

Abstract
Melanoma is the most lethal form of skin cancer. There is a lack of effective treatments for individuals with advanced disease. Many melanomas exhibit high levels of radioresistance. The direct consequence of γ-irradiation for most melanoma cells is growth arrest at the G2-M phase of cell cycle. However, radiation-induced signaling pathways may affect numerous additional targets in cancer cells. We show in the present study that γ-irradiation, as well as α-particle exposure, dramatically increases the susceptibility of melanoma cells to recombinant tumor necrosis factor–related apoptosis-inducing ligand (TRAIL)-mediated apoptosis via up-regulation of surface TRAIL-receptor 1/receptor 2 (DR4/DR5) levels and to Fas ligand–mediated apoptosis via up-regulation of surface Fas levels. Additionally, increased dynamin-2 expression after irradiation is critically important in the translocation of death receptor to the cell surface. Moreover, sodium arsenite treatment may up-regulate expression of endogenous TRAIL and induces its translocation to cell surface and further down-regulates cFLIP levels in melanoma cells. We have evaluated the effects of sequential γ-irradiation and arsenite treatment of melanoma cells for the induction of death signaling. Such treatment results in an efficient TRAIL-mediated apoptosis via a paracrine mechanism. These data highlight the efficacy of combined modality treatment involving radiation and arsenite in clinical management of this often fatal form of skin cancer.
Introduction
The incidence of melanoma has substantially increased worldwide over the last 40 years. Although melanoma accounts for only 10% of skin cancer, it is responsible for at least 80% of skin cancer death. Approximately 8,000 Americans died of melanoma in 2005 and 62,200 new cases of melanoma have been diagnosed in 2006. Most advanced melanomas respond poorly to radiotherapy and chemotherapy and no effective therapy exists to inhibit the metastatic spread of this cancer (1, 2).
The tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) has a strong antitumor potential by inducing apoptosis in a wide variety of human TRAIL-receptor 1–positive (TRAIL-R1/DR4) and TRAIL-receptor 2–positive (TRAIL-R2/DR5) cancer cells but not in most normal human cells that often contain high levels of decoy receptors TRAIL-R3 and TRAIL-R4 on cell surface, thereby preventing induction of apoptotic signaling (3, 4). However, many metastatic tumors, including advanced melanomas, acquired the secondary resistance to apoptotic signaling induced by TRAIL. Actually, f60% of tumor cell lines are resistant to TRAIL (5, 6). In an addition to the presence of decoy receptors, such resistance may occur at different points of the TRAIL-induced death signaling pathway. Suppression of surface expression of the death receptors (TRAIL-R1/DR4 and TRAIL-R2/DR5), dysfunction of death receptors due to mutations, suppression of caspase-8 or caspase-3 functions, and overexpression of endogenous inhibitors of apoptosis, such as cFLIP, cIAP, XIAP, Bcl-2, and Bcl-xL, directly correlate with resistance to TRAIL in many types of cancer, including melanomas (7). There is evidence that progression in melanoma is often linked with decreased surface expression of TRAIL-R1/R2. Furthermore, general activations of the nuclear factor-κB (NF-κB), phosphatidy-linositol 3-kinase (PI3K)-AKT, and Ras-BRAF-mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) kinase-ERK signaling pathways, which are characteristic features of advanced tumors (8, 9), often result in protection against TRAIL-mediated apoptosis via up-regulation of gene expression of anti-apoptotic proteins (10, 11).
The direct consequences of γ-irradiation of cancer cells are either growth arrest due to activation of specific signaling pathways following DNA damage or radiation-induced cell death via apoptosis or necrosis. Radiation-induced apoptotic signaling could use both p53-dependent and p53-independent pathways (12). Ionizing radiation is an important treatment modality for cancer (∼50% of all types of cancer). However, it is not effective for treatment of melanomas, which are largely radioresistant. On the other hand, rationally designed combined treatment, including g-irradiation and chemotherapy or biochemotherapy, which simultaneously targets several critical functions of cancer cells, may be effective in some cancer treatments (13).
In the present study, we address the question of whether it would be possible to increase surface expression of death receptors and to suppress, simultaneously, antiapoptotic functions in melanoma cells to effectively induce the death signaling cascade. The results obtained show that sequential treatment of melanoma cells with γ-irradiation or α-particle irradiation and recombinant TRAIL may substantially increase apoptotic response of TRAIL-resistant melanoma cells. Furthermore, many types of cancer cells, including melanomas, contain intracellular pools of Fas ligand (FasL), TRAIL, or both that could be translocated to cell surface following stress signaling (14, 15). We have previously used sodium arsenite as a stress inducer for initiation of the endogenous TRAIL translocation to cell surface (16). As a consequence, such combined treatment of melanoma cells with γ-radiation and sodium arsenite efficiently induces apoptotic signaling via paracrine TRAIL/TRAIL-receptor mechanisms.
Materials and Methods
Materials
Sodium arsenite and cycloheximide were obtained from Sigma. Human soluble FasL (recombinant) and soluble Killer-TRAIL (recombinant) were purchased from Alexis. c-Jun NH2-terminal kinase (JNK) inhibitor SP600125 was obtained from Biomol; MEK inhibitor U0126 and PI3K inhibitor LY294002 were purchased from Calbiochem. Caspase inhibitors zVAD-fmk, Ac-IETD-CHO (an inhibitor of caspase-8 and caspase-6), and Ac-LEHD-CHO (an inhibitor of caspase-9) were purchased from Calbiochem.
Cell lines
Human melanoma cell lines LU1205 (also known as 1205lu), WM9, and WM35 (17) were maintained in DMEM supplemented with 10% fetal bovine serum, L-glutamine, and antibiotics.
Irradiation procedures
3He α-particles (125 keV/μm) were delivered to the melanoma cells using 4-MV Van de Graff accelerator at the Radiological Research Accelerator Facilities of Columbia University. Control and irradiated cells were collected 24 h after irradiation. Cultures were trypsinized and counted with a Coulter counter, and aliquots of the cells were replated into 100-mm-diameter dishes for colony formation or flow cytometry assay. Cultures for clonogenic survival assays were incubated for 12 days, at which time they were fixed with formaldehyde and stained with Giemsa.
To determine sensitivity to γ-rays, the plates with melanoma cells were exposed to radiation from a GammaCell 40 137Cs irradiator (dose rate, 0.82 Gy/min) of Columbia University. Six to 24 h after irradiation, cells have been used for flow cytometry or for additional treatments.
Fluorescence-activated cell sorting analysis of TRAIL, TRAIL-R1/R2 (DR4/DR5), and Fas levels
Surface levels of TRAIL on human melanomas were determined by staining with the phycoerythrin (PE)-conjugated anti-human TRAIL monoclonal antibody (mAb; eBioscience) and subsequent flow cytometry. Surface levels of TRAIL-R1/DR4, TRAIL-R2/DR5, and Fas were determined by staining with the PE-labeled mAbs from eBioscience and BD Biosciences PharMingen. PE-conjugated nonspecific mouse IgG1 was used as an immunoglobulin isotype control. A FACSCalibur flow cytometer (Becton Dickinson) combined with the CellQuest program was used to do flow cytometric analysis. All experiments were independently repeated four to five times.
Transfection and luciferase assay
The NF-κB luciferase reporter containing two κB-binding sites, Jun2-Luc reporter and empty vector tk-Luc (18), and GAS-Luc reporter containing three repeats of GAS sites from the Ly6A/E promoter were used to determine NF-κB, activator protein-1 (AP-1), and signal transducers and activators of transcription (STAT) trans-activation, respectively. Additional reporter constructs used included the following: 2.5-kb p21-WAF1-promoter-Luc (19), 1.5-kb TRAIL-promoter-Luc (20), 1.8-kb DR4/TRAIL-R1-promoter-Luc (21), DR5/TRAIL-R2-full-Luc, which contained 1.6 kb upstream of the ATG site through intron 2 in the DR5 genomic locus (22), and 1-kb cFLIP-promoter-Luc (23, 24). Transient transfection of different reporter constructs (1 μg) together with pCMV-βgal (0.25 μg) into 5 × 105 melanoma cells was done using LipofectAMINE (Life Technologies-Invitrogen). Proteins were prepared for β-galactosidase and luciferase analysis 16 h after transfection. Luciferase activity was determined using the Luciferase Assay System (Promega) and normalized based on β-galactosidase levels.
In some experiments, melanoma cells were transfected with reporter constructs together with certain expression vectors (ratio, 1:3), including the following: pCMV-IκBαΔN (25), pcDNA3-IKKβS178E/S181E (26), ΔMEKK1 expression vector (27), pcDNA3-FLAG-MKK7β 1 (28), dominant-negative JNK1-APF, and dominant-negative form of cJun/TAM67 in the presence of pCMV-βgal. Sixteen to 24 h after transfection, luciferase activity was determined.
Apoptosis studies
Cells were exposed to soluble TRAIL (50 ng/mL), alone or in combination with cycloheximide (2 μg/mL), to soluble FasL (50 ng/mL) and sodium arsenite (5 μmol/L). Apoptosis was then assessed by quantifying the percentage of hypodiploid nuclei undergoing DNA fragmentation. Flow cytometric analysis was done on a FACSCalibur flow cytometer.
cFLIP suppression by RNA interference
The pSUPER retro RNA interference (RNAi) system (OligoEngine), which has been used for the production of small RNAi transcripts, was used to suppress cFLIP expression. Two variants of RNAi of 19 nucleotides designed to target human cFLIP were expressed using vector pSUPER.retro.puro (pSR-puro). RNAi cFLIP-92 (UGUGGUUCCACCUAAUGUC) was the most efficient in the corresponding mRNA targeting.
Western blot analysis
Total cell lysates (50 μg protein) were resolved on 10% SDS-PAGE and processed according to standard protocols. The antibodies used for Western blotting included the following: monoclonal anti-β-actin (Sigma); monoclonal anti-FLIP (NF6; Axxora); monoclonal anti-XIAP (BD Biosciences); monoclonal anti-cyclooxygenase-2 (COX-2; Cayman Chemical Co.); monoclonal anti-p21-WAF1 (Cell Signaling); monoclonal anti-dynamin-2 (Upstate); polyclonal antibodies to TRAIL (human); TRAIL-R1/DR4 and TRAIL-R2/DR5 (Axxora); polyclonal anti-Fas (BD Biosciences PharMingen); and polyclonal antibodies against phosphorylated p53 (Ser20) and total p53, phosphorylated stress-activated protein kinase/JNK (Thr183/Tyr185) and JNK, phosphorylated p44/p42 MAPK (Thr202/Tyr204) and p44/p42 MAPK, and phosphorylated AKT (Ser473) and AKT (Cell Signaling). Optimal dilutions of primary antibodies were 1:1,000 to 1:5,000. The secondary antibodies anti-mouse or anti-rabbit were conjugated to horseradish peroxidase (dilution, 1:5,000 to 1:10,000); signals were detected using the enhanced chemiluminescence system (Amersham).
Electrophoretic mobility shift assay
Electrophoretic mobility shift assay (EMSA) was done for the detection of NF-κB DNA-binding activity as previously described using the labeled double-strand oligonucleotide AGCTTGGGGACTTTCCAGCCG (binding site is italicized). Ubiquitous NF-Y DNA-binding activity was used as an internal control (29).
Results
TRAIL- and radiation-induced cell death in human melanoma lines
Human melanoma cell lines showed different levels of sensitivity to soluble recombinant TRAIL (30). Based on our previous results, we selected two contrasting human metastatic melanoma lines: WM9 that was highly sensitive to TRAIL and LU1205 (also known as 1205lu) that was relatively resistant to TRAIL (Fig. 1A). Both melanoma lines express TRAIL-R2/DR5 on cell surface, although levels of surface expression of TRAIL-R2/DR5 are ∼2-fold higher in WM9 cells (Fig. 1B). Modest and low levels of expression of TRAIL-R1/DR4 have also been detected on the surface of WM9 and LU1205 cells, respectively. We showed previously that another important difference between WM9 and LU1205 cells was linked to the levels of antiapoptotic cFLIP protein, which have been significantly diminished in WM9 cells (16). In addition, we used early (radial growth phase) WM35 melanoma cells, which express relatively low levels of DR5 on cell surface and show only a modest response to either TRAIL alone or in combination with cycloheximide (Fig. 1A and B).
Figure 1.
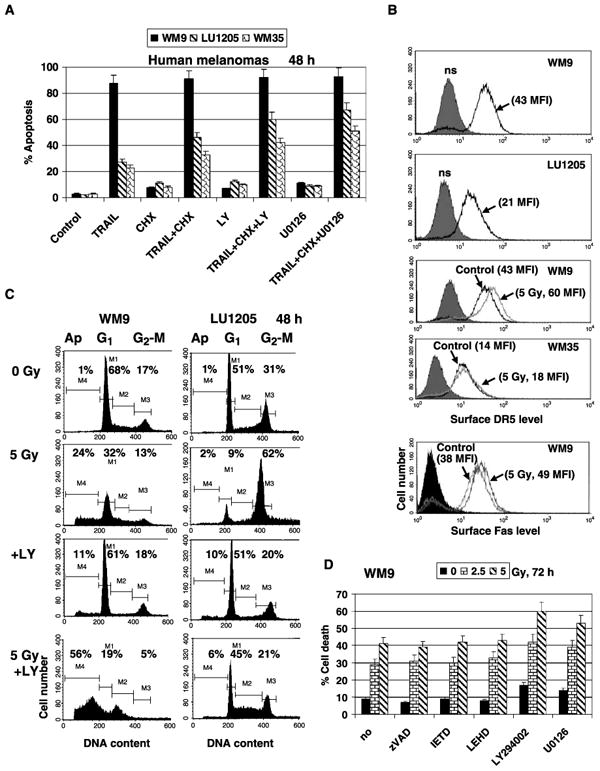
Apoptosis and cell survival in TRAIL-sensitive (WM9) and TRAIL-resistant (LU1205 and WM35) human melanoma cell lines. A, effects of combined treatment of TRAIL (50 ng/mL) and cycloheximide (CHX; 2 μg/mL), TRAIL + cycloheximide + LY294002 (LY; 50 μmol/L), or TRAIL + cycloheximide + U0126 (10 μmol/L) on melanoma apoptosis. Cells were stained by propidium iodide 48 hafter treatment. Apoptosis levels were determined as percentage of cells with hypodiploid content of DNA in the pre-G0-G1 region using flow cytometry. Columns, mean of three independent experiments; bars, SD. B, surface expression of TRAIL-R2/DR5 levels in WM9, LU1205, and WM35 cells and Fas in WM9 has been determined using PE-labeled mAbs to DR5 and Fas followed by fluorescence-activated cell sorting (FACS) analysis. MFI is indicated. WM9 and WM35 cells were γ-irradiated (5 Gy). Levels of receptors were determined 24 hafter irradiation. C, cell cycle apoptosis analysis 48 hafter γ-irradiation (5 Gy) of WM9 and LU1205 melanoma cells. LY294002 (50 μmol/L) was used as indicated. Percentages of apoptotic cells and cells at G1 and G2-M phases of cell cycle. D, effects of caspase inhibitors zVAD-fmk, IETD, and LEHD (20 μmol/L) on levels of WM9 cell deathfollowing γ-irradiation. LY294002 (50 μmol/L), an inhibitor of the PI3K-AKT, and U0126 (10 μmol/L), an inhibitor of the MEK-ERK, were also used.
Cycloheximide (2 μg/mL) notably increased TRAIL-induced apoptosis in LU1205 cells 48 h after treatment. However, apoptotic levels obtained were still lower compared with levels induced by soluble TRAIL alone in WM9 cells. An additional increase in TRAIL-mediated apoptosis could be obtained by suppression of the PI3K-AKT pathway, the main regulator of general cell survival, with a specific pharmacologic inhibitor LY294002 (50 μmol/L), or by suppression of the MEK-ERK pathway with the inhibitor U0126 (10 μmol/L; Fig. 1A). TRAIL-induced apoptosis of WM9 and LU1205 cells is sensitive to both caspase-8 and caspase-9 inhibitors (16), which indicates an involvement of mitochondrial apoptotic loop in the acceleration of TRAIL-induced apoptosis in these melanoma cells.
On the other hand, WM9 and LU1205 metastatic cell lines exhibited different degrees of radiosensitivity. In WM9 cells, a modest apoptosis, which was accompanied by the secondary necrosis, was detected 48 h after γ-irradiation (2.5–5 Gy). In contrast, LU1205 cells showed a pronounced G2-M arrest under these conditions (Fig. 1C) as was previously reported for this melanoma line (31). Radiation-induced death of WM9 cells accompanied by DNA fragmentation (Fig. 1C) was, however, insensitive to caspase inhibitors (Fig. 1D) or to anti-TRAIL inhibitory mAb (data not shown). As expected, LY294002 (50 μmol/L) and U0126 (10 μmol/L) additionally increased the level of radiation-induced death in WM9 cells (Fig. 1C and D). In contrast to WM9 cells, the effect of LY294002 (50 μmol/L) on radiosensitivity of LU1205 cells was less pronounced, inducing low-level apoptosis and a release from G2-M arrest (Fig. 1C). WM35 early melanoma cells also exhibited radioresistance similarly to LU1205 cells (data not shown). Analysis of clonogenic survival of cancer cells after γ-irradiation confirmed a higher degree of radiosensitivity of WM9 compared with LU1205 cells (data not shown). Differential response to γ-irradiation, however, was not connected with the p53 status that was wild-type for both melanoma lines (31).
Effects of γ-irradiation on cell signaling and surface expression of death receptors
In spite of the absence of pronounced apoptotic response, γ-irradiation of LU1205 melanoma cells affected many signaling pathways that activated stress-induced transcription factors, such as NF-κB and p53 and their target genes. We initially observed up-regulation of nuclear NF-κB DNA-binding activity determined by EMSA (Fig. 2A) and also increased NF-κB–dependent reporter activity (Fig. 2C). We also detected increased p53 and phosphorylated Ser20-p53 levels by Western blot analysis (Fig. 2B). Furthermore, up-regulation of the p21-WAF1 promoter activity and general AP-1–dependent reporter activities was also observed 6 h after irradiation (Fig. 2C).
Figure 2.
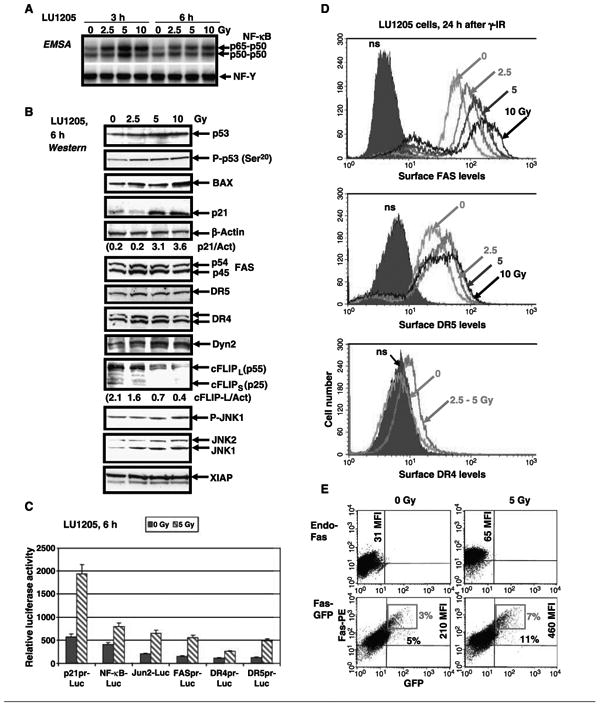
Dose-dependent effects of γ-radiation on cell signaling pathways and expression of death receptors in LU1205 melanoma cells. A, effect of γ-irradiation on nuclear NF-κB and NF-Y DNA-binding activities determined by EMSA. The lower parts of gels with free labeled probes have been removed. B, Western blot analysis of p53, phosphorylated p53 (P-p53; Ser20), BAX, p21, death receptors, XIAP, cFLIP, phosphorylated JNK1/2 (P-JNK1; Thr183/Tyr185), and total JNK1/2 levels in LU1205 cells 6 hafter γ-irradiation. Ratio of p21 to actin (p21/Act) and cFLIP-L to actin (cFLIP-L/Act). Dyn2, dynamin-2. C, LU1205 cells were γ-irradiated (5 Gy) 16 hafter transient transfection of p21pr-Luc, NF-κB-Luc, Jun2-Luc, FASpr-Luc, DR4pr-Luc, and DR5pr-Luc reporter constructs together with β-galactosidase expression vector (plasmid DNA ratio, 1:0.25). Luciferase activity was determined after an additional 6 hand normalized based on β-galactosidase activity. D, surface expression of Fas, DR5, and DR4 was determined by staining with PE-labeled anti-Fas, anti-DR5, or anti-DR4 mAbs, respectively, and FACS analysis. E, LU1205 cells were transfected by Fas-GFP expression constructs. Sixteen hours after transfection, cells have been irradiated. Surface expression of Fas-GFP has been determined after an additional 6 husing anti-Fas mAb labeled withPE. Percentage cells and MFI of surface Fas. Red quadrants, brightest Fas-positive cells.
Up-regulation of the wild-type p53 levels in both LU1205 and WM9 lines correlated with an increase in the levels of its downstream transcriptional targets, such as p21-WAF1, BAX, Fas, and TRAIL-R2/DR5 (Fig. 2B). In contrast, a rapid down-regulation of an antiapoptotic protein cFLIP (both long and short forms) was also observed in these conditions, reflecting negative regulation of cFLIP expression and stability by JNK induced by γ-irradiation (Fig. 2B; refs. 16, 32).
Hence, irradiation of melanoma cells was accompanied by notable up-regulation of the transcription and total protein levels of death receptors, Fas, DR4, and DR5, which were regulated by p53 in concert with NF-κB, AP-1, and STAT (Fig. 2B and C; refs. 21, 22). Furthermore, dynamin-2 level, a cytoskeleton protein previously shown to control Fas receptor trafficking through trans-Golgi network to cell surface (33, 34), substantially increased after γ-irradiation (Fig. 2B). This suggested that the up-regulation of surface expression of death receptors after irradiation of cancer cells was attributable to increased efficiency of protein translocation in the cell. Indeed, 24 h after γ-irradiation of LU1205 cells, a profound increase in surface Fas levels and in TRAIL-R2/DR5 levels, as well as modest increase in TRAIL-R1/DR4 surface levels, has been detected (Fig. 2D). Of note, dose-dependent increase of surface expression was observed for Fas at 2.5 to 10 Gy and for DR5 at 2.5 to 5 Gy, whereas DR4 levels increased relatively similarly after irradiation with dose 2.5 to 5 Gy (Fig. 2D). Furthermore, γ-irradiation (2.5–5 Gy) had also prominent effects in the upregulation of surface expression of Fas and DR5 in WM9 cells, whereas only a modest effect was detected in early melanoma WM35 cells (see Fig. 1B).
To determine a direct effect of γ-irradiation on Fas translocation in cells, LU1205 cells were transfected with fused Fas-green fluorescent protein (GFP) construct (Fig. 2E; ref. 35). As expected, surface expression of Fas-GFP, which was detected using PE-labeled mAb to the Fas extracellular domain, was notably increased (at least 2-fold) 6 h after irradiation based on an increase in medium fluorescence intensity (MFI) from 210 to 460 and in percentage cells with high levels of surface Fas.
Brefeldin A (100 ng/mL), an inhibitor of protein translocation from the endoplasmic reticulum to Golgi, also suppressed endogenous surface expression of Fas and DR4/DR5 induced by irradiation (data not shown). Taken together, these results show that γ-irradiation is involved not only in transcriptional control of death receptor expression but also in regulation of their translocation to cell surface.
Sequential treatment of melanoma cells with γ-irradiation and recombinant TRAIL or FasL
Changes observed in death receptor levels following γ-irradiation have a functional significance. We have used soluble recombinant TRAIL (50 ng/mL) combined with cycloheximide (2 μg/mL) to induce apoptosis in LU1205 cells 24 to 48 h after γ-irradiation (Fig. 3A and B). Detailed analysis showed a substantial increase in apoptotic levels (from 25% to 48% after an additional 24 h and from 56% to 94% after an additional 48 h of TRAIL treatment for melanoma cells), which were preexposed with γ-irradiation (Fig. 3A and B). Interestingly, an additional suppression of basal levels of cFLIP (both long and short forms) by specific RNAi (16) accelerated development of apoptosis in preirradiated melanoma cells 24 h after treatment with TRAIL and cycloheximide (48 h after γ-irradiation; Fig. 3C and D).
Figure 3.
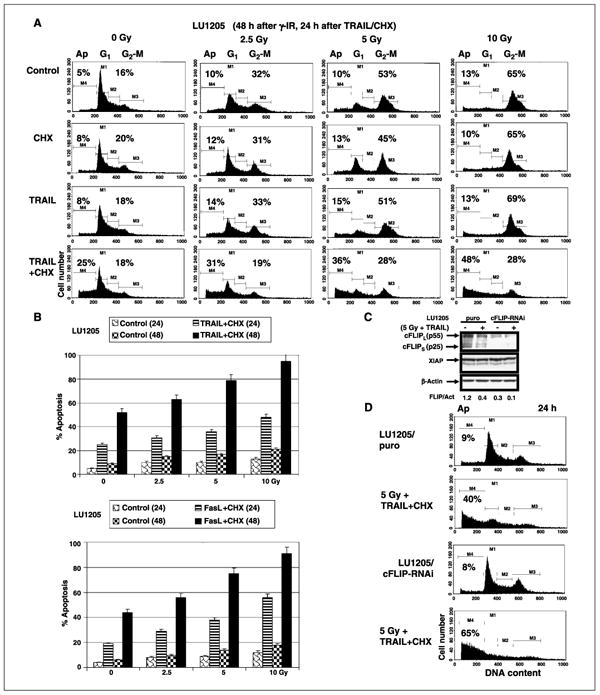
Dose-dependent effects of γ-irradiation on TRAIL- or FasL-mediated apoptosis of LU1205 human melanoma cells. A and B, cells were γ-irradiated (γ-IR) in a dose-dependent manner. Twenty-four hours after irradiation, TRAIL (50 ng/mL), cycloheximide (2 μg/mL), or their combination, FasL (50 ng/mL) + cycloheximide, was added into the medium for an additional 24 or 48 h. Apoptosis levels were determined as percentage of cells with hypodiploid content of DNA in the pre-G0-G1 region using flow cytometry. Columns, mean of three independent experiments; bars, SD. C, LU1205 cells were stably transfected with the empty vector pSR-puro or by cFLIP-RNAi expression construct. Western blot analysis of cFLIP (long and short forms) expression 16 h after γ-irradiation (5 Gy) with subsequent addition of TRAIL + cycloheximide into the medium (6 h). D, effects of sequential treatment by γ-irradiation (5 Gy, 48 h after irradiation) and TRAIL + cycloheximide (24 h) on apoptosis in control LU1205-puro and LU1205/cFLIP-RNAi cells. Apoptosis levels were determined as percentage of cells with hypodiploid content of DNA in the pre-G0-G1 region using flow cytometry. Results of a typical experiment (one from three).
Taken together, these experiments indicated that up-regulation of surface expression of death receptors and simultaneous down-regulation of antiapoptotic cFLIP protein levels may substantially increase susceptibility of resistant melanoma line to TRAIL-mediated apoptosis. Additionally, sequential treatment of irradiated LU1205 cells with recombinant FasL (50 ng/mL) in combination with cycloheximide (2 μmol/L) also induced strong Fas-mediated apoptosis in a dose-dependent manner 24 and 48 h after treatment (Fig. 3A, bottom).
Role of dynamin-2 in death receptor translocation
To further confirm a probable role of dynamin-2 expression in regulation of death receptor translocation to cell surface in irradiated cells, we took advantage of previously established LU1205 melanoma lines expressing dominant-negative dynamin-2 constructs: Dyn-2K44A and Dyn-Y231F/Y597F (Dyn-2FF; ref. 33). Dominantnegative interference of dynamin-2 (36) that partially suppressed death receptor protein translocation from trans-Golgi network to cell surface (33, 37) caused dramatic down-regulation of γ-irradiation–induced levels of DR5 (after 2.5 Gy) and Fas (2.5 Gy) in LU1205 cells 24 h after treatment (Fig. 4A and C). This was followed by decreased levels of TRAIL-mediated apoptosis in cells where dynamin-2 functions have been suppressed (Fig. 4B). We have also observed similar effects for recombinant FasL-mediated apoptosis in irradiated cells (Fig. 4D).
Figure 4.
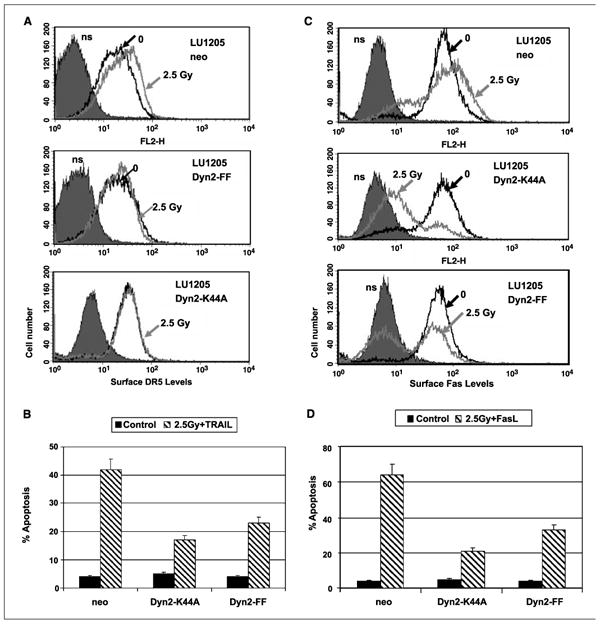
Suppression of dynamin-2 by dominant-negative Dyn-2K44A and Dyn-2FF inhibited radiation-induced up-regulation in expression of Fas and DR5 on cell surface of melanoma cells. A and C, LU1205 melanoma cells have been stably transfected with the empty vector (LU1205-neo) or with dominant-negative constructs Dyn-2K44A and Dyn-2FF. Surface expression of DR5 and Fas was detected using the correspondent mAbs and FACS analysis 18 h after γ-irradiation (2.5 Gy). B and D, cells were sequentially γ-irradiated (2.5 Gy) and, after 24 h, treated with TRAIL (50 ng/mL) + cycloheximide (2 μg/mL) for an additional 24 hor withFasL (50 ng/mL) + cycloheximide for an additional 24 h. Apoptosis levels were determined as percentage of cells with hypodiploid content of DNA in the pre-G0-G1 region using flow cytometry.
α-Particle irradiation of melanoma cells
An increased effectiveness of α-particle irradiation versus γ-irradiation has been shown in numerous studies for both initial and delayed responses to radiation, including development of apoptosis. Therefore, α-particles are very promising tool for cancer therapy, especially for isolated metastatic cells, as is observed in leukemia because of their high linear energy transfer and short effective path length. Our experiments showed that α-particle irradiation of melanoma cells, similarly to γ-irradiation, increased surface expression of death receptors in a dose-dependent manner in both WM9 (Fig. 5A) and LU1205 cells (data not shown). Furthermore, a combination of low-dose α-particle irradiation with decreased concentrations of soluble TRAIL may also effectively induce TRAIL-mediated apoptosis in WM9 cells (Fig. 5B), providing additional flexibility for rational treatment design. Indeed, exposure of WM9 cells to α-particles (1 Gy) caused accelerating apoptosis induced by lower doses of TRAIL (20 ng/mL instead 50 ng/mL; Fig. 5B).
Figure 5.
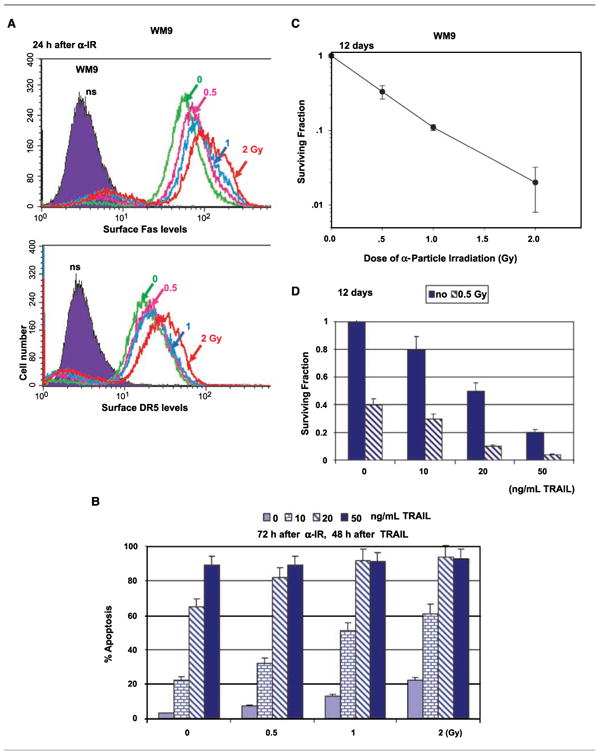
Dose-dependent effects of α-particle irradiation on TRAIL-mediated apoptosis in WM9 human melanoma cells. A, surface expression of Fas and DR5 was determined by staining withPE-labeled anti-Fas or anti-DR5 mAbs and FACS analysis 24 hafter α-irradiation (α-IR). B, 24 hafter α-particle irradiation, WM9 cells were treated withindicated concentrations of soluble TRAIL for an additional 48 h. Apoptotic levels were determined by propidium iodide staining DNA and FACS analysis. C and D, clonogenic survival of WM9 cells 12 d after α-particle irradiation. Presence of TRAIL (10–50 ng/mL) decreased cell survival after irradiation with 0.5 Gy (D).
As expected, α-particle irradiation by itself shows dose-dependent effects on cancer cell survival 12 days after exposure (Fig. 5C). Moreover, sequential treatment with low-dose α-particle irradiation (0.5 Gy) and TRAIL (10–50 ng/mL) additionally decreased cell survival and accelerated cancer cell death conditions (Fig. 5D), which might substantially improve the effectiveness of treatment.
Sodium arsenite treatment may partially substitute recombinant TRAIL
Both types of treatment (sodium arsenite and γ-irradiation) caused up-regulation of the TRAIL promoter activity in melanomas (Fig. 6A). The TRAIL promoter contains binding sites for transcription factors NF-κB, AP-1, and STAT (11, 38). Permanently active MEKK1Δ, which activates both the NF-κB and MAPK signaling pathways, strongly induced the TRAIL promoter activity in transfected melanoma cells, whereas permanently active IKKhS178E/S181E and MKK7-β1 (JNK kinase) showed average induction of the TRAIL promoter activity (Fig. 6A). In contrast, superstable inhibitor of NF-κB, IκBαΔN, dominant-negative JNK1-APF, and dominant-negative form of cJun, TAM67, partially suppressed the basal TRAIL promoter activity. Permanently active AKTmyr, which inhibited JNK activity, also strongly blocked TRAIL promoter activity (Fig. 6A).
Figure 6.
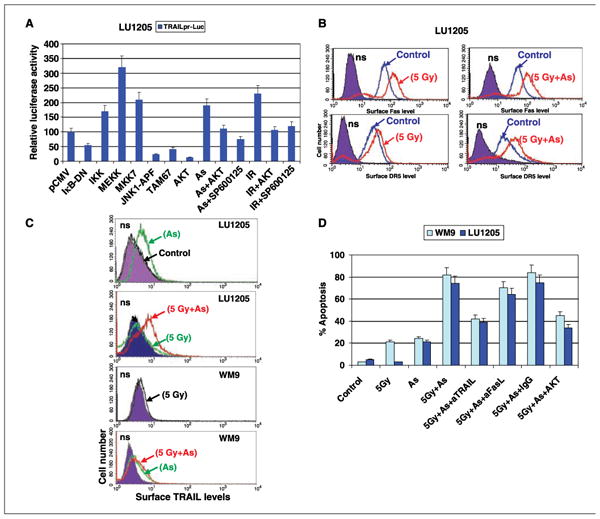
Sequential treatment with γ-irradiation and sodium arsenite significantly accelerates apoptosis of melanoma cells. A, transient transfection of TRAILpr-Luc and indicated expression vectors encoding dominant-negative and permanently active forms of protein (see Materials and Methods) in the presence of β-galactosidase expression construct (at plasmid DNA ratio of 1:3:0.5) into LU1205 cells. Sixteen hours after transfections, cells were not treated or additionally treated with γ-irradiation (IR; 5 Gy), sodium arsenite (As; 5 μmol/L), and SP600125 (10 μmol/L). Normalized luciferase activity was determined after an additional 6 h. B, LU1205 cells were γ-irradiated (5 Gy). Twenty-four hours after, cells were additionally treated with sodium arsenite (5 μmol/L, 8 h). Surface expression of Fas and DR5 was determined by staining with PE-labeled anti-Fas and anti-DR5 mAbs, respectively, and FACS analysis. C, sodium arsenite (5 μmol/L, 8 h) induced TRAIL translocation to cell surface in melanoma cells. Cells were treated by γ-irradiation (5 Gy) or by sodium arsenite alone (5 μmol/L, 8 h) or in combination with γ-irradiation (5 Gy). Surface levels of TRAIL were determined by FACS analysis using anti-TRAIL-PE mAb. D, WM9 and LU1205 cells were γ-irradiated (5 Gy). Twenty-four hours after, cells were treated with sodium arsenite (5 μmol/L) in the presence or absence of indicated mAbs (2 μg/mL). Melanoma cells stably transfected withAKTmyr were also used. Apoptosis levels were determined after an additional 24 h as percentage of cells with hypodiploid content of DNA in the pre-G0-G1 region using flow cytometry. Columns, mean of three independent experiments; bars, SD.
Effects of MEKK1Δ and IKKβ overexpression on NF-κB activity and negative effects of AKT overexpression on JNK activity in LU1205 cells have been described in our previous publications (33, 39). Arsenite-induced or γ-irradiation–induced TRAIL promoter activation is primarily mediated by JNK-cJun and could be suppressed with pharmacologic inhibitor of JNK, SP600125 (10 μmol/L; Fig. 6A). The NF-κB signaling pathway seems to play an additional role in the induction of TRAIL promoter only after γ-irradiation.
Our recent observations indicate that metastatic melanomas contain a pool of intracellular TRAIL that could be transferred to the cell surface following sodium arsenite treatment (16). Interestingly, in contrast to arsenite, γ-irradiation did not efficiently induce TRAIL translocation to cell surface in melanomas (Fig. 6C). Biochemical events in TRAIL translocation from cytoplasm to the cell surface are still largely unknown.
Because sodium arsenite induced surface expression of endogenous TRAIL, we decided to use arsenite instead of soluble TRAIL in a combined treatment with irradiation. We γ-irradiated (5 Gy) LU1205 and WM9 melanoma cells and observed increasing death receptor surface expression, which was followed after 24 h by an induction of surface expression of TRAIL using arsenite (5 μmol/L, 24 h; Fig. 6B and C). Furthermore, both types of treatment (arsenite and γ-irradiation) down-regulated cFLIP levels in LU1205 cells (Fig. 2B; ref. 16). Therefore, we detected strong synergistic effects in the development of apoptosis in melanoma lines LU1205 and WM9 after combined treatment (Fig. 6D). Introduction of anti-TRAIL mAb (2 μg/mL) in the cell medium partially protected melanoma cells against apoptosis, further highlighting a role of TRAIL-mediated signaling in the development of apoptosis (Fig. 6D). FasL-Fas pathway was only slightly involved in the mediation of apoptosis after such combined treatment of melanomas because arsenite was nonefficient inducer of FasL translocation (15). A substantial proportion of TRAIL-independent apoptosis that was observed in these conditions, however, indicated the multiple effects of sodium arsenite in acceleration of apoptosis.
On the other hand, overactivation of AKT in LU1205 cells stably transfected with AKTmyr (39) was accompanied by down-regulation of basal DR5 surface levels and suppression of an inducible increase in these levels after irradiation and arsenite treatment. AKT overexpression also partially suppressed an induction of surface expression of TRAIL following treatment of melanoma cells with arsenite (data not shown). Finally, it resulted in partial suppression of TRAIL-mediated apoptosis in melanoma cells on these conditions. Taken together, the results of our study show that sequential treatment of melanoma cells with γ-radiation (for increasing DR5 surface levels) and sodium arsenite (for induction of endogenous TRAIL translocation to the cell surface and suppression of cFLIP) efficiently induces apoptotic signaling probably via paracrine TRAIL/TRAIL-receptor mechanisms. The activation of the PI3K-AKT pathway may suppress the development of apoptotic signaling in these conditions at the multiple levels.
Discussion
The resistance to TRAIL- or FasL-mediated apoptosis in cancer cells remains a critical problem for the successful application of recombinant TRAIL and FasL in anticancer therapy (40). The strong toxicity of soluble FasL or agonistic anti-Fas mAbs rendered Fas-mediated apoptotic signaling ineffective for in vivo applications. In contrast, soluble recombinant TRAIL/APO2-L was substantially less toxic after systemic treatment as shown in numerous experiments with different types of tumors in vivo (41), although distinct recombinant versions of TRAIL showed differential hepatotoxicity (42).
Resistance of cancer cells to TRAIL can occur at different steps in the death signaling pathways. However, decreased levels of surface expression of death receptors and simultaneous over-expression of inhibitors of apoptosis, such as cFLIP or XIAP, seem to be the most frequent events (13). On the other hand, radiotherapy is widely used for cancer treatment, but the resistance to radiation is a serious problem for melanoma treatment. Our attempts to increase radiation-induced death of melanoma cells via silencing COX-2 were unsuccessful due to strong up-regulation of p21-WAF1 levels and dramatic increase in the level of G2-M arrest in melanoma cells that competed with induction of cell death pathway. Recent studies done on other cancer cell lines also showed negative effects of p21 on irradiation-induced apoptosis (43). However, in this case, the main target was cyclin-dependent kinase–mediated caspase-9 activation that does not occur in our cell lines after γ-irradiation that shows caspase independence.
Additional effects of α-particle irradiation and γ-irradiation (that were complementary to direct induction of cell death) have been observed in the present study, such as a strong up-regulation of surface expression of death receptors, Fas and DR5, and down-regulation of an antiapoptotic protein cFLIP in melanoma cells. Some of these effects have been previously shown for prostate cancer cells (44) and leukemic cells (45). In the absence of death ligands, irradiation often did not induce a rapid apoptotic signaling, leaving cells arrested at the G2-M phase that may result in slow necrotic death. However, subsequent treatment of irradiated cells with re-combinant TRAIL was very effective in the initiation of melanoma cell apoptosis similar to previous finding in prostate cancer cells (44).
It is well known that γ-irradiation can affect transcriptional activity of the promoters of death receptor genes, especially DR5 and Fas, via p53 and NF-κB activation (22, 46). JNK, however, plays opposite roles in the regulation of Fas and DR5 promoter activities, with JNK-cJun as a negative regulator of the Fas promoter (39) and JNK-SP1 as a positive regulator of the DR5 promoter (47). In contrast, PI3K-AKT pathway that suppresses JNK and STAT3 up-regulates Fas gene expression and translocation (39) but has negative effects on DR5 expression and subsequent TRAIL-mediated apoptosis as we observed in the present study.
The post-translational levels of the death receptor expression, including translocation of receptor precursors in the cell, have not been well studied. The present study further shows a role of dynamin-2, a large GTFase that supports vesicle trafficking, in the regulation of death receptor translocation not only for Fas receptor (33) but also for DR5. Treatment by γ-radiation is linked with substantial up-regulation of dynamin-2 levels in the cell (48) that probably facilitates protein transport from trans-Golgi network to cell surface and results in efficient up-regulation of surface expression of death receptors.
An important feature of many cancer cells, including advanced melanomas, is the presence of the intracellular pools of TRAIL (16) or FasL (14) that could be expressed on cell surface at the specific circumstances (e.g., under stress conditions). An implication of expression of death ligands by cancer cells and their probable role in tumor counterattack is still subject of numerous investigations (42). Based on our recent study (16), we have used a treatment with sodium arsenite that may induce translocation of endogenous TRAIL to cell surface with simultaneous strong down-regulation of cFLIP antiapoptotic protein levels. In the present study, such sequential treatment of melanoma cells with γ-irradiation and sodium arsenite resulted in an induction of apoptosis, which is partially mediated by TRAIL/TRAIL-receptor via paracrine mechanisms, in initially resistant LU1205 melanoma cells.
Arsenite is a two-sided sword: on the one hand, it is a well-established human carcinogen, and on the other hand, it has been used successfully in the treatment of acute promyelocytic leukemia by inducing apoptosis (49). However, most melanomas are resistant to sodium arsenite treatment. We have previously shown that only ∼20% of human melanoma cell lines are sensitive to apoptosis induction by arsenite alone (29). A recent clinical trial in the use of single regimen of arsenite trioxide in the treatment of metastatic melanoma failed to produce any significant improvement in clinical outcome (50). These findings suggest that multimodality regimen is required in the design of treatment strategy, a primary motivation for the present study. A better understanding in the apoptotic signaling pathways induced by arsenic treatment in melanoma cells with concurrent modulator of proapoptotic and suppression of antiapoptotic pathways will provide a useful mechanistic rationale for effective treatment design for this often fatal cancer.
Acknowledgments
Grant support: NIH grants CA 49062 and ES 11804, Superfund grant P42 ES 10349, Environmental Center grant P30 ES 09089, and National Institute of Environmental Health Sciences Center Pilot Award.
We thank Drs. Z. Ronai, R. Davis, M. Karin, W.S. El-Deiry, G.J. Gores, J. Hiscott, S-Y. Sun, A.N. Shajahan, and H. Wajant for plasmid constructs and Dr. M.A. Partridge for critical reading of the manuscript.
References
- 1.Perlis C, Herlyn M. Recent advances in melanoma biology. Oncologist. 2004;9:182–7. doi: 10.1634/theoncologist.9-2-182. [DOI] [PubMed] [Google Scholar]
- 2.Atkins MB. Cytokine-based therapy and biochemotherapy for advanced melanoma. Clin Cancer Res. 2006;12:2353–8s. doi: 10.1158/1078-0432.CCR-05-2503. [DOI] [PubMed] [Google Scholar]
- 3.Ashkenazi A, Pai RC, Fong S, et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–62. doi: 10.1172/JCI6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walczak H, Miller RE, Ariail K, et al. Tumoricidal activity of tumor necrosis factor-related apoptosisinducing ligand in vivo. Nat Med. 1999;5:157–63. doi: 10.1038/5517. [DOI] [PubMed] [Google Scholar]
- 5.Ashkenazi A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat Rev Cancer. 2002;2:420–30. doi: 10.1038/nrc821. [DOI] [PubMed] [Google Scholar]
- 6.Hersey P, Zhang XD. How melanoma cells evade trailinduced apoptosis. Nat Rev Cancer. 2001;1:142–50. doi: 10.1038/35101078. [DOI] [PubMed] [Google Scholar]
- 7.Debatin KM, Krammer PH. Death receptors in chemotherapy and cancer. Oncogene. 2004;23:2950–66. doi: 10.1038/sj.onc.1207558. [DOI] [PubMed] [Google Scholar]
- 8.Karin M. Nuclear factor-κB in cancer development and progression. Nature. 2006;441:431–6. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- 9.Dong J, Phelps RG, Qiao R, et al. BRAF oncogenic mutations correlate with progression rather than initiation of human melanoma. Cancer Res. 2003;63:3883–5. [PubMed] [Google Scholar]
- 10.Chen X, Thakkar H, Tyan F, et al. Constitutively active Akt is an important regulator of TRAIL sensitivity in prostate cancer. Oncogene. 2001;20:6073–83. doi: 10.1038/sj.onc.1204736. [DOI] [PubMed] [Google Scholar]
- 11.Ravi R, Bedi GC, Engstrom LW, et al. Regulation of death receptor expression and TRAIL/Apo2L-induced apoptosis by NF-κB. Nat Cell Biol. 2001;3:409–16. doi: 10.1038/35070096. [DOI] [PubMed] [Google Scholar]
- 12.Chipuk JE, Green DR. Dissecting p53-dependent apoptosis. Cell Death Differ. 2006;13:994–1002. doi: 10.1038/sj.cdd.4401908. [DOI] [PubMed] [Google Scholar]
- 13.Fulda S, Debatin KM. Extrinsic versus intrinsic apoptosis pathways in anticancer chemotherapy. Oncogene. 2006;25:4798–811. doi: 10.1038/sj.onc.1209608. [DOI] [PubMed] [Google Scholar]
- 14.Hahne M, Rimoldi D, Schroter M, et al. Melanoma cell expression of Fas(Apo-1/CD95) ligand: implications for tumor immune escape. Science. 1996;274:1363–6. doi: 10.1126/science.274.5291.1363. [DOI] [PubMed] [Google Scholar]
- 15.Ivanov VN, Hei TK. Dual treatment with COX-2 inhibitor and sodium arsenite leads to induction of surface Fas ligand expression and Fas-ligand-mediated apoptosis in human melanoma cells. Exp Cell Res. 2006;312:1401–17. doi: 10.1016/j.yexcr.2006.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ivanov VN, Hei TK. Sodium arsenite accelerates TRAIL-mediated apoptosis in melanoma cells through upregulation of TRAIL-R1/R2 surface levels and down-regulation of cFLIP expression. Exp Cell Res. 2006;312:4120–38. doi: 10.1016/j.yexcr.2006.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Satyamoorthy K, DeJesus E, Linnenbach AJ, et al. Melanoma cell lines from different stages of progression and their biological and molecular analyses. Melanoma Res. 1997;7(Suppl 2):S35–42. [PubMed] [Google Scholar]
- 18.van Dam H, Huguier S, Kooistra K, et al. Autocrine growth and anchorage independence: two complementing Jun-controlled genetic programs of cellular transformation. Genes Dev. 1998;12:1227–39. doi: 10.1101/gad.12.8.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang HY, Zhao K, Pizzolato JF, Fonarev M, Langer JC, Manfredi JJ. Constitutive expression of the cyclin dependent kinase inhibitor p21 is transcriptionally regulated by the tumor suppressor protein p53. J Biol Chem. 1998;273:29156–63. doi: 10.1074/jbc.273.44.29156. [DOI] [PubMed] [Google Scholar]
- 20.Baetu TM, Kwon H, Sharma S, Grandvaux N, Hiscott J. Disruption of NF-κB signaling reveals a novel role for NF-κB in the regulation of TNF-related apoptosis-inducing ligand expression. J Immunol. 2001;167:3164–73. doi: 10.4049/jimmunol.167.6.3164. [DOI] [PubMed] [Google Scholar]
- 21.Guan B, Yue P, Lotan R, Sun SY. Evidence that the human death receptor 4 is regulated by activator protein 1. Oncogene. 2002;21:3121–9. doi: 10.1038/sj.onc.1205430. [DOI] [PubMed] [Google Scholar]
- 22.Takimoto R, El-Deiry WS. Wild-type p53 transacti-vates the KILLER/DR5 gene through an intronic sequence-specific DNA-binding site. Oncogene. 2000;19:1735–43. doi: 10.1038/sj.onc.1203489. [DOI] [PubMed] [Google Scholar]
- 23.Bartke T, Siegmund D, Peters N, et al. p53 upregulates cFLIP, inhibits transcription of NF-κB-regulated genes and induces caspase-8-independent cell death in DLD-1 cells. Oncogene. 2001;20:571–80. doi: 10.1038/sj.onc.1204124. [DOI] [PubMed] [Google Scholar]
- 24.Ricci MS, Jin Z, Dews M, et al. Direct repression of FLIP expression by c-myc is a major determinant of TRAIL sensitivity. Mol Cell Biol. 2004;24:8541–55. doi: 10.1128/MCB.24.19.8541-8555.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brockman JA, Scherer DC, McKinsey TA, et al. Coupling of a signal response domain in IκBα to multiple pathways for NF-κB activation. Mol Cell Biol. 1995;15:2809–18. doi: 10.1128/mcb.15.5.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. The IκB kinase complex (IKK) contains two kinase subunits, IKKα and IKKβ, necessary for IκB phosphorylation and NF-κB activation. Cell. 1997;91:243–52. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 27.Minden A, Lin A, McMahon M, et al. Differential activation of ERK and JNK mitogen-activated protein kinases by Raf-1 and MEKK. Science. 1994;266:1719–23. doi: 10.1126/science.7992057. [DOI] [PubMed] [Google Scholar]
- 28.Tournier C, Whitmarsh AJ, Cavanagh J, Barrett T, Davis RJ. The MKK7 gene encodes a group of c-Jun NH2-terminal kinase kinases. Mol Cell Biol. 1999;19:1569–81. doi: 10.1128/mcb.19.2.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ivanov VN, Hei TK. Arsenite sensitizes human melanomas to apoptosis via tumor necrosis factor α-mediated pathway. J Biol Chem. 2004;279:22747–58. doi: 10.1074/jbc.M314131200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiao C, Yang BF, Song JH, Schulman H, Li L, Hao C. Inhibition of CaMKII-mediated c-FLIP expression sensitizes malignant melanoma cells to TRAIL-induced apoptosis. Exp Cell Res. 2005;304:244–55. doi: 10.1016/j.yexcr.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 31.Satyamoorthy K, Chehab NH, Waterman MJ, et al. Aberrant regulation and function of wild-type p53 in radioresistant melanoma cells. Cell Growth Differ. 2000;11:467–74. [PubMed] [Google Scholar]
- 32.Chang L, Kamata H, Solinas G, et al. The E3 ubiquitin ligase itch couples JNK activation to TNFα-induced cell death by inducing c-FLIP(L) turnover. Cell. 2006;124:601–13. doi: 10.1016/j.cell.2006.01.021. [DOI] [PubMed] [Google Scholar]
- 33.Ivanov VN, Ronai Z, Hei TK. Opposite roles of FAP-1 and dynamin in the regulation of Fas (CD95) translocation to the cell surface and susceptibility to Fas ligandmediated apoptosis. J Biol Chem. 2006;281:1840–52. doi: 10.1074/jbc.M509866200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cao H, Weller S, Orth JD, et al. Actin and Arf1-dependent recruitment of a cortactin-dynamin complex to the Golgi regulates post-Golgi transport. Nat Cell Biol. 2005;7:483–92. doi: 10.1038/ncb1246. [DOI] [PubMed] [Google Scholar]
- 35.Ivanov VN, Lopez Bergami P, Maulit G, Sato TA, Sassoon D, Ronai Z. FAP-1 association with Fas (Apo-1) inhibits Fas expression on the cell surface. Mol Cell Biol. 2003;23:3623–35. doi: 10.1128/MCB.23.10.3623-3635.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shajahan AN, Timblin BK, Sandoval R, Tiruppathi C, Malik AB, Minshall RD. Role of Src-induced dynamin-2 phosphorylation in caveolae-mediated endocytosis in endothelial cells. J Biol Chem. 2004;279:20392–400. doi: 10.1074/jbc.M308710200. [DOI] [PubMed] [Google Scholar]
- 37.Cao H, Thompson HM, Krueger EW, McNiven MA. Disruption of Golgi structure and function in mammalian cells expressing a mutant dynamin. J Cell Sci. 2000;113:1993–2002. doi: 10.1242/jcs.113.11.1993. [DOI] [PubMed] [Google Scholar]
- 38.Wang Q, Ji Y, Wang X, Evers BM. Isolation and molecular characterization of the 5′-upstream region of the human TRAIL gene. Biochem Biophys Res Commun. 2000;276:466–71. doi: 10.1006/bbrc.2000.3512. [DOI] [PubMed] [Google Scholar]
- 39.Ivanov VN, Krasilnikov M, Ronai Z. Regulation of Fas expression by STAT3 and c-Jun is mediated by phosphatidylinositol 3-kinase-AKT signaling. J Biol Chem. 2002;277:4932–44. doi: 10.1074/jbc.M108233200. [DOI] [PubMed] [Google Scholar]
- 40.Shankar S, Srivastava RK. Enhancement of therapeutic potential of TRAIL by cancer chemotherapy and irradiation: mechanisms and clinical implications. Drug Resist Updat. 2004;7:139–56. doi: 10.1016/j.drup.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 41.Almasan A, Ashkenazi A. Apo2L/TRAIL: apoptosis signaling, biology, and potential for cancer therapy. Cytokine Growth Factor Rev. 2003;14:337–48. doi: 10.1016/s1359-6101(03)00029-7. [DOI] [PubMed] [Google Scholar]
- 42.Wajant H. CD95L/FasL and TRAIL in tumour surveillance and cancer therapy. Cancer Treat Res. 2006;130:141–65. doi: 10.1007/0-387-26283-0_7. [DOI] [PubMed] [Google Scholar]
- 43.Sohn D, Essmann F, Schulze-Osthoff K, Janicke RU. p21 blocks irradiation-induced apoptosis downstream of mitochondria by inhibition of cyclin-dependent kinase mediated caspase-9 activation. Cancer Res. 2006;66:11254–62. doi: 10.1158/0008-5472.CAN-06-1569. [DOI] [PubMed] [Google Scholar]
- 44.Shankar S, Singh TR, Srivastava RK. Ionizing radiation enhances the therapeutic potential of TRAIL in prostate cancer in vitro and in vivo: intracellular mechanisms. Prostate. 2004;61:35–49. doi: 10.1002/pros.20069. [DOI] [PubMed] [Google Scholar]
- 45.Gong B, Almasan A. Apo2 ligand/TNF-related apoptosis-inducing ligand and death receptor 5 mediate the apoptotic signaling induced by ionizing radiation in leukemic cells. Cancer Res. 2000;60:5754–60. [PubMed] [Google Scholar]
- 46.Wang S, El-Deiry WS. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003;22:8628–33. doi: 10.1038/sj.onc.1207232. [DOI] [PubMed] [Google Scholar]
- 47.Higuchi H, Grambihler A, Canbay A, Bronk SF, Gores GJ. Bile acids up-regulate death receptor 5/TRAIL-receptor 2 expression via a c-Jun N-terminal kinase-dependent pathway involving Sp1. J Biol Chem. 2004;279:51–60. doi: 10.1074/jbc.M309476200. [DOI] [PubMed] [Google Scholar]
- 48.Qian J, Yang J, Dragovic AF, Abu-Isa E, Lawrence TS, Zhang M. Ionizing radiation-induced adenovirus infection is mediated by Dynamin 2. Cancer Res. 2005;65:5493–7. doi: 10.1158/0008-5472.CAN-04-4526. [DOI] [PubMed] [Google Scholar]
- 49.Zhao WL, Chen SJ, Shen Y, et al. Treatment of acute promyelocytic leukemia with arsenic trioxide: clinical and basic studies. Leuk Lymphoma. 2001;42:1265–73. doi: 10.3109/10428190109097751. [DOI] [PubMed] [Google Scholar]
- 50.Kim KB, Bedikian AY, Camacho LH, Papadopoulos NE, McCullough C. A phase II trial of arsenic trioxide in patients with metastatic melanoma. Cancer. 2005;104:1687–92. doi: 10.1002/cncr.21386. [DOI] [PubMed] [Google Scholar]