

1. Introduction
Since their discovery in the 1980s and 1990s, the human protein lysine acetyltransferase encoded by the paralogous p300 and CBP genes has received much interest. p300/CBP functions in regulating the expression of genes controlling several basic cellular processes, such as proliferation and homeostasis, and plays a role in a variety of human diseases, particularly solid tumors.
Numerous natural product and synthetic inhibitors of the acetyltransferase activity have been identified and used to generate a crystal structure of the active site, probe the p300/CBP enzyme kinetics, and interrogate p300/CBP cellular functions. These studies revealed that acetyl-CoA binds in a tunnel enclosed by a unique loop, while the substrate protein transiently associates with an acidic patch, following a hit-and-run kinetic mechanism. p300/CBP acetylates histones as well as other proteins including other epigenetic enzymes and transcription factors. Studies with inhibitor compounds in cells and animals have confirmed that the p300/CBP acetylation activity has roles in diverse functions including cell migration and invasion, maintenance of the differentiated state, tau-mediated neurodegeneration, and learning and memory.
Also, important components of p300/CBP are the domains flanking the acetyltransferase domain, including three cysteine/histidine-rich domains and a bromodomain. Protein ligands of these have been identified. Their roles in regulating the acetyltransferase activity and substrate specificity, as well as identification of compounds that can block or mimic ligand binding, are topics of ongoing study.
Biochemical investigation of protein acetylation has posed unique challenges, due in part to its dynamic reversibility, the weak affinity of binding modules that recognize it, and the complexity of the multiprotein interaction networks. Therefore, diverse techniques in biochemistry, molecular biology, and proteomics have coevolved with our understanding of the p300/CBP enzyme. In this Review, part of the thematic issue on Epigenetics, we summarize the current understanding of p300/CBP including the novel technologies developed for these studies.
2. Background Information
2.1. Epigenetics and Chromatin
While the central dogma of molecular biology describes how the DNA sequence dictates the cell’s functions, recent researchers have observed several intriguing phenomena that prove reality to be more complex. Identical twins inherit the same DNA sequence, but can have differences in appearance, intellect, and disease incidence. Multicellular organisms are composed of extremely diverse cell types, almost all of which contain the same DNA sequence. Mice with the same DNA sequence can have different coat colors and body weight. Calico cats have patches of fur with different colors, but the underlying skin cells have the same DNA sequences. Several disorders exist that have different outcomes depending on whether the mutation responsible was inherited from a female or a male parent. Also, the human genome encodes only twice as many genes as the fly genome, for example, so the number of genes cannot account for the greater complexity of humans.
These examples all illustrate the existence of mechanisms over and above the genome, which influence biology, and fall into the emerging field of epigenetics. The term “epigenetics” was coined by Waddington in 1942.1 As was eloquently put, “Mendel’s gene is more than a DNA moiety.”2 Several mechanisms exist in cells that contribute to epigenetic inheritance. In addition to the DNA inherited by a gamete during meiosis, the egg and embryo receive maternal RNAs. These serve various transient functions in the progeny’s cells before the RNAs are degraded. Similarly, the cytoplasm of the daughter cell contains many proteins that were made in the germline of the parent, constituting another mode of inheritance of traits not necessarily encoded by the DNA. Furthermore, the DNA inherited is not a naked sequence; it is packaged into chromatin, which is the topic of the rest of this section.
The human genome, a copy of which is contained in almost every human cell, is about 2 m in total length, if placed end-to-end, while the chomatinized genome is about 0.09 mm in total length, if placed end-to-end, which can fit into the cell nucleus, that is typically less than 0.01 mm in diameter.3 Chromatin is made up of fundamental subunits called nucleosomes. A nucleosome consists of 146 base pairs of DNA wrapped around an octamer of histone proteins. The histone octamer consists of two copies each of histones H2A, H2B, H3, and H4. Furthermore, at least 20bp of DNA between each nucleosome can be packaged with the linker histone, H1. The histones, however, are not simply inert packaging, but rather they arrange the DNA in a way that the information can be stored and retrieved.
Chromatin can be assembled in different conformations, which are influenced by a variety of factors, including DNA sequence elements, DNA modifications, histone modifications, incorporation of histone variants, nucleosome occupancy and spacing, nucleosome sliding, and association of nonhistone proteins with the chromatin. The physical structure of chromatin has functional consequences for DNA templated processes, including gene expression, DNA replication, and DNA repair. Open, permissive, active chromatin promotes DNA transcription and replication, whereas closed, silenced, repressed chromatin is turned “off” to these processes. For example, telomeres and centromeres are generally silenced, and boundary elements prevent the silencing from spreading into regions that are turned “on”. Yet these distinctions are not static: enzymes that modify DNA and histones, chaperones that allow histones to be replaced, and enzymes that consume ATP to slide nucleosomes can alter the local chromatin structure. Through these actions, chromatin structure can be altered during one’s lifetime in response to a variety of environmental and internal conditions. In this way, the expression of genes can be altered in a cell or a cell lineage without changing the DNA sequence.
In addition to duplication of the DNA sequence, aspects of the chromatin conformation are passed down as the cell divides, as modifications to the DNA and histones distribute semiconservatively to the daughter strands during replication. For example, gene X could be transcriptionally silenced in a parent cell, and the progeny cell then inherits this silenced state along with the gene sequence. Thus, chromatin embodies an important level of epigenetic inheritance.
2.2. Post-Translational Modifications of Proteins
Once made, proteins can be regulated by an astounding variety of changes. Collectively, these changes are called post-translational modifications, or PTMs. PTMs can affect enzyme activity, protein–protein interactions, or other functions of the target protein.
Certain residue side chains can undergo chemical modifications,4 which expand the building blocks of proteins from the 20 canonical amino acids to a growing list of interesting molecules, and expand the proteome to include differentially modified proteins. Without changing the atoms in a proline residue, reversible isomerization can change its structure. Arginine residue side chains can be deiminated, whereby a terminal nitrogen atom is replaced with oxygen. In rare cases, histidine can be modified to dipthamide. Several types of small groups, which are usually donated from coenzymes, can be covalently added to residue side chains. Serine, threonine, tyrosine, and histidine side chains (as well as arginine and lysine side chains in prokaryotes) can be reversibly phosphorylated. Lysine side chains’ epsilon amine (as well as protein N-terminal alpha amine and, in rare cases, serine and threonine side chains5) can be reversibly acetylated, and lysine side chains’ epsilon amine can also be formylated, crotonylated, propionylated, butyrylated, hydroxybutyrylated, glutarylated, succinylated, malonylated, or 3-phosphoglycerylated.6 Arginine side chains can be mono- or dimethylated, while lysine side chains can be reversibly mono-, di-, or trimethylated. Tyrosine side chains can be hydroxylated. Serine and threonine side chains can be O-GlcNAcylated. Cysteine side chains can be prenylated (by the addition of a farnesyl or a geranyl–geranyl moiety). Serine side chains can be octanoylated. Arginine, glutamic acid, aspartic acid, asparagine, cysteine, and phosphoserine (and sometimes lysine and diphthamide) side chains can be mono- or poly-ADP-ribosylated. There are also different redox states of certain side chains, which can give rise to cysteine oxidation, cysteine disulfide formation, and histidine oxidation. Post-translational modifications can also be larger in size: target proteins can be covalently linked at their lysine side chains to small proteins like ubiquitin, SUMO, Rubb, and Nedd, either with one such addition or linkage in a chain. Finally, proteins can be modified by the removal of part of the sequence by proteolytic cleavage.
This field of post-translational modifications is best summarized by Cohen et al.: “one of the major goals in postgenomic biology is to unravel the molecular basis and the physiological role of covalent protein modifications... This has the potential to identify novel disease pathways and thereby new therapeutic targets as well as diagnostic markers.”7
This Review is focused on lysine acetylation. The acetyl group in question is donated by the cofactor, acetyl-coenzyme A. Coenzyme A, which is found in all characterized living organisms on earth,8 is synthesized by cells from pantothenic acid (vitamin B5) and cysteine,9 and its thiol moiety can react with the carboxylic acids of acyl groups to activate those groups with an energy-rich thioester bond. Acetyltransferase enzymes catalyze the covalent transfer of the acetyl group from acetyl-CoA to the epsilon amine of the lysine side chain, with the release of the free thiol, CoASH (see Figure 1). The resultant addition of atomic bulk to the side chain and loss of positive charge can change intramolecular protein conformation or change affinity for binding partners. Thus, this small modification can have a variety of possible influences on the function of the particular substrate protein.
Figure 1.
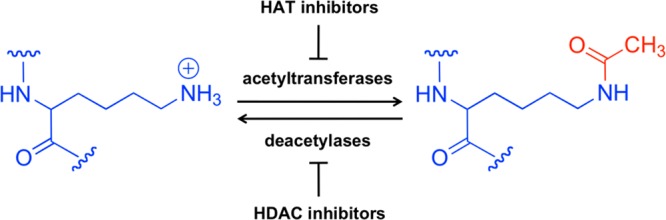
Reversible acetylation of the epsilon amine group of lysine side chains.
One method of studing post-translational modifications is to measure the function of site-specifically modified proteins, but this has encountered challenges in the case of acetylation. Acetylation and deacetylation can be mimicked using site-specific glutamine and arginine mutations (respectively), which preserve the charge of the residue they are replacing but are not substrates for acetyl transfer or deacetylation. However, these are not perfect mimics and often do not phenocopy the acetylated or deacetylated protein in cells. Proteins can be purified from cells that lack deacetylases or acetyltransferases, generating populations that are hyper- or hypo-acetylated (respectively), but these alterations typically affect more than one substrate and/or residue. Third, purified proteins can be reacted with purified acetyltransferase enzyme and acetyl-CoA to generate acetylated products, but the site-specificity of acetyltransferases is often promiscuous and reactions rarely go to completion.10 Acetyl-lyine can be installed in a site-specific manner to proteins using a nonsense-suppression methodology,11,12 but this typically generates only very small quantities of the desired protein. Fifth, acetyl-lysine can be installed in a site-specific manner to purified proteins using expressed protein ligation13 or native chemical ligation, to generate a semisynthetic product, but this is restricted to proteins with acetylation close to one of the termini (or approachable by circular permutation14), and has additional structural and sequence requirements. Finally, cysteine alkylation chemistry can be performed on purified proteins to generate an acetyl-lysine or the corresponding lysine mimic, although any other cysteines present would similarly be modified.15 This latter methodology produces an acetyl-lysine mimic that is not hydrolyzed by deacetylases (as demonstrated by reactions with two example deacetylases). This therefore addresses a further challenge in lysine acetylation research, which is that the acetyl group is rapidly hydrolyzed in experiments where deacetylase enzymes are present, such as lysates or other complex mixtures.
One group of proteins that undergo acetylation is histones. All histones, including the four canonical histones, their variants, and linker histones, can undergo acetylation, and this was first observed as early as the 1960s.16,17 Indeed, histones have served as a model system for the study of acetylation and other post-translational modifications, partially because of their abundance in cells and their high sequence conservation among eukaryotes.18
Several nonhistone proteins also have well-characterized acetylation sites,19 including the tumor suppressor protein p53 and the tubulin components of the cytoskeleton.20 The perception of the “acetylome” as being on a scale close to that of the phospho-proteome has only recently become appreciated.21 Several reports of proteome-wide study of lysine acetylation have identified thousands of sites.22−24 These proteins span many important cellular pathways, including chromatin remodeling, cell cycle, splicing, nuclear transport, actin nucleation, and mitochondrial metabolism. Interestingly, a great proportion of acetylated proteins seem to be included in large macromolecular complexes. For example, 61 of the 81 ribosomal subunits are acetylated, 11 of the 16 F1F0-ATP synthase subunits are acetylated, and 69 of the 143 spliceosome subunits are acetylated. These example complexes are found in the cytoplasm, mitochondrion, and nucleus (respectively), and this further illustrates the pervasiveness of acetylation throughout the cell.
The link between metabolism and acetylation is multifaceted. In addition to the acetylation of metabolic enzymes to regulate their activities, and regulation of the expression of metabolic enzymes by acetylation of histones and transcription factors, metabolic pathways influence the amount of acetyl-CoA available to lysine acetyltranferase enzymes (see Figure 2).25 In general, a high energy balance leads to a higher level of acetyl-CoA in the cell, due to catabolism of glucose and fatty acids, while some acetyl-CoA is available during conditions of low energy balance from acetate. Although acetyl-CoA is membrane-impermeable, it can pass through the nuclear pore complex, and pyruvate can be imported to the mitochondrion or citrate exported to the cytoplasm for acetyl-CoA production. Recently, the pyruvate dehydrogenase complex subunits were found to be present in the nucleus, providing a mechanism for nuclear acetyl-CoA production.26 Reactions that compete with lysine acetylation for the pool of acetyl-CoA include the synthesis of fatty acids, cholesterol, citrate, and ketone bodies. In fact, about 4% of cellular enzymes utilize coenzyme A and its thioesters as a cofactor.27
Figure 2.
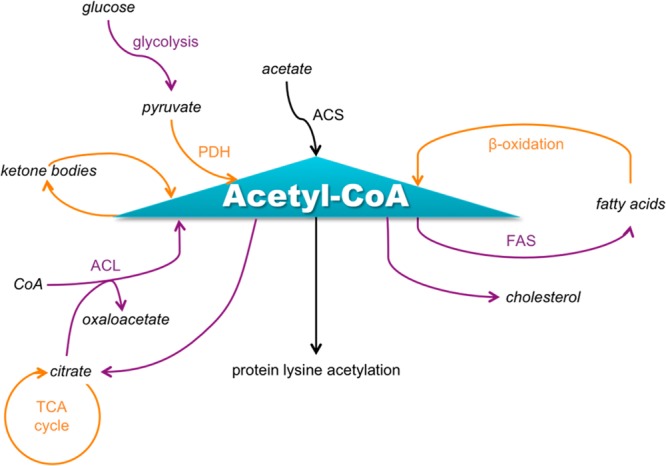
Major metabolic processes that produce or consume acetyl-CoA. Processes occurring in the cytoplasm are indicated using purple font, and processes occurring in the mitochondrion are indicated using orange font. Note that PDH can also be nuclear. This figure was adapted in part from Albaugh et al.30
A further way in which lysine acetylation is linked to energy balance is through the sirtuin deacetylase enzymes, which require a nicotinamide adenine dinucleotide (NAD) cofactor,28 and therefore low nutrient conditions can facilitate deacetylation. Acetylation is not the only PTM that is influenced by energy balance; for example, glycosylation of proteins is regulated by the availability of UDP-GlcNAc through the hexosamine pathway. These mechanisms help a cell make appropriate responses to nutrient conditions,29 but might also provide mechanisms for disruption of post-translational modification states in diseased cells that have altered metabolism.
2.3. Case Study: Histone H4 Tail
Between the 1960s and 1990s, histone lysine acetylation was found to correlate with transcriptional activation.31−34 It is now known that this correlation is especially true for histone H4 lysines 5 and 8 (as well as histone H3 lysines 9 and 14), where acetylation is considered permissive to transcription, but there are exceptions, for example, histone H4 lysine 12, where acetylation may be repressive to transcription.35
One way that the post-translational modifications on the histone tail can influence chromatin structure is by weakening the interaction of positively charged residue side chains with the negatively charged phosphodiester backbone of DNA, thereby allowing easier access of the DNA by polymerase.36 Indeed, of the 23 residues of the histone H4 tail, 10 residues are positively charged. However, recent reports reveal that disruption of these electrostatic interactions is not sufficient to cause the histone tails to dissociate from the DNA.37
In addition to altering affinity for DNA, protein function can be regulated by acetylation through altering affinity for other proteins. Bromodomains are domains with the special purpose of binding acetylated proteins, bearing different flanking sequence specificities.38 Bromodomains are contained either singly or doubly in more than 40 human proteins. Although the binding affinity of each bromodomain with each acetylated lysine is generally weak, it is believed that it has major functional relevance in the cell, due in part to some proteins possessing more than one bromodomain in tandem that synergize to increase binding affinity, and also due to histones possessing multiple acetylated residue that can act combinatorially. In the example of the histone H4 tail, at least three bromodomain-containing proteins have been identified that interact with the various acetylated residues. Gcn5 has one bromodomain, while TAF1 and BRDT each have double bromodomains in tandem.
Acetylation is not the only post-translational modification that attracts proteins with a specifically tailored recognition domain. The list of proteins with domains that bind methylated, phosphorylated, or other modified residues is growing.39 For example, with respect to the histone H4 tail, di-/trimethylated lysine 20 is specifically recognized by the tandem tudor domains of 53BP1, the tandem tudor domains of Crb2,40 and the tandem tudor domains of JMJD2A,41 while mono-/dimethylated lysine 20 is specifically recognized by the three MBT repeats of L3MBTL2.42,43 An interesting phenomenon of cooperativity in binding between two nearby histone molecules is illustrated by the protein BPTF, which has a bromodomain that binds acetylated histone H4 as well as a PHD-finger domain that binds methylated histone H3.44
Given the number of residues on each histone that can be post-translationally modified, and the variety of modifications possible, one can begin to envision the potential combinatorial complexity possible for each nucleosome. For every molecule of the histone H4 N-terminal tail, over 500 such combinations are possible! One way to frame this problem is the Allis lab’s “histone code hypothesis”, whereby the “marks” (PTMs) on histones are “written” (by enzymes like acetyltransferases and methyltransferases), “read” (by proteins with domains like bromodomains and tudor domains), and “erased” (by enzymes like deacetylases and demethylases) to encode information in each nucleosome to direct its functions.45 A further extension of this scheme is the “nucleosome code hypothesis”, whereby histone H4 could be grouped with sequence variants of H3, H2A, and H2B to create another level of the information encoded by each nucleosome.46
While many combinations are theoretically possible for each nucleosome, it has been hypothesized that only a few functionally relevant patterns are prevalent in the cell,47 and recent evidence supports this model.48 Indeed, there are documented examples of crosstalk between different modified residues, which may be the mechanisms for how these combination states are established. In the example of histone H4, methylation of R3 could increase acetylation of K5 (because of the chromodomain in the acetyltranferase, yeast Esa149), methylation of K20 could increase acetylation of K16 (because of the chromodomain of Msl3 in the acetyltransferase complex, Mof48), acetylation of K5 could increase dimethylation of R3 and decrease monomethylation of R3 (by altering the activity of the methyltransferases, PRMT5 and PRMT150), lysine SUMOylation decreases lysine acetylation and ubiquitylation (because these modifications compete for the same residues51), and acetylation of K16 decreases possible lysine ADP-ribosylation.52
Furthermore, several acetyltransferases have bromodomains,53 which could mediate further cooperativity between acetylation sites. This has been proposed to influence the histone H4 N-terminal tail acetylation by an an N-terminally progressing “zip” mechanism. In this model, K16 is the first to be acetylated, followed by K12, then K8, and finally K5. In this manner, acetylation on this tail may be an all-or-nothing phenomenon, where K16 acetylation sets off a cascade that results in hyperacetylation of all four lysines on each molecule of histone H4.54,55
In the case of histone acetylation, the balance of addition and removal of the acetyl groups is thought to be highly dynamic,56−58 which is especially true in regions of the chromatin with high levels of histone methylation,59,60 specifically histone H3 with trimethylated K4.61,62 This dynamic state is possible because of the action of histone deacetylase enzymes (HDACs), the first of which was cloned in 1996.63 We now know that HDACs can act by a simple hydrolytic mechanism (the classical HDACs, which contain a crucial zinc ion), or a complex NAD-requiring mechanism (the sirtuins). At least one sirtuin, SIRT2, is known to deacetylate histone H4.
Several acetyltransferases are known to acetylate histone H4. Lysine acetyltransferase enzymes are classified according to sequence conservation into four families, GNAT family (Gcn5 and Pcaf), p300/CBP, Rtt109 (not present in humans), and the MYST family (includes MOZ/Morf, Ybf2/Sas3, Sas2, Tip60), and examples from more than one class are thought to acetylate histone H4, including p300/CBP. Specifically, when recombinantly expressed and purified, Pcaf can acetylate histone H4 K864 and Tip60 can acetylate histone H4 K5/8/12/16.65 Recombinantly expressed and purified p300 acetyltransferase domain efficiently acetylates H4 N-terminal tail when supplied as a synthetic peptide.66 Purified full-length p300 also efficiently acetylates H4 N-terminal tail when supplied as full-length free histones or mononucleosomes,67 with a preference for K5/K8.68 To extend this to histones in a more native chromatin conformation, Cos7 cells were transiently transfected with p300 or CBP and analyzed for histone acetylation by immunofluorescence microscopy. In cells with increased p300/CBP levels, increased H4 K5/K8 acetylation was seen, as well as other sites.69 In cells with p300/CBP knockout, reduced H4 acetylation at certain gene sequences was observed by chromatin IP with antibodies against acetylated K5, K8, K12, and K16.70
In the field of cancer therapeutic discovery, development of acetyltransferase inhibitors has lagged behind other epigenetic target classes. The deacetylase inhibitors Vorinostat and Romidepsin have proved useful in the clinic for treating cutaneous T cell lymphoma, and compounds that block the acetyl-binding pocket of BET bromodomains (in proteins like Brd2/3/4) are currently in clinical trials for midline carcinoma and other cancers.71 To our knowledge, there have been no acetyltransferase inhibitors that have progressed to clinical trials, despite the sense that they might prove equally, or more, valuable as compared to deacetylase and bromodomain inhibitors. Acetyltransferase enzymes, in particular p300/CBP, their involvement in disease, and their inhibitor development is the topic of the rest of this Review.
2.4. Discovery of p300/CBP
p300 and CBP are two acetyltransferase enzymes in humans and most higher eukaryotes. p300 (also called EP300 or KAT3B) is so-named because it is about 300 kDa in size (with 2414 amino acids). p300 was reported in 198572 and 198973 (and cloned in 199474) in studies looking for proteins that bind E1A, an adenoviral oncogenic transcription factor. Meanwhile, CBP (also called CREBBP or KAT3A) was first reported and cloned in 1993 in a study of proteins that bind CREB,75 a transcription factor that binds cAMP response elements (CREs). CBP is composed of 2441 amino acids, and because of the high sequence homology observed between it and p300,76 the two proteins are now collectively referred to as p300/CBP. They are in a family of their own, with little sequence homology between them and other acetyltransferases in the human genome.77
Soon after the first known histone acetyltransferases (HATs) were cloned in 1995–1996,78−80 p300/CBP was found to have HAT activity as well.81,82 Characterization of this acetyltransferase activity has been of ongoing interest since then, and significant contributions have been made by many laboratories.
The search for p300/CBP orthologs in a variety of living organisms has yielded several interesting observations. First, no orthologs have been found in prokaryotes, and within the domain of eukaryotes, only the “higher” eukaryotes appear to have p300/CBP orthologs.83 Because nuclear84 and chromatin85 structures are largely conserved between “lower” and “higher” eukaryotes, this argues that the evolution of p300/CBP was not triggered by the evolution of the nucleus and chromatin, but later when metazoa evolved. Thus, p300/CBP function may be especially important in aspects of growth and development that are prevalent in multicellular organisms, such as cell-to-cell communication or organ morphogenesis.86 That being said, crystallography studies have unveiled that p300/CBP has a structural homologue in yeast, called Rtt109,87 although there is little sequence or apparent functional similarity between the two acetyltransferases.
Four genes that may be orthologs of p300/CBP have been identified in the plant Arabidopsis.88 Within the animal kingdom, many different species have been found to possess p300/CBP, but with different copy numbers. Evolutionarity distant animals such as flies and roundworms possess one p300/CBP each, although flatworms seem to have acquired their own duplication of p300/CBP. Sea squirts, which are in the phylum chordata but are not in the subphylum vertebrates, also seem to possess one p300/CBP. When vertebrates evolved, the chromosomal region corresponding to p300/CBP, as well as eight nearby genes, seems to have undergone a duplication that resulted in the p300 gene (on human chromosome 22, at the locus 22q13) and the CBP gene (on human chromosome 16, at the locus 16p13).89 Thus, chickens, opossums, mice, and humans all have one p300 gene and one CBP gene. However, even among the vertebrates there is some variation in copy number, as frogs appear to have lost their p300 gene, zebrafish appear to have lost their CBP gene, and puffer-fish appear to have duplicated their CBP gene.
After the duplication event, p300 and CBP have diverged in sequence somewhat, with 61% overall sequence identity. Interestingly, some domains have retained more similarity than others. Not surprisingly, sequence variation in the catalytic acetyltransferase domain has been selected against, resulting in 86% amino acid residue identity, in the region corresponding to the acetyltransferase domain and its two flanking domains, when comparing human p300 protein and human CBP protein sequence.90 The acetyltransferase domain spans residues 1284–1673.91 This conservation suggests an evolutionarily optimized function of the catalytic domain and underscores the importance of the acetyltransferase roles of p300 and CBP.
Furthermore, there is significant sequence homology in other noncatalytic domains of p300/CBP, including in several types of protein–protein interacting motifs (see Figure 3). At the N-terminal side there is a nuclear receptor interacting domain (NRID or RID), which is also one of two SPC domains (SPC-1 and SPC-2), that may bind PXXP motifs. SPC-2 is also a Sin3 interacting domain (SID). There are three cysteine/histidine-rich (C/H) domains. The C/H1 and C/H3 domains contain transcriptional adaptor zinc fingers (TAZ1 and TAZ2), and the C/H3 also contains a ZZ zinc finger. The C/H2, which is part of the catalytic domain, contains a plant homeo domain (PHD). Between the C/H1 and C/H2, there are an interferon binding homology domain (IHD), KIX domain, and a bromodomain. At the C-terminal side of p300/CBP there is an interferon binding domain (IBiD), which contains a nuclear coactivator binding domain (NCBD) and a glutamine-rich domain, followed by a proline-containing PxP motif.92,93
Figure 3.

Domain structure of p300/CBP. Exon–intron gene diagrams are shown for p300 and CBP (top). Below are example protein structures for the bromodomain (PDB 3I3J, 2.33 Å), catalytic HAT domain (PDB 3BIY, 1.7 Å), ZZ zinc finger (PDB 1TOT), and TAZ2 domain (PDB 3IO2, 2.5 Å). All structures were produced using purified p300, except the ZZ zinc finger, which used purified CBP. p300/CBP proteins are colored with a rainbow, with blue at the N-terminus and red at the C-terminus, and residues included in the structure are listed below each. Zinc ions are black spheres. All structures are based on X-ray crystallography, except the ZZ zinc finger structures from solution NMR. The p300 bromodomain structure shown here is remarkably similar to an independently generated CBP bromodomain structure (not shown, PDB 3DWY, 1.98 Å). Below is a model for full-length p300/CBP with all domains shown, and is a compilation based on several recent analyses.:94,95 three cysteine/histidine-rich (C/H) domains are shown in turquoise, three zinc fingers are shown in yellow, and the catalytic acetyltransferase domain is shown in orange, with its autoacetylated regulatory loop drawn above, which corresponds to residues 1523–1554. A few examples of proteins that bind p300/CBP are listed below the protein model, with the particular domain involved in binding indicated with a black line. Below that, amino acid similarity is indicated, for comparing p300 and CBP sequences within either the catalytic BHC region (from the bromodomain to the C/H3 domain) or the entire protein. At the bottom, commonly purified active p300 variants are indicated, including p300 acetyltransferase/HAT domain, BHC enzyme (bromodomain-HAT-C/H3), and full-length protein. It should be noted that p300 HAT has a deletion in residues 1529–1560.
Sequence homology in these domains helps explain why p300 and CBP have many overlapping functions. For example, p300 was first identified because of its ability to bind E1A, but CBP can also do this,96 for which they both use their mutually conserved C/H3-TAZ2 region.97 Also, CBP was first identified because of its capacity to bind CREB, but p300 also has this ability,98 for which they both use their mutually conserved KIX domain.99 However, despite these similarities, p300 and CBP appear to also have a handful of distinct functions, which may be due to slight differences in substrate specificity and/or in a subset of protein–protein binding interactions.100
2.5. p300/CBP Function in Signaling Pathways
As discussed in section 2.1, the cell must turn different subsets of genes on, in response to intracellular or extracellular signals, to regulate cellular functions, and this is accomplished partially by the acetylation of histone proteins to open the chromatin conformation and promote transcription. An intriguing collection of cellular signaling pathways have been dissected, and one striking finding is that p300/CBP is almost always a player. Examples of signals that ultimately use p300/CBP as downstream effectors include calcium signaling, response to hypoxia, Notch signaling, and NFκB signaling (see Figure 4).
Figure 4.
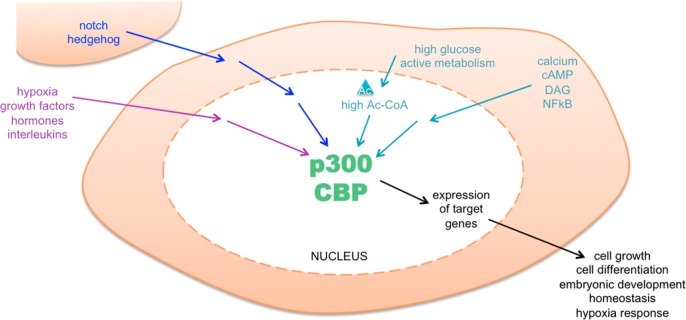
p300/CBP is central to many important signaling pathways. These include pathways that respond to intracellular signals (turquoise), extracellular signals (purple), and intercellular signals (blue). These pathways control the key cellular functions via altering expression of target genes, through the action of p300/CBP in the nucleus.
Notch is a transmembrane protein that is involved in intercellular signaling. When neighboring cell proteins engage and activate Notch, this induces cleavage of Notch and release of the active intracellular domain (ICD). The ICD then binds CSL and Mastermind. CSL targets the complex to DNA elements, while Mastermind recruits coactivators including p300/CBP and Pcaf, and together this complex activates transcription of target genes.101 For a more detailed description of this example, see section 5.2.
Another example is the cAMP pathway, which was mentioned because of its historical role in the first identification and naming of CBP. In this signaling pathway, growth factors or other extracellular signals are received by transmembrane G protein-coupled receptors (GPCRs), which activate heterotrimeric G proteins. The G proteins can then activate adenylyl cyclase, which produces cAMP, and this releases the active protein kinase A (PKA) from a complex with an inhibitory subunit. PKA then diffuses into the nucleus and phosphorylates CREB at serine 133. CREB dimers bind cAMP-response elements (CREs) in the genomic DNA and also recruit p300/CBP to activate transcription of an estimated 100 target genes.102
A third example is the estrogen pathway, which is important to hormone-dependent cancers (see section 6). Estrogen is a steroid hormone that diffuses into cells and binds to intracellular estrogen receptors. Estrogen receptor dimers bind estrogen-response elements (EREs) in the genomic DNA. Upon ligand binding, the complex recruits p300/CBP to activate transcription of around 1000 target genes.103
p300/CBP is also involved in stress response pathways, in which adaptation to conditions such as hyperosmolarity involves changes in gene expression.104 For example, osmotic shock induces several kinase cascades, including those involving the kinases p38, JNK, and ERK.105 Phosphorylation of p300/CBP can regulate its activity in different ways, depending on the phosphorylation site,106 and thereby regulate transcription of target genes.
There are also several links between p300/CBP and the DNA damage response pathway. The DNA damage response is orchestrated mainly by the protein p53, and its expression and post-translational regulation are modulated by p300/CBP activity. p53 and other proteins in the DNA damage response are substrates for acetylation by p300/CBP.107,108 p300/CBP is also involved in the activation of genes following DNA damage, such as p21.109
One way in which p300/CBP is able to respond to signaling pathways is by the post-translational modifications of p300/CBP itself. Phosphorylation of p300/CBP occurs at several sites, catalyzed by kinases including PKC,110 cyclin E/CDK-2, CaMKIV, IKK, and AKT,111 that are reported to negatively or positively regulate p300/CBP acetyltransferase activity, protein–protein interactions, or protein stability, depending on the site.112−114 p300/CBP also has two sites of methylation near the KIX domain, and one effect of an arginine methylation event by CARM1 is the decreased recruitment by CREB115 but the increased recruitment by estrogen receptor.116 p300/CBP can also be SUMOylated at three lysines near the bromodomain, which may influence p300/CBP subnuclear localization,117 is antagonistic with acetylation at the same sites, and negatively regulates p300/CBP activity.118 Acetylation of p300/CBP occurs on 17 lysine residues of a regulatory loop within the acetyltransferase domain, probably by a highly efficient and cooperative intermolecular reaction,119 and stimulates its acetyltransferase activity120,121 and binding of proteins.122 One regulatory mechanism that this autoacetylation could serve is by relieving the steric hindrance in the substrate binding site, with the regulatory loop acting as a pseudosubstrate.
Another way in which p300/CBP is able to respond to signaling pathways is due to the ability of p300/CBP to bind a multitude of different proteins, using its various protein-interacting domains. The number of proteins that p300/CBP can bind to, also referred to as the p300/CBP interactome, is a list that is ever growing. In 2006, the count was at 312,123 and today at least 411 proteins are implicated in binding p300/CBP (see Supporting Information Table 2).124 Some of these proteins interact directly with DNA, sometimes in response to a signal, recruiting p300/CBP to the target gene element. Genomic profiling of p300/CBP recruitment to DNA sequences has been analyzed by several groups using chromatin immunoprecipitation. At these DNA elements, p300/CBP nucleates a complex of many proteins that together allow for transcriptional activation. Transcriptome profiling of genes regulated by p300/CBP has been analyzed by one study, using treatment with a p300/CBP inhibitor and microarray analysis, which implicated 615 as being regulated by p300/CBP.125 One limitation of these interactome and transcriptome studies of p300/CBP is that cryptic protein partners, DNA elements, and regulated genes might be missing from the lists, if certain signaling pathways need to be activated before p300/CBP can engage in a specific binding partnership, recruitment, or transcriptional event.
Many of the protein–protein interactions making up transcription complexes are mediated by the acetylation of lysines on the proteins, by p300/CBP or by other associated acetyltransferases, and the resultant binding to other proteins with bromodomains. To date, ∼100 proteins have been implicated as p300/CBP acetylation substrates (see Supporting Information Table 1).126 Thus, p300/CBP helps supply acetylation as a glue to hold the complex together, while also serving as a scaffold to bring together many proteins, and a bridge to connect one protein-bound DNA element with another (see Figure 5).127,128
Figure 5.

p300/CBP functions as a scaffold, bridge, and acetyltransferase. The acetyltransferase reactions are illustrated by turquoise arrows, indicating acetylation of histone and nonhistone substrates (in yellow), as well as autoacetylation of the p300/CBP acetyltransferase domain. The bridge function is illustrated by turquoise squares, representing DNA-binding proteins that bring DNA elements into proximity with p300/CBP through their interactions. The scaffold function is illustrated by orange squares, representing a protein complex being recruited by p300/CBP. These functions together allow for gene expression.
In light of the astounding number of possibilities of complexes in which p300/CBP could participate, one may wonder how this is regulated. The first clue is that certain domains, such as the C/H3 domain, have many potential protein binding partners that compete for the same recognition sequence within p300/CBP. Another clue is CBP haploinsufficiency, where one wild-type CBP copy is not enough for normal mouse development, and, conversely, CBP overexpression is also lethal in the fruitfly Drosophila melanogaster, indicating a tightly controlled, limiting pool of p300/CBP in cells.129 Therefore, in our current model, which protein any given molecule of p300/CBP might interact with would depend upon the relative nuclear concentrations of the potential partners and upon relative binding affinities of the potential partners. Therefore, information from an intracellular or extracellular signal would be relayed through a pathway to produce a change in a certain effector protein’s nuclear concentration or affinity for p300/CBP, altering p300/CBP cellular complexes. For example, upstream signaling pathways may enhance p300/CBP recruitment to a specific target gene by translocation of a certain protein to the nucleus, or post-translational modification of the protein to increase the binding affinity to p300/CBP.
What emerges is a fascinating picture of different signaling pathways competing for p300/CBP.130 This hypothesis has been supported by several interesting studies. In one report, tumor necrosis factor-α (TNF-α) was used to stimulate the RelA subunit of NFκB binding to p300/CBP (via its zinc finger) and resultant induction of HIV gene expression, but this was inhibited when interferon-α (IFN-α) was used to stimulate STAT2 binding to p300/CBP (via the same zinc finger).131 In another study, when AP-1 was bound to p300/CBP (via its C-terminal region132), this resulted in induction of TRE genes (involved in the antioxidant response), but this was inhibited when p53 was bound to p300/CBP (via the C/H1 and/or C/H3, resulting in induction of p53-regulated promoters),133 and also inhibited when glucocorticoids were used to stimulate glucocorticoid receptor binding to p300/CBP (via the N-terminal nuclear receptor interacting domain).134 Similarly, genotoxic stress stimulates p53 binding to p300/CBP and resultant induction of p53-dependent genes (resulting in cell cycle arrest), but this is inhibited when overexpressed E2F-1 is bound to p300/CBP (via its TAZ2 domain,135 resulting in apoptosis).136 In another study, cAMP stimulates CREB binding to p300/CBP (via its KIX domain) and resultant induction of cAMP-responsive genes, but this is inhibited when insulin or growth factors were used to stimulate S6 kinase pp90RSK binding to p300/CBP (via its C/H3 domain), which resulted in induction of Ras-responsive genes.137
Taken together, these and other studies demonstrate that p300/CBP is not only essential to many cellular signaling pathways, but utilizes its protein–protein interactions to integrate the different signals and determine how the cell will respond. In this manner, p300/CBP has a hand in controlling virtually all of the major cellular functions, including proliferation, differentiation, and response to stress (see Figure 4).
3. p300 Acetyltransferase Activity
p300/CBP has a central acetyltransferase (also called HAT) domain that catalyzes the reaction in which the acetyl group from acetyl-CoA is transferred to a protein lysine side chain. p300/CBP has the ability to acetylate a wide variety of proteins (see Supporting Information Table 1). These substrate proteins are located principally in the nucleus (such as histones) but are also present in other parts of the cell (such as the cytosolic GAPDH). Immunofluorescence microscopy and subcellular fractionation studies have localized p300/CBP predominantly to the nucleus. It is not completely understood how p300/CBP shuttles in and out of the nucleus, and part of the answer may be its binding interactions with protein ligands that are themselves targeted to the nucleus or elsewhere.
3.1. p300 Acetyltransferase Domain Structure
One of the challenges for biochemical characterization of p300/CBP is the difficulty to express and purify active enzyme. This may be due to the large size of full-length protein (see Figure 3) and/or inappropriate hyperacetylation when the active acetyltransferase domain is overexpressed. This can be overcome by coexpression with the deacetylase Sir2. Alternately, for the acetyltransferase domain, it is possible to express an inactive protein followed by ligation to a synthetic peptide to generate the active enzyme.138,139 Purified p300 containing its acetyltransferase domain plus its two flanking domains can be prepared efficiently from insect cells.140 The effect of the flanking domains on acetyltransferase activity will be discussed in further detail in section 5.1.
As transferase enzymes, acetyltransferases have two substrates (acetyl-CoA and a lysine-containing protein) and two products (CoASH and an acetyl-lysine-containing protein). For an enzyme with two substrates, a rational strategy to create a high potency inhibitor is to incorporate the major structural features of both substrates, and thereby allow binding to be tighter than for either substrate on its own. This idea sparked the generation of a variety of acetyltransferase inhibitors, starting with the minimalist bisubstrate analog Lys-CoA, and including various peptide sequences appended to the lysine. Discovery of Lys-CoA, in addition to enzymology applications that will be discussed in section 3.2, allowed for the crystallization of the p300 acetyltransferase domain active site in complex with the bisubstrate, with the structure solved at 1.7 Å resolution (see Figure 6).141
Figure 6.
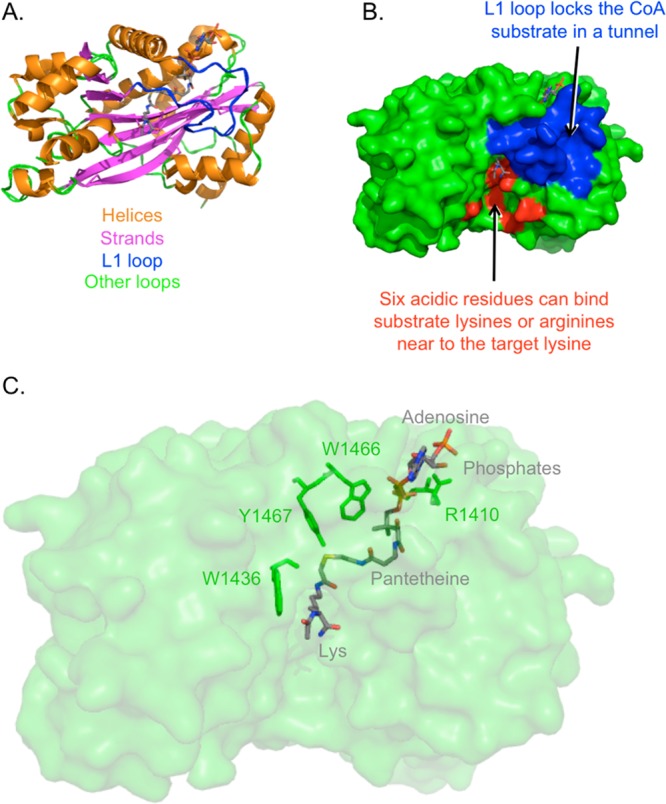
p300 acetyltransferase domain structure bound to Lys-CoA. (A) Secondary structures of p300 acetyltransferase domain. (B) L1 loop and an acidic surface. (C) Parts of Lys-CoA bisubstrate analog (gray) and four p300 residues of interest (green). Generated in PyMol based on Protein Databank entry 3BIY, published by Liu et al.143
This crystal structure revealed six nucleophilic residues in sufficient proximity to be able to attack the carbonyl carbon of acetyl-CoA, but mutational analysis and the use of an electrophilic CoA labeling reagent142 refuted a mechanism that p300 catalysis proceeds through a covalent enzyme intermediate.
In contrast to other crystallized acetyltransferases like Gcn5 and yeast Esa1, the crystal structure of p300 revealed a shallow, acidic surface about 10 Å distance from the substrate lysine (see Figure 6). This surface is thought to interact with the substrate flanking sequence, and mutational analysis confirmed that E1505 and D1628, both found in this surface, are important for catalysis.137 This provides insight into the broad peptide substrate specificity of p300, and the preference for nearby basic residues (usually a lysine or arginine, within 3 or 4 residues of the modified lysine). This contribution to catalysis was analyzed for histone H4, where the K8 substrate is flanked by basic residues at K5 and K12. Replacing K5 and K12 with alanine impaired acetylation at K8, but this was rescued if either alanine was changed to arginine to preserve the positive charge. Acetylation was not rescued if K5A was changed to citrulline, which can still be a hydrogen-bond donor but loses the potential for an electrostatic interaction associated with lysine, arguing that the positive charge is important in substrate recognition and catalysis.65
In addition to revealing this acidic surface, the crystal structure highlighted the importance of many residues involved in Lys-CoA binding (see Figures 6 and 7). Mutationally sensitive Y1467 was implicated in a hydrogen bond between its side chain hydroxyl and the sulfur of Lys-CoA. This may allow the Y1467 to function as a general acid, facilitating the protonation of the leaving group during catalysis. W1436 was implicated in a hydrogen bond between its main chain oxygen and epsilon nitrogen atom of the substrate lysine, as well as van der Waals interactions between its hydrophobic indole side chain and the aliphatic portion of the substrate lysine. This forms a hydrophobic tunnel in combination with two tyrosines, which may reduce the substrate lysine pKa, such that the neutral amine required for attack is more favored. p300 acetyltransferase catalytic activity was sharply reduced by W1436A mutation. As the pH–rate profile of p300 reveals an important pKa of 8.4,137 it is proposed that this pKa may correspond to this substrate amine in complex with p300. In addition, R1410 showed salt bridging interactions with the phosphates of CoA, whose importance was confirmed using 3′-dephospho-Lys-CoA and mutagenesis.137 Interestingly, W1436 and R1410 p300 point mutations have been observed in patients with lung cancer, suggesting that p300 can be a tumor suppressor.
Figure 7.

Acetyl transfer catalysis by p300. (A) The p300 active site is drawn in green, and histone H4 substrate in blue, with important residues indicated. CoA is drawn in black, and binds in a specific tunnel. (B) Four steps in a proposed p300 mechanism. acetyl-CoA binds, then peptidyl-lysine binds. The hydrophobic indole of W1436 promotes an uncharged lysine and positions it for attack. The lysine attacks the carbonyl of acetyl-CoA, while Y1467 acts as a general acid to protonate the leaving group. Acetyl-lysine-containing product leaves quickly, then CoASH departs slowly.
The crystal structure of the p300 acetyltransferase domain also showed that p300 has structural similarity, and hence a possibly ancient common ancestor, with the fungal acetyltransferase Rtt109.144 Furthermore, although p300/CBP has little sequence similarity with other acetyltransferases, p300 has a conserved CoA binding region in common with other crystallized acetyltransferases like Gcn5, yeast Esa1, and yeast Rtt109.145 In this CoA binding region, the pantetheine arms of CoA adopts similar positions surrounded by several integral beta strands and alpha helices that overlap in superposition models, but the adenosine rings of CoA occupy differing conformations, with Gcn5146 and Esa1147 structures being more flexible. In the p300 structure, a specialized substrate-binding loop, called the L1 loop, interacts with the entire length of the lysine and CoA, and locks them in place (see Figure 6).137 This is consistent with the tight binding observed with p300 and acetyl-CoA, as well as the reduced inhibition seen for Lys-CoA varieties with longer peptide sequences, as there may be a steric clash between the peptide and the L1 loop. Assays with bisubstrate analogs containing various length linkers between the peptidyl-Lys and the CoA showed that linker length correlated with potency, although these compounds never exceeded the potency of the original Lys-CoA. This suggests that the crystal structure is of p300 trapped in the conformation of a late step in catalysis, where the acetylated peptide substrate is rapidly ejected from the active site.148
3.2. p300 Acetyltransferase Reaction Chemistry
There are several kinetic mechanisms possible for a two-substrate two-product “bi bi” scheme like the reaction catalyzed by p300/CBP. We now know that most acetyltransferases follow an ordered binding, ternary complex mechanism, but p300 appears to proceed through a Theorell–Chance mechanism, in which there is no stable ternary complex, and the peptide substrate associates only very transiently with the enzyme, leaving as soon as the reaction is complete.149
One of the first insights into the catalysis by p300 was derived from solvent viscosity studies. Initial velocities were measured in the presence of different concentrations of sucrose, and a significant dose-dependent loss in catalysis was observed. Moreover, substrate analogs that were poor substrates did not have as large a solent viscosity effect. These data argue that release of at least one of the products is the rate-limiting step in the reaction with a normal substrate, as opposed to the chemical step(s).65 Binding studies with CoASH and substrate analog acetonyl-CoA helped characterize the tight binding properties. p300 binds acetyl-CoA with an affinity in the high nanomolar or low micromolar range.140
To distinguish among several possible p300 kinetic mechanisms, two-substrate kinetics was performed and reported in 2001.65 The steady-state kinetic parameters, using initial velocities, were determined for acetyl-CoA at different concentrations of a H4–20 peptide substrate. In double-reciprocal plots (E/V versus 1/[acetyl-CoA]), parallel lines were observed, which was originally interpreted as support for a ping-pong kinetic mechanism rather than a ternary complex mechanism involving direct transfer of the acetyl group from acetyl-CoA to a lysine-containing peptide.65,150
These steady-state kinetics of p300 confounded researchers for some time, because a covalent enzyme intermediate could not be identified, and because the bisubstrate analog Lys-CoA was a potent p300 inhibitor. Once the crystal structure of the p300 acetyltransferase domain was elucidated, a Theorell–Chance, also known as hit-and-run, catalytic mechanism was considered. Implicit in this model151 was a relatively high affinity for acetyl-CoA, which was consistent with the observed low Ki of the substrate analog acetonyl-CoA. The Theorell–Chance mechanism was further investigated using product inhibition studies. This included four separate experiments, with inhibition by either CoASH or Ac-peptide (K8Ac H4–12 with K5R and K12R), and varied substrate as either acetyl-CoA or peptide (H4–12 with K5R and K12R). The observed pattern was fully consistent with a Theorell–Chance mechanism, but not ping-pong or ordered bi bi mechanisms.137
The Theorell–Chance mechanism helps account for why Lys-CoA is a ∼20-fold better p300 inhibitor than the longer peptide-CoA conjugate H4-CoA-20, even though Ac-Lys is a ∼15-fold worse p300 substrate than H4–20. In contrast, Gcn5 and Pcaf acetyltransferases obey classical ternary complex mechanisms, and are more potently inhibited by the longer peptide-CoA conjugate bisubstrate analogs.152 A further application of Lys-CoA and the cocrystal structure was to facilitate in silico modeling of small molecules in the p300 active site. The understanding of the features that distinguish p300/CBP from other acetyltransferases, including the Theorell–Chance catalytic mechanism and the key role that acetyl-CoA binding plays in the reaction, led to interest in designing inhibitors that would bind in the same tunnel as acetyl-CoA, acting as competitive inhibitors that would be expected to be specific for p300/CBP versus other acetyltransferases. These studies resulted in the discovery of C646, a small molecule inhibitor of p300/CBP that is relatively potent, specific, and cell permeable. In agreement with the rationale behind this inhibitor, it is competitive for acetyl-CoA and selective for p300/CBP.153 This and other inhibitors of p300/CBP will be discussed in further detail in section 4.
4. Inhibitors of p300 Acetyltransferase Activity
Especially for the p300/CBP field, a challenge faced interpreting gene knockdowns or knockouts is that the resultant phenotypes can be attributed to the disruption of either the enzymatic acetylation activity of p300/CBP or the protein–protein binding activities. One way that the two properties of p300/CBP can be deciphered in cells is through the application of inhibitors that only block the catalytic activity and not the protein–protein binding. Acetyltransferase inhibitors may therefore be useful as tool compounds in understanding acetyltransferase functions and also have the potential to treat a variety of diseases (which will be described in section 6). Therefore, the identification and characterization of pharmacologic acetyltransferase inhibitors are ongoing.
4.1. Bisubstrate Acetyltransferase Inhibitors
The first potent acetyltransferase inhibitor identified is Lys-CoA,154 which is a covalent fusion between an acetyl-lysine and coenzyme A, with an extra methylene in between (see Figure 8). Structure–activity relationship studies have subsequently attempted to improve potency of Lys-CoA and probe p300 catalysis.155−157 Lys-CoA proved a powerful tool in studies of the p300 mechanism by crystallography and kinetics.65,137,158
Figure 8.

Bisubstrate inhibitors of acetyltransferases.
The rationale behind the bisubstrate design is that features of the acetyltransferase that engage each individual substrate (the lysine and the CoA) can be simultaneously engaged by a compound that mimics both. A similar bisubstrate strategy has also been employed to generate potent inhibitors of carnitine acetyltransferase,159 gentamycin acetyltransferase,160 spermidine acetyltransferase,161,162 serotonin acetyltransferase,163−165 GNAT acetyltransferases,166 aminoglycoside acetyltransferase,167−170 ghrelin O-acyltransferase,171,172 and protein kinases.173,174 Lys-CoA is potent against p300, with a Ki of 19 nM,175 and is between 50- and 400-fold tighter binding than CoASH alone.176,177 Because the inhibition by Lys-CoA follows slow, tight-binding kinetics,137 the measured IC50 can vary depending on whether a preincubation step with enzyme is included.
Lys-CoA is specific for p300 (and CBP, which has a near-identical acetyltransferase domain sequence). This selectivity can be explained by its Theorell–Chance kinetic mechanism, whereby p300 interacts with the substrate protein weakly using a broad, promiscuous surface (see Figures 6 and 7). This is in contrast to other acetyltransferases, many of which employ a ternary complex mechanism,178−182 in which protein substrate interactions are more important for enzyme engagement. For non-p300/CBP acetyltransferases, it can be deduced that the simple Lys-CoA molecule does not engage enough of their active site to confer high affinity. This explains why Lys-CoA inhibits other acetyltransferases with lower potency as compared to p300.
However, with its Theorell–Chance kinetic mechanism, p300 does not naturally engage the substrate protein for a long period of time, ejecting it from the active site soon after acetyl transfer. This explains why adding flanking peptide sequences in the Lys-CoA design actually reduces potency against p300. Even though H2A, H3, and H4 tail peptides are substrates for p300, embedding Lys-CoA in these sequences reduces potency against p300.183
By appending different peptide sequences flanking the lysine residue in Lys-CoA, bisubstrate inhibitors were generated with varying specificity profiles with other acetyltransferases (see Figure 8). These acetyltransferases are more substrate selective, and thus the inhibitor selectivity comes from matching the flanking sequences to that of the natural substrate. For example, H3-CoA-20 (20-mer peptide with K14 as the site of CoA conjugation) is potent (Ki of around 28 nM) and selective for Pcaf.184,185 An H3-CoA-20 derivative proved useful in generating a cocrystal structure with Gcn5.186 Additional studies on peptide-CoA conjugates have explored the broader potential of these bisubstrate analogs against a range of acetyltransferases.187
One major limitation of these bisubstrate inhibitors is that they are not efficiently cell permeable. Cell delivery by microinjection allowed the study of p300 acetyltransferase functions in a variety of systems, including the nematode Caenorhabditis elegans,188 frog oocytes,189 and cultured mammalian cells,190,191 although these methods are labor-intensive. Delivery by cell permeabilization allowed the introduction of Lys-CoA into muscle cells192 and melanocytes,193 but the lipid-based detergent used has some cytotoxicity.
Because the negative charges on the three phosphates of the phosphoadenosine moiety may be responsible for reduced cell permeability, analogs were explored that lack some of these groups, but most had no detectable p300 inhibition, and the best was 3′-dephospho-Lys-CoA, which had a 32-fold worse potency (IC50 of 1.6 μM).194 Prodrug strategies have been suggested to mask the phosphate groups.193 Some success has been achieved by appending positively charged arginines and lysines in peptides appended to the target lysine moiety. Appending the Tat peptide sequence (see Figure 8), which originates from the well-established cell-penetrating HIV Tat protein, resulted in a mere 5-fold worse IC50 for p300, but also made it less selective for p300, with 91-fold better potency for Pcaf (IC50 of 2.2 μM for Pcaf, which is only 8.8-fold that of p300).195 A similar Tat-conjugate was employed to generate H3-CoA-20-Tat, which retained 300-fold selectivity for Pcaf over p300, and these were used to study the functional interaction of acetyltransferases and the protein PZLF in cells.196
4.2. Natural Product Acetyltransferase Inhibitors
There is much interest in the identification of nonpeptidic small molecule inhibitors of acetyltransferases. Many natural products have been isolated and tested for acetyltransferase inhibitory properties. It should be noted that as these were studied by different laboratories employing varying methodologies, the IC50 numbers included here might not all be directly comparable. It is also notable that most of those described here are conveniently commercially available. The first natural product that was found to have acetyltransferase inhibitory properties is anacardic acid (see Figure 9).197
Figure 9.

Natural products implicated as modulators of acetyltransferases.
Anacardic acid can be isolated from cashew nutshell oil, mangos, and geraniums. It consists of a salicylic acid substituted with an alkyl chain. It is partially miscible in ethanol and ether, but immiscible in water. It is known to be toxic to Gram-positive bacteria and causes an allergic skin rash in humans.198 A PubMed search for “anacardic acid” yields 117 articles, spanning the years 1976–2013.
The inhibition of acetyltransferases by anacardic acid was first described in 2003, and it appeared to be fairly potent against p300 (IC50 of 8.5 μM) and Pcaf (IC50 of 5 μM), showing noncompetitive inhibition kinetics.199 However, an independent analysis of anacardic acid suggested inhibitory action against Tip60 (IC50 of 64 μM) and MOF (IC50 of 43 μM) but not against p300 or Pcaf (IC50 > 200 μM),200 while a third study measured IC50 above 200 μM for yeast Esa1, Tip60, p300, and Pcaf.201 Certain benzamide analogs of anacardic acid have increased potency against acetyltransferases.202 Intriguingly, modification of anacardic acid by adding a phenyl ring, to produce the benzamide analog called CTPB (see Figure 9), was reported to confer p300 activation.203−205 In addition to its likely modulation of other nonacetyltransferase targets in the cell, a major limitation of anacardic acid is its poor solubility.206
Another natural product found to have acetyltransferase inhibitory properties is curcumin (see Figure 9).207 Curcumin can be isolated from tumeric, which is a root spice in the ginger family. It consists of two phenols connected by two carbonyls. It is soluble in oil and DMSO, but not in water. It is a commonly used yellow food dye, and it is nontoxic: a daily dose of 12 g for 3 months is safe.208 A PubMed search for “curcumin” yields an astounding 5400 articles, spanning the years 1949–2013. The use of curcumin in food and ethnic medicines has a long history.209 In addition to acetyltransferases, curcumin is thought to affect a variety of other targets in cells, including several kinases, COX2, phosphatases, and Bcl proteins.210−213
Its inhibition of acetyltransferases includes p300 and acetyltransferases in the MYST family,214 and may occur via the electrophilic unsaturated ketone in curcumin being attacked by a acetyltransferase cysteine thiol group, to produce a covalent adduct.215 The IC50 against p300 is 25 μM.216 Despite many clinical observations of a variety of positive effects, curcumin showed unacceptably poor absorption, rapid metabolism, and elimination in Phase I clinical trials.217,218 Several curcumin analogs have been described,219,220 but a pharmacologically useful acetyltransferase-specific inhibitor has yet to be derived from curcumin.
In the same year that curcumin was found to have acetyltransferase inhibitory properties, the natural product garcinol was also found to have acetyltransferase-inhibitory properties (see Figure 9).221,222 Garcinol can be isolated from Garcinia indica, also known as kokum, which is a plant in the mangosteen family that produces edible fruit. Garcinol consists of a polyisoprenylated benzophenone. It is soluble in DMSO, ethanol, or DMF, but not in water. A PubMed search for “garcinol” yields 76 articles, spanning the years 1987–2013. It is a fairly potent acetyltransferase inhibitor, hitting p300 (IC50 of 7 μM) and Pcaf (IC50 of 5 μM), and has mixed-type inhibition kinetics.223 Unfortunately, garcinol (and the related isogarcinol) is fairly toxic in cells. An analog of garcinol, called LTK-14 (see Figure 9), improves toxicity and selectivity, with retained p300 potency (IC50 around 6 μM) but has no detectable inhibition of Pcaf.224,225 Garcinol and LTK-14 are poorly soluble and unstable, and this has been attempted to be corrected through further investigation of structural analogs.226−228 One intriguing garcinol analog is nemorosone, which was found to have a modest (1.5–2-fold at 10 μM) activation of p300.227
Also in 2004, certain γ-butyrolactone analogs, including one named MB-3 (see Figure 9), were found to have acetyltransferase inhibitory properties.229 Although the γ-butyrolactones used in that study were rationally designed and of synthetic origin, γ-butyrolactones can be isolated from wine, and have therefore been included in this section on natural products. MB-3 is soluble in DMSO or methanol. γ-Butyrolactones were developed with varying specificities against Pcaf and a Gcn5-like acetyltransferase, although potencies were modest (for example, the Gcn5 inhibitor has an IC50 of 100 μM228) and cellular activity remains to be demonstrated.230
Another natural product that was found to have acetyltransferase-inhibitory properties is plumbagin (see Figure 9).231 Plumbagin can be isolated from the plant genera Plumbago (leadwort), Drosera, and Nepenthes. Plumbagin is a napthoquinone derivative. It is soluble in DMSO and ethanol, but only slightly in water. A PubMed search for “plumbagin” yields 370 articles, spanning the years 1968–2013. Plumbagin is fairly safe at low doses but toxic at high doses.232 Plumbagin is thought to undergo redox cycling, which has effects on reactive oxygen species and the chelating of metals in cells. It also affects the drug efflux mechanism in cells and the conjugative transfer of antibiotic-resistance plasmids in bacteria.233 The effect of plumbagin on acetyltransferases was revealed in 2009, with modest inhibition of p300 (IC50 of 25 μM) and Pcaf (IC50 of 50 μM). Further analysis of structural analogs of plumbagin did not result in improved potency or specificity with p300 or Pcaf.234
Also in 2009, the natural product epigallocatechin-3-gallate (EGCG) was found to have acetyltransferase inhibitory properties (see Figure 9).235 EGCG can be isolated from tea plants. It consists of an epigallocatechin linked to a gallic acid via an ester linkage. It is soluble in water, DMSO, ethanol, and DMF. A PubMed search for “epigallocatechin-3-gallate” yields 3434 articles, spanning the years 1985–2013. EGCG appears to be a broad-spectrum acetyltransferase inhibitor, with modest potency against p300 (IC50 of 30 μM), CBP (IC50 of 50 μM), Pcaf (IC50 of 60 μM), and Tip60 (IC50 of 70 μM), and uncompetitive inhibition kinetics.234 EGCG has several targets in cells, including topoisomerase.236−239 EGCG appears to negate the effects of other compounds when combination therapies were attempted, suggesting potentially detrimental contraindications.240,241 EGCG, like other topoisomerase poisons, may be carcinogenic.242
Also in 2009, the natural product quercetin was found to have acetyltransferase-inhibitory properties (see Figure 9).243 Quercetin can be isolated from a wide variety of plants, including the oak tree for which it is named, Quercus, as well as tea leaves, honey, fruits, and vegetables. Quercetin is a polyphenolic flavonoid, is insoluble in water, and is soluble in DMSO. A PubMed search for “quercetin” yields an astounding 9568 articles, spanning the years 1948–2013. Quercetin also has other effects in cells, including scavenging reactive oxygen species244 and inhibiting cytochromes p450,245 monoamine-oxidase,246 and DNA gyrase.247 Although the effects of quercetin on acetyltransferases have not yet been fully analyzed, an IC50 for p300 was measured at 100 μM. However, they isolated p300 from cells that were treated with the compound, and tested the purified enzyme for acetyltransferase activity, and therefore the effects of quercetin on p300 activity could be indirectly mediated by another factor, such as by altering p300 phosphorylation.248
Another natural product found to have acetyltransferase inhibitory properties is ochratoxin A. Ochratoxin A is produced by certain bacteria in the Aspergillus and Penicillium genuses, it is poisonous, and humans are believed to be exposed to substantial amounts of ochratoxin A in their environment and food.249 It is a bicyclic lactone and is soluble in water and organic solvents. A PubMed search for “ochratoxin A” yields 2410 articles, spanning the years 1965–2013. Although detailed analysis of its acetyltransferase target or its mode of action has not been done, it appears to inhibit acetyltransferase(s) with moderate potency (IC50 of 25 μM).250
The final natural product discussed here, which was found to have acetyltransferase-inhibitory properties, is a prostaglandin, specifically Δ12-PG-J2 (see Figure 9).251 Prostaglandins are fatty acids with important functions in the human body, and Δ12-PG-J2 is a decomposition product of PG-D2, which is a major prostaglandin produced by mast cells, and is found in mast cells and brain cells. The “Δ12” indicates that it is unsaturated at the twelfth carbon along the chain, and the “J” refers to the α,β-unsaturated cyclopentanone moiety. Prostaglandins are slightly soluble in ethanol and insoluble in water. PG-J2 derivatives are natural activating ligands of the nuclear receptor PPARγ.252 The electrophilic carbon in the cyclopentanone moiety appears to be attacked by the p300 cysteine 1438 to form a covalent adduct with reduced acetyltransferase activity.253 It is relatively potent against p300 (IC50 of 750 nM) but not Pcaf (no detectable inhibition at 5 μM), and also inhibits acetylation in cells (ED50 of 5 μM by immunoblotting against acetyl-K9/K14 H3 in HepG2 cells).
4.3. Synthetic Acetyltransferase Inhibitors
Apart from natural products, several small molecules and other synthetic compounds have been found to have acetyltransferase-inhibitory activity through various chemical screens. Isothiazolones with acetyltransferase-inhibitory properties were identified in 2005 (see Figure 10).254 A series of isothiazolone analogs were compared for potency against Pcaf (which was as low as an IC50 of 1 μM255,256) and p300 (which was as low as an IC50 of ∼30 μM), and many were Pcaf-specific. Interestingly, no inhibition was observed in the presence of 1 mM DTT,253 implying that these compounds may not work well in reducing intracellular conditions, and also implying a possible mechanism. The nitrogen–sulfur bond is thought to be weak and susceptible to cleavage by thiols, such as via reaction with an acetyltransferase cysteine, to produce a disulfide adduct.257 Consistent with this, isothiazolones also inhibit the cysteine protease cathepsin B, although certain structural analogs exhibit >20-fold selectivity for Pcaf over cathepsin B.258 Intriguingly, the isothiazolone Kathon CG, an ingredient in cosmetics, was found to have potent inhibition of Pcaf and caused decreased cell growth.259
Figure 10.

Synthetic inhibitors of acetyltransferases.
Also in 2005, quinoline compounds were found to have acetyltransferase-inhibitory properties (see Figure 10). Inhibition of Gcn5 by the quinoline MC1626 was first implicated by chemical genetic studies,260 but the effects observed were subsequently thought to occur via a Gcn5-independent mechanism in cells.261 However, several quinoline analogs were determined to have bona fide modest potency against p300 and CBP (IC50 as low as 25 μM) but little or no inhibition of Pcaf or Gcn5.262,263 Intriguingly, the quinoline HCQ (hydroxychloroquine, an antimalarial drug) and a structurally unrelated all-trans retinoic acid were found to stimulate Pcaf activity (1.5–2-fold at 1 μM).264 A larger molecule that contains a quinoline moiety, called montelukast, which is a leukotriene receptor antagonist and used to treat asthma and allergies, was found to inhibit cellular NFkB-associated acetyltransferase activity with submicromolar potency, although potentially indirectly.265
Another class of compounds found to have acetyltransferase-inhibitory properties is the thiazoles (see Figure 10), including the compound CPTH2, which has weak potency (high-mM IC50) against Gcn5.266 A compound that bears some structural similarities to the thiazoles is the so-called compound a (see Figure 10), found by the Zheng lab to have acetyltransferase-inhibitory properties.267 This compound appears to have weak potency against several acetyltransferases, including Tip60 (IC50 of 149 μM), yeast Esa1 (IC50 of 190 μM), and p300 (IC50 of 150 μM).
Virtual screening of a commercially available library of ∼500 000 compounds started with in silico docking to the acetyl-CoA binding pocket of the p300 acetyltransferase domain.152 Compounds with predicted binding affinity were then screened by various assays with purified p300 acetyltransferase domain. This led to the identification of three inhibitors of p300, which all shared a linear arrangement of 3 or 4 aromatic rings terminating in a benzoic acid. Despite this apparent structural similarity, the steady-state kinetic mechanism of each inhibitor was distinct. Compound C646 appeared to act by competition with acetyl-CoA but not with peptide substrate, which was consistent with the original docking model and further validated by site-directed mutagenesis (of the residues highlighted in Figure 11). Although C646 contains a potentially electrophilic group, its inhibition of p300 appeared to be noncovalent. C646 was fairly potent against p300 (Ki of 400 nM) and had at least 8-fold selectivity versus Pcaf, Gcn5, yeast Rtt109, Sas histone acetyltransferase, MOZ histone acetyltransferase, and serotonin N-acetyltransferase. C646 was also proven to inhibit acetylation in cells, making it an attractive probe for p300/CBP acetyltransferase studies.
Figure 11.

C646 modeled in the acetyltransferase active site. (A) C646 is shown in magenta, computationally docked in the crystal structure of the acetyltransferase active site, which was generated as a cocrystal with Lys-CoA. Several residues that coordinate CoA binding are predicted to similarly coordinate C646 binding, as shown in aqua stick representations of the side chains. (B) The structure of C646, shown in an orientation similar to that in the docked model above.
An analog of C646, C107, retains potency as a p300 acetyltransferase inhibitor, as measured against purified enzyme and in cells by acid-urea gel analysis. In assays where C646-induced fluorescence interferes with measurements, C107 appears to be a valid substitute as it lacks the intrinsic fluorescence properties of C646.140 It also lacks the pharmacologic liabilities commonly associated with furan rings, but still retains most of the same structural features of C646, including a five-membered heterocycle that may cause off-target effects.268,269
5. Protein Ligands of p300/CBP
5.1. Structures of p300/CBP with Bound Protein Ligands
Another factor in the functions, catalysis, and inhibition of p300 is interactions of domains that flank that catalytic domain. On the N-terminal side of the p300 catalytic domain is a bromodomain, and on the C-terminal side is a cysteine-histidine rich (C/H3) domain. Many proteins have been identified that bind to these domains (see Supporting Information Table 2). These ligands include acetylated proteins that interact with the p300 bromodomain, such as histones (for example, histone H4 acetylated at K20,270 and H3 in the context of chromatin271), MyoD (acetylated at K99, K102),272,273 STAT3 (acetylated at K49, K87),274 p53 (acetylated at K382),275 CtBP,276 and HIV Tat (acetylated at K50). Many proteins bind the C/H3 domain, including NUT,277 adenoviral E1A, FOS, Pcaf,278 Gcn5, TFIIB, p53, FOXO3a, STAT1, and MEF2.279 There are some ligands that seem to bind both the bromodomain and the C/H3 domain, such as p53 and MyoD. Furthermore, there may be crosstalk between ligand-binding domains, for example, between the p300 bromodomain and PHD finger in the C/H2 domain.280
Some of the p300–ligand interactions have been studied in structural detail by cocrystal X-ray diffraction281 and solution binding NMR282−286 analysis. Bromodomain binding typically occurs on one side of the four-helical bundle, and is mediated by the two loops between helices, although one crystal structure had an additional ligand bound against the side of the helix (see Figure 12). A recent study reports that the histone chaperone Asf1 binds the p300/CBP bromodomain in a noncanonical way.287 A recent structure from Daniel Panne and colleagues reveals a potential intramolecular regulatory interaction between the p300 bromodomain and its neighboring acetyltransferase domain.288 Structural features of these protein ligand interactions have been replicated by several recent synthetic ligands for the p300/CBP bromodomain, which will be discussed in section 5.3.
Figure 12.
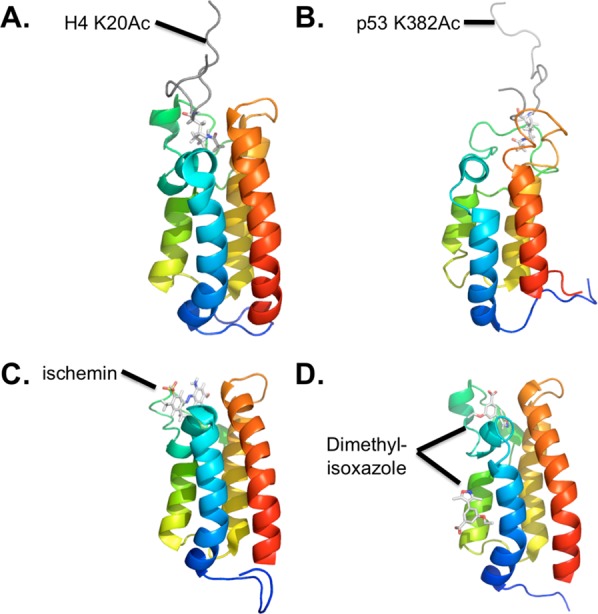
Structures of CBP bromodomain bound to ligands. The purified CBP bromodomain (residues 1081–1197, shown in a rainbow blue to red) is shown bound to (A) histone H4 residues 14–28 acetylated at K20 (PDB 2RNY); (B) p53 residues 367–386 acetylated at K382 (PDB 1JSP); (C) the compound ischemin (PDB 2L84); and (D) the compound dimethylisoxazole (PDB 3SVH). Peptide ligands are shown in gray (A,B) or stick models colored by atom (C,D and acetyl-lysines in A,B). All structures are based on solution NMR except for (D), which is from X-ray crystallography (1.8 Å).
The TAZ2 region of the C/H3 domain appears to bind ligands in diverse conformations (see Figure 13). C/H3 binding to the N-terminal region of MEF2 appears to be via any of three distinct surfaces on the TAZ2 domain. The crystal structure had a MEF2 dimer with three bound TAZ2 domains, each in a different conformation.289 C/H3 binding to the p53 TAD1 appears to be via extended interactions across a large part of the TAZ2 domain.290 C/H3 binding to E1A appears to induce a helical structure in the CR1 region of E1A, which is otherwise unstructured.291 A structure of C/H3 TAZ2 binding to STAT1 helps explain why TAZ2 binds STAT1 while TAZ1 binds STAT2, which occurs when the STAT1/STAT2 heterodimer engages p300 through both contacts.292
Figure 13.
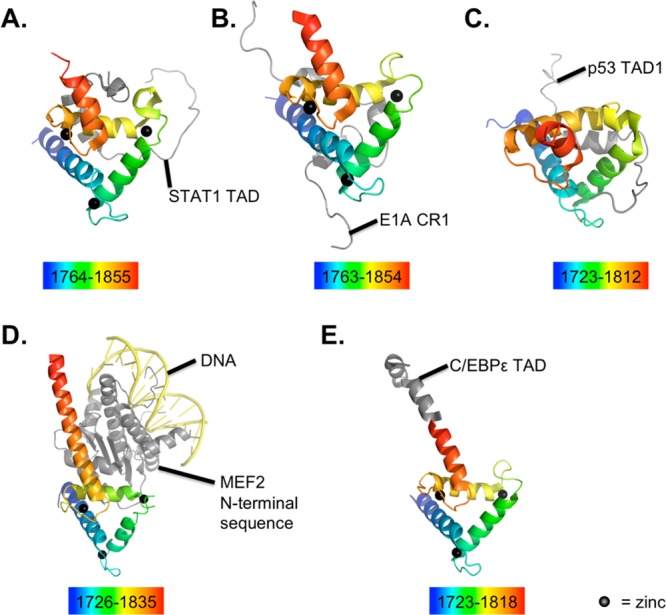
Structures of p300/CBP TAZ2 domain bound to ligands. The TAZ2 domain is shown colored in a rainbow (blue to red, residues included listed below each) bound to various ligands: STAT1 (A, PDB 2KA6); E1A (B, PDB 2KJE); p53 (C, PDB 2K8F); MEF2-DNA complex (D, PDB 3P57), and C/EBPε (PDB 3T92). All structures are based on solution NMR except for two from X-ray crystallography: that in (D) (2.192 Å) and that in (E) (1.5 Å). All structures were produced using purified p300, except the (A) and (B), which used purified CBP. Zinc ions are black spheres, protein ligands are gray, and DNA is yellow. The crystal structure with MEF2 revealed binding in three possible conformations with TAZ2, and one example is shown here.
5.2. Flanking Domains on p300/CBP Acetyltransferase Activity
One well-documented effect of these p300–ligand binding interactions is to facilitate the formation of a transcriptionally activating complex. DNA-binding ligands could recruit p300 to gene elements. p300 could nucleate complex formation by serving as a scaffold, also interacting with general transcriptional machinery. Some p300 ligands are effector proteins that modulate chromatin or have other activities, which are brought into proximity with their chromatin substrate by the bridging p300.293 Some p300 ligands have activities that are inhibitory to transcription, for example, HDAC1, which binds the p300 C/H3 domain,294 and these corepressors could be relieved by competing interaction with an alternate ligand. By these mechanisms, regulated ligand binding changes the subnuclear targeting of p300 and other proteins, without necessarily influencing the catalytic function of p300 (see Figure 14).
Figure 14.
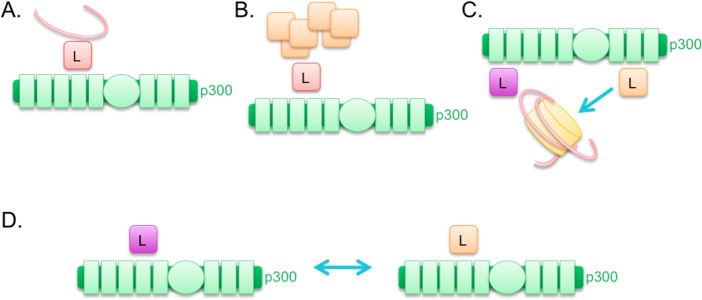
Models for targeting influenced by p300-ligand binding. In these models, p300 is shown in green, the histone octamer is shown in yellow, DNA is shown with a red strand, and p300 ligands are indicated with an “L”. In (A), a ligand targets p300 to a gene or other DNA element due to the DNA binding affinity of the ligand. In (B), a ligand targets p300 to a protein complex due to the protein binding affinity of the ligand. In (C), two ligands bridged by p300 allow for chromatin (bound by the purple ligand) to come into proximity with a chromatin-modifying enzyme (the orange ligand). In (D), two ligands compete for the same site within p300, and the one ligand could be seen as a competitive inhibitor for the p300 association with the other.
One naturally occurring p300 ligand of interest is the protein Mastermind. Mastermind binds to the C/H3 domain of p300.295 Mastermind received its name from researchers studying the Drosophila gene, and when describing the human homologue, it can also be known as Mastermind-like protein 1, MAM, MAM1, MAML, or MAML1, but these are all the same gene. Mastermind first became a gene of interest in C. elegans (where it has been called sel-8, lag-3, or pqn-17) when its mutation was found to suppress a mutation in lin-12 (human Notch3)296 and subsequently found to function in the Notch pathway by binding LAG-1 (human CSL, a DNA-binding protein) and activating transcription.297 When protein sequence homology was found between the human genes and those in model organisms,298 this allowed researchers to understand how the Notch pathway works in humans.299,300 The first crystal structure of Mastermind was from C. elegans, and was in complex with Notch, CSL, and DNA, in which the Mastermind N-terminal region forms a long α helix resembling an arm, with a bend or elbow in the middle, that hugs around Notch and CSL.301 Subsequent crystallography of the human proteins showed a comparable structure.302−304
We now know that when activated, Notch binds CSL and Mastermind, and this complex associates with DNA via CSL (see Figure 15). The core complex then recruits transcriptional coactivators, including p300 and Pcaf. In addition to nucleating a transcriptional complex and being recruited to chromatin associated with Notch regulation, it was hypothesized that there are additional synergistic effects of the Mastermind–p300 interaction.
Figure 15.
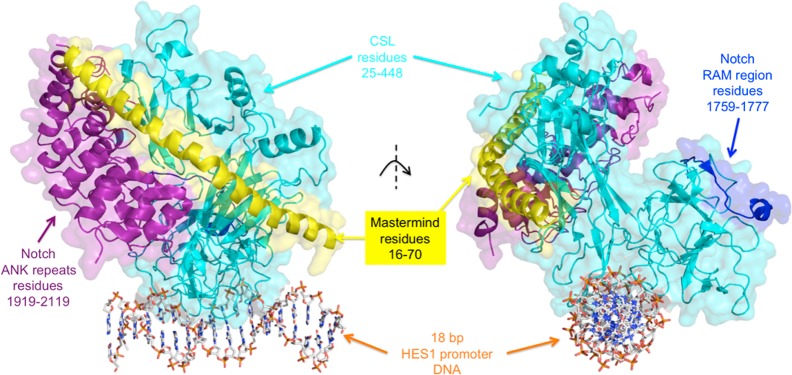
Mastermind-Notch-CSL-DNA core complex. The complex formed by DNA, CSL, Notch (ANK repeats and RAM region purified separately), and Mastermind N-terminal helix is shown with two different view angles. The X-ray crystal structure was generated at 3.85 Å, and this figure was produced in PyMol using Protein Databank entry 3V79. Proteins are shown as ribbons, with the surfaces at 70% transparency. DNA is shown as stick models colored by atom.
Interestingly, it was shown that Mastermind can activate p300 acetyltransferase catalysis.305 The effect of Mastermind on p300 acetyltransferase catalysis was dose-dependent and appeared to enhance the maximal acetylation rate of p300 and not the binding of p300 to peptide substrate. In cells, Mastermind increased p300 autoacetylation, measured using immunoblotting, and increased transcriptional coactivation, measured using a luciferase reporter. Mastermind and p300 were recruited to nuclear bodies,306 and these nuclear bodies had enhanced histone H3 and H4 acetylation. A remaining question is how the events are timed. A simple model would be that first Mastermind binds to the C/H3 domain of p300, and this promotes activation of p300 acetyl transfer, leading to its increased autoacetylation, acetylation of histones, acetylation of Mastermind, p300–Mastermind targeting to nuclear bodies, and transcriptional coactivation (see Figure 16). This is supported by the loss of Mastermind activation of p300 autoacetylation when the C/H3 domain is deleted.313 A further observation is that the activation loop (residues 1523–1554) is not required for Mastermind effects on p300 autoacetylation (at lysine 1499) or transcriptional activation. However, some of the data argue against this simple model; for example, p300 lacking the C/H3 domain still gets targeted to nuclear bodies by Mastermind.313 It is also still unknown how and when these steps are reversed, and one clue is that acetylation decreases the binding affinity between Mastermind and p300.307 Therefore, Mastermind may first associate with p300, turn it on, and then dissociate.308
Figure 16.
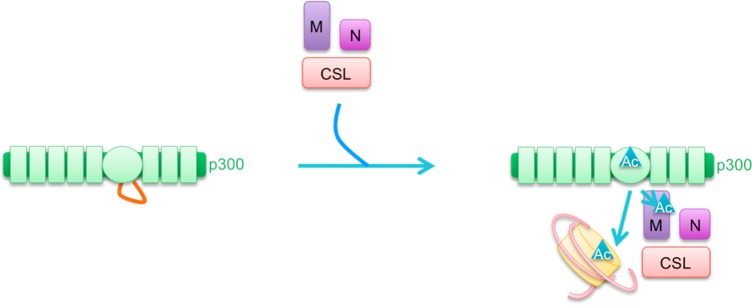
Model for Mastermind activation of p300. In this model, p300 (green) initially has inhibited acetyltransferase activity due to an autoinhibitory loop (orange, left). This is relieved upon recruitment by the Notch-Mastermind-CSL complex. Mastermind (purple) binds to the p300 C/H3 domain, and also to Notch intracellular domain (magenta) and CSL (red). p300 autoacetylation, Mastermind acetylation, and histone acetylation are then catalyzed by p300 (turquoise ▲).
It also remains to be seen whether these trends observed with Mastermind will extend to other p300 ligands. Because there are more than one ligand that interact with the C/H3 domain, there may be a common mechanism by which they influence p300 catalysis, or it may vary entirely depending on the specific ligand in question. Interestingly, a ligand of the p300 bromodomain, called CtBP, appears to change p300 acetyltransferase activity via steric hindrance that blocks other substrates.309 Furthermore, many ligands of other p300 domains, and their effects on p300 catalysis, are still being uncovered and investigated. It could be hypothesized that ligand binding could influence substrate specificity through direct competition for the same binding site or conformational changes in the binding pockets of p300/CBP or the multiprotein complex. Ligand binding could also theoretically change the catalysis itself via long-range conformational changes in the region that binds acetyl-CoA or the residues involved in catalysis.
In purified p300, the flanking bromodomain and C/H3 domain have a significant effect on catalysis in the absence of ligands. p300 BHC (a construct containing the bromodomain, catalytic acetyltransferase domain, and C/H3 domain) is considerably more efficient as a histone acetyltransferase than the p300 acetyltransfarase domain, with a 5-fold higher kcat.140 Even more dramatic, p300 BHC has a 10-fold lower Km for acetyl-CoA. Interestingly, the small molecule inhibitors C646 and C107 appeared to show about 4–5-fold reduced potency against BHC acetyltransferase activity versus the isolated acetyltransferase domain. However, another study showed different results.310 The bromodomain-HAT crystal structure suggests the potential for intramolecular interactions that can be communicated between the domains.311
5.3. Compounds That Block or Mimic p300/CBP Ligand Binding
Recent progress has been made in developing inhibitors of bromodomain interactions (reviewed by Filippakopoulos et al.312). Several compounds have been identified that inhibit BET bromodomains, such as I-BET762/GSK525762A,313 I-BET151/GSK1210151A,314,315 JQ1,316 PFI-1,317,318 and various isoxazoles.319−321 These have proven useful in a variety of exciting biological applications, including in HIV,322−325 merkel cell polyomavirus,326 leukemia,327−331 lymphoma,332 multiple myeloma,333 lung adenocarcinoma,334 inflammation,335−337 kidney disease,338 and fertility339 research. Low micromolar inhibitors of other bromodomains have also been reported.340 In general, bromodomain inhibitors may block the binding interactions of many different protein ligands, often complicating analysis of single ligand interactions in cells.341−345
Compounds that target the p300/CBP bromodomain have been identified by the Structural Genomics Consortium (SGC) and Ming–Ming Zhou’s group (see Figure 17). MS7972 appears to inhibit the p300/CBP bromodomain with an affinity of around 20 μM and undetermined specificity for other bromodomain-containing proteins.346 The compound ischemin was identified that also appears to inhibit the p300/CBP bromodomain with affinity of around 20 μM and undetermined specificity for other bromodomain-containing proteins.347 Cyclic peptides containing acetyl-lysine were designed to inhibit the p300/CBP bromodomain with an affinity of as low as 8 μM and undetermined specificity for other bromodomain-containing proteins.348 Further compounds were identified that have an affinity for the p300/CBP bromodomain of around 0.39 μM and affinity for a BET bromodomain of around 1.4 μM.349 The Brennan group tested a collection of [1,2,4]triazolo[4,3-a]phthalazines against a panel of bromodomains, and found several with submicomolar affinity for p300/CBP bromodomain, although all had relatively strong affinities for BET bromodomains.350 The widely used analgesic acetaminophen (paracetamol) was also found to bind the CBP bromodomain, probably by a weak nonspecific interaction.351 A series of 3,5-dimethylisoxazoles were found to have varying micomolar inhibitory properties against the p300/CBP bromodomain, although all still retained relatively potent inhibitory activity against BET bromodomains.352 There is structural evidence that several of these inhibitors bind the bromodomain in a manner similar to that of the natural protein ligands (see Figure 12).
Figure 17.
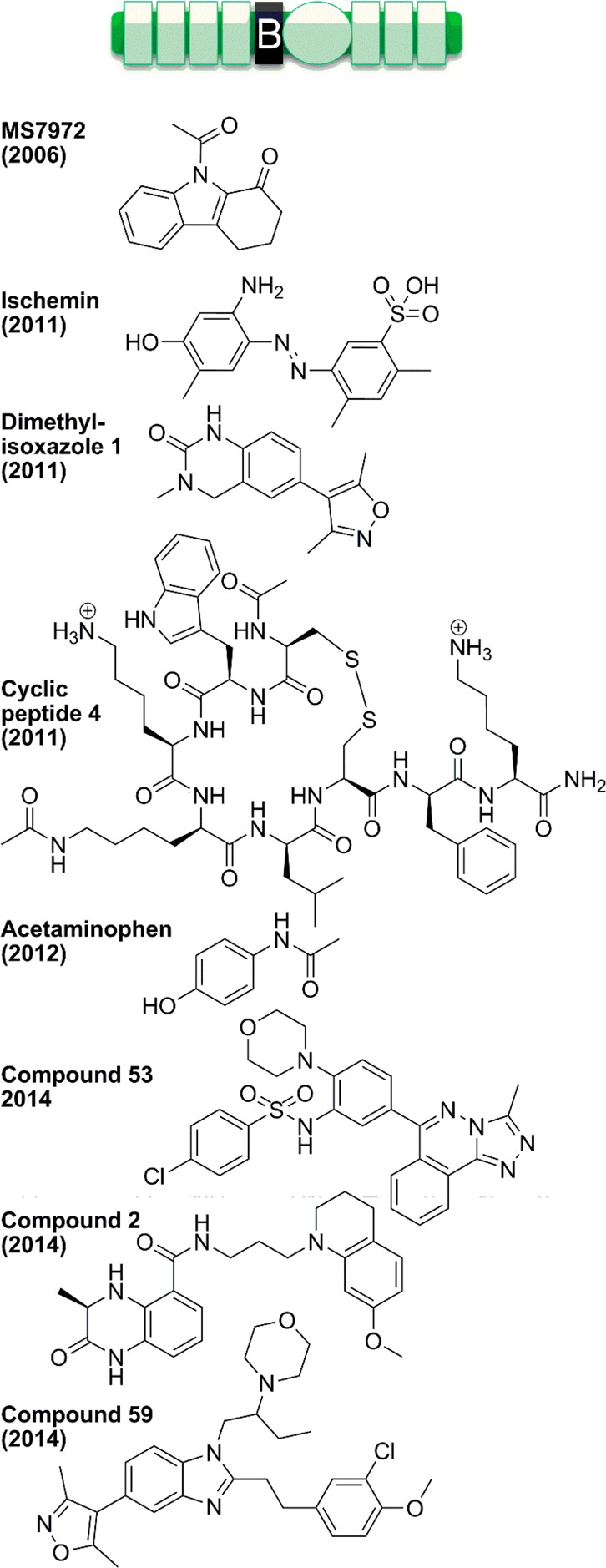
Inhibitors of the p300/CBP bromodomain.
More recently, the compound named 59 was identified with affinity of around 32 and 21 nM for p300 and CBP bromodomains, respectively, and reported 40-fold or greater selectivity versus all 10 other bromodomains tested. Although promising, this compound did show some pan-inhibitory properties against several unrelated enzymes including adrenergic receptors, phosphodiesterase-5, and platelet-activating factor.353
For the C/H3 domain, no inhibitors have been carefully studied to our knowledge, although there is some evidence to suggest that the drug Roscovitine (see Figure 18), also known as Seliciclib, which is an inhibitor of cyclin-dependent kinase (CDK), may in fact inhibit the interaction of p300 C/H3 and CDK.354
Figure 18.
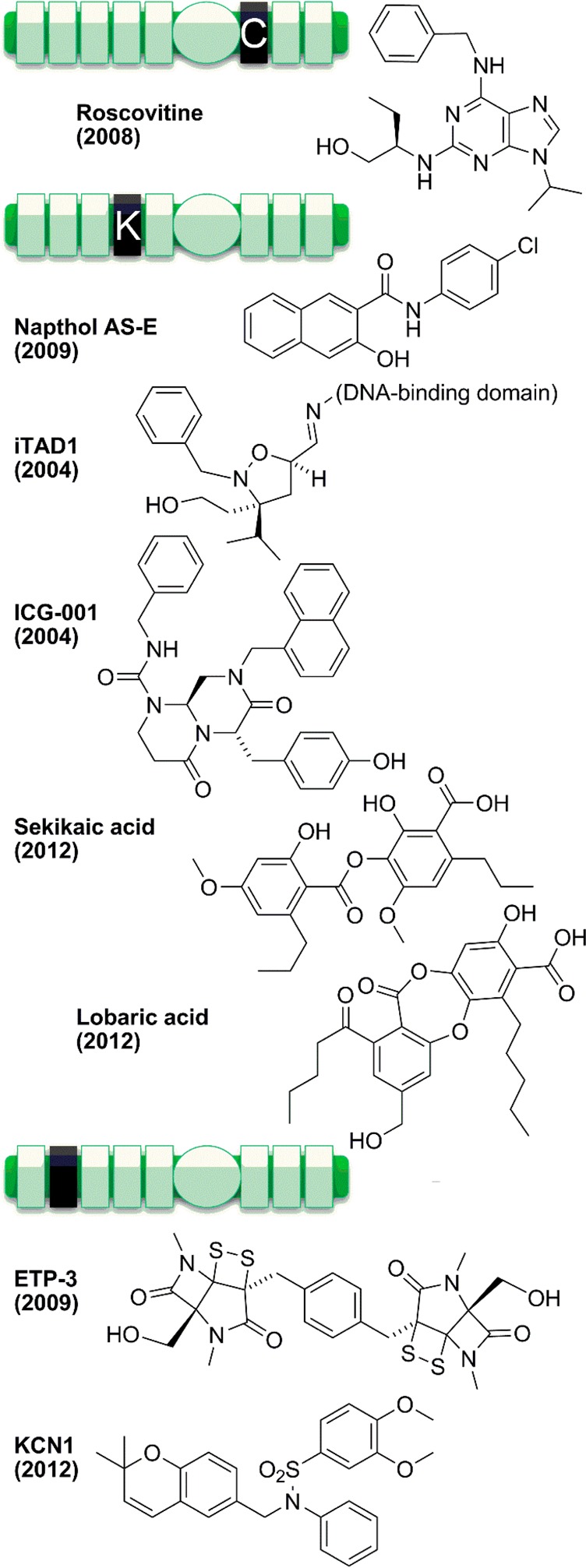
Inhibitors of the other p300/CBP domains.
The KIX domain (located before the N-terminus of the bromodomain) is the target of several small molecules. The compound napthol AS-E (see Figure 18) appears to bind the KIX domain with an affinity of 8.6 μM and undetermined specificity.355 The NMR structure was obtained of the KIX domain in complex with the phosphorylated compound, known as KG-501 (identical to napthol AS-E except the hydroxyl group is replaced with a phosphate group). This compound was the first KIX inhibitor identified,356 and was subsequently reported to be converted to more-active napthol AS-E in the cell medium prior to cellular entry.362 In the costructure, KG-501 does not directly compete for ligand binding. Rather, it binds at a site that is known to dynamically regulate binding interactions at the distal groove.363 It is likely that the active dephosphorylated compound binds in a similar manner. The effect of binding allosterically inhibits the binding of the CREB KID domain, inhibits CREB-mediated gene induction, but appears to increase histone acetylation.362,363 At around the same time, a series of isoxazolidine-containing molecules named iTADs were described357 that bind in the same binding site of KIX as transactivation domains of proteins such as MLL, c-Jun, and Tat.358 iTAD1 (see Figure 18) binds KIX with an affinity of around 38 μM and promiscuously binds other proteins known to also interact with transactivation domains, such as TRRAP and Med23. Binding of iTAD to KIX competitively inhibits endogenous transactivation domain-containing ligands from binding the same site, while activating CBP activities that are normally induced by protein ligand binding.359 The addition of a DNA-binding moiety to iTAD can allow activation of the associated gene by recruiting the active CBP.360 The CBP KIX domain may also be the target of the compound ICG-001 (see Figure 18), which appears to block the association of CBP with β-catenin, and decrease expression of genes usually induced in the Wnt/β-catenin/CBP pathway. Although the specificity of this compound is not known, nor the exact binding mechanism, it does show promising effects in animal models of colon cancer, myocardial infarction, pulmonary fibrosis, and fibrotic kidney disease.361−364
More recently, two natural products from lichen, sekikaic acid and lobaric acid (see Figure 18), were found to have KIX-binding properties, in addition to their other known activities.365 Sekikaic acid and lobaric acid inhibit the KIX interaction with the ligand MLL with IC50 values of 34 and 17 μM, respectively. Interestingly, they also inhibit the binding of the KID domain of CREB, at a distinct binding site within KIX, with IC50 values of 64 and 25 μM, respectively. Furthermore, the interaction appears to have some specificity, because the binding of Med15 and its cognate transactivation domain within VP16 was not inhibited by sekikaic acid. The effect is a downregulation of p300/CBP-mediated gene expression, including the Cyclin D1 gene normally induced by c-Jun through its interaction with KIX.
The binding of the C/H1 region of p300/CBP with the protein ligand HIF-1α has also been the target of inhibition studies. Several epidithiodiketopiperazine (ETP) compounds have been found to inhibit this interaction, including the fungal natural products chaetocin and chetomin,366 as well as the synthetic ETP 3 (see Figure 18).367 Chetomin and ETP 3 both bind to C/H1 (KD values of 540 and 750 nM, respectively) and inhibit HIF-1α binding (IC50 values of 540 and 1500 nM, respectively), probably by disrupting the global folding of the domain, which prevented hypoxia-induced gene induction in cultured cells. The specificity of these compounds for the p300/CBP C/H1 has not yet been studied, but it will be interesting to see whether the promising effects that these compounds have on animal models are due to the p300/CBP inhibition or another target. Another compound, called KCN1 (see Figure 18), appears to inhibit p300/CBP interaction with HIF-1α, although by an unknown mechanism.368
6. Potential Applications for Inhibitors and Activators of p300/CBP
6.1. Loss of p300/CBP Activity in Human Disease
Given the pivotal role of p300/CBP in essential cellular functions, it is not surprising that p300 homozygous knockout, CBP homozygous knockout, p300/CBP double homozygous knockout, and p300/CBP double heterozygous knockout are lethal in mammals.369,370 These gene dosage effects provide further evidence for the existence of a limiting pool of p300/CBP that may allow for differential p300/CBP recruitment depending on signaling pathway activation.
Furthermore, single heterozygous loss-of-function mutations, in p300 or CBP, are one of the known causes of Rubinstein–Taybi syndrome (abbreviated RTS or RSTS), a congenital developmental disorder. The frequency of RTS is approximately one affected individual out of every 100 000 newborns.371 p300/CBP mutations that cause RTS are usually de novo mutations (i.e., not inherited from either parent), but in the extremely rare cases of vertical transmission, inheritance is Mendelian autosomal dominant.372
p300/CBP mutations that cause RTS are found in the germline of the patient, can occur in any of several p300/CBP exons (and disrupt any of several p300/CBP protein domains), and are usually point mutations or small deletions or insertions (which can cause truncations371,373), but can also be translocations, inversions, or large deletions.374
40% of RTS patients either have unknown RTS etiology, or may be more appropriately diagnosed with a different (but phenotypically similar) syndrome, such as Saethre–Chotzen syndrome (caused by mutations in TWIST1) or Mowat–Wilson syndrome (caused by mutations in ZEB2).375 Approximately 55% of RTS patients have germline mutations in CBP, and approximately 5% of RTS patients have germline mutations in p300.374 RTS patients with p300 mutations often have milder phenotypes than those with CBP mutations.376−378 In RTS patients, CBP mutations are more common than p300 mutations, but in the human population, single-nucleotide polymorphisms in general are more common in the p300 gene than the CBP gene.379 It has therefore been hypothesized that p300 germline mutations could be associated with a disorder or phenotype other than classical RTS, which has yet to be found.
RTS is characterized by postnatal growth deficiency, mental retardation, skeletal and cardiac abnormalities, psychomotor development delay, and a dysmorphology involving broad thumbs and big toes, short stature, and characteristic facial features.380 Cells taken from these patients have reduced acetylation of histones, particularly H2A and H2B, as compared to control cells,381 further implicating the loss of function of p300/CBP in the disease etiology. Another phenotype of RTS is an increased likelihood to develop cancer. About 5% of children with RTS develop tumors of neural crest origin.382 It has been hypothesized that such tumors might have somatic mutations in the remaining p300/CBP allele.383
Apart from RTS-associated tumors, loss-of-function in p300/CBP has been implicated in several additional cancers with somatic mutations in p300 or CBP. One study, which screened 103 cancer cell lines, examined loss of heterozygosity (LOH) and found that 51% of the cell lines had LOH at the p300 locus and 35% had LOH at the CBP locus (with 19% of the cell lines having LOH at both loci),384 and these numbers are consistent with another study of 203 cancer samples, which found 35–50% of primary ovarian, colon, and breast tumors had LOH at the p300 locus.385 In another study, 222 cancer samples were examined, and while only 1% of the samples had mutations in CBP (heterozygous truncations), 3.6% of the samples had LOH at the p300 locus (either by mutation or by silencing of one p300 allele) combined with a truncation in the remaining p300 allele. Similarly, in a study of colorectal and gastric carcinomas, LOH at the p300 locus was combined with missense mutations in the remaining p300 allele.386 These data are consistent with a “two-hit” inactivation model, which is characteristic of tumor suppressors. However, other genes are also located within the regions lost in many of these large deletions, so that LOH may only be part of the story.
Furthermore, p300 mutations identified in primary tissues were found in solid tumors of colorectal, breast, and gastric tissue, while p300 mutations identified in cell lines were found in colorectal, breast, ovarian, pancreatic, oral squamous cell, and cervical cancers.387 A recent study of B-cell non-Hodgkin’s lymphoma identified CBP (or more rarely p300) heterozygous loss of function mutations in 40% of samples,388 while another study of 154 lung cancer samples identified mutations in 5–10% of samples.389 These cancer-associated p300/CBP mutations were found in several p300/CBP exons, without any obvious connection between the p300/CBP protein domain disrupted in each case and the phenotypes observed.
While these mutation screening studies may be largely correlative, several experiments have helped establish the p300/CBP mutation as being causative. In one study, carcinoma cell lines with p300 truncation mutations (but not cancer cell lines without p300 mutations) acquired a reduction in proliferation when transfected with full-length, wild-type p300 (although not CBP), further implicating the p300 mutation in the cancerous phenotypes of this cancer.390 In a recent study of acute lymphoblastic leukemia (ALL), cells were taken from 71 ALL patients at the time of diagnosis and again at the time of relapse, and CBP loss of function mutation was found to correlate with patients acquiring resistance to therapy.391
In addition to p300/CBP mutation, another mechanism by which p300/CBP may experience loss of function in cancer is through decreased expression, selective degradation, or both. This was explored in a study in which prolonged activation of Ras signaling and platelet-derived growth factor (PDGF) signaling, which are known to cause cancer, also causes reduced p300/CBP expression and proteasomal degradation. These experiments were performed in NIH 3T3 fibroblasts, but the decreased p300/CBP protein levels might be relevant in cancers involving Ras, PDGF, and possibly other pathways.392 Another observation of decreased p300/CBP expression was made in certain breast and colorectal carcinomas (but not in bladder, renal, or ovarian tumors)393 and in a similar study finding decreased p300 expression in breast and prostate cancer (but not in cervical cancer).394 A third example was observed in instances of acute myeloid leukemia that were caused by a translocation in which MOZ is fused with TIF2,395,396 and this oncogenic chimeric protein bound to p300/CBP and resulted in decreased p300/CBP protein levels by an unknown mechanism.397
In summary, p300/CBP partial loss of function can contribute to cancer in humans. This result may seem paradoxical, becausep300/CBP total loss of function prevents cell growth, but one explanation is that p300/CBP acts in concert with tumor suppressor proteins. One such example is in the TGF-β pathway, which involves the tumor suppressors TGF-β, Smad2, Smad4, and RUNX.398 In this pathway, TGF-β signaling results in the transcriptional activation of target genes, and this transcriptional regulation requires p300/CBP in cooperation with Smads and RUNX. Another example of a tumor suppressor protein is p53,399 which is acetylated by p300/CBP, contributing to its stability. In addition, p53 binds to p300/CBP to form a coactivator complex with DNA, and utilizes the acetyltransferase activity of p300/CBP to activate target gene expression. Another tumor suppressor protein that is regulated by p300/CBP is retinoblastoma protein Rb, whose acetylation decreases its phosphorylation,400 and the acetylation of the associated E2F increases its ubiquitylation.401
To further explore p300/CBP loss-of-function in RTS and cancer, several mouse models have been developed. Mouse models of RTS with heterozygous truncation in CBP402 or p300403 have phenotypes that mimic RTS, and consistent with observations in humans, the p300 mutants had milder phenotypes. Mice with heterozygous knockout of CBP also had hematopoeitic failure and hematological malignancy.404 The role for p300/CBP in hematopoiesis is not surprising, considering that over 56 proteins that bind p300/CBP are known to be essential for T and B cell production in mice.405 Furthermore, in these malignant cells, there was often somatic loss of heterozygosity (LOH) at the CBP locus, which inactivated the wild-type allele, similar to LOH seen in human cancers.
Although embryonic lethal, homozygotes were successfully generated from these mice using a chimera strategy, and p300 or CBP knockout caused hematological malignancy. Interestingly, the type of hematological malignancy was different depending on whether p300 or CBP was mutated.406 Another strategy to generate homozygous knockouts of p300 and/or CBP utilized flanking LoxP sites to create conditional deletions, dependent on the presence of Cre recombinase, that was used to inactivate p300 and/or CBP in thymocytes. In this study, loss of CBP (but not p300) occasionally caused T cell lymphoma, but neither loss consistently resulted in drastic T-cell phenotypes, while loss of both p300 and CBP caused T cells to die.407
Taken together, these studies using mouse genetics validate the role of p300/CBP loss-of-function in RTS and cancer, confirm our understanding of p300/CBP gene dosage and a limiting pool of p300/CBP protein, and provide further evidence of subtle functional differences between the p300 and CBP paralogs. Diseases involving loss-of-function of p300/CBP could theoretically be treated using compounds that chemically complement point mutations in the acetyltransferase active site,408 stimulate p300/CBP activity, or inhibit deacetylase activity. Studies of loss-of-function serve as a caution that activity inhibitors might be contraindicated in certain patients, and correct dosing would need to be optimized to minimize harmful consequences of too little p300/CBP activity.
6.2. Overexpression, Inappropriate Activation, or Mistargeting of p300/CBP
While loss-of-function in p300/CBP is deleterious, gain-of-function in p300/CBP also contributes to human disease, because the correct balance of acetylation and deacetylation, in the right time and place, is required for normal cell functioning. This section explores cases of gain-of-function in p300/CBP, either by direct mutation or overexpression of p300/CBP or by the involvement of p300/CBP in the disease etiology, as well as additional instances where disruption of p300/CBP activity would be therapeutically advantageous (see Figure 19).
Figure 19.
Diseases of potential therapeutic application for a p300/CBP inhibitor. No p300/CBP inhibitor has yet made it into clinical trials, but the biology of p300/CBP action and documented effects of p300/CBP disruption lead us to hypothesize a beneficial therapeutic potential for a p300/CBP inhibitor in many diseases.
There are several instances where p300/CBP overexpression has been found to correlate with cancer. In one study of 209 normal nasopharyngeal mucosa and nasopharyngeal carcinoma samples, p300 expression correlated with malignancy, aggressive behavior of the carcinoma, and poor patient prognosis.409 Furthermore, in a study of 123 hepatocellular carcinoma samples, plus 123 matching adjacent nonmalignant liver tissue samples, p300 expression correlated with malignancy, tumor size, poor differentiation, tumor progression, and poor prognosis.410
Overexpression of p300 is also present in many hormone-dependent cancers. In one study of 183 breast cancer samples,411 and another that included matched normal breast tissue,412 p300 expression correlated with malignancy, activation of the hypoxia response, and dominant-negative mutant p53 levels. In a study of 95 prostate cancer samples,413 and another of 92 prostate cancer samples,414 p300 expression correlated with phenotypes of malignancy, including proliferation, tumor volume, extraprostatic extension, seminal vesicle involvement, tumor progression, nuclear alterations, and poor prognosis. Furthermore, p300 knockdown resulted in decreased prostate cancer cell proliferation. Targeting p300 in these cancers could also decrease hormone receptor acetylation, which would modulate sensitivity to hormones and drugs like tamoxifen.415
Genomic profiling in 101 melanoma samples provides evidence for p300 overexpression in this cancer.416 Subsequently, profiling of samples from 358 melanoma patients revealed that p300 levels were increased in the cytoplasm while decreased in the nucleus,417 and that the increase in the cytoplasm correlates with melanoma progression, tumor size, and ulceration status.418 Furthermore, in normal melanocytes, decreased p300/CBP protein levels correlate with activation of replicative senescence, and because senescence is broadly tumor-suppressive, this prompted the study of p300/CBP inhibitors in melanoma. Indeed, inhibition of p300/CBP reduced proliferation and induced senescence in melanoma cells,419 as well as sensitizing cells to DNA damage, which increases the interest in potential combination therapies of p300/CBP inhibition plus DNA damaging agents.420
This advantageous combination with DNA damage has also been demonstrated for colorectal cancer. In one report using colorectal cancer cells, p300 knockout strains were generated, and they responded to DNA damage by undergoing apoptosis at a greater rate than cells expressing p300. Moreover, xenografts of the p300 knockout cells were hypersensitive to doxorubicin, a cancer therapeutic that induces DNA damage.421
In the case of certain hematological malignancies, part of p300 or CBP is translocated such that it becomes fused with another protein, creating an oncogenic fusion protein with inappropriate p300/CBP activity. These translocations are more common for CBP than p300, and this is thought to be due to an unstable genomic element near the 5′ end of the CBP gene,422 although breakpoints in the second intron have also been described as occurring often.423 Truncations near the 5′ end of the CBP gene result in loss of the regulatory N-terminal portion of CBP, may activate the catalytic acetyltransferase domain, and may target the fusion protein to inappropriate genes or substrates.
One example fuses the monocytic leukemia zing-finger (MOZ) gene with CBP (or in rare cases, with p300),424−426 and is found in acute myeloid leukemia (AML). Another example fuses the gene for MOZ-related factor (MORF), with CBP, and is also found in AML, but less frequently than for MOZ.427−430 A third example fuses the mixed lineage leukemia (MLL) gene with p300 or CBP,431−434 is found in patients with infantile acute leukemia and patients with treatment-related leukemia, and has been studied using a conditional knockin mouse model.435
A further avenue of gain-of-function in p300/CBP is through the action of oncogenes that misappropriate p300/CBP in their transformation of cells. One such example is in acute myeloid leukemia, which can be caused by an oncogenic fusion of AML1 and ETO genes. Leukemogenesis induced by AML1-ETO involves binding of this fusion protein with p300/CBP, acetylation of the fusion protein by p300/CBP, and the activation of target genes.436 Another example is in NUT midline carcinoma, which is caused by oncogenic fusions of the NUT protein, including BRD4-NUT and BRD3-NUT. Malignant cell transformation by BRD4-NUT involves binding of this fusion protein with p300/CBP, translocation of this complex to nuclear foci, and stimulation of p300/CBP catalytic activity at these foci.437 Another way that p300/CBP can be required for the activity of oncogenes is by controlling the transcription of those oncogenes, which is true for the immediate early proto-oncogenes c-jun and c-fos.438 These are oncogenic transcription factors, and they, as well as c-myb,439 utilize the action of p300/CBP in activating target genes in their oncogenic pathways.
Another example of gain-of-function in p300/CBP is in viral transformation. Transformation occurs when a virus usurps the cell’s p300/CBP to promote the viral life cycle, and in so doing confers inappropriate activities on p300/CBP, and as a result the cell becomes immortalized. p300 was first identified because of its ability to bind the adenoviral E1A protein, and subsequent experiments have elucidated the essential role of the interaction with p300/CBP in transformation of the host cell. Likewise, the papillomaviral E6 protein and the viral SV40 large T antigen protein require interaction with p300/CBP for cellular transformation.440
Chromatin modifiers, including p300/CBP, are also being explored as therapeutic targets for treating retroviral infections, such as HIV. Part of this is because the viral genome expression is impacted by host chromatin dynamics,441 including in HIV LTR transcription initiation and the reversal of viral latency.442 To that end, targets that have been investigated include the lysine demethylase Lsd1,443 lysine deacetylases, and lysine acetyltransferases. Furthermore, p300/CBP might be dually useful for HIV treatment because the HIV lifecycle involves p300/CBP acetylating the HIV Tat444 and integrase proteins.445,446 There is also evidence that HIV Tat expression could cause the induction of p300 overexpression,447 further demonstrating the potential benefit for testing p300/CBP inhibition in HIV treatment.
Genetic studies suggest a role for p300/CBP in platelet cell production, where disruption of p300/CBP can cause a high platelet count in a normal animal, or stimulate platelet production in a diseased animal. This is likely due to the involvement of p300/CBP in transcriptional control of genes involved in platelet and megakaryocyte production. p300/CBP disruption has proved a promising therapeutic option for several animal models of different types of thrombocytopenia.448,449
Additionally, p300/CBP inhibition may prove therapeutic for heart disease. As mentioned above, patients with RTS and mice with p300 deleted have cardiac abnormalities, which is consistent with the role of p300/CBP in transcriptional control by cardiac factors such as MyoD, MEF2, and GATA4/5/6.450 On the other hand, overexpression of p300 has been shown to induce hypertrophy in cardiomyocytes. Treatment of cardiomyocytes with the hypertrophic agent phenylephrine has been shown to activate p300, and genetic knockdown of p300 or CBP inhibited the hypertrophy.451
p300/CBP is also a potential therapeutic target in diabetes. Hepatic gluconeogenesis is controlled at the transcriptional level by p300/CBP as cAMP signaling results in the activation of genes that drive glucose production. In the fed state, insulin signaling leads to p300/CBP phosphorylation, preventing its association with CREB and down-regulating glucose production.452 In diabetic patients with decreased insulin or insulin insensitivity, disruption of p300/CBP activity could therefore reduce the transcription of CREB-controlled genes, and result in lower glucose production. In a related pathway that leads to type 2 diabetes, obesity, and nonalcoholic fatty liver disease, p300/CBP plays a role in transcriptional activation of genes in lipogenesis and fat accumulation in the liver. High glucose levels result in activation of p300/CBP, acetylation of carbohydrate-responsive element-binding protein (ChREBP), formation of a complex including p300/CBP and ChREBP, acetylation of promoter histone H4, and activation of target gene transcription.453 p300 overexpression results in fat accumulation, insulin resistance, and inflammation of the liver, thus providing a further potential role for p300/CBP inhibition in the treatment of type 2 diabetes, obesity, and nonalcoholic fatty liver disease.454
In summary, p300/CBP is involved in a variety of different human diseases, including several where p300/CBP acetyltransferase inhibition may be of therapeutic value. Thus, understanding p300/CBP function, developing assays to measure p300/CBP activity in cells, and developing small molecule agents that target p300/CBP are of pharmacologic interest.
6.3. Cell-Based Assays of p300/CBP Activity and Inhibition
This section deals with studies in cultured cells of inhibitor target validation, pharmacodynamics, and studying the phenotypes downstream of p300/CBP modulation. A variety of techniques have been developed to study post-translational modifications; however, the understanding of acetylation has lagged behind some of the better-characterized PTMs, such as ubiquitylation and phosphorylation. In many cases, this may be due to technical challenges faced when studying acetylation.
For the separation and/or detection of acetylation states of proteins from cells and cell lysates, typical methods can involve acetyl-specific antibodies, specially formulated acid-urea gel electrophoresis, or mass spectrometry. Acid-urea gel electrophoresis and mass spectrometry require fixed time points of bulk populations of cells, and are conducted after cells are killed and lysed. Assays that involve antibodies include immunoblotting (also known as western blotting), immunoprecipitation (also known as pulldowns), chromatin immunoprecipitation (also known as ChIP), and immunohistochemistry (also known as immunofluorescence microscopy). Several antibodies have been developed that recognize either acetyl-lysine in general, or acetyl-lysine in the context of specific neighboring sequences. These antibodies have been, and continue to be, useful for a variety of studies, but their poor affinities and imperfect specificities are often a limitation. Of these techniques, only immunocytochemistry makes single cell analysis possible, which allows analysis of heterogeneity within a cell population, and also makes it possible to visualize subcellular spatial distribution of the signal. For conventional immunofluorescence, the cells must be fixed and permeabilized, which are known to be associated with disruptions to the sensitive structures in the cell.455
Each of these traditional techniques involves several steps between harvesting the cells and analyzing the signal, during which time the acetylation state could theoretically be altered. Acetyl-lysine-specific antibodies could be used in living cells via two possible methods. First, the antibody can be labeled with a dye and introduced into cells by microinjection or bead loading,456−800 which are disruptive to cells, although less drastic than killing and lysing cells. Bead loading of antibodies or antibody fragments is less laborious and results in higher sample numbers than microinjection. In a second method for using antibodies in living cells, genetically encoded single-chain antibodies can be fused to fluorescent proteins,460,461 which is a promising strategy that has yet to be demonstrated for the study of p300/CBP. Both of these methods would theoretically be able to track the localization of acetylated proteins and any redistribution that might occur when p300/CBP is disrupted, but would not be capable of measuring changes in the total amount of acetyl-protein or the ratio of acetyl- to unmodified-protein, because the fluorescence level is constitutive.
To follow acetyltransferase-deacetylase activity in living cells, one potential method that has been applied to methylation is using bimolecular fluorescence complementation (BiFC).462 Another approach is with bimolecular Förster resonance energy transfer (FRET).463,464 One group tagged bromodomain-containing proteins BRD2, TAFII250, and Pcaf with YFP, tagged histones with CFP, and measured the CFP–YFP interaction by flow cytometry.465 The major limitation of this flow cytometry approach is that changes in acetylation dynamics within a single cell cannot be monitored in real-time. Also, the apparent changes in acetylation could be on an untagged substrate, providing it is in close enough proximity to the tagged subunit, and therefore the target site determination requires inference from mutagenesis studies.
As an improvement on these techniques, the Minoru Yoshida and colleagues developed a unimolecular FRET-based assay that allows the analysis of acetylation and deacetylation in single, living cells.140,466,467 This methodology has proved useful for the dynamic analysis of other post-translational modifications,468−472 including on histones,473,474 and has only recently been expanded to study acetylation, by the creation of reporters based on histone H4 lysine substrates and BRDT (binds acetyl-K5/K8) or BRD2 (binds acetyl-K12) bromodomains. These FRET experiments can show dynamics in real-time; for example, a baseline with normal acetyltransferase/deacetylase activity levels is established, followed by addition of drug, and the time-course of drug response is thereby followed in each cell. As a first demonstration of the technique, dose-dependent FRET decreases of about 20% (Y/C emission) after 2 h of treatment with trichostatin A (TSA), a deacetylase inhibitor,475 were observed for the K5/K8 and K12 reporters. This could be reversed upon washout and the response of the K12 reporter (and to a lesser degree the K5/K8 reporter) could be blocked by BIC1, a compound that weakly disrupts the bromodomain that the reporter depends upon. Dose-dependent FRET increases of about 10% after 5 min of treatment with 20 μM C107, a p300/CBP acetyltransferase inhibitor, were observed for the K5/K8 reporter.140 Similarly, FRET was increased by about 20% in cells with stably knocked down p300 or CBP as compared to control cells, for the K5/K8 reporter. Limitations of this assay include potential signal from intermolecular FRET such as in dense chromatin, disruptions to the cells from transfection agents and expression of a tagged histone, potential phototoxicity and photobleaching after long time-courses, and the inability to do time-courses for genetic disruption of p300/CBP as opposed to pharmacologic disruption. An exciting extension of this methodology would be to adapt it for high-throughput drug screening, such as by epifluorescence imaging in 96-well glass-bottomed culture dish format, with robotics to control the field of view in each well, and employing a microfluidic device for precise timing of drug delivery in each well.
In addition to target validation, an important question in drug discovery is whether disease phenotypes can be reversed by pharmacologic inhibition of p300/CBP. As discussed in section 6.2, overexpression or dysregulation of p300/CBP can contribute to cell transformation. Hallmarks of cancer include increased malignant cell growth (and evasion of pathways that could block growth or induce cell death such as apoptosis), increased blood vessel growth (angiogenesis), ability to multiply indefinitely (immortality), movement to distant sites and invasion of surrounding tissues (metastasis), aberrant metabolism, evasion of the immune system, chromosome abnormalities and DNA instability, and inflammation.476,477 Genetic knockout or knockdown of p300 has implicated p300 in pathways that favor growth and angiogenesis. However, as discussed in section 2.5, p300 has diverse functions as a scaffold and bridge, and thus it is unclear how significantly the acetyltransferase function of p300 contributes to these cellular phenotypes. p300 inhibitors can therefore also be used as tools to dissect the role of the catalytic activity in cellular pathways.
For these reasons, the various acetyltransferase inhibitors have been utilized in a variety of experiments aimed at measuring their biological effects. In fact, the anticancer properties of several natural products were described before their acetyltransferase-inhibitory properties were known. Because, to date, C646 is the most potent, specific, cell-permeable, small molecule inhibitor of p300/CBP that has been reported, we will focus on its applications in this section, but it should be noted that many of the effects have been phenocopied in studies with other acetyltransferase inhibitors such as curcumin.
One biological effect of acetyltransferase inhibition that can be measured is changes in gene expression, including genes involved in cancer. One study of a carcinoma caused by a BRD4-NUT fusion protein found that C646 interferes with the changes in gene expression seen with different expression levels of BRD4-NUT.478 Another study found that C646 interferes with the c-jun and c-fos oncogene expression induced by growth factor.479 A study of prostate cancer cell lines found that C646 interfered with expression of an NFκB subunit,480 and a study of a hepatocellular carcinoma cell line found that C646 interfered with expression of microRNA-224.481 When examining melanoma cell lines, C646 interfered with the expression of several genes involved in cell cycle regulation, DNA damage repair, and nucleosome assembly, while enhancing expression of the tumor suppressor protein p53.482
Other cancerous phenotypes have also been examined in the presence of p300 inhibition. C646 was observed to induce apoptosis and increase prostate-specific antigen secretion in prostate cancer cell lines.483 C646 was observed to decrease the growth of leukemia cells but not control cells, and in a mouse model of leukemia, C646 was observed to improve white blood cell count, anemia, thrombocytopenia, and survival.484 Inhibition of p300/CBP by genetic or small molecule approaches blocked regulatory T cells and enhanced immune surveillance of tumors.485
Other effects, unrelated to cancer, of C646 treatment in cells have been recorded. C646 was observed to interfere with tau phosphorylation, an event involved in neurodegeneration.486 C646 was observed to interfere with the reprogramming of fibroblasts to produce induced pluripotent stem (iPS) cells.487 In explants, C646 was observed to decrease hepatoblast production, while at the same time changing gene expression levels that relate to liver and pancreas cell fate.488 In mice, C646 was observed to enhance the consolidation of fear extinction memory and long-term memory potentiation.489
6.4. Overlap between Genetic and Pharmacologic Disruption of p300/CBP
In contrast to small molecules, which have been developed that target less than 5% of the proteome,490 RNAi strategies have allowed for comparatively straightforward specific targeting of virtually every gene in the transcriptome, and, more recently, CRISPR/TALEN technology has made genomic editing even more accessible. As discussed in sections 6.1 and 6.2, genetic studies of p300/CBP have been useful in identifying phenotypes of p300/CBP knockdown or overexpression and understanding human diseases where p300/CBP has loss- or gain-of-function. However, there are examples in the biochemical literature where genetics and pharmacology specifically targeting the same gene or protein have led to different phenotypes.489
Phenomena that can cause apparent discrepancy between genetic and pharmacologic effects include protein level thresholds, compound allosteric effects, protein–protein binding independent of enzyme activity, and cellular compensation over time.489 All of these could be expected to play a part in p300/CBP function, and it could be predicted that p300/CBP would present such a dichotomy. For example, inhibitors target a particular function or activity, while genetic knockdowns and knockout target the total protein levels.
However, in general, the phenotypes of cells treated with p300/CBP inhibitors, RNAi-based knockdowns, and conditional knockouts all have common features. For the live-cell studies of histone hyperacetylation dynamics, a similar FRET increase was observed for C107 and shRNA against p300 or CBP. The real-time response to C107 was rapid and peaked at around 45 min after addition of C107, while the response to knockdown was measured after several passages of cells stably expressing the shRNA and compared to empty vector control cells.140
Cells with stable knockdown could compensate for prolonged p300/CBP deficiency by overexpressing another acetyltransferase with overlapping activity,491 by downregulating deacetylase activity to restore the acetylation balance, or through other mechanisms. Indeed, the Brindle lab observed partial compensation through CRTC2 overexpression in their p300/CBP knockout cells.492 Another acetyltransferase that could compensate is Gcn5, because mice with heterozygous mutation in Gcn5 and heterozygous mutation in p300 have greater lethality and phenotypic defects than either heterozygous mutation alone, arguing for functional overlap between Gcn5 and p300.493 However, we observed that a chronic defect in acetylation was present in cells with p300 or CBP stable knockdown.
7. Future Directions
Although many substrates, protein ligands, and target genes have been identified for p300/CBP, it is still unclear what fraction of the acetylome is catalyzed by p300/CBP and under what circumstances each occurs. Mutagenesis and methods to artificially introduce acetylated lysines (or their mimics) into proteins have been used to study some specific acetylation events, but others still have unknown functions. Another area of current investigation is the flanking domain interactions of p300/CBP, and determining how specific ligands regulate acetyltransferase activity and/or substrate specificity. Further studies with natural and synthetic ligands will prove useful to determine the effects of various protein ligand interactions. Finally, it remains possible that there are additional protein lysine acetyltransferases that have yet to be discovered, the identification of which may require high throughput screening and CoA capture compounds.
Recent progress has been made toward developing high-throughput assays to measure p300/CBP inhibition against purified enzyme and in cells, screening of natural product and synthetic compound libraries, and developing candidate molecules that could be active in vivo. Although several small molecule probes are available for research, there remains a need for better compounds with varied structures and improved pharmacokinetics that can be used in live animals.
Of the many theoretical medical applications of p300/CBP inhibitors, a few have been supported by encouraging data using cultured cells, such as certain models of solid tumors. A new era of p300/CBP biomedical research will emerge as new p300/CBP inhibitors are tested in animal disease models. Important questions will be what level of inhibition of p300/CBP defines an effective therapeutic window, and what the main toxicities of this inhibition will be.
It will be interesting to see whether future p300-specific and CBP-specific inhibitors have therapeutic value in distinct and/or overlapping disease states. Developing inhibitors for other acetyltransferases besides p300/CBP is also an area of potential significance to biomedicine.
Because epigenetic marks can function combinatorially (see section 2.3), it is possible that acetyltransferase inhibitors will be highly useful in combination therapies with other epigenetic modulators, in specific cases. In the cell, different post-translational modifications on the same segment of chromatin can combine to fine-tune the functional readout, and PTM reader domains are often found together in a large macromolecular complex. Furthermore, multiple PTM-altering abnormalities can be present in the same disease state. To exploit this, combination therapies have been explored for other epigenetic modulators,494−496 but not yet for acetyltransferases. Also, along the lines of combination therapies, there is reason to believe that p300/CBP inhibitors will synergize in cancer cells with drugs that act by inducing DNA damage.497 Furthermore, bifunctional drugs or “codrugs” that have two individual active moieties linked covalently may offer therapeutic advantages.498
8. Conclusion
Three decades of research into p300/CBP, since the first report in 1985, have revealed its importance to normal cell health and etiology of disease. p300/CBP is a key enzyme in higher eukaryotes, where it acts as an effector in myriad major cellular signaling pathways, which modulate protein functions and gene expression in response to a variety of signals. This is accomplished by binding of over 400 protein ligands to its various protein interaction-mediating domains and the acetylation of ∼100 protein substrates. The unusual hit-and-run kinetic mechanism has permitted the identification of p300/CBP-specific acetyltransferase inhibitors, which have shown promising effects in studies in living cells. The protein acetylation and protein binding functions of p300/CBP are intricately interrelated, and both are targets of pharmacological interventions that may have considerable therapeutic applications.
Acknowledgments
We thank the National Institutes of Health (NIH) and Flight Attendant Medical Research Institute (FAMRI) Foundation for funding.
Glossary
Abbreviations
- Ac
acetyl
- CBP
CREB (cAMP response element-binding) binding protein
- CoA
coenzyme A
- ED50
half-maximal effective dose in a cell, tissue, or organism
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- IC50
half-maximal inhibitory concentration in solution with a purified enzyme
- K
lysine
- NAD
nicotinamide adenine dinucleotide
- p300
E1A binding protein p300
- PTM
post-translational modification
- TSA
trichostatin A
Biographies

Beverley M. Dancy, originally from Cape Town, was introduced to research in the laboratory of Greg J. Hermann at Lewis & Clark College. Her graduate studies with Philip A. Cole, centered on p300 acetyltransferase inhibition, earned her a Ph.D. in Pharmacology and Molecular Sciences from the Johns Hopkins University School of Medicine in 2013. As a postdoctoral fellow in the Morgan-Sedensky laboratory, she investigated enzymes and pathways involved in mitochondrial disease, and in 2014 she will begin a fellowship at the National Institutes of Health Laboratory of Cardiac Energetics, with Robert S. Balaban. Dr. Dancy is interested in developing molecular tools and imaging technologies for understanding the consequences of disease at the molecular, cellular, and organismal levels. In addition to research, she is passionate about bridging the gap between science research and a broader audience through communication and engagement.

Philip A. Cole was born in Paterson, NJ and graduated from Yale University with a B.S. in Chemistry in 1984 and then spent a year as a Churchill Scholar at the University of Cambridge, England. Cole went on to obtain M.D. and Ph.D. degrees from Johns Hopkins University where he pursued research in bioorganic chemistry in 1991. Cole then entered postdoctoral fellowship training at Harvard Medical School prior to joining the Rockefeller University in 1996 as a junior lab head. In 1999, Cole moved back to Johns Hopkins as the Marshall-Maren professor and director of pharmacology. His research interests are in the area of protein post-translational modifications and chemical biology.
Supporting Information Available
Table of 98 protein acetylation substrates of p300/CBP and table of 411 protein binding partners of p300/CBP. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† National Heart, Lung, and Blood Institute Laboratory for Cardiac Energetics, National Institutes of Health, Building 10, Room B1D400, MSC 1061, 10 Center Drive, Bethesda, Maryland 20892, United States.
The authors declare the following competing financial interest(s): P.A.C. is a cofounder, advisor, and equity owner in the company Acylin Therapeutics that is developing HAT inhibitors. B.M.D. declares no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Waddington C. H. Endeavour 1942, 18. [Google Scholar]
- Klar A. J. Trends Genet. 1998, 14, 299. [DOI] [PubMed] [Google Scholar]
- Redon C.; Pilch D.; Rogakou E.; Sedelnikova O.; Newrock K.; Bonner W. Curr. Opin. Genet. Dev. 2002, 12, 162. [DOI] [PubMed] [Google Scholar]
- Dawson M. A.; Kouzarides T. Cell 2012, 150, 12. [DOI] [PubMed] [Google Scholar]
- Mukherjee S.; Hao Y. H.; Orth K. Trends Biochem. Sci. 2007, 32, 210. [DOI] [PubMed] [Google Scholar]
- Moellering R. E.; Cravatt B. F. Science 2013, 341, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen I.; Poręba E.; Kamieniarz K.; Schneider R. Genes Cancer 2011, 2, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing S.; Poirier Y. Trends Plant Sci. 2012, 17, 423. [DOI] [PubMed] [Google Scholar]
- Daugherty M.; Polanuyer B.; Farrell M.; Scholle M.; Lykidis A.; de Crécy-Lagard V.; Osterman A. J. Biol. Chem. 2002, 277, 21431. [DOI] [PubMed] [Google Scholar]
- Robinson P. J.; An W.; Routh A.; Martino F.; Chapman L.; Roeder R. G.; Rhodes D. J. Mol. Biol. 2008, 381, 816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H.; Peak-Chew S. Y.; Chin J. W. Nat. Chem. Biol. 2008, 4, 232. [DOI] [PubMed] [Google Scholar]
- Guo J.; Wang J.; Lee J. S.; Schultz P. G. Angew. Chem., Int. Ed. 2008, 47, 6399. [DOI] [PubMed] [Google Scholar]
- Muir T. W.; Sondhi D.; Cole P. A. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karukurichi K. R.; Wang L.; Uzasci L.; Manlandro C. M.; Wang Q.; Cole P. A. J. Am. Chem. Soc. 2010, 132, 1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang R.; Holbert M. A.; Tarrant M. K.; Curtet S.; Colquhoun D. R.; Dancy B. M.; Dancy B. C.; Hwang Y.; Tang Y.; Meeth K.; Marmorstein R.; Cole R. N.; Khochbin S.; Cole P. A. J. Am. Chem. Soc. 2010, 132, 9986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns E. W.; Phillips D. M.; Simson P.; Butler J. A. Biochem. J. 1961, 80, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips D. M. Biochem. J. 1963, 87, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen I.; Poręba E.; Kamieniarz K.; Schneider R. Genes Cancer 2011, 2, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Close P.; Creppe C.; Gillard M.; Ladang A.; Chapelle J. P.; Nguyen L.; Chariot A. Cell. Mol. Life Sci. 2010, 67, 1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glozak M. A.; Sengupta N.; Zhang X.; Seto E. Gene 2005, 363, 15. [DOI] [PubMed] [Google Scholar]
- Norris K. L.; Lee J. Y.; Yao T. P. Sci. Signaling 2009, 2, pe76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C.; Kumar C.; Gnad F.; Nielsen M. L.; Rehman M.; Walther T. C.; Olsen J. V.; Mann M. Science 2009, 325, 834. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Zhao W.; Yang J. S.; Cheng Z.; Luo H.; Lu Z.; Tan M.; Gu W.; Zhao Y. Mol. Cell. Proteomics 2012, 11, 1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S.; Xu W.; Jiang W.; Yu W.; Lin Y.; Zhang T.; Yao J.; Zhou L.; Zeng Y.; Li H.; Li Y.; Shi J.; An W.; Hancock S. M.; He F.; Qin L.; Chin J.; Yang P.; Chen X.; Lei Q.; Xiong Y.; Guan K. L. Science 2010, 327, 1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albaugh B. N.; Arnold K. M.; Denu J. M. ChemBioChem 2011, 12, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutendra G.; Kinnaird A.; Dromparis P.; Paulin R.; Stenson T. H.; Haromy A.; Hashimoto K.; Zhang N.; Flaim E.; Michelakis E. D. Cell 2014, 158, 84. [DOI] [PubMed] [Google Scholar]
- Begley T. P.; Kinsland C.; Strauss E. Vitam. Horm. 2001, 61, 157. [DOI] [PubMed] [Google Scholar]
- Xing S.; Poirier Y. Trends Plant Sci. 2012, 17, 423. [DOI] [PubMed] [Google Scholar]
- Wellen K. E.; Thompson C. B. Nat. Rev. Mol. Cell Biol. 2012, 13, 270. [DOI] [PubMed] [Google Scholar]
- Albaugh B. N.; Arnold K. M.; Denu J. M. ChemBioChem 2011, 12, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allfrey V. G.; Faulkner R.; Mirsky A. E. Proc. Natl. Acad. Sci. U.S.A. 1964, 51, 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebbes T. R.; Thorne A. W.; Crane-Robinson C. EMBO J. 1988, 7, 1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebbes T. R.; Clayton A. L.; Thorne A. W.; Crane-Robinson C. EMBO J. 1994, 13, 1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunstein M.; Rose A. B.; Holmes S. G.; Allis C. D.; Broach J. R. Genes Dev. 1993, 7, 592. [DOI] [PubMed] [Google Scholar]
- Turner B. M.; Birley A. J.; Lavender J. Cell 1992, 69, 375. [DOI] [PubMed] [Google Scholar]
- Allfrey V. G.; Faulkner R.; Mirsky A. E. Proc. Natl. Acad. Sci. U.S.A. 1964, 51, 786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutskov V.; Gerber D.; Angelov D.; Ausio J.; Workman J.; Dimitrov S. Mol. Cell. Biol. 1998, 18, 6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L.; Zhou M. M. FEBS Lett. 2002, 513, 124. [DOI] [PubMed] [Google Scholar]
- Taverna S. D.; Li H.; Ruthenburg A. J.; Allis C. D.; Patel D. J. Nat. Struct. Mol. Biol. 2007, 14, 1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botuyan M. V.; Lee J.; Ward I. M.; Kim J. E.; Thompson J. R.; Chen J.; Mer G. Cell 2006, 127, 1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Thompson J. R.; Botuyan M. V.; Mer G. Nat. Struct. Mol. Biol. 2008, 15, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y.; Nady N.; Qi C.; Allali-Hassani A.; Zhu H.; Pan P.; Adams-Cioaba M. A.; Amaya M. F.; Dong A.; Vedadi M.; Schapira M.; Read R. J.; Arrowsmith C. H.; Min J. Nucleic Acids Res. 2009, 37, 2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Fischle W.; Wang W.; Duncan E. M.; Liang L.; Murakami-Ishibe S.; Allis C. D.; Patel D. J. Mol. Cell 2007, 28, 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Ilin S.; Wang W.; Duncan E. M.; Wysocka J.; Allis C. D.; Patel D. J. Nature 2006, 442, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T.; Allis C. D. Science 2001, 293, 1074. [DOI] [PubMed] [Google Scholar]
- Bernstein E.; Hake S. B. Biochem. Cell Biol. 2006, 84, 505. [DOI] [PubMed] [Google Scholar]
- Schreiber S. L.; Bernstein B. E. Cell 2002, 111, 771. [DOI] [PubMed] [Google Scholar]
- Ernst J.; Kellis M. Nat. Biotechnol. 2010, 28, 817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenuwein T.; Allis C. D. Science 2001, 293, 1074. [DOI] [PubMed] [Google Scholar]
- Feng Y.; Wang J.; Asher S.; Hoang L.; Guardiani C.; Ivanov I.; Zheng Y. G. J. Biol. Chem. 2011, 286, 20323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan D.; Ingvarsdottir K.; Sterner D. E.; Bylebyl G. R.; Dokmanovic M.; Dorsey J. A.; Whelan K. A.; Krsmanovic M.; Lane W. S.; Meluh P. B.; Johnson E. S.; Berger S. L. Genes Dev. 2006, 20, 966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messner S.; Altmeyer M.; Zhao H.; Pozivil A.; Roschitzki B.; Gehrig P.; Rutishauser D.; Huang D.; Caflisch A.; Hottiger M. O. Nucleic Acids Res. 2010, 38, 6350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhalluin C.; Carlson J. E.; Zeng L.; He C.; Aggarwal A. K.; Zhou M. M. Nature 1999, 399, 491. [DOI] [PubMed] [Google Scholar]
- Thorne A. W.; Kmiciek D.; Mitchelson K.; Sautiere P.; Crane-Robinson C. Eur. J. Biochem. 1990, 193, 701. [DOI] [PubMed] [Google Scholar]
- Zhang K.; Williams K. E.; Huang L.; Yau P.; Siino J. S.; Bradbury E. M.; Jones P. R.; Minch M. J.; Burlingame A. L. Mol. Cell. Proteomics 2002, 1, 500. [DOI] [PubMed] [Google Scholar]
- Clayton A. L.; Hazzalin C. A.; Mahadevan L. C. Mol. Cell 2006, 23, 289. [DOI] [PubMed] [Google Scholar]
- Waterborg J. H. Biochem. Cell Biol. 2002, 80, 363. [DOI] [PubMed] [Google Scholar]
- Katan-Khaykovich Y.; Struhl K. Genes Dev. 2002, 16, 743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annunziato A. T.; Eason M. B.; Perry C. A. Biochemistry 1995, 34, 2916. [DOI] [PubMed] [Google Scholar]
- Hendzel M. J.; Davie J. R. Biochem. J. 1991, 273, 753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazzalin C. A.; Mahadevan L. C. PLoS Biol. 2005, 3, e393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump N. T.; Hazzalin C. A.; Bowers E. M.; Alani R. M.; Cole P. A.; Mahadevan L. C. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taunton J.; Hassig C. A.; Schreiber S. L. Science 1996, 272, 408. [DOI] [PubMed] [Google Scholar]
- Schiltz R. L.; Mizzen C. A.; Vassilev A.; Cook R. G.; Allis C. D.; Nakatani Y. J. Biol. Chem. 1999, 274, 1189. [DOI] [PubMed] [Google Scholar]
- Kimura A.; Horikoshi M. Genes Cells 1998, 3, 789. [DOI] [PubMed] [Google Scholar]
- Thompson P. R.; Kurooka H.; Nakatani Y.; Cole P. A. J. Biol. Chem. 2001, 276, 33721. [DOI] [PubMed] [Google Scholar]
- Ogryzko V. V.; Schiltz R. L.; Russanova V.; Howard B. H.; Nakatani Y. Cell 1996, 87, 953. [DOI] [PubMed] [Google Scholar]
- Schiltz R. L.; Mizzen C. A.; Vassilev A.; Cook R. G.; Allis C. D.; Nakatani Y. J. Biol. Chem. 1999, 274, 1189. [DOI] [PubMed] [Google Scholar]
- McManus K. J.; Hendzel M. J. Mol. Cell. Biol. 2003, 23, 7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper L. H.; Lerach S.; Wang J.; Wu S.; Jeevan T.; Brindle P. K. EMBO J. 2010, 29, 3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson M. A.; Kouzarides T. Cell 2012, 150, 12. [DOI] [PubMed] [Google Scholar]
- Yee S. P.; Branton P. E. Virology 1985, 147, 142. [DOI] [PubMed] [Google Scholar]
- Whyte P.; Williamson N. M.; Harlow E. Cell 1989, 56, 67. [DOI] [PubMed] [Google Scholar]
- Eckner R.; Ewen M. E.; Newsome D.; Gerdes M.; DeCaprio J. A.; Lawrence J. B.; Livingston D. M. Genes Dev. 1994, 8, 869. [DOI] [PubMed] [Google Scholar]
- Chrivia J. C.; Kwok R. P.; Lamb N.; Hagiwara M.; Montminy M. R.; Goodman R. H. Nature 1993, 365, 855. [DOI] [PubMed] [Google Scholar]
- Arany Z.; Sellers W. R.; Livingston D. M.; Eckner R. Cell 1994, 77, 799. [DOI] [PubMed] [Google Scholar]
- Heery D. M.; Fischer P. M. Drug Discovery Today 2007, 12, 88. [DOI] [PubMed] [Google Scholar]
- Brownell J. E.; Zhou J.; Ranalli T.; Kobayashi R.; Edmondson D. G.; Roth S. Y.; Allis C. D. Cell 1996, 84, 843. [DOI] [PubMed] [Google Scholar]
- Brownell J. E.; Allis C. D. Proc. Natl. Acad. Sci. U.S.A. 1995, 92, 6364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleff S.; Andrulis E. D.; Anderson C. W.; Sternglanz R. J. Biol. Chem. 1995, 270, 24674. [DOI] [PubMed] [Google Scholar]
- Bannister A. J.; Kouzarides T. Nature 1996, 384, 641. [DOI] [PubMed] [Google Scholar]
- Ogryzko V. V.; Schiltz R. L.; Russanova V.; Howard B. H.; Nakatani Y. Cell 1996, 87, 953. [DOI] [PubMed] [Google Scholar]
- Eckner R. Biol. Chem. 1996, 377, 685. [PubMed] [Google Scholar]
- Taddei A.; Gasser S. M. Genetics 2012, 192, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando O. J.; Chang H. Y. Annu. Rev. Biochem. 2009, 78, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordoli L.; Netsch M.; Lüthi U.; Lutz W.; Eckner R. Nucleic Acids Res. 2001, 29, 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y.; Holbert M. A.; Wurtele H.; Meeth K.; Rocha W.; Gharib M.; Jiang E.; Thibault P.; Verreault A.; Cole P. A.; Marmorstein R. Nat. Struct. Mol. Biol. 2008, 15, 998. [Google Scholar]
- Bordoli L.; Netsch M.; Lüthi U.; Lutz W.; Eckner R. Nucleic Acids Res. 2001, 29, 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles R. H.; Dauwerse H. G.; van Ommen G. J.; Breuning M. H. Am. J. Hum. Genet. 1998, 63, 1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan H. M.; La Thangue N. B. J. Cell Sci. 2001, 114, 2363. [DOI] [PubMed] [Google Scholar]
- Ogryzko V. V.; Schiltz R. L.; Russanova V.; Howard B. H.; Nakatani Y. Cell 1996, 87, 953. [DOI] [PubMed] [Google Scholar]
- Yang X. J.; Seto E. Mol. Cell 2008, 31, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford D. C.; Brindle Ph. Aging (N. Y.) 2012, 4, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. J.; Seto E. Mol. Cell 2008, 31, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford D. C.; Brindle P. K. Aging (N. Y.) 2012, 4, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany Z.; Newsome D.; Oldread E.; Livingston D. M.; Eckner R. Nature 1995, 374, 81. [DOI] [PubMed] [Google Scholar]
- Ferreon J. C.; Martinez-Yamout M. A.; Dyson H. J.; Wright P. E. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundblad J. R.; Kwok R. P.; Laurance M. E.; Harter M. L.; Goodman R. H. Nature 1995, 374, 85. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan I.; Pérez-Alvarado G. C.; Parker D.; Dyson H. J.; Montminy M. R.; Wright P. E. Cell 1997, 91, 741. [DOI] [PubMed] [Google Scholar]
- Kalkhoven E. Biochem. Pharmacol. 2004, 68, 1145. [DOI] [PubMed] [Google Scholar]
- Nam Y.; Sliz P.; Song L.; Aster J. C.; Blacklow S. C. Cell 2006, 124, 973. [DOI] [PubMed] [Google Scholar]
- Mayr B.; Montminy M. Nat. Rev. Mol. Cell Biol. 2001, 2, 599. [DOI] [PubMed] [Google Scholar]
- Bourdeau V.; Deschênes J.; Métivier R.; Nagai Y.; Nguyen D.; Bretschneider N.; Gannon F.; White J. H.; Mader S. Mol. Endocrinol. 2004, 18, 1411. [DOI] [PubMed] [Google Scholar]
- Saito H.; Posas F. Genetics 2012, 192, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh-Hamad D.; Di Mari J.; Suki W. N.; Safirstein R.; Watts B. A. III; Rouse D. J. Biol. Chem. 1998, 273, 1832. [DOI] [PubMed] [Google Scholar]
- Chen Y. J.; Wang Y. N.; Chang W. C. J. Biol. Chem. 2007, 282, 27215. [DOI] [PubMed] [Google Scholar]
- Jang E. R.; Choi J. D.; Lee J. S. FEBS Lett. 2011, 585, 47. [DOI] [PubMed] [Google Scholar]
- Sakaguchi K.; Herrera J. E.; Saito S.; Miki T.; Bustin M.; Vassilev A.; Anderson C. W.; Appella E. Genes Dev. 1998, 12, 2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer N. G.; Chin S. F.; Ozdag H.; Daigo Y.; Hu D. E.; Cariati M.; Brindle K.; Aparicio S.; Caldas C. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 7386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L. W.; Gambee J. E. J. Biol. Chem. 2000, 275, 40946. [DOI] [PubMed] [Google Scholar]
- Yang X. J.; Seto E. Mol. Cell 2008, 31, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legube G.; Trouche D. EMBO Rep. 2003, 4, 944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz C.; Beck K.; Mink S.; Schmolke M.; Budde B.; Wenning D.; Klempnauer K. H. EMBO J. 2003, 22, 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poizat C.; Puri P. L.; Bai Y.; Kedes L. Mol. Cell. Biol. 2005, 25, 2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W.; Chen H.; Du K.; Asahara H.; Tini M.; Emerson B. M.; Montminy M.; Evans R. M. Science 2001, 294, 2507. [DOI] [PubMed] [Google Scholar]
- Ceschin D. G.; Walia M.; Wenk S. S.; Duboé C.; Gaudon C.; Xiao Y.; Fauquier L.; Sankar M.; Vandel L.; Gronemeyer H. Genes Dev. 2011, 25, 1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan C. M.; Kindle K. B.; Collins H. M.; Heery D. M. Biochem. Biophys. Res. Commun. 2010, 391, 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo H. Y.; Chang C. C.; Jeng J. C.; Hu H. M.; Lin D. Y.; Maul G. G.; Kwok R. P.; Shih H. M. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 16973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanam B.; Jiang L.; Wang L.; Kelleher N. L.; Cole P. A. J. Biol. Chem. 2006, 281, 40292. [DOI] [PubMed] [Google Scholar]
- Karukurichi K. R.; Wang L.; Uzasci L.; Manlandro C. M.; Wang Q.; Cole P. A. J. Am. Chem. Soc. 2010, 132, 1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson P. R.; Wang D.; Wang L.; Fulco M.; Pediconi N.; Zhang D.; An W.; Ge Q.; Roeder R. G.; Wong J.; Levrero M.; Sartorelli V.; Cotter R. J.; Cole P. A. Nat. Struct. Mol. Biol. 2004, 11, 308. [DOI] [PubMed] [Google Scholar]
- Karanam B.; Wang L.; Wang D.; Liu X.; Marmorstein R.; Cotter R.; Cole P. A. Biochemistry 2007, 46, 8207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper L. H.; Fukuyama T.; Biesen M. A.; Boussouar F.; Tong C.; de Pauw A.; Murray P. J.; van Deursen J. M.; Brindle P. K. Mol. Cell. Biol. 2006, 26, 789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Personal communication with Paul Brindle, see: http://www.stjude.org/stjude/v/index.jsp?vgnextoid=c30215f204294210VgnVCM1000001e0215acRCRD.
- Yan G.; Eller M. S.; Elm C.; Larocca C. A.; Ryu B.; Panova I. P.; Dancy B. M.; Bowers E. M.; Meyers D.; Lareau L.; Cole P. A.; Taverna S. D.; Alani R. M. J. Invest. Dermatol. 2013, 133, 2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen I.; Poręba E.; Kamieniarz K.; Schneider R. Genes Cancer 2011, 2, 631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan H. M.; La Thangue N. B. J. Cell Sci. 2001, 114, 2363. [DOI] [PubMed] [Google Scholar]
- Pan C. Q.; Sudol M.; Sheetz M.; Low B. C. Cell. Signalling 2012, 24, 2143. [DOI] [PubMed] [Google Scholar]
- Tanaka Y.; Naruse I.; Maekawa T.; Masuya H.; Shiroishi T.; Ishii S. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei Y.; Xu L.; Heinzel T.; Torchia J.; Kurokawa R.; Gloss B.; Lin S. C.; Heyman R. A.; Rose D. W.; Glass C. K.; Rosenfeld M. G. Cell 1996, 85, 403. [DOI] [PubMed] [Google Scholar]
- Hottiger M. O.; Felzien L. K.; Nabel G. J. EMBO J. 1998, 17, 3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X.; Warner D. R.; Roberts E. A.; Pisano M. M.; Greene R. M. Biochem. Biophys. Res. Commun. 2005, 329, 1010. [DOI] [PubMed] [Google Scholar]
- Avantaggiati M. L.; Ogryzko V.; Gardner K.; Giordano A.; Levine A. S.; Kelly K. Cell 1997, 89, 1175. [DOI] [PubMed] [Google Scholar]
- Kamei Y.; Xu L.; Heinzel T.; Torchia J.; Kurokawa R.; Gloss B.; Lin S. C.; Heyman R. A.; Rose D. W.; Glass C. K.; Rosenfeld M. G. Cell 1996, 85, 403. [DOI] [PubMed] [Google Scholar]
- Goodman R. H.; Smolik S. Genes Dev. 2000, 14, 1553. [PubMed] [Google Scholar]
- Lee C. W.; Sørensen T. S.; Shikama N.; La Thangue N. B. Oncogene 1998, 16, 2695. [DOI] [PubMed] [Google Scholar]
- Nakajima T.; Fukamizu A.; Takahashi J.; Gage F. H.; Fisher T.; Blenis J.; Montminy M. R. Cell 1996, 86, 465. [DOI] [PubMed] [Google Scholar]
- Thompson P. R.; Wang D.; Wang L.; Fulco M.; Pediconi N.; Zhang D.; An W.; Ge Q.; Roeder R. G.; Wong J.; Levrero M.; Sartorelli V.; Cotter R. J.; Cole P. A. Nat. Struct. Mol. Biol. 2004, 11, 308. [DOI] [PubMed] [Google Scholar]
- Muir T. W.; Sondhi D.; Cole P. A. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancy B. M.; Crump N. T.; Peterson D. J.; Mukherjee C.; Bowers E. M.; Ahn Y. H.; Yoshida M.; Zhang J.; Mahadevan L. C.; Meyers D. J.; Boeke J. D.; Cole P. A. ChemBioChem 2012, 13, 2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Wang L.; Zhao K.; Thompson P. R.; Hwang Y.; Marmorstein R.; Cole P. A. Nature 2008, 451, 846. [DOI] [PubMed] [Google Scholar]
- Hwang Y.; Thompson P. R.; Wang L.; Jiang L.; Kelleher N. L.; Cole P. A. Angew. Chem., Int. Ed. 2007, 46, 7621. [DOI] [PubMed] [Google Scholar]
- Liu X.; Wang L.; Zhao K.; Thompson P. R.; Hwang Y.; Marmorstein R.; Cole P. A. Nature 2008, 451, 846. [DOI] [PubMed] [Google Scholar]
- Tang Y.; Holbert M. A.; Wurtele H.; Meeth K.; Rocha W.; Gharib M.; Jiang E.; Thibault P.; Verreault A.; Cole P. A.; Marmorstein R. Nat. Struct. Mol. Biol. 2008, 15, 998. [Google Scholar]
- Wang L.; Tang Y.; Cole P. A.; Marmorstein R. Curr. Opin. Struct. Biol. 2008, 18, 741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poux A. N.; Cebrat M.; Kim C. M.; Cole P. A.; Marmorstein R. Proc. Natl. Acad. Sci. U.S.A. 2009, 99, 14065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H.; Rossetto D.; Mellert H.; Dang W.; Srinivasan M.; Johnson J.; Hodawadekar S.; Ding E. C.; Speicher K.; Abshiru N.; Perry R.; Wu J.; Yang C.; Zheng Y. G.; Speicher D. W.; Thibault P.; Verreault A.; Johnson F. B.; Berger S. L.; Sternglanz R.; McMahon S. B.; Côté J.; Marmorstein R. EMBO J. 2012, 31, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karukurichi K. R.; Cole P. A. Bioorg. Chem. 2011, 39, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleland W. W. Biochim. Biophys. Acta 1963, 67, 104. [DOI] [PubMed] [Google Scholar]
- Cleland W. W. Biochim. Biophys. Acta 1963, 67, 104. [DOI] [PubMed] [Google Scholar]
- Segel I.Enzyme Kinetics; Wiley Interscience: New York, 1975. [Google Scholar]
- Zheng Y.; Thompson P. R.; Cebrat M.; Wang L.; Devlin M. K.; Alani R. M.; Cole P. A. Methods Enzymol. 2004, 376, 188. [DOI] [PubMed] [Google Scholar]
- Bowers E. M.; Yan G.; Mukherjee C.; Orry A.; Wang L.; Holbert M. A.; Crump N. T.; Hazzalin C. A.; Liszczak G.; Yuan H.; Larocca C.; Saldanha S. A.; Abagyan R.; Sun Y.; Meyers D. J.; Marmorstein R.; Mahadevan L. C.; Alani R. M.; Cole P. A. Chem. Biol. 2010, 17, 471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau O. D.; Kundu T. K.; Soccio R. E.; Ait-Si-Ali S.; Khalil E. M.; Vassilev A.; Wolffe A. P.; Nakatani Y.; Roeder R. G.; Cole P. A. Mol. Cell 2000, 5, 589. [DOI] [PubMed] [Google Scholar]
- Sagar V.; Zheng W.; Thompson P. R.; Cole P. A. Bioorg. Med. Chem. 2004, 12, 3383. [DOI] [PubMed] [Google Scholar]
- Karukurichi K. R.; Cole P. A. Bioorg. Chem. 2011, 39, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebrat M.; Kim C. M.; Thompson P. R.; Daugherty M.; Cole P. A. Bioorg. Med. Chem. 2003, 11, 3307. [DOI] [PubMed] [Google Scholar]
- Lau O. D.; Kundu T. K.; Soccio R. E.; Ait-Si-Ali S.; Khalil E. M.; Vassilev A.; Wolffe A. P.; Nakatani Y.; Roeder R. G.; Cole P. A. Mol. Cell 2000, 5, 589. [DOI] [PubMed] [Google Scholar]
- Chase J. F.; Tubbs P. K. Biochem. J. 1969, 111, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J. W.; Northrop D. B. J. Antibiot. 1979, 32, 1147. [DOI] [PubMed] [Google Scholar]
- Cullis P. M.; Wolfenden R.; Cousens L. S.; Alberts B. M. J. Biol. Chem. 1982, 257, 12165. [PubMed] [Google Scholar]
- Erwin B. G.; Persson L.; Pegg A. E. Biochemistry 1984, 23, 4250. [DOI] [PubMed] [Google Scholar]
- Khalil E. M.; Cole P. A. J. Am. Chem. Soc. 1998, 120, 6195. [Google Scholar]
- Khalil E. M.; De Angelis J.; Ishii M.; Cole P. A. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C. M.; Cole P. A. J. Med. Chem. 2001, 44, 2479. [DOI] [PubMed] [Google Scholar]
- Yu M.; Magalhães M. L.; Cook P. F.; Blanchard J. S. Biochemistry 2006, 45, 14788. [DOI] [PubMed] [Google Scholar]
- Gao F.; Yan X.; Baettig O. M.; Berghuis A. M.; Auclair K. Angew. Chem., Int. Ed. 2005, 44, 6859. [DOI] [PubMed] [Google Scholar]
- Gao F.; Yan X.; Shakya T.; Baettig O. M.; Ait-Mohand-Brunet S.; Berghuis A. M.; Wright G. D.; Auclair K. J. Med. Chem. 2006, 49, 5273. [DOI] [PubMed] [Google Scholar]
- Gao F.; Yan X.; Zahr O.; Larsen A.; Vong K.; Auclair K. Bioorg. Med. Chem. Lett. 2008, 18, 5518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalhães M. L.; Vetting M. W.; Gao F.; Freiburger L.; Auclair K.; Blanchard J. S. Biochemistry 2008, 47, 579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnett B. P.; Hwang Y.; Taylor M. S.; Kirchner H.; Pfluger P. T.; Bernard V.; Lin Y. Y.; Bowers E. M.; Mukherjee C.; Song W. J.; Longo P. A.; Leahy D. J.; Hussain M. A.; Tschöp M. H.; Boeke J. D.; Cole P. A. Science 2010, 330, 1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor M. S.; Hwang Y.; Hsiao P. Y.; Boeke J. D.; Cole P. A. Methods Enzymol. 2012, 514, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parang K.; Till J. H.; Ablooglu A. J.; Kohanski R. A.; Hubbard S. R.; Cole P. A. Nat. Struct. Biol. 2001, 8, 37. [DOI] [PubMed] [Google Scholar]
- Shen K.; Hines A. C.; Schwarzer D.; Pickin K. A.; Cole P. A. Biochim. Biophys. Acta 2005, 1754, 65. [DOI] [PubMed] [Google Scholar]
- Lau O. D.; Kundu T. K.; Soccio R. E.; Ait-Si-Ali S.; Khalil E. M.; Vassilev A.; Wolffe A. P.; Nakatani Y.; Roeder R. G.; Cole P. A. Mol. Cell 2000, 5, 589. [DOI] [PubMed] [Google Scholar]
- Wu J.; Xie N.; Wu Z.; Zhang Y.; Zheng Y. G. Bioorg. Med. Chem. 2009, 17, 1381. [DOI] [PubMed] [Google Scholar]
- Zheng Y.; Thompson P. R.; Cebrat M.; Wang L.; Devlin M. K.; Alani R. M.; Cole P. A. Methods Enzymol. 2004, 376, 188. [DOI] [PubMed] [Google Scholar]
- De Angelis J.; Gastel J.; Klein D. C.; Cole P. A. J. Biol. Chem. 1998, 273, 3045. [DOI] [PubMed] [Google Scholar]
- Lau O. D.; Courtney A. D.; Vassilev A.; Marzilli L. A.; Cotter R. J.; Nakatani Y.; Cole P. A. J. Biol. Chem. 2000, 275, 21953. [DOI] [PubMed] [Google Scholar]
- Tanner K. G.; Langer M. R.; Denu J. M. Biochemistry 2000, 39, 15652. [DOI] [PubMed] [Google Scholar]
- Tanner K. G.; Langer M. R.; Kim Y.; Denu J. M. J. Biol. Chem. 2000, 275, 22048. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Barlev N. A.; Haley R. H.; Berger S. L.; Marmorstein R. Mol. Cell 2000, 6, 1195. [DOI] [PubMed] [Google Scholar]
- Wu J.; Xie N.; Wu Z.; Zhang Y.; Zheng Y. G. Bioorg. Med. Chem. 2009, 17, 1381. [DOI] [PubMed] [Google Scholar]
- Lau O. D.; Kundu T. K.; Soccio R. E.; Ait-Si-Ali S.; Khalil E. M.; Vassilev A.; Wolffe A. P.; Nakatani Y.; Roeder R. G.; Cole P. A. Mol. Cell 2000, 5, 589. [DOI] [PubMed] [Google Scholar]
- Lau O. D.; Courtney A. D.; Vassilev A.; Marzilli L. A.; Cotter R. J.; Nakatani Y.; Cole P. A. J. Biol. Chem. 2000, 275, 21953. [DOI] [PubMed] [Google Scholar]
- Poux A. N.; Cebrat M.; Kim C. M.; Cole P. A.; Marmorstein R. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 14065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.; Xie N.; Wu Z.; Zhang Y.; Zheng Y. G. Bioorg. Med. Chem. 2009, 17, 1381. [DOI] [PubMed] [Google Scholar]
- Victor M.; Bei Y.; Gay F.; Calvo D.; Mello C.; Shi Y. EMBO Rep. 2002, 3, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z. Q.; Li J.; Sachs L. M.; Cole P. A.; Wong J. EMBO J. 2003, 22, 2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo A.; Merlo P.; Pediconi N.; Fulco M.; Sartorelli V.; Cole P. A.; Fontemaggi G.; Fanciulli M.; Schiltz L.; Blandino G.; Balsano C.; Levrero M. Mol. Cell 2002, 9, 175. [DOI] [PubMed] [Google Scholar]
- Kaehlcke K.; Dorr A.; Hetzer-Egger C.; Kiermer V.; Henklein P.; Schnoelzer M.; Loret E.; Cole P. A.; Verdin E.; Ott M. Mol. Cell 2003, 12, 167. [DOI] [PubMed] [Google Scholar]
- Polesskaya A.; Naguibneva I.; Fritsch L.; Duquet A.; Ait-Si-Ali S.; Robin P.; Vervisch A.; Pritchard L. L.; Cole P.; Harel-Bellan A. EMBO J. 2001, 20, 6816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay D.; Okan N. A.; Bales E.; Nascimento L.; Cole P. A.; Medrano E. E. Cancer Res. 2002, 62, 6231. [PubMed] [Google Scholar]
- Cebrat M.; Kim C. M.; Thompson P. R.; Daugherty M.; Cole P. A. Bioorg. Med. Chem. 2003, 11, 3307. [DOI] [PubMed] [Google Scholar]
- Zheng Y.; Balasubramanyam K.; Cebrat M.; Buck D.; Guidez F.; Zelent A.; Alani R. M.; Cole P. A. J. Am. Chem. Soc. 2005, 127, 17182. [DOI] [PubMed] [Google Scholar]
- Guidez F.; Howell L.; Isalan M.; Cebrat M.; Alani R. M.; Ivins S.; Hormaeche I.; McConnell M. J.; Pierce S.; Cole P. A.; Licht J.; Zelent A. Mol. Cell. Biol. 2005, 25, 5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanyam K.; Swaminathan V.; Ranganathan A.; Kundu T. K. J. Biol. Chem. 2003, 278, 19134. [DOI] [PubMed] [Google Scholar]
- Rosen T.; Fordice D. B. South Med. J. 1994, 87, 543. [DOI] [PubMed] [Google Scholar]
- Balasubramanyam K.; Swaminathan V.; Ranganathan A.; Kundu T. K. J. Biol. Chem. 2003, 278, 19134. [DOI] [PubMed] [Google Scholar]
- Ghizzoni M.; Wu J.; Gao T.; Haisma H. J.; Dekker F. J.; George Zheng Y. Eur. J. Med. Chem. 2012, 47, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J.; Xie N.; Wu Z.; Zhang Y.; Zheng Y. G. Bioorg. Med. Chem. 2009, 17, 1381. [DOI] [PubMed] [Google Scholar]
- Chandregowda V.; Kush A.; Reddy G. C. Eur. J. Med. Chem. 2009, 44, 2711. [DOI] [PubMed] [Google Scholar]
- Balasubramanyam K.; Swaminathan V.; Ranganathan A.; Kundu T. K. J. Biol. Chem. 2003, 278, 19134. [DOI] [PubMed] [Google Scholar]
- Devipriya B.; Parameswari A. R.; Rajalakshmi G.; Palvannan T.; Kumaradhas P. Indian J. Biochem. Biophys. 2010, 47, 364. [PubMed] [Google Scholar]
- Mantelingu K.; Kishore A. H.; Balasubramanyam K.; Kumar G. V.; Altaf M.; Swamy S. N.; Selvi R.; Das C.; Narayana C.; Rangappa K. S.; Kundu T. K. J. Phys. Chem. B 2007, 111, 4527. [DOI] [PubMed] [Google Scholar]
- Varier R. A.; Swaminathan V.; Balasubramanyam K.; Kundu T. K. Biochem. Pharmacol. 2004, 68, 1215. [DOI] [PubMed] [Google Scholar]
- Balasubramanyam K.; Varier R. A.; Altaf M.; Swaminathan V.; Siddappa N. B.; Ranga U.; Kundu T. K. J. Biol. Chem. 2004, 279, 51163. [DOI] [PubMed] [Google Scholar]
- Goel A.; Kunnumakkara A. B.; Aggarwal B. B. Biochem. Pharmacol. 2008, 75, 787. [DOI] [PubMed] [Google Scholar]
- Maheshwari R. K.; Singh A. K.; Gaddipati J.; Srimal R. C. Life Sci. 2006, 78, 2081. [DOI] [PubMed] [Google Scholar]
- Aggarwal B. B.; Shishodia S. Biochem. Pharmacol. 2006, 71, 1397. [DOI] [PubMed] [Google Scholar]
- Zhou H.; Beevers C. S.; Huang S. Curr. Drug Targets 2011, 12, 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X.; Xu B.; Beevers C. S.; Odaka Y.; Chen L.; Liu L.; Luo Y.; Zhou H.; Chen W.; Shen T.; Huang S. Carcinogenesis 2012, 33, 868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S. C.; Prasad S.; Kim J. H.; Patchva S.; Webb L. J.; Priyadarsini I. K.; Aggarwal B. B. Nat. Prod. Rep. 2011, 28, 1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heery D. M.; Fischer P. M. Drug Discovery Today 2007, 12, 88. [DOI] [PubMed] [Google Scholar]
- Marcu M. G.; Jung Y. J.; Lee S.; Chung E. J.; Lee M. J.; Trepel J.; Neckers L. Med. Chem. 2006, 2, 169. [DOI] [PubMed] [Google Scholar]
- Balasubramanyam K.; Varier R. A.; Altaf M.; Swaminathan V.; Siddappa N. B.; Ranga U.; Kundu T. K. J. Biol. Chem. 2004, 279, 51163. [DOI] [PubMed] [Google Scholar]
- Anand P.; Kunnumakkara A. B.; Newman R. A.; Aggarwal B. B. Mol. Pharmaceutics 2007, 4, 807. [DOI] [PubMed] [Google Scholar]
- Sharma R. A.; Euden S. A.; Platton S. L.; Cooke D. N.; Shafayat A.; Hewitt H. R.; Marczylo T. H.; Morgan B.; Hemingway D.; Plummer S. M.; Pirmohamed M.; Gescher A. J.; Steward W. P. Clin. Cancer Res. 2004, 10, 6847. [DOI] [PubMed] [Google Scholar]
- Mishra S.; Kapoor N.; Mubarak Ali A.; Pardhasaradhi B. V.; Kumari A. L.; Khar A.; Misra K. Free Radical Biol. Med. 2005, 38, 1353. [DOI] [PubMed] [Google Scholar]
- Weber W. M.; Hunsaker L. A.; Abcouwer S. F.; Deck L. M.; Vander Jagt D. L. Bioorg. Med. Chem. 2005, 13, 3811. [DOI] [PubMed] [Google Scholar]
- Varier R. A.; Swaminathan V.; Balasubramanyam K.; Kundu T. K. Biochem. Pharmacol. 2004, 68, 1215. [DOI] [PubMed] [Google Scholar]
- Balasubramanyam K.; Altaf M.; Varier R. A.; Swaminathan V.; Ravindran A.; Sadhale P. P.; Kundu T. K. J. Biol. Chem. 2004, 279, 33716. [DOI] [PubMed] [Google Scholar]
- Balasubramanyam K.; Altaf M.; Varier R. A.; Swaminathan V.; Ravindran A.; Sadhale P. P.; Kundu T. K. J. Biol. Chem. 2004, 279, 33716. [DOI] [PubMed] [Google Scholar]
- Mantelingu K.; Reddy B. A.; Swaminathan V.; Kishore A. H.; Siddappa N. B.; Kumar G. V.; Nagashankar G.; Natesh N.; Roy S.; Sadhale P. P.; Ranga U.; Narayana C.; Kundu T. K. Chem. Biol. 2007, 14, 645. [DOI] [PubMed] [Google Scholar]
- Arif M.; Pradhan S. K.; Thanuja G. R.; Vedamurthy B. M.; Agrawal S.; Dasgupta D.; Kundu T. K. J. Med. Chem. 2009, 52, 267. [DOI] [PubMed] [Google Scholar]
- Baggett S.; Protiva P.; Mazzola E. P.; Yang H.; Ressler E. T.; Basile M. J.; Weinstein I. B.; Kennelly E. J. J. Nat. Prod. 2005, 68, 354. [DOI] [PubMed] [Google Scholar]
- Gartner M.; Müller T.; Simon J. C.; Giannis A.; Sleeman J. P. ChemBioChem 2005, 6, 171. [DOI] [PubMed] [Google Scholar]
- Dal Piaz F.; Tosco A.; Eletto D.; Piccinelli A. L.; Moltedo O.; Franceschelli S.; Sbardella G.; Remondelli P.; Rastrelli L.; Vesci L.; Pisano C.; De Tommasi N. ChemBioChem 2010, 11, 818. [DOI] [PubMed] [Google Scholar]
- Biel M.; Kretsovali A.; Karatzali E.; Papamatheakis J.; Giannis A. Angew. Chem., Int. Ed. 2004, 43, 3974. [DOI] [PubMed] [Google Scholar]
- Heery D. M.; Fischer P. M. Drug Discovery Today 2007, 12, 88. [DOI] [PubMed] [Google Scholar]
- Ravindra K. C.; Selvi B. R.; Arif M.; Reddy B. A.; Thanuja G. R.; Agrawal S.; Pradhan S. K.; Nagashayana N.; Dasgupta D.; Kundu T. K. J. Biol. Chem. 2009, 284, 24453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandur S. K.; Ichikawa H.; Sethi G.; Ahn K. S.; Aggarwal B. B. J. Biol. Chem. 2006, 281, 17023. [DOI] [PubMed] [Google Scholar]
- Padhye S.; Dandawate P.; Yusufi M.; Ahmad A.; Sarkar F. H. Med. Res. Rev. 2012, 32, 1131. [DOI] [PubMed] [Google Scholar]
- Ravindra K. C.; Selvi B. R.; Arif M.; Reddy B. A.; Thanuja G. R.; Agrawal S.; Pradhan S. K.; Nagashayana N.; Dasgupta D.; Kundu T. K. J. Biol. Chem. 2009, 284, 24453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K. C.; Jung M. G.; Lee Y. H.; Yoon J. C.; Kwon S. H.; Kang H. B.; Kim M. J.; Cha J. H.; Kim Y. J.; Jun W. J.; Lee J. M.; Yoon H. G. Cancer Res. 2009, 69, 583. [DOI] [PubMed] [Google Scholar]
- Neukam K.; Pastor N.; Cortés F. Mutat. Res. 2008, 654, 8. [DOI] [PubMed] [Google Scholar]
- Berger S. J.; Gupta S.; Belfi C. A.; Gosky D. M.; Mukhtar H. Biochem. Biophys. Res. Commun. 2001, 288, 101. [DOI] [PubMed] [Google Scholar]
- Suzuki K.; Yahara S.; Hashimoto F.; Uyeda M. Biol. Pharm. Bull. 2001, 24, 1088. [DOI] [PubMed] [Google Scholar]
- Bandele O. J.; Osheroff N. Chem. Res. Toxicol. 2008, 21, 936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golden E. B.; Lam P. Y.; Kardosh A.; Gaffney K. J.; Cadenas E.; Louie S. G.; Petasis N. A.; Chen T. C.; Schönthal A. H. Blood 2009, 113, 5927. [DOI] [PubMed] [Google Scholar]
- Ge J.; Tan B. X.; Chen Y.; Yang L.; Peng X. C.; Li H. Z.; Lin H. J.; Zhao Y.; Wei M.; Cheng K.; Li L. H.; Dong H.; Gao F.; He J. P.; Wu Y.; Qiu M.; Zhao Y. L.; Su J. M.; Hou J. M.; Liu J. Y. J. Mol. Med. 2011, 89, 595. [DOI] [PubMed] [Google Scholar]
- Strick R.; Strissel P. L.; Borgers S.; Smith S. L.; Rowley J. D. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 4790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz P. A.; Braune A.; Hölzlwimmer G.; Quintanilla-Fend L.; Haller D. J. Nutr. 2007, 137, 1208. [DOI] [PubMed] [Google Scholar]
- Chen J.; Han J.; Wang J. Toxicol. Ind. Health 2013, 29, 360. [DOI] [PubMed] [Google Scholar]
- Si D.; Wang Y.; Zhou Y. H.; Guo Y.; Wang J.; Zhou H.; Li Z. S.; Fawcett J. P. Drug Metab. Dispos. 2009, 37, 629. [DOI] [PubMed] [Google Scholar]
- Saaby L.; Rasmussen H. B.; Jäger A. K. J. Ethnopharmacol. 2009, 121, 178. [DOI] [PubMed] [Google Scholar]
- Hilliard J. J.; Krause H. M.; Bernstein J. I.; Fernandez J. A.; Nguyen V.; Ohemeng K. A.; Barrett J. F. Adv. Exp. Med. Biol. 1995, 390, 59. [DOI] [PubMed] [Google Scholar]
- Xiao X.; Shi D.; Liu L.; Wang J.; Xie X.; Kang T.; Deng W. PLoS One 2011, 6, e22934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien E.; Dietrich D. R. Crit. Rev. Toxicol. 2005, 35, 33. [DOI] [PubMed] [Google Scholar]
- Czakai K.; Müller K.; Mosesso P.; Pepe G.; Schulze M.; Gohla A.; Patnaik D.; Dekant W.; Higgins J. M.; Mally A. Toxicol. Sci. 2011, 122, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravindra K. C.; Narayan V.; Lushington G. H.; Peterson B. R.; Prabhu K. S. Chem. Res. Toxicol. 2012, 25, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reginato M. J.; Krakow S. L.; Bailey S. T.; Lazar M. A. J. Biol. Chem. 1998, 273, 1855. [DOI] [PubMed] [Google Scholar]
- Ravindra K. C.; Narayan V.; Lushington G. H.; Peterson B. R.; Prabhu K. S. Chem. Res. Toxicol. 2012, 25, 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stimson L.; Rowlands M. G.; Newbatt Y. M.; Smith N. F.; Raynaud F. I.; Rogers P.; Bavetsias V.; Gorsuch S.; Jarman M.; Bannister A.; Kouzarides T.; McDonald E.; Workman P.; Aherne G. W. Mol. Cancer Ther. 2005, 4, 1521. [DOI] [PubMed] [Google Scholar]
- Dekker F. J.; Ghizzoni M.; van der Meer N.; Wisastra R.; Haisma H. J. Bioorg. Med. Chem. 2009, 17, 460. [DOI] [PubMed] [Google Scholar]
- Gorsuch S.; Bavetsias V.; Rowlands M. G.; Aherne G. W.; Workman P.; Jarman M.; McDonald E. Bioorg. Med. Chem. 2009, 17, 467. [DOI] [PubMed] [Google Scholar]
- Gorsuch S.; Bavetsias V.; Rowlands M. G.; Aherne G. W.; Workman P.; Jarman M.; McDonald E. Bioorg. Med. Chem. 2009, 17, 467. [DOI] [PubMed] [Google Scholar]
- Wisastra R.; Ghizzoni M.; Maarsingh H.; Minnaard A. J.; Haisma H. J.; Dekker F. J. Org. Biomol. Chem. 2011, 9, 1817. [DOI] [PubMed] [Google Scholar]
- Dekker F. J.; Ghizzoni M.; van der Meer N.; Wisastra R.; Haisma H. J. Bioorg. Med. Chem. 2009, 17, 460. [DOI] [PubMed] [Google Scholar]
- Ornaghi P.; Rotili D.; Sbardella G.; Mai A.; Filetici P. Biochem. Pharmacol. 2005, 70, 911. [DOI] [PubMed] [Google Scholar]
- Smith A. T.; Livingston M. R.; Mai A.; Filetici P.; Queener S. F.; Sullivan W. J. Jr. Antimicrob. Agents Chemother. 2007, 51, 1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai A.; Rotili D.; Tarantino D.; Ornaghi P.; Tosi F.; Vicidomini C.; Sbardella G.; Nebbioso A.; Miceli M.; Altucci L.; Filetici P. J. Med. Chem. 2006, 49, 6897. [DOI] [PubMed] [Google Scholar]
- Mai A.; Rotili D.; Tarantino D.; Nebbioso A.; Castellano S.; Sbardella G.; Tini M.; Altucci L. Bioorg. Med. Chem. Lett. 2009, 19, 1132. [DOI] [PubMed] [Google Scholar]
- Rahim R.; Strobl J. S. Anticancer Drugs 2009, 20, 736. [DOI] [PubMed] [Google Scholar]
- Tahan F.; Jazrawi E.; Moodley T.; Rovati G. E.; Adcock I. M. Clin. Exp. Allergy 2008, 38, 805. [DOI] [PubMed] [Google Scholar]
- Chimenti F.; Bizzarri B.; Maccioni E.; Secci D.; Bolasco A.; Chimenti P.; Fioravanti R.; Granese A.; Carradori S.; Tosi F.; Ballario P.; Vernarecci S.; Filetici P. J. Med. Chem. 2009, 52, 530. [DOI] [PubMed] [Google Scholar]
- Wu J.; Wang J.; Li M.; Yang Y.; Wang B.; Zheng Y. G. Bioorg. Chem. 2011, 39, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitty A. Future Med. Chem. 2011, 3, 797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. J. Med. Chem. 2010, 53, 2719. [DOI] [PubMed] [Google Scholar]
- Mujtaba S.; Zeng L.; Zhou M. M. Oncogene 2007, 26, 5521. [DOI] [PubMed] [Google Scholar]
- Manning E. T.; Ikehara T.; Ito T.; Kadonaga J. T.; Kraus W. L. Mol. Cell. Biol. 2001, 21, 3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L.; Jamonnak N.; Choy J.; Wang Z.; Zheng W. Biochem. Biophys. Res. Commun. 2008, 368, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polesskaya A.; Naguibneva I.; Duquet A.; Bengal E.; Robin P.; Harel-Bellan A. Mol. Cell. Biol. 2001, 21, 5312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou T.; Ray S.; Lee C.; Brasier A. R. J. Biol. Chem. 2008, 283, 30725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mujtaba S.; He Y.; Zeng L.; Yan S.; Plotnikova O.; Sachchidanand; Sanchez R.; Zeleznik-Le N. J.; Ronai Z.; Zhou M. M. Mol. Cell 2004, 13, 251. [DOI] [PubMed] [Google Scholar]
- Kim J. H.; Cho E. J.; Kim S. T.; Youn H. D. Nat. Struct. Mol. Biol. 2005, 12, 423. [DOI] [PubMed] [Google Scholar]
- Reynoird N.; Schwartz B. E.; Delvecchio M.; Sadoul K.; Meyers D.; Mukherjee C.; Caron C.; Kimura H.; Rousseaux S.; Cole P. A.; Panne D.; French C. A.; Khochbin S. EMBO J. 2010, 29, 2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan H. M.; La Thangue N. B. J. Cell Sci. 2001, 114, 2363. [DOI] [PubMed] [Google Scholar]
- Wang F.; Marshall C. B.; Ikura M. Cell. Mol. Life Sci. 2013, 70, 3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragvin A.; Valvatne H.; Erdal S.; Arskog V.; Tufteland K. R.; Breen K.; ØYan A. M.; Eberharter A.; Gibson T. J.; Becker P. B.; Aasland R. J. Mol. Biol. 2004, 337, 773. [DOI] [PubMed] [Google Scholar]
- He J.; Ye J.; Cai Y.; Riquelme C.; Liu J. O.; Liu X.; Han A.; Chen L. Nucleic Acids Res. 2011, 39, 4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L.; Zhang Q.; Gerona-Navarro G.; Moshkina N.; Zhou M. M. Structure 2008, 16, 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mujtaba S.; He Y.; Zeng L.; Yan S.; Plotnikova O.; Sachchidanand; Sanchez R.; Zeleznik-Le N. J.; Ronai Z.; Zhou M. M. Mol. Cell 2004, 13, 251. [DOI] [PubMed] [Google Scholar]
- Ferreon J. C.; Martinez-Yamout M. A.; Dyson H. J.; Wright P. E. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H.; Jenkins L. M.; Durell S. R.; Hayashi R.; Mazur S. J.; Cherry S.; Tropea J. E.; Miller M.; Wlodawer A.; Appella E.; Bai Y. Structure 2009, 17, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojciak J. M.; Martinez-Yamout M. A.; Dyson H. J.; Wright P. E. EMBO J. 2009, 28, 948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das C.; Roy S.; Namjoshi S.; Malarkey C. S.; Jones D. N.; Kutateladze T. G.; Churchill M. E.; Tyler J. K. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, E1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delvecchio M.; Gaucher J.; Aguilar-Gurrieri C.; Ortega E.; Panne D. Nat. Struct. Mol. Biol. 2013, 20, 1040. [DOI] [PubMed] [Google Scholar]
- He J.; Ye J.; Cai Y.; Riquelme C.; Liu J. O.; Liu X.; Han A.; Chen L. Nucleic Acids Res. 2011, 39, 4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H.; Jenkins L. M.; Durell S. R.; Hayashi R.; Mazur S. J.; Cherry S.; Tropea J. E.; Miller M.; Wlodawer A.; Appella E.; Bai Y. Structure 2009, 17, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreon J. C.; Martinez-Yamout M. A.; Dyson H. J.; Wright P. E. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 13260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojciak J. M.; Martinez-Yamout M. A.; Dyson H. J.; Wright P. E. EMBO J. 2009, 28, 948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan H. M.; La Thangue N. B. J. Cell Sci. 2001, 114, 2363. [DOI] [PubMed] [Google Scholar]
- Simone C.; Stiegler P.; Forcales S. V.; Bagella L.; De Luca A.; Sartorelli V.; Giordano A.; Puri P. L. Oncogene 2004, 23, 2177. [DOI] [PubMed] [Google Scholar]
- Saint Just Ribeiro M.; Hansson M. L.; Wallberg A. E. Biochem. J. 2007, 404, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tax F. E.; Thomas J. H.; Ferguson E. L.; Horvitz H. R. Genetics 1997, 147, 1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petcherski A. G.; Kimble J. Nature 2000, 405, 364. [DOI] [PubMed] [Google Scholar]
- Petcherski A. G.; Kimble J. Curr. Biol. 2000, 10, R471. [DOI] [PubMed] [Google Scholar]
- Kitagawa M.; Oyama T.; Kawashima T.; Yedvobnick B.; Kumar A.; Matsuno K.; Harigaya K. Mol. Cell. Biol. 2001, 21, 4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L.; Aster J. C.; Blacklow S. C.; Lake R.; Artavanis-Tsakonas S.; Griffin J. D. Nat. Genet. 2000, 26, 484. [DOI] [PubMed] [Google Scholar]
- Wilson J. J.; Kovall R. A. Cell 2006, 124, 985. [DOI] [PubMed] [Google Scholar]
- Arnett K. L.; Hass M.; McArthur D. G.; Ilagan M. X.; Aster J. C.; Kopan R.; Blacklow S. C. Nat. Struct. Mol. Biol. 2010, 17, 1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam Y.; Sliz P.; Song L.; Aster J. C.; Blacklow S. C. Cell 2006, 124, 973. [DOI] [PubMed] [Google Scholar]
- Choi S. H.; Wales T. E.; Nam Y.; O’Donovan D. J.; Sliz P.; Engen J. R.; Blacklow S. C. Structure 2012, 20, 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson M. L.; Popko-Scibor A. E.; Saint Just Ribeiro M.; Dancy B. M.; Lindberg M. J.; Cole P. A.; Wallberg A. E. Nucleic Acids Res. 2009, 37, 2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer C. J.; Lamar E.; Turbachova I.; Kintner C.; Jones K. A. Genes Dev. 2002, 16, 1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint Just Ribeiro M.; Hansson M. L.; Wallberg A. E. Biochem. J. 2007, 404, 289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black J. C.; Choi J. E.; Lombardo S. R.; Carey M. Mol. Cell 2006, 23, 809. [DOI] [PubMed] [Google Scholar]
- Kim J. H.; Cho E. J.; Kim S. T.; Youn H. D. Nat. Struct. Mol. Biol. 2005, 12, 423. [DOI] [PubMed] [Google Scholar]
- Kraus W. L.; Manning E. T.; Kadonaga J. T. Mol. Cell. Biol. 1999, 19, 8123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delvecchio M.; Gaucher J.; Aguilar-Gurrieri C.; Ortega E.; Panne D. Nat. Struct. Mol. Biol. 2013, 20, 1040. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Knapp S. Nat. Rev. Drug Discovery 2014, 13, 337. [DOI] [PubMed] [Google Scholar]
- Nicodeme E.; Jeffrey K. L.; Schaefer U.; Beinke S.; Dewell S.; Chung C. W.; Chandwani R.; Marazzi I.; Wilson P.; Coste H.; White J.; Kirilovsky J.; Rice C. M.; Lora J. M.; Prinjha R. K.; Lee K.; Tarakhovsky A. Nature 2010, 468, 1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirguet O.; Lamotte Y.; Donche F.; Toum J.; Gellibert F.; Bouillot A.; Gosmini R.; Nguyen V. L.; Delannée D.; Seal J.; Blandel F.; Boullay A. B.; Boursier E.; Martin S.; Brusq J. M.; Krysa G.; Riou A.; Tellier R.; Costaz A.; Huet P.; Dudit Y.; Trottet L.; Kirilovsky J.; Nicodeme E. Bioorg. Med. Chem. Lett. 2012, 22, 2963. [DOI] [PubMed] [Google Scholar]
- Dawson M. A.; Prinjha R. K.; Dittmann A.; Giotopoulos G.; Bantscheff M.; Chan W. I.; Robson S. C.; Chung C. W.; Hopf C.; Savitski M. M.; Huthmacher C.; Gudgin E.; Lugo D.; Beinke S.; Chapman T. D.; Roberts E. J.; Soden P. E.; Auger K. R.; Mirguet O.; Doehner K.; Delwel R.; Burnett A. K.; Jeffrey P.; Drewes G.; Lee K.; Huntly B. J.; Kouzarides T. Nature 2011, 478, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Nature 2010, 468, 1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fish P. V.; Filippakopoulos P.; Bish G.; Brennan P. E.; Bunnage M. E.; Cook A. S.; Federov O.; Gerstenberger B. S.; Jones H.; Knapp S.; Marsden B.; Nocka K.; Owen D. R.; Philpott M.; Picaud S.; Primiano M. J.; Ralph M. J.; Sciammetta N.; Trzupek J. D. J. Med. Chem. 2012, 55, 9831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picaud S.; Da Costa D.; Thanasopoulou A.; Filippakopoulos P.; Fish P. V.; Philpott M.; Fedorov O.; Brennan P.; Bunnage M. E.; Owen D. R.; Bradner J. E.; Taniere P.; O’Sullivan B.; Müller S.; Schwaller J.; Stankovic T.; Knapp S. Cancer Res. 2013, 73, 3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewings D. S.; Wang M.; Philpott M.; Fedorov O.; Uttarkar S.; Filippakopoulos P.; Picaud S.; Vuppusetty C.; Marsden B.; Knapp S.; Conway S. J.; Heightman T. D. J. Med. Chem. 2011, 54, 6761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay D.; Fedorov O.; Filippakopoulos P.; Martin S.; Philpott M.; Picaud S.; Hewings D. S.; Uttakar S.; Heightman T. D.; Conway S. J.; Knapp S.; Brennan P. E. Med. Chem. Commun. 2013, 4, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamborough P.; Diallo H.; Goodacre J. D.; Gordon L.; Lewis A.; Seal J. T.; Wilson D. M.; Woodrow M. D.; Chung C. W. J. Med. Chem. 2012, 55, 587. [DOI] [PubMed] [Google Scholar]
- Boehm D.; Calvanese V.; Dar R. D.; Xing S.; Schroeder S.; Martins L.; Aull K.; Li P. C.; Planelles V.; Bradner J. E.; Zhou M. M.; Siliciano R. F.; Weinberger L.; Verdin E.; Ott M. Cell Cycle 2013, 12, 452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z.; Guo J.; Wu Y.; Zhou Q. Nucleic Acids Res. 2013, 41, 277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J.; Gaiha G. D.; John S. P.; Pertel T.; Chin C. R.; Gao G.; Qu H.; Walker B. D.; Elledge S. J.; Brass A. L. Cell Rep. 2012, 2, 807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee C.; Archin N.; Michaels D.; Belkina A. C.; Denis G. V.; Bradner J.; Sebastiani P.; Margolis D. M.; Montano M. J. Leukocyte Biol. 2012, 92, 1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Li J.; Schowalter R. M.; Jiao J.; Buck C. B.; You J. PLoS Pathog. 2012, 8, e1003021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goupille O.; Penglong T.; Lefèvre C.; Granger M.; Kadri Z.; Fucharoen S.; Maouche-Chrétien L.; Leboulch P.; Chrétien S. Biochem. Biophys. Res. Commun. 2012, 429, 1. [DOI] [PubMed] [Google Scholar]
- Ott C. J.; Kopp N.; Bird L.; Paranal R. M.; Qi J.; Bowman T.; Rodig S. J.; Kung A. L.; Bradner J. E.; Weinstock D. M. Blood 2012, 120, 2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picaud S.; Da Costa D.; Thanasopoulou A.; Filippakopoulos P.; Fish P. V.; Philpott M.; Fedorov O.; Brennan P.; Bunnage M. E.; Owen D. R.; Bradner J. E.; Taniere P.; O’Sullivan B.; Müller S.; Schwaller J.; Stankovic T.; Knapp S. Cancer Res. 2013, 73, 3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson M. A.; Prinjha R. K.; Dittmann A.; Giotopoulos G.; Bantscheff M.; Chan W. I.; Robson S. C.; Chung C. W.; Hopf C.; Savitski M. M.; Huthmacher C.; Gudgin E.; Lugo D.; Beinke S.; Chapman T. D.; Roberts E. J.; Soden P. E.; Auger K. R.; Mirguet O.; Doehner K.; Delwel R.; Burnett A. K.; Jeffrey P.; Drewes G.; Lee K.; Huntly B. J.; Kouzarides T. Nature 2011, 478, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J.; Shi J.; Wang E.; Rappaport A. R.; Herrmann H.; Sison E. A.; Magoon D.; Qi J.; Blatt K.; Wunderlich M.; Taylor M. J.; Johns C.; Chicas A.; Mulloy J. C.; Kogan S. C.; Brown P.; Valent P.; Bradner J. E.; Lowe S. W.; Vakoc C. R. Nature 2011, 478, 524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertz J. A.; Conery A. R.; Bryant B. M.; Sandy P.; Balasubramanian S.; Mele D. A.; Bergeron L.; Sims R. J. III. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 16669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore J. E.; Issa G. C.; Lemieux M. E.; Rahl P. B.; Shi J.; Jacobs H. M.; Kastritis E.; Gilpatrick T.; Paranal R. M.; Qi J.; Chesi M.; Schinzel A. C.; McKeown M. R.; Heffernan T. P.; Vakoc C. R.; Bergsagel P. L.; Ghobrial I. M.; Richardson P. G.; Young R. A.; Hahn W. C.; Anderson K. C.; Kung A. L.; Bradner J. E.; Mitsiades C. S. Cell 2011, 146, 904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockwood W. W.; Zejnullahu K.; Bradner J. E.; Varmus H. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 19408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandukwala H. S.; Gagnon J.; Togher S.; Greenbaum J. A.; Lamperti E. D.; Parr N. J.; Molesworth A. M.; Smithers N.; Lee K.; Witherington J.; Tough D. F.; Prinjha R. K.; Peters B.; Rao A. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirguet O.; Lamotte Y.; Donche F.; Toum J.; Gellibert F.; Bouillot A.; Gosmini R.; Nguyen V. L.; Delannée D.; Seal J.; Blandel F.; Boullay A. B.; Boursier E.; Martin S.; Brusq J. M.; Krysa G.; Riou A.; Tellier R.; Costaz A.; Huet P.; Dudit Y.; Trottet L.; Kirilovsky J.; Nicodeme E. Bioorg. Med. Chem. Lett. 2012, 22, 2963. [DOI] [PubMed] [Google Scholar]
- Bamborough P.; Diallo H.; Goodacre J. D.; Gordon L.; Lewis A.; Seal J. T.; Wilson D. M.; Woodrow M. D.; Chung C. W. J. Med. Chem. 2012, 55, 587. [DOI] [PubMed] [Google Scholar]
- Zhang G.; Liu R.; Zhong Y.; Plotnikov A. N.; Zhang W.; Zeng L.; Rusinova E.; Gerona-Nevarro G.; Moshkina N.; Joshua J.; Chuang P. Y.; Ohlmeyer M.; He J. C.; Zhou M. M. J. Biol. Chem. 2012, 287, 28840–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzuk M. M.; McKeown M. R.; Filippakopoulos P.; Li Q.; Ma L.; Agno J. E.; Lemieux M. E.; Picaud S.; Yu R. N.; Qi J.; Knapp S.; Bradner J. E. Cell 2012, 150, 673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson F. M.; Fedorov O.; Chaikuad A.; Philpott M.; Muniz J. R.; Felletar I.; von Delft F.; Heightman T.; Knapp S.; Abell C.; Ciulli A. J. Med. Chem. 2013, 56, 10183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Prakash C.; Sum C.; Gong Y.; Li Y.; Kwok J. J.; Thiessen N.; Pettersson S.; Jones S. J.; Knapp S.; Yang H.; Chin K. C. J. Biol. Chem. 2012, 287, 43137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholomeeusen K.; Xiang Y.; Fujinaga K.; Peterlin B. M. J. Biol. Chem. 2012, 287, 36609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaiah B. N.; Lewis B. A.; Cherman N.; Hewitt M. C.; Albrecht B. K.; Robey P. G.; Ozato K.; Sims R. J. III; Singer D. S. Proc. Natl. Acad. Sci. U.S.A. 2012, 109, 6927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palermo R. D.; Webb H. M.; West M. J. PLoS Pathog. 2011, 7, e1002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore J. E.; Issa G. C.; Lemieux M. E.; Rahl P. B.; Shi J.; Jacobs H. M.; Kastritis E.; Gilpatrick T.; Paranal R. M.; Qi J.; Chesi M.; Schinzel A. C.; McKeown M. R.; Heffernan T. P.; Vakoc C. R.; Bergsagel P. L.; Ghobrial I. M.; Richardson P. G.; Young R. A.; Hahn W. C.; Anderson K. C.; Kung A. L.; Bradner J. E.; Mitsiades C. S. Cell 2011, 146, 904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachchidanand; Resnick-Silverman L.; Yan S.; Mutjaba S.; Liu W. J.; Zeng L.; Manfredi J. J.; Zhou M. M. Chem. Biol. 2006, 13, 81. [DOI] [PubMed] [Google Scholar]
- Borah J. C.; Mujtaba S.; Karakikes I.; Zeng L.; Muller M.; Patel J.; Moshkina N.; Morohashi K.; Zhang W.; Gerona-Navarro G.; Hajjar R. J.; Zhou M. M. Chem. Biol. 2011, 18, 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerona-Navarro G.; Yoel-Rodríguez; Mujtaba S.; Frasca A.; Patel J.; Zeng L.; Plotnikov A. N.; Osman R.; Zhou M. M. J. Am. Chem. Soc. 2011, 133, 2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney T. P.; Filippakopoulos P.; Fedorov O.; Picaud S.; Cortopassi W. A.; Hay D. A.; Martin S.; Tumber A.; Rogers C. M.; Philpott M.; Wang M.; Thompson A. L.; Heightman T. D.; Pryde D. C.; Cook A.; Paton R. S.; Müller S.; Knapp S.; Brennan P. E.; Conway S. J. Angew. Chem., Int. Ed. 2014, 53, 6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov O.; Lingard H.; Wells C.; Monteiro O. P.; Picaud S.; Keates T.; Yapp C.; Philpott M.; Martin S. J.; Felletar I.; Marsden B. D.; Filippakopoulos P.; Müller S.; Knapp S.; Brennan P. E. J. Med. Chem. 2014, 57, 462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C. W.; Dean A. W.; Woolven J. M.; Bamborough P. J. Med. Chem. 2012, 55, 576. [DOI] [PubMed] [Google Scholar]
- Hewings D. S.; Wang M.; Philpott M.; Fedorov O.; Uttarkar S.; Filippakopoulos P.; Picaud S.; Vuppusetty C.; Marsden B.; Knapp S.; Conway S. J.; Heightman T. D. J. Med. Chem. 2011, 54, 6761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay D. A.; Fedorov O.; Martin S.; Singleton D. C.; Tallant C.; Wells C.; Picaud S.; Philpott M.; Monteiro O. P.; Rogers C. M.; Conway S. J.; Rooney T. P.; Tumber A.; Yapp C.; Filippakopoulos P.; Bunnage M. E.; Müller S.; Knapp S.; Schofield C. J.; Brennan P. E. J. Am. Chem. Soc. 2014, 136, 9308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey A.; Wong E. T.; Cheok C. F.; Tergaonkar V.; Lane D. P. Cell Death Differ. 2008, 15, 263. [DOI] [PubMed] [Google Scholar]
- Li B. X.; Xiao X. ChemBioChem 2009, 10, 2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best J. L.; Amezcua C. A.; Mayr B.; Flechner L.; Murawsky C. M.; Emerson B.; Zor T.; Gardner K. H.; Montminy M. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 17622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minter A. R.; Brennan B. B.; Mapp A. K. J. Am. Chem. Soc. 2004, 126, 10504. [DOI] [PubMed] [Google Scholar]
- Buhrlage S. J.; Bates C. A.; Rowe S. P.; Minter A. R.; Brennan B. B.; Majmudar C. Y.; Wemmer D. E.; Al-Hashimi H.; Mapp A. K. ACS Chem. Biol. 2009, 4, 335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates C. A.; Pomerantz W. C.; Mapp A. K. Biopolymers 2011, 95, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe S. P.; Casey R. J.; Brennan B. B.; Buhrlage S. J.; Mapp A. K. J. Am. Chem. Soc. 2007, 129, 10654. [DOI] [PubMed] [Google Scholar]
- Henderson W. R. Jr.; Chi E. Y.; Ye X.; Nguyen C.; Tien Y. T.; Zhou B.; Borok Z.; Knight D. A.; Kahn M. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 14309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emami K. H.; Nguyen C.; Ma H.; Kim D. H.; Jeong K. W.; Eguchi M.; Moon R. T.; Teo J. L.; Kim H. Y.; Moon S. H.; Ha J. R.; Kahn M. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 12682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T.; Hwang H.; Nguyen C.; Kloner R. A.; Kahn M. PLoS One 2013, 8, e75010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S.; He W.; Li Y.; Ding H.; Hou Y.; Nie J.; Hou F. F.; Kahn M.; Liu Y. J. Am. Soc. Nephrol. 2011, 22, 1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majmudar C. Y.; Højfeldt J. W.; Arevang C. J.; Pomerantz W. C.; Gagnon J. K.; Schultz P. J.; Cesa L. C.; Doss C. H.; Rowe S. P.; Vásquez V.; Tamayo-Castillo G.; Cierpicki T.; Brooks C. L. III; Sherman D. H.; Mapp A. K. Angew. Chem., Int. Ed. 2012, 51, 11258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung A. L.; Zabludoff S. D.; France D. S.; Freedman S. J.; Tanner E. A.; Vieira A.; Cornell-Kennon S.; Lee J.; Wang B.; Wang J.; Memmert K.; Naegeli H. U.; Petersen F.; Eck M. J.; Bair K. W.; Wood A. W.; Livingston D. M. Cancer Cell 2004, 6, 33. [DOI] [PubMed] [Google Scholar]
- Block K. M.; Wang H.; Szabó L. Z.; Polaske N. W.; Henchey L. K.; Dubey R.; Kushal S.; László C. F.; Makhoul J.; Song Z.; Meuillet E. J.; Olenyuk B. Z. J. Am. Chem. Soc. 2009, 131, 18078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin S.; Kaluz S.; Devi N. S.; Jabbar A. A.; de Noronha R. G.; Mun J.; Zhang Z.; Boreddy P. R.; Wang W.; Wang Z.; Abbruscato T.; Chen Z.; Olson J. J.; Zhang R.; Goodman M. M.; Nicolaou K. C.; Van Meir E. G. Clin. Cancer Res. 2012, 18, 6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y.; Naruse I.; Maekawa T.; Masuya H.; Shiroishi T.; Ishii S. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 10215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao T. P.; Oh S. P.; Fuchs M.; Zhou N. D.; Ch’ng L. E.; Newsome D.; Bronson R. T.; Li E.; Livingston D. M.; Eckner R. Cell 1998, 93, 361. [DOI] [PubMed] [Google Scholar]
- Roelfsema J. H.; Peters D. J. Expert Rev. Mol. Med. 2007, 9, 1. [DOI] [PubMed] [Google Scholar]
- Qaksen H.; Bartsch O.; Okur M.; Temel H.; Açikgoz M.; Yilmaz C. Genet. Couns. 2009, 20, 255. [PubMed] [Google Scholar]
- Viosca J.; Lopez-Atalaya J. P.; Olivares R.; Eckner R.; Barco A. Neurobiol. Dis. 2010, 37, 186. [DOI] [PubMed] [Google Scholar]
- Roelfsema J. H.; Peters D. J. Expert Rev. Mol. Med. 2007, 9, 1. [DOI] [PubMed] [Google Scholar]
- Gervasini C.; Mottadelli F.; Ciccone R.; Castronovo P.; Milani D.; Scarano G.; Bedeschi M. F.; Belli S.; Pilotta A.; Selicorni A.; Zuffardi O.; Larizza L. Eur. J. Hum. Genet. 2010, 18, 768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley P.; Bunyan D.; Stratton J.; Dillon M.; Lynch S. A. Am. J. Med. Genet., Part A 2009, 149A, 997. [DOI] [PubMed] [Google Scholar]
- Bartholdi D.; Roelfsema J. H.; Papadia F.; Breuning M. H.; Niedrist D.; Hennekam R. C.; Schinzel A.; Peters D. J. J. Med. Genet. 2007, 44, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai A. C.; Dossett C. J.; Walton C. S.; Cramer A. E.; Eng P. A.; Nowakowska B. A.; Pursley A. N.; Stankiewicz P.; Wiszniewska J.; Cheung S. W. Eur. J. Hum. Genet. 2011, 19, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann N.; Acosta A. M.; Kohlhase J.; Bartsch O. Eur. J. Hum. Genet. 2007, 15, 837. [DOI] [PubMed] [Google Scholar]
- Bartholdi D.; Roelfsema J. H.; Papadia F.; Breuning M. H.; Niedrist D.; Hennekam R. C.; Schinzel A.; Peters D. J. J. Med. Genet. 2007, 44, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Atalaya J. P.; Gervasini C.; Mottadelli F.; Spena S.; Piccione M.; Scarano G.; Selicorni A.; Barco A.; Larizza L. J. Med. Genet. 2012, 49, 66. [DOI] [PubMed] [Google Scholar]
- Miller R. W.; Rubinstein J. H. Am. J. Med. Genet. 1995, 56, 112. [DOI] [PubMed] [Google Scholar]
- Iyer N. G.; Ozdag H.; Caldas C. Oncogene 2004, 23, 4225. [DOI] [PubMed] [Google Scholar]
- Tillinghast G. W.; Partee J.; Albert P.; Kelley J. M.; Burtow K. H.; Kelly K. Genes, Chromosomes Cancer 2003, 37, 121. [DOI] [PubMed] [Google Scholar]
- Bryan E. J.; Jokubaitis V. J.; Chamberlain N. L.; Baxter S. W.; Dawson E.; Choong D. Y.; Campbell I. G. Int. J. Cancer 2002, 102, 137. [DOI] [PubMed] [Google Scholar]
- Muraoka M.; Konishi M.; Kikuchi-Yanoshita R.; Tanaka K.; Shitara N.; Chong J. M.; Iwama T.; Miyaki M. Oncogene 1996, 12, 1565. [PubMed] [Google Scholar]
- Iyer N. G.; Ozdag H.; Caldas C. Oncogene 2004, 23, 4225. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L.; Dominguez-Sola D.; Chiarenza A.; Fabbri G.; Grunn A.; Trifonov V.; Kasper L. H.; Lerach S.; Tang H.; Ma J.; Rossi D.; Chadburn A.; Murty V. V.; Mullighan C. G.; Gaidano G.; Rabadan R. Nature 2011, 471, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto M.; Kohno T.; Okudela K.; Otsuka A.; Sasaki H.; Tanabe C.; Sakiyama T.; Hirama C.; Kitabayashi I.; Minna J. D.; Takenoshita S.; Yokota J. Clin. Cancer Res. 2005, 11, 512. [PubMed] [Google Scholar]
- Suganuma T.; Kawabata M.; Ohshima T.; Ikeda M. A. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 13073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullighan C. G.; Zhang J.; Kasper L. H.; Lerach S.; Payne-Turner D.; Phillips L. A.; Heatley S. L.; Holmfeldt L.; Collins-Underwood J. R.; Ma J.; Buetow K. H.; Pui C. H.; Baker S. D. Nature 2011, 471, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Molina S.; Oliva J. L.; García-Vargas S.; Valls E.; Rojas J. M.; Martínez-Balbás M. A. Biochem. J. 2006, 398, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer N. G.; Ozdag H.; Caldas C. Oncogene 2004, 23, 4225. [DOI] [PubMed] [Google Scholar]
- Fan S.; Ma Y. X.; Wang C.; Yuan R. Q.; Meng Q.; Wang J. A.; Erdos M.; Goldberg I. D.; Webb P.; Kushner P. J.; Pestell R. G.; Rosen E. M. Cancer Res. 2002, 62, 141. [PubMed] [Google Scholar]
- Liang J.; Prouty L.; Williams B. J.; Dayton M. A.; Blanchard K. L. Blood 1998, 92, 2118. [PubMed] [Google Scholar]
- Carapeti M.; Aguiar R. C.; Goldman J. M.; Cross N. C. Blood 1998, 91, 3127. [PubMed] [Google Scholar]
- Kindle K. B.; Troke P. J.; Collins H. M.; Matsuda S.; Bossi D.; Bellodi C.; Kalkhoven E.; Salomoni P.; Pelicci P. G.; Minucci S.; Heery D. M. Mol. Cell. Biol. 2005, 25, 988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y.; Miyazono K. Curr. Opin. Genet. Dev. 2003, 13, 43. [DOI] [PubMed] [Google Scholar]
- Grossman S. R. Eur. J. Biochem. 2001, 268, 2773. [DOI] [PubMed] [Google Scholar]
- Iyer N. G.; Xian J.; Chin S. F.; Bannister A. J.; Daigo Y.; Aparicio S.; Kouzarides T.; Caldas C. Oncogene 2007, 26, 21. [DOI] [PubMed] [Google Scholar]
- Galbiati L.; Mendoza-Maldonado R.; Gutierrez M. I.; Giacca M. Cell Cycle 2005, 4, 930. [DOI] [PubMed] [Google Scholar]
- Oike Y.; Hata A.; Mamiya T.; Kaname T.; Noda Y.; Suzuki M.; Yasue H.; Nabeshima T.; Araki K.; Yamamura K. Hum. Mol. Genet. 1999, 8, 387. [DOI] [PubMed] [Google Scholar]
- Viosca J.; Lopez-Atalaya J. P.; Olivares R.; Eckner R.; Barco A. Neurobiol. Dis. 2010, 37, 186. [DOI] [PubMed] [Google Scholar]
- Kung A. L.; Rebel V. I.; Bronson R. T.; Ch’ng L. E.; Sieff C. A.; Livingston D. M.; Yao T. P. Genes Dev. 2000, 14, 272. [PMC free article] [PubMed] [Google Scholar]
- Kasper L. H.; Fukuyama T.; Biesen M. A.; Boussouar F.; Tong C.; de Pauw A.; Murray P. J.; van Deursen J. M.; Brindle P. K. Mol. Cell. Biol. 2006, 26, 789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebel V. I.; Kung A. L.; Tanner E. A.; Yang H.; Bronson R. T.; Livingston D. M. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 14789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper L. H.; Fukuyama T.; Biesen M. A.; Boussouar F.; Tong C.; de Pauw A.; Murray P. J.; van Deursen J. M.; Brindle P. K. Mol. Cell. Biol. 2006, 26, 789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Y.; Molina H.; Pandey A.; Zhang J.; Cole P. A. Science 2006, 311, 1293. [DOI] [PubMed] [Google Scholar]
- Liao Z. W.; Zhou T. C.; Tan X. J.; Song X. L.; Liu Y.; Shi X. Y.; Huang W. J.; Du L. L.; Tu B. J.; Lin X. D. J. Transl. Med. 2012, 10, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M.; Luo R. Z.; Chen J. W.; Cao Y.; Lu J. B.; He J. H.; Wu Q. L.; Cai M. Y. J. Transl. Med. 2011, 9, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vleugel M. M.; Shvarts D.; van der Wall E.; van Diest P. J. Hum. Pathol. 2006, 37, 1085. [DOI] [PubMed] [Google Scholar]
- Hudelist G.; Czerwenka K.; Kubista E.; Marton E.; Pischinger K.; Singer C. F. Breast Cancer Res. Treat. 2003, 78, 193. [DOI] [PubMed] [Google Scholar]
- Debes J. D.; Sebo T. J.; Lohse C. M.; Murphy L. M.; Haugen D. A.; Tindall D. J. Cancer Res. 2003, 63, 7638. [PubMed] [Google Scholar]
- Isharwal S.; Miller M. C.; Marlow C.; Makarov D. V.; Partin A. W.; Veltri R. W. Prostate 2008, 68, 1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heery D. M.; Fischer P. M. Drug Discovery Today 2007, 12, 88. [DOI] [PubMed] [Google Scholar]
- Lin W. M.; Baker A. C.; Beroukhim R.; Winckler W.; Feng W.; Marmion J. M.; Laine E.; Greulich H.; Tseng H.; Gates C.; Hodi F. S.; Dranoff G.; Sellers W. R.; Thomas R. K.; Meyerson M.; Golub T. R.; Dummer R.; Herlyn M.; Getz G.; Garraway L. A. Cancer Res. 2008, 68, 664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotte A.; Bhandaru M.; Cheng Y.; Sjoestroem C.; Martinka M.; Li G. PLoS One 2013, 8, e75405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandaru M.; Ardekani G. S.; Zhang G.; Martinka M.; McElwee K. J.; Li G.; Rotte A. BMC Cancer 2014, 14–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay D.; Okan N. A.; Bales E.; Nascimento L.; Cole P. A.; Medrano E. E. Cancer Res. 2002, 62, 6231. [PubMed] [Google Scholar]
- Yan G.; Eller M. S.; Elm C.; Larocca C. A.; Ryu B.; Panova I. P.; Dancy B. M.; Bowers E. M.; Meyers D.; Lareau L.; Cole P. A.; Taverna S. D.; Alani R. M. J. Invest. Dermatol. 2013, 133, 2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer N. G.; Chin S. F.; Ozdag H.; Daigo Y.; Hu D. E.; Cariati M.; Brindle K.; Aparicio S.; Caldas C. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 7386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles R. H.; Petrij F.; Dauwerse H. G.; den Hollander A. I.; Lushnikova T.; van Ommen G. J.; Goodman R. H.; Deaven L. L.; Doggett N. A.; Peters D. J.; Breuning M. H. Genomics 1997, 42, 96. [DOI] [PubMed] [Google Scholar]
- Rozman M.; Camós M.; Colomer D.; Villamor N.; Esteve J.; Costa D.; Carrió A.; Aymerich M.; Aguilar J. L.; Domingo A.; Solé F.; Gomis F.; Florensa L.; Montserrat E.; Campo E. Genes, Chromosomes Cancer 2004, 40, 140. [DOI] [PubMed] [Google Scholar]
- Borrow J.; Stanton V. P. Jr.; Andresen J. M.; Becher R.; Behm F. G.; Chaganti R. S.; Civin C. I.; Disteche C.; Dubé I.; Frischauf A. M.; Horsman D.; Mitelman F.; Volinia S.; Watmore A. E.; Housman D. E. Nat. Genet. 1996, 14, 33. [DOI] [PubMed] [Google Scholar]
- Chaffanet M.; Gressin L.; Preudhomme C.; Soenen-Cornu V.; Birnbaum D.; Pébusque M. J. Genes, Chromosomes Cancer 2000, 28, 138. [DOI] [PubMed] [Google Scholar]
- Kitabayashi I.; Aikawa Y.; Yokoyama A.; Hosoda F.; Nagai M.; Kakazu N.; Abe T.; Ohki M. Leukemia 2001, 15, 89. [DOI] [PubMed] [Google Scholar]
- Panagopoulos I.; Fioretos T.; Isaksson M.; Samuelsson U.; Billström R.; Strömbeck B.; Mitelman F.; Johansson B. Hum. Mol. Genet. 2001, 10, 395. [DOI] [PubMed] [Google Scholar]
- Vizmanos J. L.; Larráyoz M. J.; Lahortiga I.; Floristán F.; Alvarez C.; Odero M. D.; Novo F. J.; Calasanz M. J. Genes, Chromosomes Cancer 2003, 36, 402. [DOI] [PubMed] [Google Scholar]
- Uppal G. K.; Leighton J.; Da Costa D.; Czulewicz A.; Palazzo I. E. Hematol. Rep. 2011, 3, e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima K.; Kaneda K.; Yoshida C.; Dansako H.; Fujii N.; Yano T.; Shinagawa K.; Yasukawa M.; Fujita S.; Tanimoto M. Br. J. Haematol. 2003, 120, 271. [DOI] [PubMed] [Google Scholar]
- Taki T.; Sako M.; Tsuchida M.; Hayashi Y. Blood 1997, 89, 3945. [PubMed] [Google Scholar]
- Sobulo O. M.; Borrow J.; Tomek R.; Reshmi S.; Harden A.; Schlegelberger B.; Housman D.; Doggett N. A.; Rowley J. D.; Zeleznik-Le N. J. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 8732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake N.; Ishida Y.; Otoh Y.; Hinohara S.; Kobayashi H.; Sakashita A.; Maseki N.; Kaneko Y. Genes, Chromosomes Cancer 1997, 20, 60. [PubMed] [Google Scholar]
- Ida K.; Kitabayashi I.; Taki T.; Taniwaki M.; Noro K.; Yamamoto M.; Ohki M.; Hayashi Y. Blood 1997, 90, 4699. [PubMed] [Google Scholar]
- Wang J.; Iwasaki H.; Krivtsov A.; Febbo P. G.; Thorner A. R.; Ernst P.; Anastasiadou E.; Kutok J. L.; Kogan S. C.; Zinkel S. S.; Fisher J. K.; Hess J. L.; Golub T. R.; Armstrong S. A.; Akashi K.; Korsmeyer S. J. EMBO J. 2005, 24, 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Gural A.; Sun X. J.; Zhao X.; Perna F.; Huang G.; Hatlen M. A.; Vu L.; Liu F.; Xu H.; Asai T.; Xu H.; Deblasio T.; Menendez S.; Voza F.; Jiang Y.; Cole P. A.; Zhang J.; Melnick A.; Roeder R. G.; Nimer S. D. Science 2011, 333, 765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynoird N.; Schwartz B. E.; Delvecchio M.; Sadoul K.; Meyers D.; Mukherjee C.; Caron C.; Kimura H.; Rousseaux S.; Cole P. A.; Panne D.; French C. A.; Khochbin S. EMBO J. 2010, 29, 2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump N. T.; Hazzalin C. A.; Bowers E. M.; Alani R. M.; Cole P. A.; Mahadevan L. C. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattabiraman D. R.; Sun J.; Dowhan D. H.; Ishii S.; Gonda T. J. Mol. Cancer Res. 2009, 7, 1477. [DOI] [PubMed] [Google Scholar]
- Iyer N. G.; Ozdag H.; Caldas C. Oncogene 2004, 23, 4225. [DOI] [PubMed] [Google Scholar]
- Varier R. A.; Kundu T. K. Curr. Pharm. Des. 2006, 12, 1975. [DOI] [PubMed] [Google Scholar]
- Mujtaba S.; Zhou M. M. Methods 2011, 53, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakane N.; Kwon H. S.; Pagans S.; Kaehlcke K.; Mizusawa Y.; Kamada M.; Lassen K. G.; Chan J.; Greene W. C.; Schnoelzer M.; Ott M. PLoS Pathog. 2011, 7, e1002184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mujtaba S.; Zhou M. M. Methods 2011, 53, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allouch A.; Di Primio C.; Alpi E.; Lusic M.; Arosio D.; Giacca M.; Cereseto A. Cell Host Microbe 2011, 9, 484. [DOI] [PubMed] [Google Scholar]
- Terreni M.; Valentini P.; Liverani V.; Gutierrez M. I.; Di Primio C.; Di Fenza A.; Tozzini V.; Allouch A.; Albanese A.; Giacca M.; Cereseto A. Retrovirology 2010, 7, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W.; Wang Z.; Liu Y.; Fan Y.; Zhou B. Y.; Yang X. F.; He J. J. Glia 2010, 58, 1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppi M.; Murphy J. M.; de Graaf C. A.; Hyland C. D.; Greig K. T.; Metcalf D.; Hilton A. A.; Nicola N. A.; Kile B. T.; Hilton D. J.; Alexander W. S. Blood 2008, 112, 3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton D. J.; Kile B. T.; Alexander W. S. Blood 2009, 113, 5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanazume T.; Morimoto T.; Wada H.; Kawamura T.; Hasegawa K. Mol. Cell. Biochem. 2003, 248, 115. [DOI] [PubMed] [Google Scholar]
- Gusterson R. J.; Jazrawi E.; Adcock I. M.; Latchman D. S. J. Biol. Chem. 2003, 278, 6838. [DOI] [PubMed] [Google Scholar]
- Zhou X. Y.; Shibusawa N.; Naik K.; Porras D.; Temple K.; Ou H.; Kaihara K.; Roe M. W.; Brady M. J.; Wondisford F. E. Nat. Med. 2004, 10, 633. [DOI] [PubMed] [Google Scholar]
- Cha-Molstad H.; Saxena G.; Chen J.; Shalev A. J. Biol. Chem. 2009, 284, 16898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricambert J.; Miranda J.; Benhamed F.; Girard J.; Postic C.; Dentin R. J. Clin. Invest. 2010, 120, 4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell U.; Dijk F.; Sjollema K. A.; Giepmans B. N. Nat. Methods 2012, 9, 152. [DOI] [PubMed] [Google Scholar]
- McNeil P. L. J. Cell Sci. 1987, 88, 669. [DOI] [PubMed] [Google Scholar]
- Hayashi-Takanaka Y.; Yamagata K.; Wakayama T.; Stasevich T. J.; Kainuma T.; Tsurimoto T.; Tachibana M.; Shinkai Y.; Kurumizaka H.; Nozaki N.; Kimura H. Nucleic Acids Res. 2011, 39, 6475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi-Takanaka Y.; Yamagata K.; Nozaki N.; Kimura H. J. Cell Biol. 2009, 187, 781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen K. M.; Johansen J. Chromosome Res. 2006, 14, 393. [DOI] [PubMed] [Google Scholar]
- Stasevich T. J.; Hayashi-Takanaka Y.; Sato Y.; Maehara K.; Ohkawa Y.; Sakata-Sogawa K.; Tokunaga M.; Nagase T.; Nozaki N.; McNally J. G.; Kimura H. Nature 2014, 516, 272. [DOI] [PubMed] [Google Scholar]
- Rothbauer U.; Zolghadr K.; Tillib S.; Nowak D.; Schermelleh L.; Gahl A.; Backmann N.; Conrath K.; Muyldermans S.; Cardoso M. C.; Leonhardt H. Nat. Methods. 2006, 3, 887. [DOI] [PubMed] [Google Scholar]
- Sato Y.; Mukai M.; Ueda J.; Muraki M.; Stasevich T. J.; Horikoshi N.; Kujirai T.; Kita H.; Kimura T.; Hira S.; Okada Y.; Hayashi-Takanaka Y.; Obuse C.; Kurumizaka H.; Kawahara A.; Yamagata K.; Nozaki N.; Kimura H. Sci. Rep. 2013, 3, 2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincenz C.; Kerppola T. K. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 16572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster T. H. Ann. Phys. 1948, 437, 55. [Google Scholar]
- Helms V.Fluorescence Resonance Energy Transfer. Principles of Computational Cell Biology; Wiley-VCH: Weinheim, 2008; p 202, ISBN 978-3-527-31555-0. [Google Scholar]
- Kanno T.; Kanno Y.; Siegel R. M.; Jang M. K.; Lenardo M. J.; Ozato K. Mol. Cell 2004, 13, 33. [DOI] [PubMed] [Google Scholar]
- Sasaki K.; Ito T.; Nishino N.; Khochbin S.; Yoshida M. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 16257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T.; Umehara T.; Sasaki K.; Nakamura Y.; Nishino N.; Terada T.; Shirouzu M.; Padmanabhan B.; Yokoyama S.; Ito A.; Yoshida M. Chem. Biol. 2011, 18, 495. [DOI] [PubMed] [Google Scholar]
- Carrillo L. D.; Krishnamoorthy L.; Mahal L. K. J. Am. Chem. Soc. 2006, 128, 14768. [DOI] [PubMed] [Google Scholar]
- Newman R. H.; Zhang J. Mol. BioSyst. 2008, 4, 496. [DOI] [PubMed] [Google Scholar]
- Nagai Y.; Miyazaki M.; Aoki R.; Zama T.; Inouye S.; Hirose K.; Iino M.; Hagiwara M. Nat. Biotechnol. 2000, 18, 313. [DOI] [PubMed] [Google Scholar]
- Ting A. Y.; Kain K. H.; Klemke R. L.; Tsien R. Y. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 15003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurokawa K.; Mochizuki N.; Ohba Y.; Mizuno H.; Miyawaki A.; Matsuda M. J. Biol. Chem. 2001, 276, 31305. [DOI] [PubMed] [Google Scholar]
- Lin C. W.; Jao C. Y.; Ting A. Y. J. Am. Chem. Soc. 2004, 126, 5982. [DOI] [PubMed] [Google Scholar]
- Lin C. W.; Ting A. Y. Angew. Chem., Int. Ed. 2004, 43, 2940. [DOI] [PubMed] [Google Scholar]
- Yoshida M.; Kijima M.; Akita M.; Beppu T. J. Biol. Chem. 1990, 265, 17174. [PubMed] [Google Scholar]
- Hanahan D.; Weinberg R. A. Cell 2000, 100, 57. [DOI] [PubMed] [Google Scholar]
- Hanahan D.; Weinberg R. A. Cell 2011, 144, 646. [DOI] [PubMed] [Google Scholar]
- Reynoird N.; Schwartz B. E.; Delvecchio M.; Sadoul K.; Meyers D.; Mukherjee C.; Caron C.; Kimura H.; Rousseaux S.; Cole P. A.; Panne D.; French C. A.; Khochbin S. EMBO J. 2010, 29, 2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump N. T.; Hazzalin C. A.; Bowers E. M.; Alani R. M.; Cole P. A.; Mahadevan L. C. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santer F. R.; Höschele P. P.; Oh S. J.; Erb H. H.; Bouchal J.; Cavarretta I. T.; Parson W.; Meyers D. J.; Cole P. A.; Culig Z. Mol. Cancer Ther. 2011, 10, 1644. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Toh H. C.; Chow P.; Chung A. Y.; Meyers D. J.; Cole P. A.; Ooi L. L.; Lee C. G. FASEB J. 2012, 26, 3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan G.; Eller M. S.; Elm C.; Larocca C. A.; Ryu B.; Panova I. P.; Dancy B. M.; Bowers E. M.; Meyers D.; Lareau L.; Cole P. A.; Taverna S. D.; Alani R. M. J. Invest. Dermatol. 2013, 133, 2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santer F. R.; Höschele P. P.; Oh S. J.; Erb H. H.; Bouchal J.; Cavarretta I. T.; Parson W.; Meyers D. J.; Cole P. A.; Culig Z. Mol. Cancer Ther. 2011, 10, 1644. [DOI] [PubMed] [Google Scholar]
- Wang L.; Gural A.; Sun X. J.; Zhao X.; Perna F.; Huang G.; Hatlen M. A.; Vu L.; Liu F.; Xu H.; Asai T.; Xu H.; Deblasio T.; Menendez S.; Voza F.; Jiang Y.; Cole P. A.; Zhang J.; Melnick A.; Roeder R. G.; Nimer S. D. Science 2011, 333, 765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Wang L.; Predina J.; Han R.; Beier U. H.; Wang L. C.; Kapoor V.; Bhatti T. R.; Akimova T.; Singhal S.; SBrindle P. K.; Cole P. A.; Albelda S. M.; Hancock W. W. Nat. Med. 2013, 19, 1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min S. W.; Cho S. H.; Zhou Y.; Schroeder S.; Haroutunian V.; Seeley W. W.; Huang E. J.; Shen Y.; Masliah E.; Mukherjee C.; Meyers D.; Cole P. A.; Ott M.; Gan L. Neuron 2010, 67, 953.Erratum in: Neuron2010, 68, 801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P.; Chou B. K.; Yen J.; Ye Z.; Zou J.; Dowey S.; Brodsky R. A.; Ohm J. E.; Yu W.; Baylin S. B.; Yusa K.; Bradley A.; Meyers D. J.; Mukherjee C.; Cole P. A.; Cheng L. Stem Cells 2010, 28, 713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C. R.; Cole P. A.; Meyers D. J.; Kormish J.; Dent S.; Zaret K. S. Science 2011, 332, 963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek R.; Coelho C. M.; Sullivan R. K.; Baker-Andresen D.; Li X.; Ratnu V.; Dudley K. J.; Meyers D.; Mukherjee C.; Cole P. A.; Sah P.; Bredy T. W. J. Neurosci. 2011, 31, 7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight Z. A.; Shokat K. M. Cell 2007, 128, 425. [DOI] [PubMed] [Google Scholar]
- Kasper L. H.; Fukuyama T.; Biesen M. A.; Boussouar F.; Tong C.; de Pauw A.; Murray P. J.; van Deursen J. M.; Brindle P. K. Mol. Cell. Biol. 2006, 26, 789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper L. H.; Lerach S.; Wang J.; Wu S.; Jeevan T.; Brindle P. K. EMBO J. 2010, 29, 3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan H. M.; Xu A. W.; Coco C.; Srajer G.; Wyszomierski S.; Evrard Y. A.; Eckner R.; Dent S. Y. Dev. Dyn. 2005, 233, 1337. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Vasilatos S. N.; Boric L.; Shaw P. G.; Davidson N. E. Breast Cancer Res. Treat. 2012, 131, 777–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H.; Yang X.; Pandiyan K.; Liang G. PLoS One 2013, 8, e75136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusevich P.; Kalin J. H.; Ming S. A.; Basso M.; Givens J.; Li X.; Hu J.; Taylor M. S.; Cieniewicz A. M.; Hsiao P. Y.; Huang R.; Roberson H.; Adejola N.; Avery L. B.; Casero R. A. Jr.; Taverna S. D.; Qian J.; Tackett A. J.; Ratan R. R.; McDonald O. G.; Feinberg A. P.; Cole P. A. ACS Chem. Biol. 2014, 9, 1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan G.; Eller M. S.; Elm C.; Larocca C. A.; Ryu B.; Panova I. P.; Dancy B. M.; Bowers E. M.; Meyers D.; Lareau L.; Cole P. A.; Taverna S. D.; Alani R. M. J. Invest. Dermatol. 2013, 133, 2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das N.; Dhanawat M.; Dash B.; Nagarwal R. C.; Shrivastava S. K. Eur. J. Pharm. Sci. 2010, 41, 571. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.