishable from the native structure. Several analogues of H2B-K120ub-containing nucleosomes were synthesized to test if Dot1 is stimulated by the canonical protein–protein interaction hotspots on nucleosomes: the H2A acidic patch (E64, N68), a basic stretch in the H4 tail (R17,19), and the ubiquitin hydrophobic patch (L8, I44). Methyltransferase assays revealed that Dot1 engages ubiquitylated nucleosomes through surfaces orthogonal to the acidic and hydrophobic patches of H2A and ubiquitin, respectively. Mutation of H4R17 and R19 to alanine, however, reduced Dot1 activity both on ubiquitylated and on unmodified nucleosomes, suggesting that Dot1 binds the H4 tail regardless of stimulation.
Figure 50.

Streamlined semisyntheses of ubiquitylated histones. (a) Synthesis of H2B-K120ub containing the G76A mutation in ubiquitin. This mutation enables introduction of residue 76 of ubiquitin as a cysteine and subsequent NCL with a ubiquitin α-thioester. Finally, desulfurization converts Cys76 into an alanine residue. (b) Synthesis of H2A-K119ub containing the G76A mutation in ubiquitin. A penicillamine moiety permits NCL at a valine residue. (c) Disulfide-based conjugation of ubiquitin to H2B-K120C. In this approach, the ubiquitin α-thioester is reacted with cysteamine (top) and coupled to histones activated as disulfides (below), thus yielding disulfide-bonded analogues of H2B-K120ub (H2B-K120ubSS).
The size of ubiquitin as a PTM raises the question if exact placement is essential for its diverse biochemical functions. To tackle this question, a semisynthesis of H2A-K119ub, a PTM associated with polycomb-dependent gene repression, was developed.453,454 Borrowing from the H2B-K120ub strategy, Fierz et al. ligated a ubiquitin α-thioester (residues 1–75) to a cysteine residue attached to H2A-K119 on a synthetic peptide via an isopeptide bond (Figure 50b). The second ligation to complete the H2A sequence was mediated by a penicillamine residue in place of Val104. Upon desulfurization, this amino acid is converted to valine, restoring the native H2A sequence, and leaving the benign G76A ligation “scar” at the C-terminus of ubiquitin.454 H2A-K119ub, in contrast to H2B-K120ub, did not stimulate Dot1-catalyzed methylation at H3K79.453 The ubiquitin marks had only minor effects on the activity of PRC2 containing the core subunits,453 although the presence of additional PRC2 modules increases HMT activity upon H2A ubiquitylation.455
To probe position-dependent biochemical effects of histone ubiquitylation in more detail, the synthesis of these reagents was further accelerated through the use of a disulfide-directed histone ubiquitylation scheme (Figure 50c).456 In this approach, ubiquitin is furnished with a C-terminal sulfhydryl group by treatment of an intein-derived ubiquitin α-thioester with cysteamine. In parallel, Lys 120 of H2B was mutated to a cysteine, and this residue was activated with 2,2′-dithiobis(5-nitropyridine), enabling attachment of the thiolated ubiquitin to yield H2B-K120ubSS. The slightly elongated attachment handle did not affect Dot1 stimulation. Repositioning of the ubiquitin mark to side-chains adjacent to H2B-K120, using the appropriate histone cysteine mutants, revealed considerable plasticity in the regulation of Dot1. Disulfide-based transplantation of the ubiquitin mark to H2B-125 and H2A-22 was well-tolerated, but transfer to residues 108 or 116 of H2B, both of which lie closer to the methylation site at H3K79, was detrimental to stimulation (Figure 51).
Figure 51.
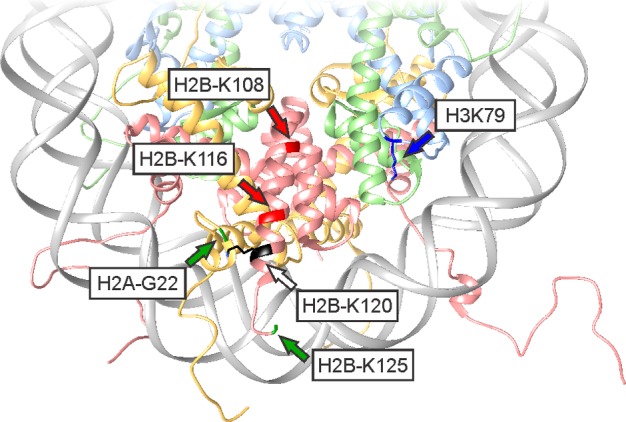
Plasticity in the stimulation of Dot1 by histone ubiquitylation. The canonical ubiquitylation site of H2B is indicated in black (K120, white arrow), permissive sites in green (H2A-G22 and H2B-K125), prohibitive sites in red (H2B-K108 and H2B-K116). The substrate residue (H3K79) of Dot1 is highlighted in blue.
What are the biophysical consequences of attaching an entire protein like ubiquitin to a nucleosome? On the mononucleosome level, semisynthetic H2A-K119ub and H2B-K120ub containing the G76A mutation are only marginally destabilizing compared to unmodified histones.454 The impact of histone ubiquitylation on nucleosome arrays is much more dramatic.457 Analytical ultracentrifugation revealed that H2B-K120ubSS inhibits array compaction. The mechanism of this effect was scrutinized using a homo-FRET-based compaction assay. H2A was fluorescently labeled at an engineered cysteine residue at position 110 with maleimide chemistry. In open chromatin, fluorescence polarization is high due to the long rotational correlation time of large arrays. Upon Mg2+-induced compaction, labeled H2A moieties approach each other, and homo-FRET occurs. Because of the relative orientation of the fluorophores, homo-FRET results in a decrease in polarization, monitored by the steady-state anisotropy (Figure 52).458 H2B-K120ubSS specifically interfered with the later stages of compaction, but did not alter chromatin structure at low concentrations of MgCl2.457 This effect could be completely reversed by treating H2B-K120ubSS-containing arrays with DTT. Interestingly, acetylated H4, generated by NCL, inhibited chromatin compaction even at low ionic strength, indicating that these modifications alter chromatin structure through different mechanisms. In addition to their impact on array compaction, these PTMs reduce the propensity for interstrand interactions.407,413,457
Figure 52.
Homo-FRET assay to monitor chromatin compaction. In the extended conformation, fluorescein labels (yellow stars) are far apart, thus limiting the amount of homo-FRET. Upon compaction, the distance between fluorophores is decreased, resulting in homo-FRET, which is detected by a reduction in the steady-state anisotropy (SSA) of the system.
Besides the canonical H2B ubiquitylation at K120, this PTM also occurs on K34, K46, K108, and K116.459 Brik and co-workers developed a synthetic protocol to generate H2B site-specifically ubiquitylated at K34 to shed light on this lesser known modification.460 This tour-de-force strategy entailed the convergent assembly of H2B-K34ub from five fragments (Figure 53). First, H2B containing a thiolated lysine analogue at position 34 was assembled. The synthesis was initiated by the ligation of an HA-tagged N-terminal fragment (residues 1–20) to residues 21–57. The latter fragment contained an A21C mutation and an o-nitrobenzyl protected δ-mercaptolysine at position 34. Lys57 was Nvoc protected to preclude lactamization upon oxidation of the C-terminal acylhydrazide moiety. Concurrently, the C-terminal fragment (residues 97–125, A97C) was condensed with residues 58–96, where A58 was replaced by a thiazolidine-protected cysteine residue. Upon deprotection of Cys58, residues 58–125 were coupled to residues 1–57, activated as an acyl azide. Photolysis liberated the δ-mercaptolysine residue, which was reacted with a ubiquitin α-thioester. Finally, desulfurization afforded native H2B-K34ub.
Figure 53.

Stepwise total synthesis of H2B-K34ub. This convergent ligation strategy involving four NCL steps commences with the synthesis of orthogonally protected histone H2B (top to bottom right) and finishes with ubiquitin conjugation and desulfurization (bottom left).
To complement SPPS-based approaches, Virdee et al. reported biosynthetic incorporation of N-ε-protected δ-mercaptolysine residues into proteins.435 Upon deprotection by photolysis, this residue can be reacted with ubiquitin α-thioesters by NCL. Subsequent desulfurization results in traceless, site-specifically ubiquitylated proteins.
Chemical synthesis is a reliable method to grant access to diversely modified histones. With state of the art semisynthesis methods, any chemically stable PTM can be homogeneously incorporated at any position within histones, although modifications at the termini are preferable. Notably, histone semisynthesis allows the installation of many PTMs into a single protein, in particular if the sites are clustered at the termini. Semisynthetic nucleosomes have served to unravel the biochemical and biophysical consequences of a broad variety of histone modifications. Histone ubiquitylation in particular has been fertile ground for chemical explorations, resulting in an assortment of technologies that promise to find use far beyond chromatin biochemistry.
4.4. Synthesis of Chromatin with Defined PTM Patterns
In cells, histone PTMs rarely manifest in isolation.461,462 Instead, patterns of modifications co-occur within spatially defined chromatin states.463,464 Thus, chromatin domains display global enrichment of specific histone PTMs and their combinations. Within each domain, however, histone modifications are not uniformly distributed but occur in specific patterns that help guide DNA-templated processes.465 For example, methylation of H3K4 is focused at transcription start sites, while the abundance of H3K79me2 and H3K36me3 peaks early and late, respectively, within the coding sequence of actively transcribed regions.466,467 These observations prompted the in vitro preparation of chromatin templates that carry selected patterns of histone PTMs.
4.4.1. Multivalent Recognition of Histone PTMs
Individual reader domains are often sensitive to histone PTMs adjacent to their cognate mark.165 To integrate multiple signals present on distal sites within the same tail or on separate histones, many nuclear proteins harbor several binding modules, or multiply their interaction capabilities through oligomerization.464,468 Moreover, additional sensor domains are also frequently contributed by partners in multisubunit complexes. In some cases, cooperative binding of effectors to several PTMs can be measured using histone peptide substrates. Examples include the recognition of polyacetylated histone tails by certain bromodomains53,54 (see section 3.1.2) or the interaction between a chromodomain dimer of CMT3 with H3 tails trimethylated at K9 and K27.469 However, when PTMs are distributed to different histones, the use of site-specifically modified nucleosomes to investigate the binding mechanism becomes imperative.
The coupled PHD-BD of BPTF (introduced in section 3.2.3) is the paradigm for this mode of multivalent PTM recognition.470 Individually, the PHD finger specifically binds to H3K4me3, but the bromodomain promiscuously interacts with several acetylated H4 peptides on SPOT arrays and in solution. To test if coupling of the domains would impart specificity on the BD, Ruthenburg et al. assembled nucleosomes containing combinations of unmodified histones, semisynthetic H3K4me3, and H4 acetylated at K12, K16, or K20 (Figure 54). The BD alone was insufficient for binding any of the nucleosomes, but in the presence of the PHD and H3K4me3, selective interaction with H4K16ac-containing nucleosomes was observed. Synergistic binding depended on proper orientation of the PHD-BD: disruption of the helical linker or insertion of additional residues to alter the relative rotation between the domains abolished the cooperativity. This landmark study demonstrated that coupled binding modules are indeed capable of recognizing histone PTM patterns, which dramatically increases the depth of information that can be administered on nucleosomes.
Figure 54.
Bivalent recognition of doubly modified mononucleosomes by BPTF. The PHD finger of BPTF binds to H3K4me3 (gray arrows). In a nucleosomal context, this binding is reinforced through the recognition of H4K16ac by the adjacent bromodomain (black arrow).
4.4.2. Asymmetric Nucleosomes
Histone octamers are inherently symmetric entities, but DNA sequence can render nucleosomes asymmetric. Upon post-translational modification of nucleosomes, one histone copy is most likely targeted first, providing an additional level of asymmetry. Little is known about the biological significance and functional consequence of asymmetrically modified nucleosomes, but recent progress based on studying laboratory manufactured asymmetric nucleosomes in vitro has provided insight into their recognition, and enabled the development of a pipeline to quantify symmetry of nucleosomes from cells.
Pioneering work on dissecting the contribution of single tails to the biochemistry of nucleosome acetylation was performed in the Shogren-Knaak laboratory.409,471 Prompted by the observation that H3 acetylation by the yeast SAGA complex was cooperative on nucleosome arrays and mononucleosome substrates but not histone peptides, this group prepared asymmetrically modified nucleosomes.471 Histone octamers were refolded in the presence of a 10-fold excess of unmodified H3 over a modified His-tagged H3 variant (Figure 55a). Subsequent purification by metal affinity chromatography yielded complexes with one copy of wild-type H3 and either tail-less, Lys-to-Ala mutant (residues 9, 14, 18, 23), or tetra-acetylated H3. Histone acetyltransferase assays with SAGA revealed that cooperative acetylation hinges upon the presence of two acetylatable H3 tails (Figure 55b). Preacetylation of one H3 tail increased the affinity of SAGA for its substrate, and the bromodomain of GCN5 (the active subunit of the complex) was required for this effect.409 Thus, the coupling of a BD with a histone acetyltransferase (HAT) domain leads to burst-like nucleosome acetylation to aid in transcription activation.
Figure 55.

Preparation and application of asymmetric mononucleosomes. (a) Synthesis of asymmetrically modified nucleosomes using a tagged, modified copy of H3 and an excess of an unmodified version. (b) The SAGA-complex is stimulated by its own mark. Nucleosomes that can only be acetylated on one H3 tail (tail-less and Lys9,14,18,23Ala) are poor SAGA substrates (gray arrows). Asymmetrically acetylated nucleosomes (right) recruit SAGA (dashed arrow) to promote acetylation of the unmodified H3 tail.
Asymmetrically modified histones have also shed light on the mechanism of PTM binding and crosstalk. Partially ubiquitylated histones, for instance, revealed that each H2B-K120ub molecule stimulates Dot1 methylation on only one nucleosome face, presumably in an orthosteric fashion.452 In addition, HP1 dimers bound mononucleosomes with only one H3K9me3 mark equally well as doubly modified variants.331 It is therefore likely that each of the dimer’s CDs engages a distinct nucleosome to effect compaction of chromatin regions demarked with the heterochromatin mark H3K9me3.334,472
Asymmetric nucleosomes produced in vitro also represent a gateway to quantify nucleosome asymmetry in vivo.473 Voigt et al. harnessed tandem affinity purification of H3/H4 tetramers, where one copy of H3 is furnished with a trimethyllysine analogue at position 27 and a Strep tag, while the wild-type copy of H3 contains a His-tag (Figure 56a). Purification by Ni-NTA chromatography, followed by a streptactin column, provided access to mononucleosomes containing one copy of each H3 version. Immunoprecipitation of asymmetrically modified mononucleosomes, as well as unmodified and doubly methylated congeners with K27me3-specific antibodies and subsequent analysis of H3 variants by MS, confirmed the expected composition of each nucleosome batch: Asymmetric nucleosomes contained equal proportions of wild-type and K27me3, while symmetrically trimethylated nucleosomes contained only K27me3. Unmodified nucleosomes were not enriched by the pull-down step. When the same analytics workflow was applied to mononucleosomes extracted from embryonic stem cells, the presence of symmetrically and asymmetrically modified nucleosomes was established on the basis of the relative amounts of K27me2/3 and K27me0/1 before and after immunoprecipitation (Figure 56b). Approximately one-half of the total mononucleosomes contain K27 in the me0/1 methylation state in both H3 copies, and nucleosomes containing K27me2/3 on both tails are overrepresented as compared to asymmetric versions. A particularly interesting finding concerns so-called bivalent domains. These regions of chromatin are common in stem cells and contain H3K4me3 and H3K27me3, archetypal activating and repressive PTMs, respectively. Analysis of the symmetry of bivalent nucleosomes failed to detect histones that are simultaneously trimethylated at H3K4 and H3K27, suggesting that these marks are present on different tails within one nucleosome (Figure 56c). Consistent with this observation, nucleosome arrays containing cysteine-derived trimethyllysine analogues at H3K4 in only one H3 copy were substrates for the H3K27-specific methyltransferase PRC2. In contrast, arrays homogeneously carrying trimethylated thialysine at H3K4 could not be further methylated by PRC2 at H3K27. These results demonstrate that symmetrically and asymmetrically modified nucleosomes exist in vivo, and might exert unique biochemical downstream effects.
Figure 56.
Nucleosome asymmetry in vivo. (a) Assembly of asymmetric H3/H4 tetramers using a tandem affinity tag strategy. (b) Distribution of H3K27 methyl marks in ES cells into symmetric and asymmetric mononucleosomes. (c) Bivalent domains consist of asymmetric nucleosomes with one H3 tail di- or trimethylated at Lys4, and another tail marked with K27me2/3.
4.4.3. Synthesis of Sequence-Specific Oligonucleosome Arrays
Many functional features of histone PTMs can be recapitulated at the mononucleosome level or with homogeneous arrays, but certain phenomena are dependent on the presence of neighboring sites with specific properties. Examples of such chromatin transactions include the multivalent recognition of heterotypic PTMs distributed on different nucleosomes, or deposition of homo- or heterotypic modifications upon stimulation in trans. Sequence-defined hetero-oligomers of chromatin are therefore indispensable to study the effect of histone PTMs on distinct nucleosomes. Their preparation and applications are detailed below.
More than 10 years ago, Zheng and Hayes used asymmetric dinucleosomes that contain a phenylazide photo-cross-linking group on the N-terminal histone tails on one of the nucleosomes and a reporter on the DNA of the other to investigate internucleosomal contacts.299 To construct these reagents, homogeneously modified mononucleosomes were linked using T4 DNA ligase (Figure 57a). Specificity in the linkage was configured by nonpalindromic single-stranded overhangs present on the mononucleosome starting material. Following UV irradiation and DNA cleavage, cross-linked products were detected by gel shift experiments, which informed on contacts between histone tails and DNA on a neighboring nucleosome. The results showed that the N-terminal tails of H2A and H2B, but not H3 and H4, partook in internucleosomal interactions in this dinucleosome system. Extending the nucleosome ligation technology to tetranucleosome arrays, Blacketer et al. investigated the contribution of H4 tails on interstrand association.474 Installation of one to four nucleosomes lacking the H4 tails into sequence-defined tetranucleosome arrays revealed that the H4 tails cooperate to mediate chromatin self-assembly.
Figure 57.

Oligonucleosome arrays. (a) Synthesis of asymmetric dinucleosomes using nonpalindromic DNA overhangs and their application in studying histone-DNA contacts. Cross-links are detected through a gel-shift of the 32P-labeled DNA. (b) BPTF binds its marks (H3K4me3, blue flag, and H4K16ac, green circle) in a mononucleosomal context (black arrow). (c) H2B-K120ub stimulates Dot1 in cis (black arrow), but not toward methylation of adjacent nucleosomes. (d) Clr4/Suv39-mediated spreading of the heterochromatin-associated H3K9me3 mark (red flag). (e) Rpd3s deacetylation is stimulated by H3K36 methylation (orange flag) in an intra- and internucleosomal fashion. Note that Rpd3S recognizes dinucleosomes more readily than similarly modified mononucleosomes.
Sequence-defined nucleosome arrays have also been harnessed to analyze the geometric preference of PTM binding and histone modifying enzymes.324,445,470,475 For example, comparative assays using mono- and heterotypic dinucleosomes established that BPTF preferentially engages H3K4me3 and H4K16ac on the same nucleosome (Figure 57b).470 Similarly, Dot1 was determined to act in an intranucleosomal cross-talk, but cannot methylate nucleosomes adjacent to H2B-K120ub (Figure 57c).445
By contrast, several histone-modifying enzymes are controlled by histone PTMs in an internucleosomal fashion. For instance, the histone methyltransferase Clr4 (the yeast homologue of the human Suv39h1) mediates heterochromatin spreading330,476 by propagating a methyl mark in trans.324 This enzyme uses a SET domain to methylate H3K9,70 and a chromodomain to bind to its product, H3K9me3, thus generating a positive feedback loop.477 Narlikar and co-workers used dinucleosomes composed of one octamer with unmodified H3 and one octamer containing H3K9Cme3 to study Clr4 activation (Figure 57d).324 The presence of the K9me3 mark stimulated methylation of the adjacent mononucleosome, but not when the different mononucleosomes were incubated in trans. Kinetic characterization demonstrated that this circuit operates by enhancing catalysis by Clr4, rather than binding to the hemimodified dinucleosomes. Another example of internucleosomal effects on enzymatic activity is provided by the histone deacetylase, Rpd3s.478,479 This enzyme binds thialysine analogues of H3K36me3, preferentially in a dinucleosomal context.478 In model ligated dinucleosomes, K36 methylation stimulated deacetylation both intra- and internucleosomally (Figure 57e),479 thereby allowing Rpd3s to produce a hypo-acetylated microenvironment surrounding H3K36me3 marks to suppress aberrant transcription initiation within coding sequences.480
Biochemical and biophysical analyses using ordered nucleosome arrays have thus provided insight into the geometric properties of a variety of chromatin-associated systems. Additional levels of sophistication in terms of size and composition of arrays to reflect the heteropolymeric complexity of chromatin in vivo will increase the resolution and diversity of future chromatin-related challenges that can be addressed in vitro.
4.5. Increasing the Throughput of Chromatin Biochemistry
In the last 10 years, methods to produce site-specifically modified histones have burgeoned. Armed with these tools, chemists and biologists have captured the mechanistic essence of a broad range of phenomena operating on chromatin. Despite this tremendous progress, crafting “designer” chromatin and deploying these precious reagents in biochemical and biophysical investigations has remained a challenging pursuit. Therefore, strategies geared toward parallelized interrogation of the features that define the molecular circuits operating on chromatin and the nuclear proteome are extremely valuable.
4.5.1. Identification of PTM-Specific Chromatin Binding Proteins
SILAC-based approaches (see also sections 3.2.3 and 3.11.2) represent appealing strategies to identify factors that interact with nucleosomes in a PTM-dependent fashion and on a proteome-wide level. Kouzarides and co-workers explored the possibility for crosstalk in the interactions mediated by histone and DNA methylation.481 A set of three modified H3 variants was assembled by ligating peptide α-thioesters containing H3K4me3, K9me3, or K27me3 to a tail-less recombinant fragment with an N-terminal cysteine in place of Thr32. Subsequently, the semisynthetic histones were incorporated into nucleosomes in the presence or absence of methylation at C5 of cytosine in CpG dinucleotide sequences, artificially installed with a prokaryotic DNA methyltransferase. Initially, the approach was validated by screening known interactors of specific Kme3 marks such as HP1 for binding H3K9me3 or the PRC2 subunit Suz12 for (indirectly) engaging H3K27me3. By comparing the SILAC profiles between nucleosomes containing methylated DNA and/or methylated histones, some novel interactions were observed. For instance, several members of the origin recognition complex associated specifically with the heterochromatin marks H3K9me3 and H3K27me3. In certain cases, synergies and antagonisms between histone and DNA methylation were detected (Figure 58a). The former behavior is exemplified by UHRF where binding to H3K9me3-containing nucleosomes (discussed in detail in section 3.12.1) was reinforced by DNA methylation. By contrast, the interaction of PRC2 components with H3K27me3 was diminished when CpG groups were methylated.
Figure 58.

Identification of PTM-binding factors using modified chromatin as bait. (a) Synergies and antagonisms between DNA and histone methylation recognition. (b) Peptide- and chromatin-based probes reveal partially overlapping interactors. Here only a few examples are shown: BPTF and CHD4 are associated with chromatin remodeling, ING1 is a transcriptional regulator, TFIID is a general transcription factor complex, SIN3 is a histone deacetylase, PCAF and CDYL are histone acetyltransferases, HP1 is a “glue” for heterochromatin, UHRF is a recruiter of DNA methyltransferase, and NUP93 is a member of the nuclear pore. (c) Examples of H2B-K120ub-binding complexes. (d) Synthesis of a hydrolase-resistant H2B-K120ub analogue.
Nikolov et al. used site-specifically modified nucleosome arrays for SILAC-based identification of PTM-specific binders, and directly compared the results to screens performed with peptide-based affinity matrixes.482 These analyses expanded the list of potential readers for the H3K4me3 and H3K9me3 marks, including the spindle-associated protein Spindlin1. The H3K4me3 binding properties of Spindlin1 have been corroborated by a contemporary study relying on biochemical analyses with nucleosomes containing methyllysine analogues and structural data.483 Of note, the candidate list generated by using methylated chromatin partially overlaps with the predictions from mononucleosome and peptide-based SILAC assays, but each format yielded a remarkable set of unique proteins (Figure 58b). Possibly, the high concentrations of peptides that can be employed enable the isolation of weakly interacting modules. In contrast, chromatin-based templates provide additional contact opportunities, particularly in the case of nucleosome arrays, which are inherently polyvalent.
In related proteomic studies, arrays containing H2B-K120ub were used as a bait to discover specific binding proteins for this PTM.484 Resilience against deubiquitination was imparted by methylation of the N-ε-amino group of Lys120 according to the method of Brik and co-workers.485 Specifically, this modification was installed by on-resin alkylation of the N-ε-amino group of lysine 120 within a synthetic H2B peptide (residues 118–125), enabled by orthogonal protection of this residue. Subsequently, the secondary amine was acylated with the C-terminal Gly residue of ubiquitin, followed by SPPS of the C-terminal portion of ubiquitin. Iterative NCL and ensuing desulfurization provided the final, deubiquitinase resistant H2B-K120ub analogue. As anticipated, SILAC analysis confirmed known H2B-K120ub interacting modules, and suggested that several complexes associated with diverse chromatin transactions (gene expression, DNA replication, chromatin remodeling, etc.) recognize this moiety (Figure 58c). Follow-up experiments confirmed that ubiquitylated H2B interacts with SWI/SNF family chromatin remodelers, and that this association is important for gene regulation.
By further altering the linkage between histones and ubiquitin, Long et al. were able to generate deubiquitinase-resistant H2A-K119ub and H2B-K120ub isoforms.486 The authors joined a G76C mutant of ubiquitin to H2B-K120C or H2A-K119C via a dichloroacetone cross-link (Figure 58d).486 This non-native linkage is hydrolase-resistant due to the absence of an isopeptide bond, the increased length of the module, and the presence of a carboxyl group. Notably, because of the hydrolytic stability of these compounds, H2A-K119ub/H2B or H2A/H2B-K120ub dimer baits could be used to isolate the deubiquitinase Usp15 from HeLa cell nuclear extracts. This enzyme was able to cleave semisynthetic H2B-K120ub containing the native isopeptide linkage, preferentially in the form of histone octamers rather than nucleosomes.
Thus, the union of histone and chromatin substrates with modern proteomic approaches such as SILAC has provided substantial insight into how PTMs are recognized in a chromatin context on a proteome-wide level. Biochemical and genetic follow-up studies have confirmed several predicted interaction pairs, attesting to the general utility of these sophisticated screens.
4.5.2. Chromatin Biochemistry with DNA-Barcoded Nucleosome Libraries
Peptide libraries have greatly increased the throughput in the analysis of signaling through histone modifications (section 3.12). Can similar strategies be harnessed to accelerate chromatin biochemistry at the nucleosome level? To realize this goal, two challenges must be overcome. Specifically, synthetic protocols to obtain nucleosome libraries and analytical methods to read out the desired biochemical properties of the library members need to be implemented.
Nguyen et al. have recently solved these issues and developed a versatile platform to perform chromatin biochemistry with increased throughput.487 Nucleosome libraries containing over 50 unique combinations of acetylation, methylation, and ubiquitylation signatures were assembled from semisynthetic histones on a microgram scale (Figure 59). Importantly, each nucleosome contained a unique hexanucleotide barcode that specified the histone variants from which the particular nucleosome is constructed. Library members were subjected to various biochemical assays, and desired variants isolated by affinity- or immunoprecipitation (IP) and analyzed by next generation DNA sequencing, which provides exquisite sensitivity. This workflow was applied to profile the specificity of PTM-specific antibodies, histone binding proteins, and histone modifying enzymes. For example, the coupled BD-PHD of BPTF (see also section 4.4.1) was found to display a marked preference for nucleosomes containing H3K4me3 and polyacetylated H4. Similarly, polyacetylated H4 mediated the recruitment of p300 to nucleosomes. This interaction stimulated p300-dependent acetylation of H3, as determined by performing pull-downs with antibodies selective for the p300 product H3K18ac. This positive feedback loop was also observed when nucleosome libraries were incubated with nuclear extracts: H4 acetylation promoted H3 acetylation.
Figure 59.

Schematic overview of a screening platform based on DNA-barcoded nucleosome libraries. Recombinant and semisynthetic histones are refolded into >50 different octamers in parallel, and assembled into mononucleosomes with barcoded DNA. Upon pooling, aliquots of the library are subjected to biochemical assays involving a pull-down step to enrich variants that exhibit certain traits. Subsequently, nucleosomal DNA is isolated and analyzed by next generation sequencing to provide a semiquantitative readout of hundreds to thousands of experiments.
DNA barcoding enables storage of the synthetic and biochemical history of each MN variant in an easily interpretable format, which facilitates ultrasensitive and versatile readout of the molecular properties of the corresponding nucleosome. Accordingly, Nguyen et al. were able to decipher how multiple PTMs on chromatin are interpreted and converted into orthogonal signals. While shown for binding studies, acetyltransferase and methyltransferase reactions, DNA-barcoded nucleosome libraries may find application in many more areas of chromatin biochemistry and biophysics.
5. Summary and Future Perspectives
Peptide and protein chemistry have become an integral part of chromatin research. Methods ranging from solid-phase synthesis to recombinant technology are available to construct site-specifically modified histone peptides and chromatin templates with distinct patterns. In particular, a plethora of histones carrying PTMs and their analogues have been generated in a chemically defined fashion (Figure 60). These reagents can directly feed into cutting edge biochemical and biophysical pipelines, including transcription assays341 or structural studies based on X-ray crystallography,282,488,489 electron microscopy,283,490 or NMR spectroscopy.491,492 Given these advances, what are the remaining challenges and opportunities for protein chemists to further contribute to unraveling the mechanism of histone-based signaling?
Figure 60.

Diagram of PTMs and their analogues that have been site-specifically incorporated into histones (as of September 2014). For clarity, connections are shown only to one copy of each histone.
5.1. Tackling the Combinatorial Complexity
Several peptide- and mononucleosome based approaches have been developed to biochemically address the enormous combinatorial possibilities of histone PTM combinations. Nevertheless, methods to synthesize hundreds of proteins in parallel are still lacking. Innovative purification schemes or reliable fragment condensation protocols that allow bypassing of individual workup steps are needed to attain the level of throughput that peptide synthesis can achieve. Furthermore, can the resulting histone libraries be incorporated into templates that more closely reflect the heteropolymeric nature of chromatin fibers? Such arrays will enable dissection of spatial components that underlie the control of chromatin-templated processes, both on a biophysical and on a biochemical level. Specific aspects that remain largely unanswered include questions concerning how, which, and if defined PTM patterns alter structures synergistically, and whether these structural perturbations are propagated along the chromatin fiber beyond the actual installation site. Transcription is a vectorial process, and chromatin architecture contributes to defining the coordinates of the origin and direction of polymerase action. At which level do histone PTM gradients facilitate guidance of the transcription machinery, and in what ways do these modification patterns also contribute to local memory of transcriptional states? We believe that some of these issues can be addressed with next-generation chromatin biochemistry on the foundation of designer histones.
5.2. Beyond Histones
The vast majority of contributions that protein chemistry has so far made in the chromatin biochemistry area have revolved around histone modifications. Yet, many other chromatin-associated proteins are hubs for PTMs, in particular RNA polymerase and coactivators such as p53. In addition, many enzymes characterized as histone methyl- and acetyltransferases also act on nonhistone targets. Thus, elucidating the mechanism of these processes requires that the chemical toolkit, originating in basic research, and since refined for histone synthesis, be extended to the manufacture of other cellular factors that are considerably larger than histones. Semisyntheses of p53 (ref (493)), a bacterial RNA polymerase,494 as well as the p300 HAT domain495 have already been achieved, laying the groundwork for systems-wide analysis of how PTM-based nuclear signaling affects transcription.
5.3. Synthetic Chromatin Chemistry in Live Cells
Modified histones have contributed immensely to biochemical and biophysical analyses in vitro. What are the prospects of implementing these reagents in vivo to elucidate the mechanism of their action? Currently, access to specifically modified histones in vivo is mainly limited to genetic strategies. Typical examples include site-directed mutagenesis of a target histone residue, such as Lys-to-Gln or Lys-to-Arg substitutions to mimic acetyllysine side-chains and preclude methylation or acetylation at that position, respectively.290,496 Alternatively, overexpression of a histone-modifying enzyme can result in global accumulation of a desired PTM. Upon targeting enzymes to specific genomic sites (using the Gal4 system, or perhaps CRISPR-CAS9), perturbations can be localized to genetic reporters, enabling more defined functional assignments of histone modifying activities. In conjunction with reversible dimerization modules, these targeting strategies can shed light on the kinetics of the formation and interpretation of histone PTMs.497 Artificial expansion of the genetic code further diversifies the scope of genetic approaches to study the effect of histone modifications, as exemplified by the recent success in tracking structural consequences of mitotic histone phosphorylation.380 A multitude of bioorthogonal reactions,498 as well as the ability to perform protein trans-splicing in vivo,499,500 might also aid in generating designer chromatin in living cells.
Fueled by diverse success stories since the late 1960s, the journey for chemical biologists into the chromatin field continues. Many exciting milestones still lie ahead, with key challenges involving the vastness of the combinatorial landscape of histone modifications and the complexity of their interpretation in a cellular context. Whether these routes lead to high-throughput biochemistry or meander into cells, the journey promises to be extremely fruitful. These efforts will likely contribute to the rich tradition at the intersection of peptide and protein chemistry with histone biology, and target a systems-level understanding of how cellular signaling converges on chromatin and is relayed into functional outputs.
Acknowledgments
Research in the Muir lab discussed in this Review was supported by the U.S. National Institutes of Health (grants R37-GM086868 and R01 GM107047). A postdoctoral fellowship from the Swiss National Science Foundation to M.M.M. is gratefully acknowledged. We thank Drs. Galia Debelouchina and Zachary Brown for helpful discussions and Geoff Dann and Luis Guerra for careful proofreading of this document.
Glossary
Abbreviations
- AA
amino acid
- A, Ala
alanine
- C, Cys
cysteine
- D, Asp
aspartic acid
- E, Glu
glutamic acid
- F, Phe
phenylalanine
- G, Gly
glycine
- H, His
histidine
- I, Ile
isoleucine
- K, Lys
lysine
- L, Leu
leucine
- M, Met
methionine
- N, Asn
asparagine
- P, Pro
proline
- Q, Gln
glutamine
- R, Arg
arginine
- S, Ser
serine
- T, Thr
threonine
- V, Val
valine
- W, Trp
tryptophan
- Xaa
any amino acid
- Yaa
any amino acid
- Nle
norleucine
- aaRS
amino-acyl tRNA synthetase
- AB
antibody
- Ac
acetyl
- ACF
ATP-utilizing chromatin assembly and remodeling factor 1
- ADD
type of zinc finger found in ATRX, DNMT3, DNMT3L
- ADP
adenosine diphosphate
- AIRE
autoimmune regulator, DNA binding protein that binds unmodified H3 tails (Arg2, Lys4)
- AMP
adenosine monophosphate
- APB
4-azidophenacylbromide
- ASF1
antisilencing function protein 1, a histone chaperone
- ATP
adenosine triphosphate
- ATRX
alpha thalassemia/mental retardation syndrome X-linked, transcription regulator, and chromatin remodeler
- BAH
bromo-adjacent homology, methyllysine binding domain
- BD
bromodomain, acetyllysine binding domain
- BHC80
PHD finger protein 21A, member of a deacetylase complex
- Boc
tert-butoxycarbonyl
- Boc-OSu
N-(tert-butoxycarbonyloxy)succinimide
- BPTF
bromodomain and PHD finger-containing transcription factor, component of the nucleosome-remodeling factor (NURF) complex
- BRD4
bromodomain-containing protein 4, associated with transmission of epigenetic memory
- BRE1A
ring finger protein 20, E3 ubiquitin ligase, mediates H2B–K120 ubiquitylation
- CAF
chromatin assembly factor
- CD
chromodomain, methyllysine binding domain
- Chd1
chromodomain-helicase-DNA-binding protein 1, chromatin remodeler, component of the SAGA complex
- ChIP
chromatin immunoprecipitation
- DIC
diisopropylcarbodiimide
- Dim-5
H3K9-specific methyltransferase from Neurospora crassa
- DIPG
diffuse intrinsic pontine glioma, a brainstem tumor that occurs predominantly in children
- DMS
dimethylsulfide
- DTD
double tudor domain
- DTT
dithiothreitol
- EPL
expressed protein ligation
- EPR
electron paramagnetic resonance spectroscopy
- EZH2
enhancer of zeste 2, active subunit of PRC2
- FAD
flavin adenine dinucleotide
- Fmoc
9-fluorenylmethoxycarbonyl
- Fpr4
FK506-binding protein 4, peptidyl-prolyl cis–trans isomerase
- FRET
Förster resonance energy transfer
- G9a
H3K9 specific histone methyltransferase, also known as EHMT2
- GCN5
general control of amino acid synthesis protein 5, a histone acetyltransferase subunit of the SAGA complex
- GlcNac
N-acetylglucosamine
- GST
glutathione-S-transferase
- HAT
histone acetyltransferase
- HATU
O-(7-azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate
- HBTU
O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- HCTU
O-(6-chlorobenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- HMT
histone methyltransferase
- HOBt
hydroxybenzotriazole
- HP1
heterochromatin protein 1, binds H3K9me2/3
- HPLC
high-pressure liquid chromatography
- HST2p
homologous to SIR2 protein 2, a histone deacetylases
- ING2
protein inhibitor of growth, uses a PHD finger to recognize H3K4
- Isw2
imitation switch protein 2, chromatin remodeler
- ITC
isothermal titration calorimetry
- JmjD2a
Jumonji-domain-containing protein 2A, H3K36-specific histone demethylase
- LSD1
lysine-specific demethylase 1, demethylates H3K4me1/2
- MAR
mono-ADP-ribosylation
- MBT
malignant brain tumor repeat, methyllysine binding domain
- Me
methyl
- MLL
myeloid/lymphoid or mixed-lineage leukemia, H3K4-specific methyltransferase
- MS
mass spectrometry
- MSL3
male-specific lethal 3 homologue, member of the MSL histone acetyltransferase complex, binds H4K20me1/2
- MST1
mammalian STE20-like protein kinase 1, phosphorylates S14 of H2B
- MTSL
(1-oxyl-2,2,5,5-tetramethylpyrroline-3-methyl)-methanethiosulfonate
- NAP1
nuclear assembly protein 1
- NCL
native chemical ligation
- NSD1
nuclear SET domain-containing protein 1, a H3K36 and H4K20-specific methyltransferase
- NSD2
nuclear SET domain-containing protein 2, a H3K36-specific methyltransferase
- ORC
origin of recognition complex
- Oxyma
ethyl (hydroxyimino)cyanoacetate
- p300
histone acetyltransferase
- p53
tumor suppressor
- PAD4
peptidyl arginine deiminase 4
- PAR
poly-ADP-ribosylation
- PCAF
p300/CBP-associated factor, histone acetyltransferase
- PELDOR
pulsed electron double resonance spectroscopy
- Pg
protecting group
- Ph
phosphoryl
- PHD
plant homeodomain, contains a zinc-finger like motif
- POI
protein of interest
- PRC2
polycomb repressor complex, mediates methylation of H3K27
- PSIP1
PC4 and SFRS1 interacting protein 1, a transcriptional coactivator
- PTM
post-translational modification
- PWWP
methyllysine binding domain, contains a conserved Pro-Trp-Trp-Pro sequence
- PyBOP
(benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate
- RAG2
V(D)J recombination activating protein 2, recognizes K4me
- RbAp48
retinoblastoma binding protein 4, a histone-binding component of PRC2 and CAF
- rDNA
rRNA encoding DNA
- RNA
ribonucleic acid
- RP
reverse phase
- SAGA
Spt-Ada-Gcn5 acetyltransferase
- SET
Su(var.)3–9, enhancer-of-zeste and Trithorax, methyltransferase domain
- SET2
H3K36-specific methyltransferase
- SET7/9
H3K4-specific histone methyltransferase
- SILAC
stable isotope labeling by amino acids in cell culture
- SP140
speckled 140 kDa, component of the nuclear body
- SPOT
synthesis of peptides on a membrane support
- SPPS
solid-phase peptide synthesis
- SUMO
small ubiquitin-like modifier
- SUZ12
suppressor of zeste 12, subunit of PRC2
- SWI/SNF
switch/sucrose nonfermentable, a nucleosome remodeling complex
- TFA
trifluoroacetic acid
- TFMSA
trifluoromethanesulfonic acid
- TRIM28
transcription intermediary factor 1-beta, corepressor
- tRNA
tRNA
- TROSY
transverse relaxation-optimized spectroscopy
- TTD
tandem tudor domain
- Ub
ubiquityl
- UHRF
ubiquitin-like PHD and RING finger domain-containing protein, binds H3K9me3 and associates with DNA methyltransferases
- Ulp1
ubiquitin-like-specific protease, a deubiquitylase
- V(D)J
variable, diverse, joining; recombination mechanism during B- and T-cell maturation
Biographies

Manuel M. Müller received his masters in Biochemistry from ETH Zürich, Switzerland, in 2006. After a summer research stay with Professor Andrew D. Ellington at the University of Texas, Austin, he returned to the Laboratory of Organic Chemistry at ETH Zürich to pursue his doctoral studies under the supervision of Professor Donald Hilvert as an SSCI fellow. His Ph.D. thesis involving model systems for primordial enzymes was awarded with the ETH medal in 2011. In 2010, he joined the laboratory of Professor Tom W. Muir, then at Rockefeller University in New York, now at Princeton University, where he is developing new tools for chromatin biochemistry and applying them to investigate the (mis-)regulation of histone-modifying enzymes.

Tom W. Muir received his B.Sc. (Hons, 1st class) in Chemistry from the University of Edinburgh in 1989 and his Ph.D. in Chemistry from the same institute in 1993 under the direction of Professor Robert Ramage. After postdoctoral studies with Stephen B. H. Kent at the Scripps Research Institute, he joined the faculty at the Rockefeller University in New York City in 1996, where he was, until recently, the Richard E. Salomon Family Professor and Director of the Pels Center of Chemistry, Biochemistry and Structural Biology. In 2011, Dr. Muir joined the Princeton Faculty as the Van Zandt Williams Jr. Class of 1965 Professor of Chemistry. He has published over 170 scientific articles in the area of chemical biology and is best known for developing methods for the preparation of proteins containing unnatural amino acids, posttranslational modifications, and spectroscopic probes. These approaches are now widely employed in academia and industry. His current interests lie in the area of epigenetics, where he tries to illuminate how chemical changes to chromatin are linked to different cellular phenotypes. Professor Muir has won a number of honors for his research, including the Burroughs-Wellcome Fund New Investigator Award, the Pew Award in the Biomedical Sciences, the Alfred P. Sloan Research Fellow Award, the Leonidas Zervas Award from the European Peptide Society, the Irving Sigal Award from the Protein Society, the 2008 Vincent du Vigneaud Award in Peptide Chemistry, the 2008 Blavatnik Award from the New York Academy of Sciences, the 2008 Distinguished Teaching Award from The Rockefeller University, the 2012 Jeremy Knowles Award from the Royal Society of Chemistry, and a 2013 Arthur C. Cope Scholar Award from the American Chemical Society. Dr. Muir is a Fellow of American Association for the Advancement of Science and the Royal Society of Edinburgh.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Kornberg R. D. Annu. Rev. Biochem. 1977, 46, 931. [DOI] [PubMed] [Google Scholar]
- Woodcock C. L.; Ghosh R. P. Cold Spring Harbor Perspect. Biol. 2010, 2, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T.; Allis C. D. Science 2001, 293, 1074. [DOI] [PubMed] [Google Scholar]
- Badeaux A. I.; Shi Y. Nat. Rev. Mol. Cell Biol. 2013, 14, 211. [PubMed] [Google Scholar]
- Epigenetics; Allis C. D., Jenuwein T., Reinberg D., Eds.; Cold Spring Harbor Press: Cold Spring Harbor, NY, 2007. [Google Scholar]
- Ahmad K.; Henikoff S. Cell 2002, 111, 281. [DOI] [PubMed] [Google Scholar]
- Kornberg R. D.; Lorch Y. Curr. Opin. Genet. Dev. 1999, 9, 148. [DOI] [PubMed] [Google Scholar]
- Bird A. Genes Dev. 2002, 16, 6. [DOI] [PubMed] [Google Scholar]
- Paro R. Trends Genet. 1995, 11, 295. [DOI] [PubMed] [Google Scholar]
- Chi P.; Allis C. D.; Wang G. G. Nat. Rev. Cancer 2010, 10, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn M.; Dambacher S.; Schotta G. J. Appl. Physiol. 2010, 109, 232. [DOI] [PubMed] [Google Scholar]
- Schneider R.; Bannister A. J.; Kouzarides T. Trends Biochem. Sci. 2002, 27, 396. [DOI] [PubMed] [Google Scholar]
- Maze I.; Noh K.-M. M.; Soshnev A. A.; Allis C. D. Nat. Rev. Genet. 2014, 15, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Genes Dev. 2003, 17, 2733. [DOI] [PubMed] [Google Scholar]
- Bannister A. J.; Zegerman P.; Partridge J. F.; Miska E. A.; Thomas J. O.; Allshire R. C.; Kouzarides T. Nature 2001, 410, 120. [DOI] [PubMed] [Google Scholar]
- Maison C.; Almouzni G. Nat. Rev. Mol. Cell Biol. 2004, 5, 296. [DOI] [PubMed] [Google Scholar]
- Uversky V. N.; Gillespie J. R.; Fink A. L. Proteins 2000, 41, 415. [DOI] [PubMed] [Google Scholar]
- Van Roey K.; Uyar B.; Weatheritt R. J.; Dinkel H.; Seiler M.; Budd A.; Gibson T. J.; Davey N. E. Chem. Rev. 2014, 114, 6733. [DOI] [PubMed] [Google Scholar]
- Strahl B. D.; Allis C. D. Nature 2000, 403, 41. [DOI] [PubMed] [Google Scholar]
- Sampath S. C.; Marazzi I.; Yap K. L.; Sampath S. C.; Krutchinsky A. N.; Mecklenbräuker I.; Viale A.; Rudensky E.; Zhou M.-M. M.; Chait B. T.; Tarakhovsky A. Mol. Cell 2007, 27, 596. [DOI] [PubMed] [Google Scholar]
- Marazzi I.; Ho J. S.; Kim J.; Manicassamy B.; Dewell S.; Albrecht R. A.; Seibert C. W.; Schaefer U.; Jeffrey K. L.; Prinjha R. K.; Lee K.; García-Sastre A.; Roeder R. G.; Tarakhovsky A. Nature 2012, 483, 428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.; Zhou B. P. Front. Biol. 2012, 8, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stedman E.; Stedman E. Philos. Trans. R. Soc., B 1951, 235, 565. [Google Scholar]
- Huang R.-c. C.; Bonner J. Proc. Natl. Acad. Sci. U.S.A. 1962, 48, 1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allfrey V. G.; Littau V. C.; Mirsky A. E. Proc. Natl. Acad. Sci. U.S.A. 1963, 49, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips D. M. Biochem. J. 1963, 87, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allfrey V. G.; Faulkner R.; Mirsky A. E. Proc. Natl. Acad. Sci. U.S.A. 1964, 51, 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershey E. L.; Vidali G.; Allfrey V. G. J. Biol. Chem. 1968, 243, 5018. [PubMed] [Google Scholar]
- Akira I.; Daisaburo F. Biochem. Biophys. Res. Commun. 1969, 36, 146.5796748 [Google Scholar]
- DeLange R. J.; Fambrough D. M.; Smith E. L.; Bonner J. J. Biol. Chem. 1969, 244, 319. [PubMed] [Google Scholar]
- Ogawa Y.; Quagliarotti G.; Jordan J.; Taylor C. W.; Starbuck W. C.; Busch H. J. Biol. Chem. 1969, 244, 4387. [PubMed] [Google Scholar]
- Merrifield R. B. J. Am. Chem. Soc. 1963, 85, 2149. [Google Scholar]
- Gutte B.; Merrifield R. B. J. Biol. Chem. 1971, 246, 1922. [PubMed] [Google Scholar]
- Krieger D. E.; Vidali G.; Erickson B. W.; Allfrey V. G.; Merrifield R. B. Bioorg. Chem. 1979, 8, 409. [Google Scholar]
- Krieger D. E.; Levine R.; Merrifield R. B.; Vidali G.; Allfrey V. G. J. Biol. Chem. 1974, 249, 332. [PubMed] [Google Scholar]
- Marglin A.; Merrifield R. B. Annu. Rev. Biochem. 1970, 39, 841. [DOI] [PubMed] [Google Scholar]
- Merrifield R. B.; Stewart J. M. Nature 1965, 207, 522. [DOI] [PubMed] [Google Scholar]
- Merrifield R. B. Science 1965, 150, 178. [DOI] [PubMed] [Google Scholar]
- Chahal S. S.; Matthews H. R.; Bradbury E. M. Nature 1980, 287, 76. [DOI] [PubMed] [Google Scholar]
- Shahbazian M. D.; Grunstein M. Annu. Rev. Biochem. 2007, 76, 75. [DOI] [PubMed] [Google Scholar]
- Cary P. D.; Crane-Robinson C.; Bradbury E. M.; Dixon G. H. Eur. J. Biochem. 1982, 127, 137. [DOI] [PubMed] [Google Scholar]
- Hong L.; Schroth G. P.; Matthews H. R.; Yau P.; Bradbury E. M. J. Biol. Chem. 1993, 268, 305. [PubMed] [Google Scholar]
- Yau P.; Thorne A. W.; Imai B. S.; Matthews H. R.; Bradbury E. M. Eur. J. Biochem. 1982, 129, 281. [DOI] [PubMed] [Google Scholar]
- Tamkun J. W.; Deuring R.; Scott M. P.; Kissinger M.; Pattatucci A. M.; Kaufman T. C.; Kennison J. A. Cell 1992, 68, 561. [DOI] [PubMed] [Google Scholar]
- Haynes S. R.; Dollard C.; Winston F.; Beck S.; Trowsdale J.; Dawid I. B. Nucleic Acids Res. 1992, 20, 2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein R.; Berger S. L. Gene 2001, 272, 1. [DOI] [PubMed] [Google Scholar]
- Dhalluin C.; Carlson J. E.; Zeng L.; He C.; Aggarwal A. K.; Zhou M. M. Nature 1999, 399, 491. [DOI] [PubMed] [Google Scholar]
- Owen D. J.; Ornaghi P.; Yang J. C.; Lowe N.; Evans P. R.; Ballario P.; Neuhaus D.; Filetici P.; Travers A. A. EMBO J. 2000, 19, 6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mujtaba S.; Zeng L.; Zhou M. M. Oncogene 2007, 26, 5521. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Knapp S. FEBS Lett. 2012, 586, 2692. [DOI] [PubMed] [Google Scholar]
- Thompson M.; Chandrasekaran R. Anal. Biochem. 2008, 374, 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young N. L.; DiMaggio P. A.; Plazas-Mayorca M. D.; Baliban R. C.; Floudas C. A.; Garcia B. A. Mol. Cell. Proteomics 2009, 8, 2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson R. H.; Ladurner A. G.; King D. S.; Tjian R. Science 2000, 288, 1422. [DOI] [PubMed] [Google Scholar]
- Morinière J.; Rousseaux S.; Steuerwald U.; Soler-López M.; Curtet S.; Vitte A.-L.; Govin J.; Gaucher J.; Sadoul K.; Hart D. J.; Krijgsveld J.; Khochbin S.; Müller C. W.; Petosa C. Nature 2009, 461, 664. [DOI] [PubMed] [Google Scholar]
- Johnson D. S.; Mortazavi A.; Myers R. M.; Wold B. Science 2007, 316, 1497. [DOI] [PubMed] [Google Scholar]
- Muller S.; Isabey A.; Couppez M.; Plaue S.; Sommermeyer G.; Van Regenmortel M. H. Mol. Immunol. 1987, 24, 779. [DOI] [PubMed] [Google Scholar]
- Hebbes T. R.; Thorne A. W.; Crane-Robinson C. EMBO J. 1988, 7, 1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer U.; Ferrari N.; Vidali G. J. Biol. Chem. 1986, 261, 2496. [PubMed] [Google Scholar]
- Turner B. M.; Fellows G. Eur. J. Biochem. 1989, 179, 131. [DOI] [PubMed] [Google Scholar]
- Turner B. M.; Oneill L. P.; Allan I. M. FEBS Lett. 1989, 253, 141. [DOI] [PubMed] [Google Scholar]
- Suka N.; Suka Y.; Carmen A. A.; Wu J.; Grunstein M. Mol. Cell 2001, 8, 473. [DOI] [PubMed] [Google Scholar]
- Smith B. C.; Denu J. M. J. Am. Chem. Soc. 2007, 129, 5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith B. C.; Denu J. M. J. Biol. Chem. 2007, 282, 37256. [DOI] [PubMed] [Google Scholar]
- Starbuck W. C.; Mauritzen C. M.; Taylor C. W.; Saroja I. S.; Busch H. J. Biol. Chem. 1968, 243, 2038. [PubMed] [Google Scholar]
- Murray K. Biochemistry 1964, 3, 10. [DOI] [PubMed] [Google Scholar]
- Rice J. C.; Allis C. D. Curr. Opin. Cell Biol. 2001, 13, 263. [DOI] [PubMed] [Google Scholar]
- Peters A. H.; Kubicek S.; Mechtler K.; O’Sullivan R. J.; Derijck A. A.; Perez-Burgos L.; Kohlmaier A.; Opravil S.; Tachibana M.; Shinkai Y.; Martens J. H.; Jenuwein T. Mol. Cell 2003, 12, 1577. [DOI] [PubMed] [Google Scholar]
- Santos-Rosa H.; Schneider R.; Bannister A. J.; Sherriff J.; Bernstein B. E.; Emre N. C.; Schreiber S. L.; Mellor J.; Kouzarides T. Nature 2002, 419, 407. [DOI] [PubMed] [Google Scholar]
- Martin C.; Zhang Y. Nat. Rev. Mol. Cell Biol. 2005, 6, 838. [DOI] [PubMed] [Google Scholar]
- Rea S.; Eisenhaber F.; O’Carroll D.; Strahl B. D.; Sun Z. W.; Schmid M.; Opravil S.; Mechtler K.; Ponting C. P.; Allis C. D.; Jenuwein T. Nature 2000, 406, 593. [DOI] [PubMed] [Google Scholar]
- Lachner M.; O’Carroll D.; Rea S.; Mechtler K.; Jenuwein T. Nature 2001, 410, 116. [DOI] [PubMed] [Google Scholar]
- Strahl B. D.; Ohba R.; Cook R. G.; Allis C. D. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 14967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama J.; Rice J. C.; Strahl B. D.; Allis C. D.; Grewal S. I. Science 2001, 292, 110. [DOI] [PubMed] [Google Scholar]
- van Attikum H.; Gasser S. M. Trends Cell Biol. 2009, 19, 207. [DOI] [PubMed] [Google Scholar]
- Sanders S. L.; Portoso M.; Mata J.; Bahler J.; Allshire R. C.; Kouzarides T. Cell 2004, 119, 603. [DOI] [PubMed] [Google Scholar]
- Nguyen A. T.; Zhang Y. Genes Dev. 2011, 25, 1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y.; Lan F.; Matson C.; Mulligan P.; Whetstine J. R.; Cole P. A.; Casero R. A.; Shi Y. Cell 2004, 119, 941. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Whetstine J. R. Mol. Cell 2007, 25, 1. [DOI] [PubMed] [Google Scholar]
- Chang C.-D.; Meienhofer J. Int. J. Pept. Protein Res. 1978, 11, 246. [DOI] [PubMed] [Google Scholar]
- Knorr R.; Trzeciak A.; Bannwarth W.; Gillessen D. Tetrahedron Lett. 1989, 30, 1927. [Google Scholar]
- Carpino L. A. J. Am. Chem. Soc. 1993, 115, 4397. [Google Scholar]
- Hood C. A.; Fuentes G.; Patel H.; Page K.; Menakuru M.; Park J. H. J. Pept. Sci. 2008, 14, 97. [DOI] [PubMed] [Google Scholar]
- Coste J.; Le-Nguyen D.; Castro B. Tetrahedron Lett. 1990, 31, 205. [Google Scholar]
- Subirós-Funosas R.; Prohens R.; Barbas R.; El-Faham A.; Albericio F. Chem.—Eur. J. 2009, 15, 9394. [DOI] [PubMed] [Google Scholar]
- Huang Z.-P.; Du J.-T.; Zhao Y.-F.; Li Y.-M. Int. J. Pept. Res. Ther. 2006, 12, 187. [Google Scholar]
- Huang Z. P.; Du J. T.; Su X. Y.; Chen Y. X.; Zhao Y. F.; Li Y. M. Amino Acids 2007, 33, 85. [DOI] [PubMed] [Google Scholar]
- Perez-Burgos L.; Peters A. H.; Opravil S.; Kauer M.; Mechtler K.; Jenuwein T. Methods Enzymol. 2004, 376, 234. [DOI] [PubMed] [Google Scholar]
- Kouzarides T. Cell 2007, 128, 693. [DOI] [PubMed] [Google Scholar]
- Lu X.; Simon M. D.; Chodaparambil J. V.; Hansen J. C.; Shokat K. M.; Luger K. Nat. Struct. Mol. Biol. 2008, 15, 1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna S. D.; Li H.; Ruthenburg A. J.; Allis C. D.; Patel D. J. Nat. Struct. Mol. Biol. 2007, 14, 1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James T. C.; Elgin S. C. Mol. Cell. Biol. 1986, 6, 3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paro R.; Hogness D. S. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D. O.; Cowell I. G.; Singh P. B. BioEssays 2000, 22, 124. [DOI] [PubMed] [Google Scholar]
- Blus B. J.; Wiggins K.; Khorasanizadeh S. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs S. A.; Khorasanizadeh S. Science 2002, 295, 2080. [DOI] [PubMed] [Google Scholar]
- Nielsen P. R.; Nietlispach D.; Mott H. R.; Callaghan J.; Bannister A.; Kouzarides T.; Murzin A. G.; Murzina N. V.; Laue E. D. Nature 2002, 416, 103. [DOI] [PubMed] [Google Scholar]
- Ball L. J.; Murzina N. V.; Broadhurst R. W.; Raine A. R.; Archer S. J.; Stott F. J.; Murzin A. G.; Singh P. B.; Domaille P. J.; Laue E. D. EMBO J. 1997, 16, 2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes R. M.; Wiggins K. R.; Khorasanizadeh S.; Waters M. L. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 11184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.; Blus B. J.; Chandra V.; Huang P.; Rastinejad F.; Khorasanizadeh S. Nat. Struct. Mol. Biol. 2010, 17, 1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore S. A.; Ferhatoglu Y.; Jia Y.; Al-Jiab R. A.; Scott M. J. J. Biol. Chem. 2010, 285, 40879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botuyan M. V.; Lee J.; Ward I. M.; Kim J. E.; Thompson J. R.; Chen J.; Mer G. Cell 2006, 127, 1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Fischle W.; Wang W.; Duncan E. M.; Liang L.; Murakami-Ishibe S.; Allis C. D.; Patel D. J. Mol. Cell 2007, 28, 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J. C.; Dougherty D. A. Chem. Rev. 1997, 97, 1303. [DOI] [PubMed] [Google Scholar]
- Meyer E. A.; Castellano R. K.; Diederich F. Angew. Chem., Int. Ed. 2003, 42, 1210. [DOI] [PubMed] [Google Scholar]
- Dougherty D. A. Acc. Chem. Res. 2013, 46, 885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs S. A.; Taverna S. D.; Zhang Y.; Briggs S. D.; Li J.; Eissenberg J. C.; Allis C. D.; Khorasanizadeh S. EMBO J. 2001, 20, 5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingerman L. A.; Cuellar M. E.; Waters M. L. Chem. Commun. 2010, 46, 1839. [DOI] [PubMed] [Google Scholar]
- Tabet S.; Douglas S. F.; Daze K. D.; Garnett G. A.; Allen K. J.; Abrioux E. M.; Quon T. T.; Wulff J. E.; Hof F. Bioorg. Med. Chem. 2013, 21, 7004. [DOI] [PubMed] [Google Scholar]
- Allen H. F.; Daze K. D.; Shimbo T.; Lai A.; Musselman C. A.; Sims J. K.; Wade P. A.; Hof F.; Kutateladze T. G. Biochem. J. 2014, 459, 505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocka J.; Swigut T.; Milne T. A.; Dou Y.; Zhang X.; Burlingame A. L.; Roeder R. G.; Brivanlou A. H.; Allis C. D. Cell 2005, 121, 859. [DOI] [PubMed] [Google Scholar]
- Wysocka J.; Swigut T.; Xiao H.; Milne T. A.; Kwon S. Y.; Landry J.; Kauer M.; Tackett A. J.; Chait B. T.; Badenhorst P.; Wu C.; Allis C. D. Nature 2006, 442, 86. [DOI] [PubMed] [Google Scholar]
- Tsukiyama T.; Wu C. Cell 1995, 83, 1011. [DOI] [PubMed] [Google Scholar]
- Li H.; Ilin S.; Wang W.; Duncan E. M.; Wysocka J.; Allis C. D.; Patel D. J. Nature 2006, 442, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocka J. Methods 2006, 40, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong S.-E.; Blagoev B.; Kratchmarova I.; Kristensen D. B.; Steen H.; Pandey A.; Mann M. Mol. Cell. Proteomics 2002, 1, 376. [DOI] [PubMed] [Google Scholar]
- Vermeulen M.; Eberl H. C.; Matarese F.; Marks H.; Denissov S.; Butter F.; Lee K. K.; Olsen J. V.; Hyman A. A.; Stunnenberg H. G.; Mann M. Cell 2010, 142, 967. [DOI] [PubMed] [Google Scholar]
- Szewczuk L. M.; Culhane J. C.; Yang M.; Majumdar A.; Yu H.; Cole P. A. Biochemistry 2007, 46, 6892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culhane J. C.; Wang D.; Yen P. M.; Cole P. A. J. Am. Chem. Soc. 2010, 132, 3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.; Luo M. Curr. Opin. Chem. Biol. 2013, 17, 729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lorenzo A.; Bedford M. T. FEBS Lett. 2011, 585, 2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.; Zheng W.; Yu H.; Deng H.; Luo M. J. Am. Chem. Soc. 2011, 133, 7648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D.; Ma H.; Hong H.; Koh S. S.; Huang S. M.; Schurter B. T.; Aswad D. W.; Stallcup M. R. Science 1999, 284, 2174. [DOI] [PubMed] [Google Scholar]
- Schurter B. T.; Koh S. S.; Chen D.; Bunick G. J.; Harp J. M.; Hanson B. L.; Henschen-Edman A.; Mackay D. R.; Stallcup M. R.; Aswad D. W. Biochemistry 2001, 40, 5747. [DOI] [PubMed] [Google Scholar]
- Bedford M. T.; Clarke S. G. Mol. Cell 2009, 33, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliori V.; Phalke S.; Bezzi M.; Guccione E. Epigenomics 2010, 2, 119. [DOI] [PubMed] [Google Scholar]
- Guccione E.; Martinato F.; Finocchiaro G.; Luzi L.; Tizzoni L.; Dall’ Olio V.; Zardo G.; Nervi C.; Bernard L.; Amati B. Nat. Cell Biol. 2006, 8, 764. [DOI] [PubMed] [Google Scholar]
- Guccione E.; Bassi C.; Casadio F.; Martinato F.; Cesaroni M.; Schuchlautz H.; Luscher B.; Amati B. Nature 2007, 449, 933. [DOI] [PubMed] [Google Scholar]
- Iberg A. N.; Espejo A.; Cheng D.; Kim D.; Michaud-Levesque J.; Richard S.; Bedford M. T. J. Biol. Chem. 2008, 283, 3006. [DOI] [PubMed] [Google Scholar]
- Ramón-Maiques S.; Kuo A. J.; Carney D.; Matthews A. G. W.; Oettinger M. A.; Gozani O.; Yang W. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 18993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté J.; Richard S. J. Biol. Chem. 2005, 280, 28476. [DOI] [PubMed] [Google Scholar]
- Zhao Q.; Rank G.; Tan Y. T.; Li H.; Moritz R. L.; Simpson R. J.; Cerruti L.; Curtis D. J.; Patel D. J.; Allis C. D.; Cunningham J. M.; Jane S. M. Nat. Struct. Mol. Biol. 2009, 16, 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Daniel J.; Espejo A.; Lake A.; Krishna M.; Xia L.; Zhang Y.; Bedford M. T. EMBO Rep. 2006, 7, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. Z.; Lu Y.; Espejo A.; Wu J. C.; Xu W.; Liang S. D.; Bedford M. T. Mol. Cell 2010, 40, 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espejo A.; Côté J.; Bednarek A.; Richard S.; Bedford M. T. Biochem. J. 2002, 367, 697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K.; Guo Y.; Liu H.; Bian C.; Lam R.; Liu Y.; Mackenzie F.; Rojas L. A.; Reinberg D.; Bedford M. T.; Xu R. M.; Min J. PLoS One 2012, 7, e30375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikorsky T.; Hobor F.; Krizanova E.; Pasulka J.; Kubicek K.; Stefl R. Nucleic Acids Res. 2012, 40, 11748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K.; Chen C.; Guo Y.; Lam R.; Bian C.; Xu C.; Zhao D. Y.; Jin J.; MacKenzie F.; Pawson T.; Min J. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 18398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James L. I.; Beaver J. E.; Rice N. W.; Waters M. L. J. Am. Chem. Soc. 2013, 135, 6450. [DOI] [PubMed] [Google Scholar]
- Bannister A. J.; Schneider R.; Kouzarides T. Cell 2002, 109, 801. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Wysocka J.; Sayegh J.; Lee Y.-H.; Perlin J. R.; Leonelli L.; Sonbuchner L. S.; McDonald C. H.; Cook R. G.; Dou Y.; Roeder R. G.; Clarke S.; Stallcup M. R.; Allis C. D.; Coonrod S. A. Science 2004, 306, 279. [DOI] [PubMed] [Google Scholar]
- Raijmakers R.; Zendman A. J. W.; Egberts W. V.; Vossenaar E. R.; Raats J.; Soede-Huijbregts C.; Rutjes F. P. J. T.; van Veelen P. A.; Drijfhout J. W.; Pruijn G. J. M. J. Mol. Biol. 2007, 367, 1118. [DOI] [PubMed] [Google Scholar]
- Hidaka Y.; Hagiwara T.; Yamada M. FEBS Lett. 2005, 579, 4088. [DOI] [PubMed] [Google Scholar]
- Kearney P. L.; Bhatia M.; Jones N. G.; Yuan L.; Glascock M. C.; Catchings K. L.; Yamada M.; Thompson P. R. Biochemistry 2005, 44, 10570. [DOI] [PubMed] [Google Scholar]
- Cuthbert G. L.; Daujat S.; Snowden A. W.; Erdjument-Bromage H.; Hagiwara T.; Yamada M.; Schneider R.; Gregory P. D.; Tempst P.; Bannister A. J.; Kouzarides T. Cell 2004, 118, 545. [DOI] [PubMed] [Google Scholar]
- Christophorou M. A.; Castelo-Branco G.; Halley-Stott R. P.; Oliveira C. S.; Loos R.; Radzisheuskaya A.; Mowen K. A.; Bertone P.; Silva J. C.; Zernicka-Goetz M.; Nielsen M. L.; Gurdon J. B.; Kouzarides T. Nature 2014, 507, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsook H.; Dubnoff J. W. J. Biol. Chem. 1941, 141, 717. [Google Scholar]
- Singh R.; Kabbaj M.-H. M.; Paik J.; Gunjan A. Nat. Cell Biol. 2009, 11, 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao A.; Li H.; Shechter D.; Ahn S. H.; Fabrizio L. A.; Erdjument-Bromage H.; Ishibe-Murakami S.; Wang B.; Tempst P.; Hofmann K.; Patel D. J.; Elledge S. J.; Allis C. D. Nature 2009, 457, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson M. A.; Bannister A. J.; Gottgens B.; Foster S. D.; Bartke T.; Green A. R.; Kouzarides T. Nature 2009, 461, 819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook P. J.; Ju B. G.; Telese F.; Wang X.; Glass C. K.; Rosenfeld M. G. Nature 2009, 458, 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan N.; Jeong D. G.; Jung S.-K.; Ryu S. E.; Xiao A.; Allis C. D.; Kim S. J.; Tonks N. K. J. Biol. Chem. 2009, 284, 16066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R. K.; Gunjan A. Epigenetics 2011, 6, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakim B. T.; Aswad G. D. J. Biol. Chem. 1994, 269, 2722. [PubMed] [Google Scholar]
- Cieśla J.; Frączyk T.; Rode W. Acta Biochim. Pol. 2011, 58, 137. [PubMed] [Google Scholar]
- Chen C. C.; Smith D. L.; Bruegger B. B.; Halpern R. M.; Smith R. A. Biochemistry 1974, 13, 3785. [DOI] [PubMed] [Google Scholar]
- Besant P. G.; Attwood P. V. Biochem. Soc. Trans. 2012, 40, 290. [DOI] [PubMed] [Google Scholar]
- Perich J. W. Methods Enzymol. 1997, 289, 245. [DOI] [PubMed] [Google Scholar]
- McMurray J. S.; Coleman D. R. I.; Wang W.; Campbell M. L. Biopolymers 2001, 60, 3. [DOI] [PubMed] [Google Scholar]
- Attwood P. V.; Piggott M. J.; Zu X. L.; Besant P. G. Amino Acids 2007, 32, 145. [DOI] [PubMed] [Google Scholar]
- Kee J.-M.; Muir T. W. ACS Chem. Biol. 2012, 7, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann F. T.; Lindemann C.; Salia H.; Adamitzki P.; Karanicolas J.; Seebeck F. P. Chem. Commun. 2011, 47, 10335. [DOI] [PubMed] [Google Scholar]
- Kee J.-M.; Villani B.; Carpenter L. R.; Muir T. W. J. Am. Chem. Soc. 2010, 132, 14327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister T. E.; Nix M. G.; Webb M. E. Chem. Commun. 2011, 47, 1297. [DOI] [PubMed] [Google Scholar]
- Kee J.-M.; Oslund R. C.; Perlman D. H.; Muir T. W. Nat. Chem. Biol. 2013, 9, 416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W.; Wang Y.; Allis C. D. Nature 2003, 425, 475. [DOI] [PubMed] [Google Scholar]
- Fischle W.; Tseng B. S.; Dormann H. L.; Ueberheide B. M.; Garcia B. A.; Shabanowitz J.; Hunt D. F.; Funabiki H.; Allis C. D. Nature 2005, 438, 1116. [DOI] [PubMed] [Google Scholar]
- Hirota T.; Lipp J. J.; Toh B.-H.; Peters J.-M. Nature 2005, 438, 1176. [DOI] [PubMed] [Google Scholar]
- Macdonald N.; Welburn J. P.; Noble M. E.; Nguyen A.; Yaffe M. B.; Clynes D.; Moggs J. G.; Orphanides G.; Thomson S.; Edmunds J. W.; Clayton A. L.; Endicott J. A.; Mahadevan L. C. Mol. Cell 2005, 20, 199. [DOI] [PubMed] [Google Scholar]
- Aitken A. Trends Cell Biol. 1996, 6, 341. [DOI] [PubMed] [Google Scholar]
- Zweidler A. Biochemistry 1992, 31, 9205. [DOI] [PubMed] [Google Scholar]
- Hart G. W.; Copeland R. J. Cell 2010, 143, 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakabe K.; Wang Z.; Hart G. W. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 19915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Roche K.; Nasheuer H.-P.; Lowndes N. F. J. Biol. Chem. 2011, 286, 37483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki R.; Hashiba W.; Sekine H.; Yokoyama A.; Chikanishi T.; Ito S.; Imai Y.; Kim J.; He H. H.; Igarashi K.; Kanno J.; Ohtake F.; Kitagawa H.; Roeder R. G.; Brown M.; Kato S. Nature 2011, 480, 557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rexach J. E.; Clark P. M.; Hsieh-Wilson L. C. Nat. Chem. Biol. 2008, 4, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cecioni S.; Vocadlo D. J. Curr. Opin. Chem. Biol. 2013, 17, 719. [DOI] [PubMed] [Google Scholar]
- Agard N. J.; Bertozzi C. R. Acc. Chem. Res. 2009, 42, 788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y.; Chen J.; Wan Q.; Wilson R. M.; Danishefsky S. J. Biopolymers 2010, 94, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burzio L. O.; Riquelme P. T.; Koide S. S. J. Biol. Chem. 1979, 254, 3029. [PubMed] [Google Scholar]
- Ueda K.; Hayaishi O. Annu. Rev. Biochem. 1985, 54, 73. [DOI] [PubMed] [Google Scholar]
- Timinszky G.; Till S.; Hassa P. O.; Hothorn M.; Kustatscher G.; Nijmeijer B.; Colombelli J.; Altmeyer M.; Stelzer E. H.; Scheffzek K.; Hottiger M. O.; Ladurner A. G. Nat. Struct. Mol. Biol. 2009, 16, 923. [DOI] [PubMed] [Google Scholar]
- Hassa P. O.; Haenni S. S.; Elser M.; Hottiger M. O. Microbiol. Mol. Biol. Rev. 2006, 70, 789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feijs K. L.; Verheugd P.; Luscher B. FEBS J. 2013, 280, 3519. [DOI] [PubMed] [Google Scholar]
- van der Heden van Noort G. J.; van der Horst M. G.; Overkleeft H. S.; van der Marel G. A.; Filippov D. V. J. Am. Chem. Soc. 2010, 132, 5236. [DOI] [PubMed] [Google Scholar]
- Moyle P. M.; Muir T. W. J. Am. Chem. Soc. 2010, 132, 15878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehrson J. R.; Fried V. A. Science 1992, 257, 1398. [DOI] [PubMed] [Google Scholar]
- Minsky N.; Shema E.; Field Y.; Schuster M.; Segal E.; Oren M. Nat. Cell Biol. 2008, 10, 483. [DOI] [PubMed] [Google Scholar]
- Chatterjee C.; McGinty R. K.; Pellois J.-P.; Muir T. W. Angew. Chem., Int. Ed. 2007, 46, 2814. [DOI] [PubMed] [Google Scholar]
- Tan M.; Luo H.; Lee S.; Jin F.; Yang J.; Montellier E.; Buchou T.; Cheng Z.; Rousseaux S.; Rajagopal N.; Lu Z.; Ye Z.; Zhu Q.; Wysocka J.; Ye Y.; Khochbin S.; Ren B.; Zhao Y. Cell 2011, 146, 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen A. S.; Olsen C. A. Angew. Chem., Int. Ed. 2012, 51, 9083. [DOI] [PubMed] [Google Scholar]
- Feldman J. L.; Baeza J.; Denu J. M. J. Biol. Chem. 2013, 288, 31350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L.; Peng C.; Montellier E.; Lu Z.; Chen Y.; Ishii H.; Debernardi A.; Buchou T.; Rousseaux S.; Jin F.; Sabari B. R.; Deng Z.; Allis C. D.; Ren B.; Khochbin S.; Zhao Y. Nat. Chem. Biol. 2014, 10, 365. [DOI] [PubMed] [Google Scholar]
- Tessarz P.; Santos-Rosa H.; Robson S. C.; Sylvestersen K. B.; Nelson C. J.; Nielsen M. L.; Kouzarides T. Nature 2014, 505, 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch K. R.; Denizio J. E.; Black B. E.; Garcia B. A. Front. Genet. 2013, 4, 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur M. W.; Thornton J. M. J. Mol. Biol. 1991, 218, 397. [DOI] [PubMed] [Google Scholar]
- Andreotti A. H. Biochemistry 2003, 42, 9515. [DOI] [PubMed] [Google Scholar]
- Nelson C. J.; Santos-Rosa H.; Kouzarides T. Cell 2006, 126, 905. [DOI] [PubMed] [Google Scholar]
- Lummis S. C.; Beene D. L.; Lee L. W.; Lester H. A.; Broadhurst R. W.; Dougherty D. A. Nature 2005, 438, 248. [DOI] [PubMed] [Google Scholar]
- Torbeev V. Y.; Hilvert D. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 20051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoulders M. D.; Satyshur K. A.; Forest K. T.; Raines R. T. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G.; Broniscer A.; McEachron T. A.; Lu C.; Paugh B. S.; Becksfort J.; Qu C.; Ding L.; Huether R.; Parker M.; Zhang J.; Gajjar A.; Dyer M. A.; Mullighan C. G.; Gilbertson R. J.; Mardis E. R.; Wilson R. K.; Downing J. R.; Ellison D. W.; Zhang J.; Baker S. J. Nat. Genet. 2012, 44, 251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzentruber J.; Korshunov A.; Liu X. Y.; Jones D. T.; Pfaff E.; Jacob K.; Sturm D.; Fontebasso A. M.; Quang D. A.; Tonjes M.; Hovestadt V.; Albrecht S.; Kool M.; Nantel A.; Konermann C.; Lindroth A.; Jager N.; Rausch T.; Ryzhova M.; Korbel J. O.; Hielscher T.; Hauser P.; Garami M.; Klekner A.; Bognar L.; Ebinger M.; Schuhmann M. U.; Scheurlen W.; Pekrun A.; Fruhwald M. C.; Roggendorf W.; Kramm C.; Durken M.; Atkinson J.; Lepage P.; Montpetit A.; Zakrzewska M.; Zakrzewski K.; Liberski P. P.; Dong Z.; Siegel P.; Kulozik A. E.; Zapatka M.; Guha A.; Malkin D.; Felsberg J.; Reifenberger G.; von Deimling A.; Ichimura K.; Collins V. P.; Witt H.; Milde T.; Witt O.; Zhang C.; Castelo-Branco P.; Lichter P.; Faury D.; Tabori U.; Plass C.; Majewski J.; Pfister S. M.; Jabado N. Nature 2012, 482, 226. [DOI] [PubMed] [Google Scholar]
- Khuong-Quang D.-A.; Buczkowicz P.; Rakopoulos P.; Liu X.-Y.; Fontebasso A.; Bouffet E.; Bartels U.; Albrecht S.; Schwartzentruber J.; Letourneau L.; Bourgey M.; Bourque G.; Montpetit A.; Bourret G.; Lepage P.; Fleming A.; Lichter P.; Kool M.; von Deimling A.; Sturm D.; Korshunov A.; Faury D.; Jones D.; Majewski J.; Pfister S.; Jabado N.; Hawkins C. Acta Neuropathol. 2012, 124, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontebasso A. M.; Gayden T.; Nikbakht H.; Neirinck M.; Papillon-Cavanagh S.; Majewski J.; Jabado N.. Acta Neuropathol. 2014. [DOI] [PMC free article] [PubMed]
- Margueron R.; Reinberg D. Nature 2011, 469, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis P. W.; Müller M. M.; Koletsky M. S.; Cordero F.; Lin S.; Banaszynski L. A.; Garcia B. A.; Muir T. W.; Becher O. J.; Allis C. D. Science 2013, 340, 857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venneti S.; Garimella M. T.; Sullivan L. M.; Martinez D.; Huse J. T.; Heguy A.; Santi M.; Thompson C. B.; Judkins A. R. Brain Pathol. 2013, 23, 558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K.-M.; Fang D.; Gan H.; Hashizume R.; Yu C.; Schroeder M.; Gupta N.; Mueller S.; James C. D.; Jenkins R.; Sarkaria J.; Zhang Z. Genes Dev. 2013, 27, 985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender S.; Tang Y.; Lindroth A. M.; Hovestadt V.; Jones D. T.; Kool M.; Zapatka M.; Northcott P. A.; Sturm D.; Wang W.; Radlwimmer B.; Hojfeldt J. W.; Truffaux N.; Castel D.; Schubert S.; Ryzhova M.; Seker-Cin H.; Gronych J.; Johann P. D.; Stark S.; Meyer J.; Milde T.; Schuhmann M.; Ebinger M.; Monoranu C. M.; Ponnuswami A.; Chen S.; Jones C.; Witt O.; Collins V. P.; von Deimling A.; Jabado N.; Puget S.; Grill J.; Helin K.; Korshunov A.; Lichter P.; Monje M.; Plass C.; Cho Y. J.; Pfister S. M. Cancer Cell 2013, 24, 660. [DOI] [PubMed] [Google Scholar]
- Herz H.-M.; Morgan M.; Gao X.; Jackson J.; Rickels R.; Swanson S. K.; Florens L.; Washburn M. P.; Eissenberg J. C.; Shilatifard A. Science 2014, 345, 1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown Z. Z.; Müller M. M.; Jain S. U.; Allis C. D.; Lewis P. W.; Muir T. W. J. Am. Chem. Soc. 2014, 136, 13498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behjati S.; Tarpey P. S.; Presneau N.; Scheipl S.; Pillay N.; Van Loo P.; Wedge D. C.; Cooke S. L.; Gundem G.; Davies H.; Nik-Zainal S.; Martin S.; McLaren S.; Goody V.; Robinson B.; Butler A.; Teague J. W.; Halai D.; Khatri B.; Myklebost O.; Baumhoer D.; Jundt G.; Hamoudi R.; Tirabosco R.; Amary M. F.; Futreal P. A.; Stratton M. R.; Campbell P. J.; Flanagan A. M. Nat. Genet. 2013, 45, 1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner J. Annu. Rev. Biochem. 1993, 62, 483. [DOI] [PubMed] [Google Scholar]
- Mackinnon A. L.; Taunton J. Curr. Protoc. Chem. Biol. 2009, 1, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchanek M.; Radzikowska A.; Thiele C. Nat. Methods 2005, 2, 261. [DOI] [PubMed] [Google Scholar]
- Yuan W.; Wu T.; Fu H.; Dai C.; Wu H.; Liu N.; Li X.; Xu M.; Zhang Z.; Niu T.; Han Z.; Chai J.; Zhou X. J.; Gao S.; Zhu B. Science 2012, 337, 971. [DOI] [PubMed] [Google Scholar]
- Burdine L.; Gillette T. G.; Lin H. J.; Kodadek T. J. Am. Chem. Soc. 2004, 126, 11442. [DOI] [PubMed] [Google Scholar]
- Ciferri C.; Lander G. C.; Maiolica A.; Herzog F.; Aebersold R.; Nogales E. eLife 2012, 1, e00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer J. C.; Erickson-Viitanen S.; Wolfe H. R. Jr.; DeGrado W. F. J. Biol. Chem. 1986, 261, 10695. [PubMed] [Google Scholar]
- Li X.; Kapoor T. M. J. Am. Chem. Soc. 2010, 132, 2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Foley E. A.; Molloy K. R.; Li Y.; Chait B. T.; Kapoor T. M. J. Am. Chem. Soc. 2012, 134, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Foley E. A.; Kawashima S. A.; Molloy K. R.; Li Y.; Chait B. T.; Kapoor T. M. Protein Sci. 2013, 22, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filion G. J.; van Bemmel J. G.; Braunschweig U.; Talhout W.; Kind J.; Ward L. D.; Brugman W.; de Castro I. J.; Kerkhoven R. M.; Bussemaker H. J.; van Steensel B. Cell 2010, 143, 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M. Nat. Methods 2011, 8, 717. [DOI] [PubMed] [Google Scholar]
- Kharchenko P. V.; Alekseyenko A. A.; Schwartz Y. B.; Minoda A.; Riddle N. C.; Ernst J.; Sabo P. J.; Larschan E.; Gorchakov A. A.; Gu T.; Linder-Basso D.; Plachetka A.; Shanower G.; Tolstorukov M. Y.; Luquette L. J.; Xi R.; Jung Y. L.; Park R. W.; Bishop E. P.; Canfield T. K.; Sandstrom R.; Thurman R. E.; MacAlpine D. M.; Stamatoyannopoulos J. A.; Kellis M.; Elgin S. C.; Kuroda M. I.; Pirrotta V.; Karpen G. H.; Park P. J. Nature 2011, 471, 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson A. W.; Gozani O. Biochim. Biophys. Acta 2014, 1839, 669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbart S. B.; Krajewski K.; Strahl B. D.; Fuchs S. M. Methods Enzymol. 2012, 512, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X.; Kachirskaia I.; Walter K. L.; Kuo J.-H.; Lake A.; Davrazou F.; Chan S. M.; Martin D. G.; Fingerman I. M.; Briggs S. D.; Howe L.; Utz P. J.; Kutateladze T. G.; Lugovskoy A. A.; Bedford M. T.; Gozani O. J. Biol. Chem. 2007, 282, 2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews A. G.; Kuo A. J.; Ramon-Maiques S.; Han S.; Champagne K. S.; Ivanov D.; Gallardo M.; Carney D.; Cheung P.; Ciccone D. N.; Walter K. L.; Utz P. J.; Shi Y.; Kutateladze T. G.; Yang W.; Gozani O.; Oettinger M. A. Nature 2007, 450, 1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bua D. J.; Kuo A. J.; Cheung P.; Liu C. L.; Migliori V.; Espejo A.; Casadio F.; Bassi C.; Amati B.; Bedford M. T.; Guccione E.; Gozani O. PLoS One 2009, 4, e6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Galka M.; Iberg A.; Wang Z.; Li L.; Voss C.; Jiang X.; Lajoie G.; Huang Z.; Bedford M. T.; Li S. S. J. Proteome Res. 2010, 9, 5827. [DOI] [PubMed] [Google Scholar]
- Kuo A. J.; Song J.; Cheung P.; Ishibe-Murakami S.; Yamazoe S.; Chen J. K.; Patel D. J.; Gozani O. Nature 2012, 484, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbart S. B.; Krajewski K.; Nady N.; Tempel W.; Xue S.; Badeaux A. I.; Barsyte-Lovejoy D.; Martinez J. Y.; Bedford M. T.; Fuchs S. M.; Arrowsmith C. H.; Strahl B. D. Nat. Struct. Mol. Biol. 2012, 19, 1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbart S. B.; Dickson B. M.; Ong M. S.; Krajewski K.; Houliston S.; Kireev D. B.; Arrowsmith C. H.; Strahl B. D. Genes Dev. 2013, 27, 1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Chakravarty S.; Ghersi D.; Zeng L.; Plotnikov A. N.; Sanchez R.; Zhou M.-M. PLoS One 2010, 5, e8903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs S. M.; Krajewski K.; Baker R. W.; Miller V. L.; Strahl B. D. Curr. Biol. 2011, 21, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez C. A.; Ptaszek L. M.; Villa A.; Bozzi F.; Sobacchi C.; Brooks E. G.; Notarangelo L. D.; Spanopoulou E.; Pan Z. Q.; Vezzoni P.; Cortes P.; Santagata S. Mol. Cell. Biol. 2000, 20, 5653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callebaut I.; Courvalin J. C.; Mornon J. P. FEBS Lett. 1999, 446, 189. [DOI] [PubMed] [Google Scholar]
- Bicknell L. S.; Walker S.; Klingseisen A.; Stiff T.; Leitch A.; Kerzendorfer C.; Martin C. A.; Yeyati P.; Al Sanna N.; Bober M.; Johnson D.; Wise C.; Jackson A. P.; O’Driscoll M.; Jeggo P. A. Nat. Genet. 2011, 43, 350. [DOI] [PubMed] [Google Scholar]
- Bicknell L. S.; Bongers E. M.; Leitch A.; Brown S.; Schoots J.; Harley M. E.; Aftimos S.; Al-Aama J. Y.; Bober M.; Brown P. A.; van Bokhoven H.; Dean J.; Edrees A. Y.; Feingold M.; Fryer A.; Hoefsloot L. H.; Kau N.; Knoers N. V.; Mackenzie J.; Opitz J. M.; Sarda P.; Ross A.; Temple I. K.; Toutain A.; Wise C. A.; Wright M.; Jackson A. P. Nat. Genet. 2011, 43, 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajakumara E.; Wang Z.; Ma H.; Hu L.; Chen H.; Lin Y.; Guo R.; Wu F.; Li H.; Lan F.; Shi Y. G.; Xu Y.; Patel D. J.; Shi Y. Mol. Cell 2011, 43, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arita K.; Isogai S.; Oda T.; Unoki M.; Sugita K.; Sekiyama N.; Kuwata K.; Hamamoto R.; Tochio H.; Sato M.; Ariyoshi M.; Shirakawa M. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelato K. A.; Tauber M.; Ong M. S.; Winter S.; Hiragami-Hamada K.; Sindlinger J.; Lemak A.; Bultsma Y.; Houliston S.; Schwarzer D.; Divecha N.; Arrowsmith C. H.; Fischle W. Mol. Cell 2014, 56, 905. [DOI] [PubMed] [Google Scholar]
- Rothbart S. B.; Lin S.; Britton L. M.; Krajewski K.; Keogh M. C.; Garcia B. A.; Strahl B. D. Sci. Rep. 2012, 2, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrksich M. ACS Nano 2008, 2, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurard-Levin Z. A.; Kim J.; Mrksich M. ChemBioChem 2009, 10, 2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank R. J. Immunol. Methods 2002, 267, 13. [DOI] [PubMed] [Google Scholar]
- Hilpert K.; Winkler D. F.; Hancock R. E. Nat. Protoc. 2007, 2, 1333. [DOI] [PubMed] [Google Scholar]
- Bock I.; Kudithipudi S.; Tamas R.; Kungulovski G.; Dhayalan A.; Jeltsch A. BMC Biochem. 2011, 12, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nady N.; Min J.; Kareta M. S.; Chédin F.; Arrowsmith C. H. Trends Biochem. Sci. 2008, 33, 305. [DOI] [PubMed] [Google Scholar]
- Dhayalan A.; Tamas R.; Bock I.; Tattermusch A.; Dimitrova E.; Kudithipudi S.; Ragozin S.; Jeltsch A. Hum. Mol. Genet. 2011, 20, 2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhayalan A.; Rajavelu A.; Rathert P.; Tamas R.; Jurkowska R. Z.; Ragozin S.; Jeltsch A. J. Biol. Chem. 2010, 285, 26114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Picaud S.; Mangos M.; Keates T.; Lambert J. P.; Barsyte-Lovejoy D.; Felletar I.; Volkmer R.; Muller S.; Pawson T.; Gingras A. C.; Arrowsmith C. H.; Knapp S. Cell 2012, 149, 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathert P.; Dhayalan A.; Murakami M.; Zhang X.; Tamas R.; Jurkowska R.; Komatsu Y.; Shinkai Y.; Cheng X.; Jeltsch A. Nat. Chem. Biol. 2008, 4, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathert P.; Zhang X.; Freund C.; Cheng X.; Jeltsch A. Chem. Biol. 2008, 15, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudithipudi S.; Lungu C.; Rathert P.; Happel N.; Jeltsch A. Chem. Biol. 2014, 21, 226. [DOI] [PubMed] [Google Scholar]
- Dhayalan A.; Kudithipudi S.; Rathert P.; Jeltsch A. Chem. Biol. 2011, 18, 111. [DOI] [PubMed] [Google Scholar]
- Bock I.; Dhayalan A.; Kudithipudi S.; Brandt O.; Rathert P.; Jeltsch A. Epigenetics 2011, 6, 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heubach Y.; Planatscher H.; Sommersdorf C.; Maisch D.; Maier J.; Joos T. O.; Templin M. F.; Poetz O. Proteomics 2013, 13, 1010. [DOI] [PubMed] [Google Scholar]
- Su Z.; Boersma M. D.; Lee J.-H. H.; Oliver S. S.; Liu S.; Garcia B. A.; Denu J. M. Epigenet. Chromatin 2014, 7, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam K. S.; Salmon S. E.; Hersh E. M.; Hruby V. J.; Kazmierski W. M.; Knapp R. J. Nature 1991, 354, 82. [DOI] [PubMed] [Google Scholar]
- Houghten R. A.; Pinilla C.; Blondelle S. E.; Appel J. R.; Dooley C. T.; Cuervo J. H. Nature 1991, 354, 84. [DOI] [PubMed] [Google Scholar]
- Youngquist R. S.; Fuentes G. R.; Lacey M. P.; Keough T. J. Am. Chem. Soc. 1995, 117, 3900. [Google Scholar]
- Garske A. L.; Oliver S. S.; Wagner E. K.; Musselman C. A.; LeRoy G.; Garcia B. A.; Kutateladze T. G.; Denu J. M. Nat. Chem. Biol. 2010, 6, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garske A. L.; Craciun G.; Denu J. M. Biochemistry 2008, 47, 8094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver S. S.; Musselman C. A.; Srinivasan R.; Svaren J. P.; Kutateladze T. G.; Denu J. M. Biochemistry 2012, 51, 6534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang T. J.; Yuzawa S.; Suga H. Chem. Biol. 2008, 15, 1166. [DOI] [PubMed] [Google Scholar]
- Roberts R. W.; Szostak J. W. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipolito C. J.; Suga H. Curr. Opin. Chem. Biol. 2012, 16, 196. [DOI] [PubMed] [Google Scholar]
- Amstutz P.; Forrer P.; Zahnd C.; Plückthun A. Curr. Opin. Biotechnol. 2001, 12, 400. [DOI] [PubMed] [Google Scholar]
- Dyer P. N.; Edayathumangalam R. S.; White C. L.; Bao Y.; Chakravarthy S.; Muthurajan U. M.; Luger K. Methods Enzymol. 2004, 375, 23. [DOI] [PubMed] [Google Scholar]
- Luger K.; Rechsteiner T. J.; Flaus A. J.; Waye M. M.; Richmond T. J. J. Mol. Biol. 1997, 272, 301. [DOI] [PubMed] [Google Scholar]
- Luger K.; Rechsteiner T. J.; Richmond T. J. Methods Enzymol. 1999, 304, 3. [DOI] [PubMed] [Google Scholar]
- Luger K.; Mader A. W.; Richmond R. K.; Sargent D. F.; Richmond T. J. Nature 1997, 389, 251. [DOI] [PubMed] [Google Scholar]
- Davey C. A.; Sargent D. F.; Luger K.; Maeder A. W.; Richmond T. J. J. Mol. Biol. 2002, 319, 1097. [DOI] [PubMed] [Google Scholar]
- Luger K.; Dechassa M. L.; Tremethick D. J. Nat. Rev. Mol. Cell Biol. 2012, 13, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalashnikova A. A.; Porter-Goff M. E.; Muthurajan U. M.; Luger K.; Hansen J. C. J. R. Soc. Interface 2013, 10, 20121022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowary P. T.; Widom J. J. Mol. Biol. 1998, 276, 19. [DOI] [PubMed] [Google Scholar]
- Carruthers L. M.; Tse C.; Walker K. P. III; Hansen J. C. Methods Enzymol. 1999, 304, 19. [DOI] [PubMed] [Google Scholar]
- Dorigo B.; Schalch T.; Bystricky K.; Richmond T. J. J. Mol. Biol. 2003, 327, 85. [DOI] [PubMed] [Google Scholar]
- Lusser A.; Kadonaga J. T. Nat. Methods 2004, 1, 19. [DOI] [PubMed] [Google Scholar]
- Schalch T.; Duda S.; Sargent D. F.; Richmond T. J. Nature 2005, 436, 138. [DOI] [PubMed] [Google Scholar]
- Song F.; Chen P.; Sun D.; Wang M.; Dong L.; Liang D.; Xu R.-M.; Zhu P.; Li G. Science 2014, 344, 376. [DOI] [PubMed] [Google Scholar]
- Robinson P. J. J.; Fairall L.; Huynh V. A.; Rhodes D. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruithof M.; Chien F.-T. T.; Routh A.; Logie C.; Rhodes D.; van Noort J. Nat. Struct. Mol. Biol. 2009, 16, 534. [DOI] [PubMed] [Google Scholar]
- Eltsov M.; Maclellan K. M.; Maeshima K.; Frangakis A. S.; Dubochet J. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 19732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoryev S. A.; Woodcock C. L. Exp. Cell Res. 2012, 318, 1448. [DOI] [PubMed] [Google Scholar]
- Robinson P. J. J.; Rhodes D. Curr. Opin. Struct. Biol. 2006, 16, 336. [DOI] [PubMed] [Google Scholar]
- Maeshima K.; Hihara S.; Eltsov M. Curr. Opin. Cell Biol. 2010, 22, 291. [DOI] [PubMed] [Google Scholar]
- Dai J.; Hyland E. M.; Yuan D. S.; Huang H.; Bader J. S.; Boeke J. D. Cell 2008, 134, 1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon G. L.; Bruice T. W. Methods Enzymol. 1977, 47, 407. [DOI] [PubMed] [Google Scholar]
- Dorigo B.; Schalch T.; Kulangara A.; Duda S.; Schroeder R. R.; Richmond T. J. Science 2004, 306, 1571. [DOI] [PubMed] [Google Scholar]
- Sinha D.; Shogren-Knaak M. A. J. Biol. Chem. 2010, 285, 16572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebralidse K. K.; Grachev S. A.; Mirzabekov A. D. Nature 1988, 331, 365. [DOI] [PubMed] [Google Scholar]
- Mirzabekov A. D. Gene 1993, 135, 111. [DOI] [PubMed] [Google Scholar]
- Pepenella S.; Murphy K. J.; Hayes J. J. Chromosoma 2014, 123, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. M.; Hayes J. J. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 8959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan P.-Y.; Caterino T. L.; Hayes J. J. Mol. Cell. Biol. 2009, 29, 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C.; Hayes J. J. J. Biol. Chem. 2003, 278, 24217. [DOI] [PubMed] [Google Scholar]
- Zhou Y. B.; Gerchman S. E.; Ramakrishnan V.; Travers A.; Muyldermans S. Nature 1998, 395, 402. [DOI] [PubMed] [Google Scholar]
- Kassabov S. R.; Henry N. M.; Zofall M.; Tsukiyama T.; Bartholomew B. Mol. Cell. Biol. 2002, 22, 7524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenton H. J. Chem. Soc., Trans. 1894, 65, 899. [Google Scholar]
- Tullius T. D.; Dombroski B. A.; Churchill M. E.; Kam L. Methods Enzymol. 1987, 155, 537. [DOI] [PubMed] [Google Scholar]
- Flaus A.; Luger K.; Tan S.; Richmond T. J. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogaard K.; Xi L.; Wang J.-P.; Widom J. Nature 2012, 486, 496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews A. J.; Luger K. Annu. Rev. Biophys 2011, 40, 99. [DOI] [PubMed] [Google Scholar]
- Li G.; Widom J. Nat. Struct. Mol. Biol. 2004, 11, 763. [DOI] [PubMed] [Google Scholar]
- Andrews A. J.; Downing G.; Brown K.; Park Y. J.; Luger K. J. Biol. Chem. 2008, 283, 32412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews A. J.; Chen X.; Zevin A.; Stargell L. A.; Luger K. Mol. Cell 2010, 37, 834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews A. J.; Luger K. Methods Enzymol. 2011, 488, 265. [DOI] [PubMed] [Google Scholar]
- Buning R.; van Noort J. Biochimie 2010, 92, 1729. [DOI] [PubMed] [Google Scholar]
- Killian J. L.; Li M.; Sheinin M. Y.; Wang M. D. Curr. Opin. Struct. Biol. 2012, 22, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Tyl M.; Ward R.; Sobott F.; Maman J.; Murthy A. S.; Watson A. A.; Fedorov O.; Bowman A.; Owen-Hughes T.; El Mkami H.; Murzina N. V.; Norman D. G.; Laue E. D. Nat. Struct. Mol. Biol. 2013, 20, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reginsson G. W.; Schiemann O. Biochem. J. 2011, 434, 353. [DOI] [PubMed] [Google Scholar]
- Bowman A.; Ward R.; El-Mkami H.; Owen-Hughes T.; Norman D. G. Nucleic Acids Res. 2010, 38, 695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiemann O.; Prisner T. F. Q. Rev. Biophys. 2007, 40, 1. [DOI] [PubMed] [Google Scholar]
- Otting G. Annu. Rev. Biophys. 2010, 39, 387. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan V.; Finch J. T.; Graziano V.; Lee P. L.; Sweet R. M. Nature 1993, 362, 219. [DOI] [PubMed] [Google Scholar]
- Zhou B.-R.; Feng H.; Kato H.; Dai L.; Yang Y.; Zhou Y.; Bai Y. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 19390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messmore J. M.; Fuchs D. N.; Raines R. T. J. Am. Chem. Soc. 1995, 117, 8057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon M. D.; Chu F.; Racki L. R.; de la Cruz C. C.; Burlingame A. L.; Panning B.; Narlikar G. J.; Shokat K. M. Cell 2007, 128, 1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W.; Xu M.; Huang C.; Liu N.; Chen S.; Zhu B. J. Biol. Chem. 2011, 286, 7983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiau C.; Trnka M. J.; Bozicevic A.; Ortiz Torres I.; Al-Sady B.; Burlingame A. L.; Narlikar G. J.; Fujimori D. G. Chem. Biol. 2013, 20, 494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Sady B.; Madhani H. D.; Narlikar G. J. Mol. Cell 2013, 51, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellman S. H. Biochemistry 1991, 30, 6633. [DOI] [PubMed] [Google Scholar]
- Gloss L. M.; Kirsch J. F. Biochemistry 1995, 34, 3990. [DOI] [PubMed] [Google Scholar]
- Seeliger D.; Soeroes S.; Klingberg R.; Schwarzer D.; Grubmüller H.; Fischle W. ACS Chem. Biol. 2012, 7, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steger D. J.; Lefterova M. I.; Ying L.; Stonestrom A. J.; Schupp M.; Zhuo D.; Vakoc A. L.; Kim J.-E. E.; Chen J.; Lazar M. A.; Blobel G. A.; Vakoc C. R. Mol. Cell. Biol. 2008, 28, 2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barski A.; Cuddapah S.; Cui K.; Roh T. Y.; Schones D. E.; Wang Z.; Wei G.; Chepelev I.; Zhao K. Cell 2007, 129, 823. [DOI] [PubMed] [Google Scholar]
- Richards E. J.; Elgin S. C. Cell 2002, 108, 489. [DOI] [PubMed] [Google Scholar]
- Munari F.; Soeroes S.; Zenn H. M.; Schomburg A.; Kost N.; Schroder S.; Klingberg R.; Rezaei-Ghaleh N.; Stützer A.; Gelato K. A.; Walla P. J.; Becker S.; Schwarzer D.; Zimmermann B.; Fischle W.; Zweckstetter M. J. Biol. Chem. 2012, 287, 33756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munari F.; Rezaei-Ghaleh N.; Xiang S.; Fischle W.; Zweckstetter M. PloS One 2012, 8, e60887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheutin T.; McNairn A. J.; Jenuwein T.; Gilbert D. M.; Singh P. B.; Misteli T. Science 2003, 299, 721. [DOI] [PubMed] [Google Scholar]
- Canzio D.; Chang E. Y.; Shankar S.; Kuchenbecker K. M.; Simon M. D.; Madhani H. D.; Narlikar G. J.; Al-Sady B. Mol. Cell 2011, 41, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P.; Banjade S.; Cheng H.-C.; Kim S.; Chen B.; Guo L.; Llaguno M.; Hollingsworth J. V.; King D. S.; Banani S. F.; Russo P. S.; Jiang Q. X.; Nixon B. T.; Rosen M. K. Nature 2012, 483, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradeepa M. M.; Sutherland H. G.; Ule J.; Grimes G. R.; Bickmore W. A. PLoS Genet. 2012, 8, e1002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nuland R.; van Schaik F. M.; Simonis M.; van Heesch S.; Cuppen E.; Boelens R.; Timmers H. M.; van Ingen H. Epigenet. Chromatin 2013, 6, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugarinov V.; Hwang P. M.; Ollerenshaw J. E.; Kay L. E. J. Am. Chem. Soc. 2003, 125, 10420. [DOI] [PubMed] [Google Scholar]
- Li Y.; Trojer P.; Xu C.-F. F.; Cheung P.; Kuo A.; Drury W. J.; Qiao Q.; Neubert T. A.; Xu R.-M. M.; Gozani O.; Reinberg D. J. Biol. Chem. 2009, 284, 34283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitges F. W.; Prusty A. B.; Faty M.; Stützer A.; Lingaraju G. M.; Aiwazian J.; Sack R.; Hess D.; Li L.; Zhou S.; Bunker R. D.; Wirth U.; Bouwmeester T.; Bauer A.; Ly-Hartig N.; Zhao K.; Chan H.; Gu J.; Gut H.; Fischle W.; Müller J.; Thomä N. H. Mol. Cell 2011, 42, 330. [DOI] [PubMed] [Google Scholar]
- An W.; Roeder R. G. Methods Enzymol. 2004, 377, 460. [DOI] [PubMed] [Google Scholar]
- Lauberth S. M.; Nakayama T.; Wu X.; Ferris A. L.; Tang Z.; Hughes S. H.; Roeder R. G. Cell 2013, 152, 1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nuland R.; Schram A. W.; van Schaik F. M.; Jansen P. W.; Vermeulen M.; Marc Timmers H. T. PLoS One 2013, 8, e73495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z.; Chen W.-Y.; Shimada M.; Nguyen U. T.; Kim J.; Sun X.-J.; Sengoku T.; McGinty R. K.; Fernandez J. P.; Muir T. W.; Roeder R. G. Cell 2013, 154, 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F.; Allahverdi A.; Yang R.; Lua G.; Zhang X.; Cao Y.; Korolev N.; Nordenskiöld L.; Liu C.-F. Angew. Chem., Int. Ed. 2011, 50, 9611. [DOI] [PubMed] [Google Scholar]
- Huang R.; Holbert M. A.; Tarrant M. K.; Curtet S.; Colquhoun D. R.; Dancy B. M.; Dancy B. C.; Hwang Y.; Tang Y.; Meeth K.; Marmorstein R.; Cole R. N.; Khochbin S.; Cole P. A. J. Am. Chem. Soc. 2010, 132, 9986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le D. D.; Cortesi A. T.; Myers S. A.; Burlingame A. L.; Fujimori D. G. J. Am. Chem. Soc. 2013, 135, 2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Brock A.; Herberich B.; Schultz P. G. Science 2001, 292, 498. [DOI] [PubMed] [Google Scholar]
- Lang K.; Chin J. W. Chem. Rev. 2014, 114, 4764. [DOI] [PubMed] [Google Scholar]
- Neumann H.; Peak-Chew S. Y.; Chin J. W. Nat. Chem. Biol. 2008, 4, 232. [DOI] [PubMed] [Google Scholar]
- Neumann H.; Hancock S. M.; Buning R.; Routh A.; Chapman L.; Somers J.; Owen-Hughes T.; van Noort J.; Rhodes D.; Chin J. W. Mol. Cell 2009, 36, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y.; Xue Y.; Song C.; Grunstein M. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 11493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppikofer M.; Kueng S.; Martino F.; Soeroes S.; Hancock S. M.; Chin J. W.; Fischle W.; Gasser S. M. EMBO J. 2011, 30, 2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cerbo V.; Mohn F.; Ryan D.; Montellier E.; Kacem S.; Tropberger P.; Kallis E.; Holzner M.; Hoerner L.; Feldmann A.; Richter F.; Bannister A.; Mittler G.; Michaelis J.; Khochbin S.; Feil R.; Schuebeler D.; Owen-Hughes T.; Daujat S.; Schneider R. eLife 2014, 3, e01632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropberger P.; Pott S.; Keller C.; Kamieniarz-Gdula K.; Caron M.; Richter F.; Li G.; Mittler G.; Liu E. T.; Buhler M.; Margueron R.; Schneider R. Cell 2013, 152, 859. [DOI] [PubMed] [Google Scholar]
- Tropberger P.; Schneider R. Nat. Struct. Mol. Biol. 2013, 20, 657. [DOI] [PubMed] [Google Scholar]
- Kim C. H.; Kang M.; Kim H. J.; Chatterjee A.; Schultz P. G. Angew. Chem., Int. Ed. 2012, 51, 7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattner M. J.; Vrabel M.; Carell T. Chem. Commun. 2013, 49, 379. [DOI] [PubMed] [Google Scholar]
- Nguyen D. P.; Garcia Alai M. M.; Virdee S.; Chin J. W. Chem. Biol. 2010, 17, 1072. [DOI] [PubMed] [Google Scholar]
- Nguyen D. P.; Garcia Alai M. M.; Kapadnis P. B.; Neumann H.; Chin J. W. J. Am. Chem. Soc. 2009, 131, 14194. [DOI] [PubMed] [Google Scholar]
- Ai H. W.; Lee J. W.; Schultz P. G. Chem. Commun. 2010, 46, 5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Schiller S. M.; Schultz P. G. Angew. Chem., Int. Ed. 2007, 46, 6849. [DOI] [PubMed] [Google Scholar]
- Guo J.; Wang J.; Lee J. S.; Schultz P. G. Angew. Chem., Int. Ed. 2008, 47, 6399. [DOI] [PubMed] [Google Scholar]
- Okeley N. M.; Zhu Y.; van Der Donk W. A. Org. Lett. 2000, 2, 3603. [DOI] [PubMed] [Google Scholar]
- Li Y.; Kao G. D.; Garcia B. A.; Shabanowitz J.; Hunt D. F.; Qin J.; Phelan C.; Lazar M. A. Genes Dev. 2006, 20, 2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. U.; Wang Y.-S.; Pai P.-J.; Russell W. K.; Russell D. H.; Liu W. R. Biochemistry 2012, 51, 5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y.; van der Donk W. Org. Lett. 2001, 3, 1189. [DOI] [PubMed] [Google Scholar]
- Chalker J. M.; Gunnoo S. B.; Boutureira O.; Gerstberger S. C.; Fernandez-Gonzalez M.; Bernardes G. J. L.; Griffin L.; Hailu H.; Schofield C. J.; Davis B. G. Chem. Sci. 2011, 2, 1666. [Google Scholar]
- Chalker J. M.; Lercher L.; Rose N. R.; Schofield C. J.; Davis B. G. Angew. Chem., Int. Ed. 2012, 51, 1835. [DOI] [PubMed] [Google Scholar]
- Kallappagoudar S.; Dammer E. B.; Duong D. M.; Seyfried N. T.; Lucchesi J. C. Proteomics 2013, 13, 1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K.; Sachdeva A.; Cox D. J.; Wilf N. W.; Lang K.; Wallace S.; Mehl R. A.; Chin J. W. Nat. Chem. 2014, 6, 393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H.; Wang K.; Davis L.; Garcia-Alai M.; Chin J. W. Nature 2010, 464, 441. [DOI] [PubMed] [Google Scholar]
- Xiao H.; Chatterjee A.; Choi S. H.; Bajjuri K. M.; Sinha S. C.; Schultz P. G. Angew. Chem., Int. Ed. 2013, 52, 14080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson D. B.; Xu J.; Shen Z.; Takimoto J. K.; Schultz M. D.; Schmitz R. J.; Xiang Z.; Ecker J. R.; Briggs S. P.; Wang L. Nat. Chem. Biol. 2011, 7, 779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai T.; Yanagisawa T.; Ohtake K.; Wakamori M.; Adachi J.; Hino N.; Sato A.; Kobayashi T.; Hayashi A.; Shirouzu M.; Umehara T.; Yokoyama S.; Sakamoto K. Biochem. Biophys. Res. Commun. 2011, 411, 757. [DOI] [PubMed] [Google Scholar]
- Yanagisawa T.; Takahashi M.; Mukai T.; Sato S.; Wakamori M.; Shirouzu M.; Sakamoto K.; Umehara T.; Yokoyama S. ChemBioChem 2014, 15, 1830. [DOI] [PubMed] [Google Scholar]
- Park H.-S.; Hohn M. J.; Umehara T.; Guo L.-T.; Osborne E.; Benner J.; Noren C.; Rinehart J.; Söll D. Science 2011, 333, 1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D. P.; Mahesh M.; Elsässer S. J.; Hancock S. M.; Uttamapinant C.; Chin J. W. J. Am. Chem. Soc. 2014, 136, 2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbely E.; Torres-Kolbus J.; Deiters A.; Chin J. W. J. Am. Chem. Soc. 2012, 134, 11912. [DOI] [PubMed] [Google Scholar]
- Wilkins B. J.; Rall N. A.; Ostwal Y.; Kruitwagen T.; Hiragami-Hamada K.; Winkler M.; Barral Y.; Fischle W.; Neumann H. Science 2014, 343, 77. [DOI] [PubMed] [Google Scholar]
- Hendzel M. J.; Wei Y.; Mancini M. A.; VanHooser A.; Ranalli T.; Brinkley B. R.; Bazett-Jones D. P.; Allis C. D. Chromosoma 1997, 106, 348. [DOI] [PubMed] [Google Scholar]
- Hsu J.-Y.; Sun Z.-W.; Li X.; Reuben M.; Tatchell K.; Bishop D. K.; Grushcow J. M.; Brame C. J.; Caldwell J. A.; Hunt D. F.; Lin R.; Smith M. M.; Allis C. D. Cell 2000, 102, 279. [DOI] [PubMed] [Google Scholar]
- Dawson P.; Muir T.; Clark-Lewis I.; Kent S. Science 1994, 266, 776. [DOI] [PubMed] [Google Scholar]
- Dawson P. E.; Kent S. B. Annu. Rev. Biochem. 2000, 69, 923. [DOI] [PubMed] [Google Scholar]
- Marahiel M. A.; Stachelhaus T.; Mootz H. D. Chem. Rev. 1997, 97, 2651. [DOI] [PubMed] [Google Scholar]
- Castro E. A. Chem. Rev. 1999, 99, 3505. [DOI] [PubMed] [Google Scholar]
- Bracher P. J.; Snyder P. W.; Bohall B. R.; Whitesides G. M. Origins Life Evol. Biospheres 2011, 41, 399. [DOI] [PubMed] [Google Scholar]
- Muir T. W. Annu. Rev. Biochem. 2003, 72, 249. [DOI] [PubMed] [Google Scholar]
- Mende F.; Seitz O. Angew. Chem., Int. Ed. 2011, 50, 1232. [DOI] [PubMed] [Google Scholar]
- Nagalingam A. C.; Radford S. E.; Warriner S. L. Synlett 2007, 2007, 2517. [Google Scholar]
- Wang P.; Miranda L. P. Int. J. Pept. Res. Ther. 2005, 11, 117. [Google Scholar]
- Botti P.; Villain M.; Manganiello S.; Gaertner H. Org. Lett. 2004, 6, 4861. [DOI] [PubMed] [Google Scholar]
- Blanco-Canosa J. B.; Dawson P. E. Angew. Chem., Int. Ed. 2008, 47, 6851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G.-M.; Li Y.-M.; Shen F.; Huang Y.-C.; Li J.-B.; Lin Y.; Cui H.-K.; Liu L. Angew. Chem., Int. Ed. 2011, 50, 7645. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A.; Chytil M.; Verdine G. L. Chem. Biol. 1996, 3, 981. [DOI] [PubMed] [Google Scholar]
- Tolbert T. J.; Franke D.; Wong C.-H. H. Bioorg. Med. Chem. 2005, 13, 909. [DOI] [PubMed] [Google Scholar]
- Liu D.; Xu R.; Dutta K.; Cowburn D. FEBS Lett. 2008, 582, 1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarov A. G.; Linn K. M.; Devereaux J. J.; Valiyaveetil F. I. ACS Chem. Biol. 2009, 4, 1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waugh D. S. Protein Expression Purif. 2011, 80, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panavas T.; Sanders C.; Butt T. R. Methods Mol. Biol. 2009, 497, 303. [DOI] [PubMed] [Google Scholar]
- He S.; Bauman D.; Davis J. S.; Loyola A.; Nishioka K.; Gronlund J. L.; Reinberg D.; Meng F.; Kelleher N.; McCafferty D. G. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 12033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogren-Knaak M. A.; Fry C. J.; Peterson C. L. J. Biol. Chem. 2003, 278, 15744. [DOI] [PubMed] [Google Scholar]
- Lo W. S.; Trievel R. C.; Rojas J. R.; Duggan L.; Hsu J. Y.; Allis C. D.; Marmorstein R.; Berger S. L. Mol. Cell 2000, 5, 917. [DOI] [PubMed] [Google Scholar]
- Cheung P.; Tanner K. G.; Cheung W. L.; Sassone-Corsi P.; Denu J. M.; Allis C. D. Mol. Cell 2000, 5, 905. [DOI] [PubMed] [Google Scholar]
- Hackeng T. M.; Griffin J. H.; Dawson P. E. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 10068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L. Z.; Dawson P. E. J. Am. Chem. Soc. 2001, 123, 526. [DOI] [PubMed] [Google Scholar]
- Shogren-Knaak M. A.; Ishii H.; Sun J.-M.; Pazin M. J.; Davie J. R.; Peterson C. L. Science 2006, 311, 844. [DOI] [PubMed] [Google Scholar]
- Cohen I.; Poreba E.; Kamieniarz K.; Schneider R. Genes Cancer 2011, 2, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.; Shogren-Knaak M. A. J. Biol. Chem. 2009, 284, 9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira H.; Flaus A.; Owen-Hughes T. J. Mol. Biol. 2007, 374, 563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee N.; Sinha D.; Lemma-Dechassa M.; Tan S.; Shogren-Knaak M. A.; Bartholomew B. Nucleic Acids Res. 2011, 39, 8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinker H.; Mueller-Planitz F.; Yang R.; Forné I.; Liu C.-F.; Nordenskiöld L.; Becker P. B. PLoS One 2014, 9, e88411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allahverdi A.; Yang R.; Korolev N.; Fan Y.; Davey C. A.; Liu C.-F.; Nordenskiöld L. Nucleic Acids Res. 2011, 39, 1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Lu C.; Yang Y.; Fan Y.; Yang R.; Liu C.-F.; Korolev N.; Nordenskiöld L. J. Mol. Biol. 2011, 414, 749. [DOI] [PubMed] [Google Scholar]
- Chiang K. P.; Jensen M. S.; McGinty R. K.; Muir T. W. ChemBioChem 2009, 10, 2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q.; Danishefsky S. J. Angew. Chem., Int. Ed. 2007, 46, 9248. [DOI] [PubMed] [Google Scholar]
- Paulus H. Annu. Rev. Biochem. 2000, 69, 447. [DOI] [PubMed] [Google Scholar]
- Noren C. J.; Wang J.; Perler F. B. Angew. Chem., Int. Ed. 2000, 39, 450. [PubMed] [Google Scholar]
- Shah N. H.; Muir T. W. Chem. Sci. 2014, 5, 446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muir T. W.; Sondhi D.; Cole P. A. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong S. R.; Mersha F. B.; Comb D. G.; Scott M. E.; Landry D.; Vence L. M.; Perler F. B.; Benner J.; Kucera R. B.; Hirvonen C. A.; Pelletier J. J.; Paulus H.; Xu M. Q. Gene 1997, 192, 271. [DOI] [PubMed] [Google Scholar]
- Simon M.; North J. A.; Shimko J. C.; Forties R. A.; Ferdinand M. B.; Manohar M.; Zhang M.; Fishel R.; Ottesen J. J.; Poirier M. G. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North J. A.; Šimon M.; Ferdinand M. B.; Shoffner M. A.; Picking J. W.; Howard C. J.; Mooney A. M.; van Noort J.; Poirier M. G.; Ottesen J. J. Nucleic Acids Res. 2014, 42, 4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North J. A.; Javaid S.; Ferdinand M. B.; Chatterjee N.; Picking J. W.; Shoffner M.; Nakkula R. J.; Bartholomew B.; Ottesen J. J.; Fishel R.; Poirier M. G. Nucleic Acids Res. 2011, 39, 6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manohar M.; Mooney A. M.; North J. A.; Nakkula R. J.; Picking J. W.; Edon A.; Fishel R.; Poirier M. G.; Ottesen J. J. J. Biol. Chem. 2009, 284, 23312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyland E. M.; Cosgrove M. S.; Molina H.; Wang D.; Pandey A.; Cottee R. J.; Boeke J. D. Mol. Cell. Biol. 2005, 25, 10060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulyanova N. P.; Schnitzler G. R. Mol. Cell. Biol. 2005, 25, 11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zettler J.; Schutz V.; Mootz H. D. FEBS Lett. 2009, 583, 909. [DOI] [PubMed] [Google Scholar]
- Shah N. H.; Dann G. P.; Vila-Perelló M.; Liu Z.; Muir T. W. J. Am. Chem. Soc. 2012, 134, 11338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vila-Perelló M.; Liu Z.; Shah N. H.; Willis J. A.; Idoyaga J.; Muir T. W. J. Am. Chem. Soc. 2013, 135, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C. T.; Tung C. L.; Li X. Mol. BioSyst. 2013, 9, 826. [DOI] [PubMed] [Google Scholar]
- Yang R.; Pasunooti K. K.; Li F.; Liu X.-W.; Liu C.-F. J. Am. Chem. Soc. 2009, 131, 13592. [DOI] [PubMed] [Google Scholar]
- Malins L. R.; Cergol K. M.; Payne R. J. ChemBioChem 2013, 14, 559. [DOI] [PubMed] [Google Scholar]
- Thompson R. E.; Chan B.; Radom L.; Jolliffe K. A.; Payne R. J. Angew. Chem., Int. Ed. 2013, 52, 9723. [DOI] [PubMed] [Google Scholar]
- Virdee S.; Kapadnis P. B.; Elliott T.; Lang K.; Madrzak J.; Nguyen D. P.; Riechmann L.; Chin J. W. J. Am. Chem. Soc. 2011, 133, 10708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Fekner T.; Ottesen J. J.; Chan M. K. Angew. Chem., Int. Ed. 2009, 48, 9184. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Li F.; Liu C.-F. Chem. Commun. 2011, 47, 1746. [DOI] [PubMed] [Google Scholar]
- Popp M. W.; Ploegh H. L. Angew. Chem., Int. Ed. 2011, 50, 5024. [DOI] [PubMed] [Google Scholar]
- Piotukh K.; Geltinger B.; Heinrich N.; Gerth F.; Beyermann M.; Freund C.; Schwarzer D. J. Am. Chem. Soc. 2011, 133, 17536. [DOI] [PubMed] [Google Scholar]
- Shimko J. C.; North J. A.; Bruns A. N.; Poirier M. G.; Ottesen J. J. J. Mol. Biol. 2011, 408, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami T.; Akai Y.; Fujimoto H.; Kita C.; Aoki Y.; Konishi T.; Waseda M.; Takemura L.; Aimoto S. Bull. Chem. Soc. Jpn. 2013, 86, 690. [Google Scholar]
- Kawakami T.; Aimoto S. Tetrahedron 2009, 65, 3871. [Google Scholar]
- Casadio F.; Lu X.; Pollock S. B.; LeRoy G.; Garcia B. A.; Muir T. W.; Roeder R. G.; Allis C. D. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 14894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G.-M.; Wang J.-X.; Liu L. Angew. Chem., Int. Ed. 2012, 51, 10347. [DOI] [PubMed] [Google Scholar]
- McGinty R. K.; Kim J.; Chatterjee C.; Roeder R. G.; Muir T. W. Nature 2008, 453, 812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z.-W. W.; Allis C. D. Nature 2002, 418, 104. [DOI] [PubMed] [Google Scholar]
- Ng H. H.; Xu R.-M. M.; Zhang Y.; Struhl K. J. Biol. Chem. 2002, 277, 34655. [DOI] [PubMed] [Google Scholar]
- Briggs S. D.; Xiao T.; Sun Z.-W. W.; Caldwell J. A.; Shabanowitz J.; Hunt D. F.; Allis C. D.; Strahl B. D. Nature 2002, 418, 498. [DOI] [PubMed] [Google Scholar]
- Kim J.; Guermah M.; McGinty R. K.; Lee J.-S. S.; Tang Z.; Milne T. A.; Shilatifard A.; Muir T. W.; Roeder R. G. Cell 2009, 137, 459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.; Kim J.-A.; McGinty R. K.; Nguyen U. T. T.; Muir T. W.; Allis C. D.; Roeder R. G. Mol. Cell 2013, 49, 1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuermann J. C.; de Ayala Alonso A. G.; Oktaba K.; Ly-Hartig N.; McGinty R. K.; Fraterman S.; Wilm M.; Muir T. W.; Müller J. Nature 2010, 465, 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty R. K.; Kohn M.; Chatterjee C.; Chiang K. P.; Pratt M. R.; Muir T. W. ACS Chem. Biol. 2009, 4, 958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitcomb S. J.; Fierz B.; McGinty R. K.; Holt M.; Ito T.; Muir T. W.; Allis C. D. J. Biol. Chem. 2012, 287, 23718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierz B.; Kilic S.; Hieb A. R.; Luger K.; Muir T. W. J. Am. Chem. Soc. 2012, 134, 19548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalb R.; Latwiel S.; Baymaz H. I.; Jansen P. W.; Müller C. W.; Vermeulen M.; Müller J.. Nat. Struct. Mol. Biol. 2014. [DOI] [PubMed] [Google Scholar]
- Chatterjee C.; McGinty R. K.; Fierz B.; Muir T. W. Nat. Chem. Biol. 2010, 6, 267. [DOI] [PubMed] [Google Scholar]
- Fierz B.; Chatterjee C.; McGinty R. K.; Bar-Dagan M.; Raleigh D. P.; Muir T. W. Nat. Chem. Biol. 2011, 7, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan F. T.; Kaminski C. F.; Kaminski Schierle G. S. ChemPhysChem 2011, 12, 500. [DOI] [PubMed] [Google Scholar]
- Tweedie-Cullen R. Y.; Reck J. M.; Mansuy I. M. J. Proteome Res. 2009, 8, 4966. [DOI] [PubMed] [Google Scholar]
- Siman P.; Karthikeyan S. V.; Nikolov M.; Fischle W.; Brik A. Angew. Chem., Int. Ed. 2013, 52, 8059. [DOI] [PubMed] [Google Scholar]
- Garcia B. A.; Pesavento J. J.; Mizzen C. A.; Kelleher N. L. Nat. Methods 2007, 4, 487. [DOI] [PubMed] [Google Scholar]
- Young N. L.; DiMaggio P. A.; Garcia B. A. Cell. Mol. Life Sci. 2010, 67, 3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton T.; Yaffe E.; Kenigsberg E.; Bantignies F.; Leblanc B.; Hoichman M.; Parrinello H.; Tanay A.; Cavalli G. Cell 2012, 148, 458. [DOI] [PubMed] [Google Scholar]
- Ruthenburg A. J.; Li H.; Patel D. J.; Allis C. D. Nat. Rev. Mol. Cell Biol. 2007, 8, 983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheldof N.; Tabuchi T. M.; Dekker J. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 12463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther M. G.; Levine S. S.; Boyer L. A.; Jaenisch R.; Young R. A. Cell 2007, 130, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang G.; Lin J. C.; Wei V.; Yoo C.; Cheng J. C.; Nguyen C. T.; Weisenberger D. J.; Egger G.; Takai D.; Gonzales F. A.; Jones P. A. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg A. J.; Allis C. D.; Wysocka J. Mol. Cell 2007, 25, 15. [DOI] [PubMed] [Google Scholar]
- Lindroth A. M.; Shultis D.; Jasencakova Z.; Fuchs J.; Johnson L.; Schubert D.; Patnaik D.; Pradhan S.; Goodrich J.; Schubert I.; Jenuwein T.; Khorasanizadeh S.; Jacobsen S. E. EMBO J. 2004, 23, 4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg A. J.; Li H.; Milne T. A.; Dewell S.; McGinty R. K.; Yuen M.; Ueberheide B.; Dou Y.; Muir T. W.; Patel D. J.; Allis C. D. Cell 2011, 145, 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S.; Shogren-Knaak M. A. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 18243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzaz A. M.; Vitalini M. W.; Thomas A. S.; Price J. P.; Blacketer M. J.; Cryderman D. E.; Zirbel L. N.; Woodcock C. L.; Elcock A. H.; Wallrath L. L.; Shogren-Knaak M. A. J. Biol. Chem. 2014, 289, 6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt P.; LeRoy G.; Drury W. J. III; Zee B. M.; Son J.; Beck D. B.; Young N. L.; Garcia B. A.; Reinberg D. Cell 2012, 151, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacketer M. J.; Feely S. J.; Shogren-Knaak M. A. J. Biol. Chem. 2010, 285, 34597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal C.; Blacketer M. J.; Shogren-Knaak M. A. Anal. Biochem. 2014, 457, 51. [DOI] [PubMed] [Google Scholar]
- Grewal S. I.; Moazed D. Science 2003, 301, 798. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Mosch K.; Fischle W.; Grewal S. I. Nat. Struct. Mol. Biol. 2008, 15, 381. [DOI] [PubMed] [Google Scholar]
- Huh J. W.; Wu J.; Lee C. H.; Yun M.; Gilada D.; Brautigam C. A.; Li B. EMBO J. 2012, 31, 3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.-H. H.; Wu J.; Li B. Mol. Cell 2013, 52, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrozza M. J.; Li B.; Florens L.; Suganuma T.; Swanson S. K.; Lee K. K.; Shia W.-J. J.; Anderson S.; Yates J.; Washburn M. P.; Workman J. L. Cell 2005, 123, 581. [DOI] [PubMed] [Google Scholar]
- Bartke T.; Vermeulen M.; Xhemalce B.; Robson S. C.; Mann M.; Kouzarides T. Cell 2010, 143, 470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolov M.; Stützer A.; Mosch K.; Krasauskas A.; Soeroes S.; Stark H.; Urlaub H.; Fischle W.. Mol. Cell. Proteomics 2011, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Chen Z.; Mao Z.; Zhang H.; Ding X.; Chen S.; Zhang X.; Xu R.; Zhu B. EMBO Rep. 2011, 12, 1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shema-Yaacoby E.; Nikolov M.; Haj-Yahya M.; Siman P.; Allemand E.; Yamaguchi Y.; Muchardt C.; Urlaub H.; Brik A.; Oren M.; Fischle W. Cell Rep. 2013, 4, 601. [DOI] [PubMed] [Google Scholar]
- Haj-Yahya M.; Eltarteer N.; Ohayon S.; Shema E.; Kotler E.; Oren M.; Brik A. Angew. Chem., Int. Ed. 2012, 51, 11535. [DOI] [PubMed] [Google Scholar]
- Long L.; Thelen J. P.; Furgason M.; Haj-Yahya M.; Brik A.; Cheng D.; Peng J.; Yao T. J. Biol. Chem. 2014, 289, 8916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen U. T. T.; Bittova L.; Müller M. M.; Fierz B.; David Y.; Houck-Loomis B.; Feng V.; Dann G. P.; Muir T. W. Nat. Methods 2014, 11, 834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armache K.-J.; Garlick J.; Canzio D.; Narlikar G.; Kingston R. Science 2011, 334, 977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaudo N.; Fernández I. S.; McLaughlin S. H.; Peak-Chew S. Y.; Rhodes D.; Martino F. Nat. Struct. Mol. Biol. 2013, 20, 1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canzio D.; Liao M.; Naber N.; Pate E.; Larson A.; Wu S.; Marina D.; Garcia J.; Madhani H.; Cooke R.; Schuck P.; Cheng Y.; Narlikar G. Nature 2013, 496, 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H.; Gruschus J.; Ghirlando R.; Tjandra N.; Bai Y. J. Am. Chem. Soc. 2009, 131, 15104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M.; Nadaud P. S.; Bernier M. W.; North J. A.; Hammel P. C.; Poirier M. G.; Jaroniec C. P. J. Am. Chem. Soc. 2013, 135, 15278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochinyan S.; Sun L.; Ghosh I.; Barshevsky T.; Xu J.; Xu M.-Q. Q. BioTechniques 2006, 42, 63. [DOI] [PubMed] [Google Scholar]
- Severinov K.; Muir T. W. J. Biol. Chem. 1998, 273, 16205. [DOI] [PubMed] [Google Scholar]
- Liu X.; Wang L.; Zhao K.; Thompson P. R.; Hwang Y.; Marmorstein R.; Cole P. A. Nature 2008, 451, 846. [DOI] [PubMed] [Google Scholar]
- Pengelly A. R.; Copur Ö.; Jäckle H.; Herzig A.; Müller J. Science 2013, 339, 698. [DOI] [PubMed] [Google Scholar]
- Hathaway N. A.; Bell O.; Hodges C.; Miller E. L.; Neel D. S.; Crabtree G. R. Cell 2012, 149, 1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescher J. A.; Bertozzi C. R. Nat. Chem. Biol. 2005, 1, 13. [DOI] [PubMed] [Google Scholar]
- Giriat I.; Muir T. W. J. Am. Chem. Soc. 2003, 125, 7180. [DOI] [PubMed] [Google Scholar]
- Borra R.; Dong D.; Elnagar A. Y.; Woldemariam G. A.; Camarero J. A. J. Am. Chem. Soc. 2012, 134, 6344. [DOI] [PMC free article] [PubMed] [Google Scholar]