

Abstract
We show here that combining two existing genome wide association studies (GWAS) yields additional biologically relevant information, beyond that obtained by either GWAS separately. We propose Joint GWAS Analysis, a method that compares a pair of GWAS for similarity among the top SNP associations, top genes identified, gene functional clusters, and top biological pathways. We show that Joint GWAS Analysis identifies additional enriched biological pathways that would be missed by traditional Single-GWAS analysis. Furthermore, we examine the similarities of six complex genetic disorders at the SNP-level, gene-level, gene-cluster-level, and pathway-level. We make concrete hypotheses regarding novel pathway associations for several complex disorders considered, based on the results of Joint GWAS Analysis. Together, these results demonstrate that common complex disorders share substantially more genomic architecture than has been previously realized and that the meta-analysis of GWAS needs not be limited to GWAS of the same phenotype to be informative.
Keywords: Systems genetics, GWAS, Meta-analysis, Pathway enrichment, Pleiotropy
Introduction
Genome Wide Association Studies (GWAS) have resulted in many replicated single nucleotide polymorphisms (SNPs) that show modest effects on everything from human height [22] and body mass index [42] to cancer metastasis [11] and drug efficacy [8]. However, most GWAS identify only a handful of SNPs that meet multiple testing correction criteria for statistical significance, and the desire to mine additional biological data from GWAS has resulted in various adjunct statistical methods including 1) enrichment of biological pathways, termed “pathway analysis” [41] and 2) combination of multiple GWAS of the same phenotype, or GWAS meta-analysis [10], [46]. We suggest here a method we call Joint GWAS Analysis that is a combination of pathway- and meta-analysis, but one that is not limited to the analysis of GWAS of the same trait or disease. Instead, we leverage widespread pleiotropy of complex disease [35] to gain increased biological insight by comparing potentially unrelated GWAS. This is a method that can be understood as an alternative or as a complementary approach to a standard meta-analysis.
A small portion of the top SNPs in a GWAS are usually prioritized for further study or replication in additional populations [36], typically those SNPs reaching p < 5 ∗ 10^ − 8. This is a conservative strategy designed to minimize false-positive associations, while missing many true-positive associations that do not meet statistical significance. In contrast, pathway analysis methods are a companion to GWAS studies that consider much larger proportions of the top SNPs and investigate their aggregate associations to known biological groupings or metabolic pathways [12]. Pathway analysis studies have been successful in identifying additional biological insight and finding groupings of genes that represent biological disease processes [28]. These kinds of approaches have shown the value in considering many more of the top GWAS SNPs, even though those hits are more likely to include false-positive associations.
At the same time, the modern picture of the genetic architecture of common complex disorders has become much more broad-based than traditionally supposed [36], with most disorders and complex traits thought to have many variants of small effect [40], [43]. A study of the entire NHGRI GWAS catalog [16], which archives all SNP-phenotype associations from GWAS reported in the literature, identified 16% of genes and 4.6% of GWAS SNPs to be associated with more than one cataloged condition or trait [33]. Furthermore, these variants are increasingly realized to be shared across similar conditions and traits, including: height and body mass index [15]; cognitive and learning abilities [39]; autoimmune disorders [29], [32]; and cardiovascular diseases [13]. Genes have been shown to affect disparate phenotypes as well, including prostate cancer and type 2 diabetes [14], and more general studies of human gene pleiotropy have shown qualitative differences between pleiotropic genes that influence related and unrelated traits [6].
We propose that any time two diseases may have common biological causes or etiology, comparing the GWAS of the two diseases may lead to greater understanding of either disease than was possible in separate analyses. In this study we explore the comparison of two GWAS of similar and of disparate phenotypes. Our hypothesis is that by comparing the GWAS of two complex genetic diseases, those variants that exhibit moderate evidence of association with both disease phenotypes are more likely to represent genomic loci truly associated with each of the diseases, and thus provide an important source of additional biological insight. We show that this comparison does lead to novel biological pathways associated with disease phenotypes, and furthermore that the two complex disorders need not be commonly considered to have a clinical relationship to have common genetic risk factors. Our method, Joint GWAS Analysis, is based upon the enrichment of top SNPs in a pair of GWAS. We show that this method identifies increasingly more information biologically related to the phenotypes as one transitions from small-scale genomic resolution at SNPs, to genes, to gene groups, and finally to the large-scale resolution of biological pathways.
We demonstrate this using six published GWAS from the Welcome Trust Case Control Consortium (WTCCC), on six different diseases that have varying degrees of etiological similarity. We consider the genome-wide SNP data from WTCCC on different populations of 2000 patients with one of bipolar disorder (BP), coronary artery disease (CAD), Crohn's disease (CD), rheumatoid arthritis (RA), type 1 diabetes (T1D), type 2 diabetes (T2D); and 3000 common controls. We then conduct pair-wise comparisons of these six GWAS, at the SNP-level, the gene-level, gene-cluster level, and the pathway-level. We show that Joint GWAS Analysis results in increased biological insight at the pathway level for several pairs of the WTCCC diseases, above what is identifiable from a similar pathway analysis of a single GWAS.
Methods
GWAS methods
We obtained genome-wide SNP data from the Welcome Trust Consortium on six different cohorts for six common complex disorders (BP, CAD, CD, RA, T1D, and T2D) and a control cohort, all genotyped on the 500 k Affymetrix gene chip (Affymetrix). More information on the genotyping and inclusion criteria are available from the WTCCC publications (2007). We performed simple case–control GWAS on each of the six WTCCC diseases by comparing each of the disease populations (n = 2000) to the common control group (n = 3000). We followed advice from the original WTCCC GWAS publication [1] on how to filter for spurious SNP associations and control for genomic stratification, performing our GWAS after removing SNPs with Hardy–Weinberg Equilibrium (HWE) probability test scores lower than < 0.001, minor allele frequency < 0.05, missingness > 0.001, and individuals more than four standard deviations from the mean on any of the top six genotype principal components; and obtained similar results as the original authors. We then selected from each GWAS a common panel of ~ 100,000 tag-SNPs that were in less than r2 = 0.3 linkage disequilibrium. GWAS, filtering, and linkage-disequilibrium pruning were performed using PLINK [26]. Outliers with extremely low P values in each GWAS were removed by checking for nearby SNPs with similar p-values; this accomplished outlier removal similar to that described by WTCCC (2007) to remove spurious associations driven by genotyping errors.
Joint GWAS SNP list selection
For each pair of GWAS, we considered a “Joint GWAS” where one disease in the pair is the “Target Disease” and the other is the “Cross Disease” (and similarly, we refer to “Target GWAS” and “Cross GWAS”). A glossary of terms defined appears at the end of this work. We constructed a “Joint GWAS SNP list" of SNPs for each pair of GWAS by performing the following protocol (see also Fig. 1).
-
1)
We sorted the SNPs of both GWAS by their statistical association to their own phenotype in decreasing order of significance.
-
2)
We considered an increasing subset of the top M SNPs. We started by considering the top M = 1 SNPs, and increased M by one until M reached the total number of tag SNPs.
-
3)
At each size M, we identified the set of “Common SNPs” that was present in the top M SNPS of both Target and Cross GWAS. We obtained p-values for the enrichment of Common SNPs for each value of M from the hypergeometric distribution.
-
4)
The size M such that the hypergeometric p-value is a minimum over all window-sizes was chosen as the SNP rank cutoff value.
-
5)
The Joint GWAS SNP list is the set of Common SNPs when M is equal to the SNP rank cutoff value. The Joint GWAS SNP list of length Nsnp.
Fig. 1.

Schematic of Joint GWAS Analysis. In Joint GWAS Analysis, two GWAS of different diseases are compared for enrichment of top SNP hits. Common SNPs occurring prior to the point of maximum enrichment become the “Joint GWAS SNPs.” These SNPs are then mapped to genes to make the Joint GWAS gene list. From these genes, enriched pathways are computed.
We used Joint GWAS SNP lists constructed this way in the rest of the study. Fig. 1 shows a schematic of the dataflow and study design used in this work, starting with the enrichment of paired GWAS SNPs and the creation of the Joint GWAS SNP list, and following the Joint GWAS SNP list all the way to the pathway level.
SNP comparison methods
To make a comparison that demonstrates the difference between the Joint GWAS method and standard GWAS pathway analysis methods, we made a list of “Target GWAS SNPs” for the Target Disease. This was composed of the top Nsnp SNPs from the Target GWAS, where Nsnp was the size of the Joint GWAS SNP list. We used the NHGRI GWAS catalog [16] as a reference of known disease SNPs discovered by GWAS. SNPs listed in the catalog for any GWAS of the Target Disease were selected to form a reference “NHGRI Disease SNP list" for the Target Disease.
SNPs in the Joint GWAS or Target GWAS SNP lists were considered to match SNPs in the NHGRI Disease SNP list if they were within a linkage disequilibrium tolerance of r2 = 0.3. We computed SNP LD distances by using a cohort of Caucasians imputed to 1000-Genomes [2], comprising over six million imputed SNPs. Using this reference group, we checked the linkage disequilibrium between SNPs using PLINK.
Gene comparison methods
We translated the Joint GWAS SNP list to an associated “Joint GWAS gene list” by using the UCSC Genome Browser (build HG18, which corresponds to the genotyping done by WTCCC on the six GWAS). In cases where one SNP mapped to multiple genes, we included all genes. As with our comparison at the SNP level, we made a list of the Target GWAS genes to serve as a point of comparison. This “Target GWAS gene list” was composed of the top Ng genes of the Target GWAS, where Ng is the size of the Joint GWAS genes list, and the genes are ordered by the p-value of the SNP within the gene that has the lowest p-value for association with the Target Disease.
We used the genes reported in the NHGRI catalog for all GWAS fitting the Target Disease as the reference for comparison. Matching between genes in the Joint GWAS gene list or the Target GWAS gene list to the NHGRI Disease gene list was performed by checking the lists for the same gene names.
Gene cluster methods
We used the DAVID (Database for Annotation, Visualization and Integrated Discovery) pathway enrichment online tool [19] to obtain functional clusters for the genes in the Joint GWAS, Target GWAS, and NHGRI Disease gene lists. In order to use DAVID's web services interface, we first translated the gene list from canonical gene names to mRNA reference keys, which we did using a mapping from the UCSC Genome Browser [23]. This resulted in between 84.0% and 89.5% of the genes in each list being successfully mapped and identified by DAVID. We then obtained gene functional clusters from DAVID, allowing the Target GWAS gene list to cluster with genes from the NHGRI list and allowing the Joint GWAS gene list to cluster with genes from the NHGRI list. We defined the number of NHGRI Disease genes matched by the Joint GWAS gene list to be the number of NHGRI Disease genes in clusters with at least one gene from the Joint GWAS gene list; we defined the number of NHGRI genes matched by the Target GWAS gene list in a similar way. We defined any gene from the Joint GWAS gene list that was mapped to a gene cluster including at least one NHGRI Disease gene as a true-positive gene association for the Target Disease. We then computed false positive rates for the Joint GWAS gene list by comparing the number of true-positive gene associations to the size of that list (Table S3). We similarly computed the false-positive rate for the Target GWAS gene list (Table S3).
Pathway cluster methods
We used DAVID to generate enriched pathways of genes from the Joint GWAS gene list and Target GWAS gene lists for each pair of diseases. We used the default settings on DAVID for all DAVID operations, and discarded pathways with significance levels greater than 0.05 and pathway clusters with enrichment scores less than 1.0. We used the NHGRI Disease gene list to obtain enriched pathway clusters using DAVID, that we termed “NHGRI Disease pathways clusters” for the Target Disease. Pathway clusters are groups of overlapping pathways that may be very redundant if considered separately. The genes in the pathways in a single pathway cluster tend to overlap to a large extent; thus, for each pathway cluster, we counted the number of genes from the Joint GWAS gene list (or Target GWAS gene list) participating in one of their own enriched pathways, and that are also members of the NHGRI Disease pathway cluster in question. We compared these two numbers using Chi-square or Fisher's exact tests, and if there is a significant difference we called this cluster significantly “covered” for either the Joint GWAS gene list or the Target GWAS gene list. The numbers of significantly covered NHGRI Disease pathway clusters for the Joint GWAS gene list are reported in Table 5.
Table 5.
Comparison of Joint GWAS gene list pathway coverage vs. Target GWAS gene list pathway coverage. For each WTCCC disease, we compare the number of NHGRI pathway clusters with significantly increased coverage by genes in enriched pathways from the Joint GWAS gene list. Zeros do not necessarily indicate no pathways identified, just that no pathways were identified with greater coverage than obtained by the Target gene list. Results are dependent upon DAVID parameters (significant thresholds, pathway providers included in the aggregation).
| Target disease |
Cross disease (joint GWAS pathway coverage in excess of target GWAS pathway coverage) |
||||||
|---|---|---|---|---|---|---|---|
| Disease | NHGRI pathway clusters (n) | BD | CAD | CD | RA | T1D | T2D |
| BD | 6 | – | 1 | 0 | 1 | 0 | 0 |
| CAD | 12 | 0 | – | 1 | 0 | 1 | 0 |
| CD | 9 | 1 | 0 | – | 2 | 0 | 2 |
| RA | 5 | 0 | 0 | 1 | – | 0 | 1 |
| T1D | 3 | 0 | 0 | 1 | 1 | – | 1 |
| T2D | 9 | 2 | 1 | 1 | 0 | 2 | 0 |
We provide an additional comparison to a simple method of combining pathway analyses: we take the Cross and Target diseases and perform pathway analysis on them separately, then retain the pathways enriched in both analyses. Genes occurring in retained pathways are termed as the “Combined Target-Cross disease gene list” and are used to measure pathway cluster coverage as previously described. We compare the Joint GWAS gene list pathway coverage to the Combined Target-Cross disease gene list pathway coverage in Table S5.
The false-positive rate for NHGRI Disease pathway cluster coverage was computed using the number of genes in the Joint GWAS gene list that were either 1) not present in an enriched pathway in the Joint GWAS gene list or 2) not present in a pathway contained in a NHRGI Disease pathway cluster. This quantity of genes over the size of the Joint GWAS gene list becomes the false-positive rate for NHGRI Disease pathway cluster coverage of the Joint GWAS gene list (Table S3).
Novel pathway association methods
For each Joint GWAS gene list, we identified the genes that did not cover any NHGRI Disease pathway clusters, calling these the “Left-Over gene list.” Using DAVID, we generated enriched pathways from the Left-Over gene list. These pathways are potential novel associations with the Target Disease, which we checked by measuring enrichment of nominally significant SNPs (p < 0.05) in those genes from among the Target GWAS. Pathways passing a nominal significance test (Chi-sq p-value < 0.05) for enrichment were then further assessed for enrichment significance by permutation test.
The permutation test was conducted as follows. To assess the significance of a pathway, we took the genes in that pathway from the Joint GWAS gene list and then selected random sets of genes of the same size. To select genes that had a similar chance of low p-value SNPs, the random set was selected from among the top Ng genes from the target GWAS, where Ng is the total number of genes in the Joint GWAS gene list. The random set of genes was then checked for the number of low-p-value SNPs, those meeting a nominal cut off of 0.05 for association in the Target GWAS. The total number of such SNPs was compared to the expected number of such SNPs using Chi-square or Fisher's exact tests. Since the Target GWAS was already filtered for linkage disequilibrium down to SNPs with no more than r2 = 0.3, linkage disequilibrium was not an important factor in this permutation testing scheme. For each pathway, 1000 such random sets were generated and measured to get an estimate of the null distribution. Pathways with Chi-square or Fisher's exact p-values that were lower than all 1000 of the random values were considered enriched for Target Disease GWAS SNPs.
Pathways enriched for Target Disease GWAS SNPs were considered to be potential novel findings. These pathways may represent 1) novel biological associations to the target disease, 2) biological findings truly associated with this disease, and known to be so, but not previously associated with the Target Disease through a GWAS, and therefore unknown to the NHGRI GWAS catalog, or 3) false-positive results. We identified pathways matching case (2) by conducting PubMed queries containing the conjunction of the Target Disease name and a concise pathway descriptor (Table 6). In some cases, synonymous search terms were used for unusual pathway descriptors, which are indicated by vertical grouping in Table 6.
Table 6.
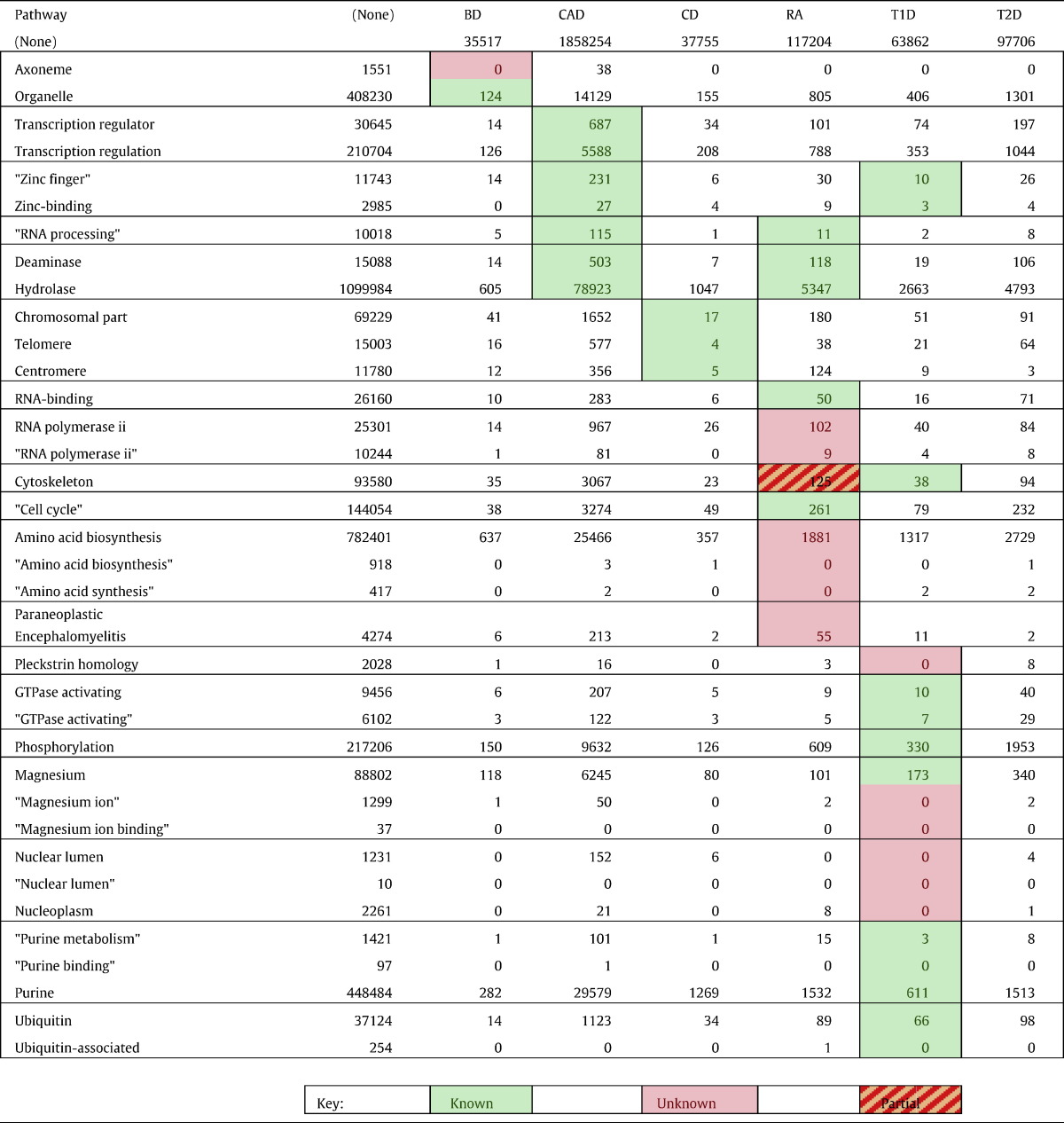
Novel GWAS pathways identified for each WTCCC disease with PubMed citations returned for the conjunction of pathway and disease search terms. Highlighted cells indicate pathways where we hypothesize an association of the pathway to the disease. Other cells are included for completeness, although no hypothesis was indicated. Green highlights indicate pathways where there is evidence in the literature for an association with the WTCCC disease, pink indicates pathways where there does not seem to be evidence of a known association; orange shading indicates pathways where there is indeterminate evidence for an association. Pathway names are summarizations generated by hand, by the authors, where pathway names in grouped rows are synonyms used for PubMed searches. Search terms in quotes were quoted in their submission to PubMed and were required to appear exactly in that order in the abstracts or titles of the research articles in question. Search terms grouped horizontally represent synonymous search terms that were used to get a broader picture of the relationship of the pathway to the six diseases.
All analysis was conducted with MATLAB and Perl code written by the authors. Software available by request.
Null simulation methods
To provide a controlled test of the Joint GWAS Association methodology, we constructed a series of null GWAS that have no biological phenotype. We also constructed a series of Vascular Endothelial Growth Factor (VEGF) Pathway-enhanced GWAS by taking the null GWAS and inserting inflated effect sizes for SNPs appearing in genes appearing in the BioCarta VEGF, Hypoxia, and Angiogenesis pathway (www.BioCarta.com). We generated 20 null-Joint GWAS Analyses by taking the WTCCC controls and randomly splitting them into four groups: cases for Null Target GWAS, controls for Null Target GWAS, cases for Null Cross GWAS, and controls for Null Cross GWAS. For each of these 20 random splits, we then performed two GWAS (the Null Target GWAS, and the Null Cross GWAS), and then performed Joint GWAS Analysis on these two GWAS. For each Null GWAS, we obtained VEGF pathway-enhanced GWAS by using the GCTA software [44] to simulate effect sizes of VEGF pathway SNPs, which we then inserted into the Null GWAS. We then performed Joint GWAS Analysis on 20 pairs of VEGF GWAS. We compare results between Joint GWAS Analysis of Null and of VEGF GWAS (see Supplemental material, Tables S7, S8 and S9).
Results
Using simple case–control designs, we conducted GWAS of each of the six diseases. We obtained similar results to the original WTCCC GWAS (2007). For each pair of the six diseases, we applied Joint GWAS Analysis (see Methods). In each Joint GWAS Analysis, the maximum enrichment occurred when considering the top 12% to 24% of SNPs (Fig. 2, Table 1). We found the strongest enrichment between rheumatoid arthritis and type 1 diabetes (Fig. 2), although this did not result in the most Common SNPs selected (Row “M”, Table 1). The general character of the enrichment for each Joint GWAS pair, as M went from 1 to approximately 100,000, showed marked similarity (Fig. 2). A simulation of 20 null GWAS showed less enrichment than each of the WTCCC Joint GWAS. At each of the SNP, Gene, and Pathway levels we assessed the extent to which the Joint GWAS SNP list revealed known associations to the Target Disease. Known associations are derived from the NHGRI GWAS catalog [16], a reference that includes all published SNP and gene associations for any trait or disease from studies that survey at least 100 k SNPs and that meet a p < 10− 5 statistical significance threshold.
Fig. 2.

Enrichment of Common SNPs for each Joint GWAS Analysis at different values of M. M ranges from zero to approximately 106 k, which represents all SNPs in the GWAS after filtering down to tag SNPs at linkage disequilibrium < 0.3. All fifteen pairs of diseases are represented. Enrichment of 20 null models is shown in gray, computed using a random split of the controls and assigned an arbitrary case/control phenotype. p-Values shown are computed using a hypergeometric distribution test. For each disease pair, the maximum enrichment is highlighted with a circle.
Table 1.
General enrichment characteristics for each Joint GWAS Analysis. Enrichment levels are chosen by peak significance of common SNP enrichment (see Fig. 2). M is the number of SNPs that maximizes that enrichment. Joint GWAS SNPs refers to the number of common SNPs in the two GWAS occurring prior to the peak significance point. Joint GWAS genes are computed from Joint GWAS SNPs by using HG18. Joint GWAS Pathways are computed from genes by using the DAVID pathway enrichment tool, including all pathways with enrichment scores better than 0.05.
| BD vs CAD | BD vs CD | BD vs RA | BD vs T1D | BD vs T2D | CAD vs CD | CAD vs RA | CAD vs T1D | CAD vs T2D | CD vs RA | CD vs T1D | CD vs T2D | RA vs T1D | RA vs T2D | T1D vs T2D | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Shared tag SNPs | 105,881 | 105,835 | 106,622 | 105,933 | 105,889 | 10,5903 | 106,722 | 106,015 | 105,957 | 106,772 | 105,974 | 105,913 | 106,773 | 106,698 | 106,005 |
| M | 13,439 | 15,041 | 15,348 | 14,182 | 19,823 | 25,246 | 14,233 | 16,175 | 16,638 | 17,121 | 14,721 | 22,039 | 16,093 | 16,716 | 18,518 |
| Joint GWAS SNPs, Nsnp | 2791 | 3315 | 3423 | 3185 | 5186 | 7711 | 3160 | 3915 | 4121 | 3963 | 3278 | 6113 | 3926 | 3950 | 4800 |
| Joint GWAS genes, Ng | 690 | 755 | 669 | 737 | 684 | 591 | 755 | 1133 | 716 | 633 | 762 | 678 | 693 | 694 | 706 |
| Joint GWAS pathways | 450 | 556 | 520 | 507 | 395 | 288 | 524 | 626 | 489 | 547 | 562 | 472 | 516 | 507 | 432 |
SNP, gene, and gene-cluster levels
For each Joint GWAS, we compared the Joint GWAS SNP list with the Target GWAS SNP list on their overlap with SNPs identified in the NHGRI GWAS catalog for the Target Disease. Our general method proceeds as follows (Figure S1). We wished to know if Joint GWAS Analysis is able to identify true disease SNPs, and how Joint analysis compares to Target GWAS testing alone. We therefore compared the Joint GWAS SNP list to the NHGRI Disease SNP list for that disease. Results, in Table 2, show that both the Joint GWAS SNP list and the Target Disease SNP list identify some of the SNPs that have been associated with the six diseases in previously published GWAS, with the Joint GWAS method identifying less SNPs in all cases than the Target Disease alone. Joint GWAS Analysis identified many SNPs (Nsnp = ~ 3000 to ~ 6000, Table 1) as potentially associated with the Target Disease, which leads to large false-positive rates (~ 99.9%) at the SNP-level; a result to be expected by including so many top SNPs, and one mirrored in the Target GWAS SNP list. Similar results are seen at the gene level (Table 3), with false-positive rates in Table S1; although in some cases the Joint GWAS gene list identified more NHGRI disease genes than the Target gene list, in particular Joint GWAS Analyses using CAD as the Cross Disease. We also note that the Joint gene and Target gene lists did not merely identify the same genes from the NHGRI database, but in most cases the Joint gene list provided additional genes not identified by the Target gene list alone (Table S3).
Table 2.
Comparison of Joint GWAS SNP list vs. Target GWAS SNP list. For each of the six WTCCC diseases, this table shows the number of SNPs identified by all published GWAS of that disease and indexed in the NHGRI catalog. For each WTCCC disease, we compare the number of NHGRI SNPs identified in the Joint GWAS SNP list (leading the slash) to the number identified in the Target GWAS SNP list (trailing the slash). SNPs within linkage disequilibrium of r2 ≥ 0.3 are considered representative of the SNP in question from the NHGRI list. In parentheses, we show how many more NHGRI SNPs were identified by the Joint GWAS SNP list than by the Target GWAS SNP list, as a percent of the total number of NHGRI SNPs. Negative numbers indicate that more NHGRI SNPs were identified by single, Top N Target GWAS than by Joint GWAS.
| Target disease |
Cross disease (joint GWAS SNP list/target GWAS SNP list, (% gain)) |
||||||
|---|---|---|---|---|---|---|---|
| Disease | NHGRI SNPs | BD | CAD | CD | RA | T1D | T2D |
| BD | 121 | 0 | 20/40 (− 16.5%) | 12/42 (− 24.8%) | 22/43 (− 17.4%) | 16/42 (− 21.5%) | 14/47 (− 27.3%) |
| CAD | 135 | 12/29 (− 12.6%) | 0 | 18/38 (− 14.8%) | 15/29 (− 10.4%) | 12/33 (− 15.6%) | 10/33 (− 17.0%) |
| CD | 151 | 19/74 (− 36.4%) | 17/82 (− 43.0%) | 0 | 16/77 (− 40.4%) | 17/74 (− 37.7%) | 16/81 (− 43.0%) |
| RA | 78 | 3/21 (− 23.1%) | 2/19 (− 21.8%) | 10/21 (− 14.1%) | 0 | 13/21 (− 10.3%) | 6/21 (− 19.2%) |
| T1D | 78 | 2/33 (− 39.7%) | 7/35 (− 35.9%) | 14/34 (− 25.6%) | 11/35 (− 30.8%) | 0 | 9/37 (− 35.9%) |
| T2D | 131 | 11/43 (− 24.4%) | 19/39 (− 15.3%) | 11/48 (− 28.2%) | 4/39 (− 26.7%) | 26/43 (− 13.0%) | 0 |
Table 3.
Comparison of Joint GWAS gene list vs. Target GWAS gene list. For each of the six WTCCC diseases, this table shows the number of genes identified by all published GWAS of that disease and indexed in the NHGRI catalog. For each WTCCC disease, we compare the number of NHGRI genes identified by the Joint GWAS gene list (leading the slash) to the number identified by the Target GWAS gene list (trailing the slash). In parentheses, we show how many more NHGRI genes were identified by the Joint GWAS gene list than by the Target GWAS gene list, as a percent of the total number of NHGRI genes. Negative numbers indicate that more NHGRI genes were identified by single, Target Disease GWAS than by Joint GWAS.
| Target disease |
Cross disease (joint GWAS gene list/target GWAS gene list, (% gain)) |
||||||
|---|---|---|---|---|---|---|---|
| Disease | NHGRI genes (n) | BD | CAD | CD | RA | T1D | T2D |
| BD | 130 | 0 | 8/6 (1.5%) | 8/6 (1.5%) | 4/6 (− 1.5%) | 7/6 (0.8%) | 4/6 (− 1.5%) |
| CAD | 92 | 6/7 (− 1.1%) | 0 | 5/12 (− 7.6%) | 7/12 (− 5.4%) | 13/13 (0.0%) | 6/12 (− 6.5%) |
| CD | 203 | 9/7 (1.0%) | 5/5 (0.0%) | 0 | 10/17 (− 3.4%) | 13/20 (− 3.4%) | 11/18 (− 3.4%) |
| RA | 66 | 5/6 (− 1.5%) | 8/5 (4.5%) | 6/7 (− 1.5%) | 0 | 6/11 (− 7.6%) | 6/11 (− 7.6%) |
| T1D | 61 | 4/3 (1.6%) | 7/4 (4.9%) | 7/7 (0.0%) | 5/7 (− 3.3%) | 0 | 2/12 (− 16.4%) |
| T2D | 105 | 13/14 (− 1.0%) | 17/12 (4.8%) | 12/14 (− 1.9%) | 14/11 (2.9%) | 15/17 (− 1.9%) | 0 |
To assess whether the genes overlapped in function rather than just in name, we used the DAVID pathway enrichment online tool to obtain functional groupings [19] for the genes in the Joint GWAS, Target GWAS, and NHGRI Disease gene lists. This use of the DAVID tool clusters genes into groups based on similar function (in contrast to clustering biological pathways into groups based on redundant genes, as employed below). We counted the number of genes from the NHGRI Disease gene list that were mapped to functional gene groups containing one of the genes from the Joint GWAS gene list, and the Target GWAS gene list (see Methods). Results (Table 4) show more pairs of diseases where the Joint Gene Analysis provides a clear improvement over just the Target GWAS, when considered against the results from the SNP- and Gene-level. When CAD is the Target Disease, Joint GWAS has significantly lower false-positive rates than the Target GWAS (Table S3). Additionally, in almost all cases, the Joint GWAS gene clusters identify some additional NHGRI disease genes that were not identified by the Target gene list clusters (Table S4). Simulation with null and VEGF-enhanced Joint GWAS shows that Joint GWAS Analysis identifies significantly more VEGF-pathway genes at both the Gene level and at the Gene Functional Group level, while having a smaller false-positive rate (Table S8).
Table 4.
Comparison of Joint GWAS gene list vs. Target GWAS gene list, considering functional overlap of NHGRI genes. For each of the six WTCCC diseases, this table shows the number of genes identified by all published GWAS of that disease and indexed in the NHGRI catalog. For each WTCCC disease, we compare the number of NHGRI genes mapped to a functional category including a gene from Joint GWAS gene list (leading the slash) to the number identified to the number mapped to a functional category including a gene from the by the Target GWAS gene list (trailing the slash). This shows the difference in identified functional gene clusters for each pair of diseases using the Joint GWAS method and the identified functional gene clusters for each Target Disease considered singly. In parentheses, we show how many more NHGRI genes were identified by the functional categories of Joint GWAS genes than by single, Target GWAS genes, as a percent of the total number of NHGRI genes. Results are dependent upon DAVID parameters (significant thresholds, pathway providers included in the aggregation). (*) indicates Joint GWAS gene lists that resulted in significantly lower false-positive rates than Target GWAS gene lists; significance assessed by Chi-square test (or Fisher's exact test in cases of low sample size).
| Target disease |
Cross disease (joint GWAS gene list/target GWAS gene list, (% gain)) |
||||||
|---|---|---|---|---|---|---|---|
| Disease | NHGRI genes (n) | BD | CAD | CD | RA | T1D | T2D |
| BD | 130 | 0 | 77/79 (− 1.5%) | 79/83 (− 3.1%) | 80/80 (0.0%) | 76/84 (− 6.2%) | 69/80 (− 8.5%) |
| CAD | 92 | 42/36 (6.5%) | 0 | 45/45 (0.0%) | 47/44 (3.3%) | 49/58 (− 9.8%) | 46/43 (3.3%) |
| CD | 203 | 121/122 (− 0.5%) | 112/97 (7.4%) | 0 | 110/132 (− 10.8%) | 114/136 (− 10.8%) | 132/134 (− 1.0%) |
| RA | 66 | 41/44 (− 4.5%) | 44/48 (− 6.1%) | 42/40 (3.0%) | 0 | 39/48 (− 13.6%) | 44/48 (− 6.1%) |
| T1D | 61 | 35/33 (3.3%) | 35/36 (− 1.6%) | 35/36 (− 1.6%) | 34/36 (− 3.3%) | 0 | 30/35 (− 8.2%) |
| T2D | 105 | 67/61 (5.7%) | 66/58 (7.6%) | 64/69 (− 4.8%) | 67/69 (− 1.9%) | 68/64 (3.8%) | 0 |
Pathway level
Given our lists of genes, we used the DAVID biological pathway aggregator to identify enriched pathways. These pathways are then grouped into similar clusters using DAVID functional annotation clustering [18]. For each pair of WTCCC GWAS, we compared the pathways enriched in the NHGRI Disease gene list to the pathways enriched in the Joint GWAS gene list and Target GWAS gene list. For each pair of diseases, we compared coverage by Joint GWAS gene list to coverage by Target GWAS gene list for each of the NHGRI Disease pathway clusters (see Methods). Results are presented in Table 5, with Tables S6–S35 showing each individual Joint GWAS pathway cluster comparison. In Supplemental material, we also present results comparing the pathway clusters showing significant coverage compared to pathway clusters covered by Combined Target-Cross disease enriched pathways (Table S5); where in 24 of 30 Joint GWAS comparisons, the Joint GWAS method identifies additional pathway clusters relative to the Combined Target-Cross disease gene lists.
In over half of the Joint GWAS comparisons (16 of 30), there was at least one additional pathway cluster identified with significantly greater gene representation in the Joint GWAS gene list over the Target GWAS gene list (Table 5). T2D in particular had one or two additional pathway clusters showing significantly better coverage by the Joint gene list in all but one Cross Disease. Interestingly, the individual pathway that was most often significantly increased in coverage was the “carbohydrates and glucose homeostasis” metabolic pathway (Tables S31, S32, S33), which seems particularly germane to diabetes etiology. In our null GWAS vs. VEGF-pathway GWAS, we identified on average 1 (of 3) possible VEGF pathway clusters with significantly greater coverage compared to null Joint GWAS (Table S9).
To assess the degree to which genes identified by Joint GWAS gene lists could represent novel biological findings, we took the Left-Over gene lists in each Joint GWAS Analysis and identified enriched pathways (see Methods). We looked for confirmation outside the NHGRI catalog that these pathways represent known associations with the Target Disease by using PubMed searches (Table 6). We see several cases of pathways with a large number of joint occurrences in the PubMed literature, that we consider to be instances of a pathway truly associated with the Target Disease, although a pathway that was not indicated by the NHGRI catalog of GWAS associations for the Target Disease. These demonstrate that this method does correctly identify known pathways associated with the Target Disease. There are also many cases of no co-occurrences of the pathway and the Target Disease; these are either cases of novel biological associations that may be validated in future work, or cases of false positives. We take a closer look at these in the Discussion section, below.
Discussion
Joint GWAS Analysis, even when combining seemingly unrelated diseases, leads to significant novel pathway association hypotheses, as well as the identification of additional known genes and pathways associated with the Target disease. That these results were the consequence of combining GWAS of disparate complex genetic disorders indicates that these diseases share genomic etiology to some extent. Consider the opinion of Wang et al.: “…joint analysis of related GWA study data sets may help reveal shared susceptibility pathways in a more powerful manner. This would be particularly relevant for diseases for which the genetic overlap is not well-understood.”[41] Our Joint GWAS Analysis method achieves this, by demonstrating that meta-analysis of GWAS need not be restricted to GWAS of the same disease.
While Joint GWAS Analysis has provided encouraging results, as presented it has some limitations, and by addressing these it is our belief that greater genomic understanding will be forthcoming. Our method is dependent upon the constituent GWASes and their execution: these must account for appropriate covariates, environmental factors, and stratification in order to provide the best input for Joint GWAS Analysis. Similarly, since we rely on GWAS data, we do not consider other forms of genomic data and variation, such as copy number variation, rare variants, gene expression, or gene epigenetics; although in principal these could be included in future versions of our methodology. In an effort to make our results as relevant as possible, we endeavored to mimic the original WTCCC GWASes as much as possible, including methods of population stratification control. We also experimented with including the top six genotype principal components as covariates, and found this resulted in some differences in the SNPs, genes, and pathways identified (+/− 20% in some cases), although the character of our results was largely unchanged. In general, we would need to perform Joint GWAS Analysis on many more pairs of GWAS to get a good understanding of what types of diseases would be most fruitful to combine, and which covariates are most important to include. We could employ alternative methods of assigning SNPs to genes, or of ranking genes by the p-values of their SNPs, of which there has been much recent discussion in the literature. The list of NHGRI Disease pathways identified from the NHGRI GWAS gene list can be considered a partial summary of the known biology of the Target Disease; not a perfect representation, but at least a summary of what has been established through GWAS. Since Joint GWAS Analysis starts with only GWAS, the NHGRI GWAS catalog makes a fitting benchmark for what is achievable through GWAS meta-analysis. On the other hand, our Joint GWAS methodology has the desirable benefit that it is not limited to GWAS that were performed on the same cohorts or with the same gene chip. In fact, the core of the method merely needs the ordering of SNPs by significance, and so does not require the original genotyping data at all. This can be an important factor in performing GWAS meta-analyses of potentially sensitive patient genomic data. Given a list of genes, there are many popular methods for determining enrichment of various biological categories or pathways.[17] The DAVID web tool aggregates pathway databases from a number of other pathway providers or categories: Gene Ontology (GO), GO Molecular Function, GO Cellular Component, KEGG Pathways, BioCarta Pathways, Swiss-Prot Keywords, BBID Pathways, SMART Domains, NIH Genetic Association DB, UniProt Sequence Features, COD/JOG Ontology, NCBI OMIM, InterPro Domains, PIR Super-Family Names, and Biological Processes.[19] In order to avoid a bias incurred by using one pathway provider exclusively (e.g., limiting ourselves to only GO or only KEGG), we used the pathways identified as enriched by DAVID, however the results presented herein are still sensitive to the pathway tool we chose to use; in principal any biological pathway aggregator could be used. In our current analysis, results may vary based on the parameters used in DAVID searches, including the particular pathway providers included in the aggregation and the significance thresholds used to identify significant pathways or pathway clusters. As our analysis of the VEGF and Null models shows, some genes may be undetectable through our method if they are too small to contain tag SNPs (see Table S8); a weakness related to the necessity of pruning the GWAS fairly stringently for linkage disequilibrium. This may result in certain pathways being hard to detect as enriched, or very large genes being much easier to detect. In future work, we will consider different methods for accounting for LD between SNPs and preserving tag SNPs for small genes.
A study of complex disease similarity at four different genomic resolutions was undertaken previously in the WTCCC GWASes by Huang et al.[20] They compared WTCCC GWASes at the SNP, gene, protein, and phenotype level, concluding that CD, RA and T1D have overlap at all four levels, while CAD, T2D, and hypertension do not exhibit similarity despite being commonly considered to be phenotypically similar. The similarity of WTCCC GWASes was also examined by Torkamani et al., who measured the correlation between gene p-values associated with each disease [38]. These authors found considerable similarity between BD, CAD, and T2D (in contrast to findings of Huang et al., above). Importantly, Torkamani et al. considered the possibility that the similarity exhibited by WTCCC GWASes might be due to WTCCC's use of a common set of control patients for each disease cohort. They proceeded to conduct a number of simulations that ruled out this possibility, stating that the similarities observed “…are not an artifact arising from WTCCC's use of common controls” for each GWAS [38]. Our Joint GWAS Analysis builds on this tradition of identifying genetic similarity in WTCCC cohorts, and goes further by leveraging this similarity to gain biological insight into each of the six diseases separately.
Methods for associating SNPs with multiple related traits have been recently reviewed [31]. The broader pursuit of examining multiple GWAS phenotypes has been explored in various ways, with some authors proposing combining traits before performing the GWAS as a way to increase power to detect pleiotropic genes [25]. Others have used multiple GWAS of the same disease to enhance biological pathway analysis [21], and statistics have been developed to identify pleiotropic SNPs from among several GWAS of related traits [7]. Our work builds on these ideas but explores a somewhat different question: that of using multiple GWAS of different phenotypes to enhance understanding of a single trait. Furthermore, it has been observed that existing methods of investigating gene pleiotropy will result in underestimates by missing SNPs with only moderate GWAS associations [35], an effect that our method addresses by virtue of its methodological similarity with pathway analysis: investigating a large proportion of the top GWAS SNPs while minimizing the multiple comparison problem.
The idea of considering the enrichment of gene sets has a rich history going back to Mootha et al.,[24] although these methods are less frequently employed in SNP comparisons, as employed in our Joint GWAS Analysis. Our method nevertheless owes much to these earlier gene set enrichment methods, particularly Subramanian et al.,[37] who considered the “leading edge” genes to be those that increased the enrichment score, and served a similar function to our maximum enrichment cut point (M, in Fig. 1). Also, other researchers have proposed methods of comparing lists of genes [9], [24], [45], however these have not been applied to lists of SNPs in the manner employed by our Joint GWAS methodology.
Where we have used DAVID pathway clusters, other researchers have used other methods of grouping pathways or pathway constituents [21]. Any such grouping of pathways is necessary to avoid problems with individual pathways, including: incomplete functional descriptions provided by ontologies such as GO [34]; enrichment results varying widely across different enrichment tools [9]; ontology inconsistencies that make counting or comparing between pathways difficult — redundant pathways, uneven hierarchical structures, and terms of varying sizes or memberships.
As might be expected for a pathway-based methodology, the results at the SNP-level were not encouraging for Joint GWAS Analysis: with the Target GWAS SNP list identifying many more known disease-associated SNPs. However, the picture improved as the analysis moved to broader scopes, to genes, to gene functional-clusters, and to pathways. There are many cases of the Joint gene list identifying more disease-associated genes than the Target gene list (Table 3); in particular in cases of CAD as the cross disease. At the level of gene functional clusters (Table 4) there are more cases of improved performance in the Joint gene list than there were at the gene-level. Although we would expect diseases to appear to be more similar as we considered broader categories of genomic resolution, we should have no similar expectation of the Joint GWAS analysis out-performing single GWAS – this occurrence is an important result. At the pathway-cluster genomic resolution, we see that there is a further increase in the number of disease pairs that show a benefit of the Joint GWAS methodology over single GWAS (Table 5). There are 17 disease pairs showing statistically increased coverage of 1 or 2 pathway clusters by the Joint GWAS method than by the Target GWAS. These results are particularly concentrated in the pathway clusters of T2D, which may be reflective of the broad metabolic basis of the disease leading to a diffuse genetic etiology. Our simple simulation of null vs. VEGF-pathway-enhanced Joint GWAS indicates that Joint GWAS Analysis does indeed identify pathways common to two GWAS, when those pathways are enriched for low-p-value GWAS SNPs. This general success of Joint GWAS Analysis at the pathway level demonstrates its ability to identify additional relevant biology, surpassing that which is identifiable from the single GWAS alone. This is also true to a lesser extent at the gene-cluster level (Table S4) and gene-level (Table S3), where genes missed by the Target GWAS are found by the Joint GWAS, although to a lesser extent in Target diseases RA and T1D (Table S4). In fact, RA and T1D may be the poor choices for Joint GWAS Analysis, since they have a large portion of their genetic basis localized to the MHC region of chromosome 6. These two are not broad-based, multi-genic disorders on the same scale as BD, T2D, CD, and CAD.
When using Joint GWAS Analysis to identify novel biological associations, we used the Left Over genes from the pathway-level analysis to generate candidate pathways associated with the Target Disease (Table 6). That several of the hypothesized pathway associations already have great support in the PubMed literature suggest that many of these hypotheses are truly associated with the Target Disease (see Table S6 for representative references). While each of these categories represents a group of proteins, some of which have known associations to Target Diseases (cells highlighted in green in Table 6), many do not have published associations with the Target Disease yet (cells highlighted in pink in Table 6). To look for novel biological hypotheses, we looked at the pathways without current PubMed literature matches: axoneme-related proteins in BD; RNA polymerase II dysfunction in RA, amino acid synthesis pathways in RA, and paraneoplastic encephalomyelitis in RA; and proteins containing Pleckstrin homologue domains in T1D, magnesium ion binding proteins in T1D, and nuclear membrane related proteins in T1D. Of these, paraneoplastic encephalomyelitis refers to an inflammatory disease that may share immune-related causative genes with RA, and may therefore not lead to new avenues of research; magnesium ion binding proteins may refer to RNA or ATP, this may actually be too broad to be of scientific use. Others of these make for very interesting hypotheses: RNA polymerase II association with rheumatoid arthritis, and axoneme proteins in bipolar disorder. These are, in our estimation, the most promising candidates for novel biological association generated by Joint GWAS Analysis.
Our results invoke the general pleiotropy of disease-causing genes and biological systems, and the general relatedness of complex disorders, as described, for example, by Barabasi et al. [3] Pleiotropy is becoming more evident as more genomic regions are associated with more conditions. A study of autoimmune disorders found many SNPs were involved with more than one disease, and this lead to new associations between genomic loci and some diseases [29]. In the WTCCC GWAS themselves, BD, T2D, and CAD were found to have slight positive correlations among their top SNPs [32]. Furthermore, the relatedness of complex disorders is evident in clinical settings, where complex diseases show significant comorbidity [4], and in patients over 65 years of age, it is common to observe more than 10 related diseases [27]. Another study looked at the co-occurrence of 161 diseases in 1.5 million clinical patients, and found a strong correlation between incidence of BD and of T2D, T1D, and RA, among others [30]. The same study found strong correlations between T2D and T1D, BD, and RA [30]. While many pleiotropy studies focus on the family of autoimmune diseases, a recent study identified seven genes with pleiotropic effects across cardiovascular- or metabolic syndrome-related conditions [13]. Others have observed that genes associated with complex disorders tend to be interconnected in interaction networks [5]. Our present work can be seen as an extension of these results.
Acknowledgments
MJM is funded by the National Institute of Health grant numbers K12-HL089990 and U01-HL065899. GLC is funded by U01-HL065899. JLS is funded by R00-HL096840. AD is funded by NIH T32 HL007427. BAR is funded by R01-HL086601 and R01-HL093976. STW is funded by P01-HL083069 and U01-HL065899.
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.gdata.2014.04.004.
Appendix A. Glossary of defined terms
Joint GWAS Analysis. Our method of combining two GWASes of different disorders for the purpose of gaining information about each disease individually.
Target disease. One of two diseases considered in the two GWAS of a Joint GWAS Analysis. For pedagogical purposes, we designate one of the two the Target Disease, and measure how much of known associations to the Target Disease are recovered in Joint GWAS Analysis.
Target GWAS. The GWAS of the Target Disease.
Cross disease. The other of the two diseases considered in the two GWAS of a Joint GWAS Analysis. For pedagogical purposes, we designate one of the two the Target Disease and the other the Cross Disease.
Cross GWAS. The GWAS of the Cross Disease.
Joint GWAS SNP list. In a Joint GWAS Analysis, the list of SNPs obtained by crossing the Target and Joint GWAS and selecting the common SNPs from among the top M ranked SNPs.
Joint GWAS gene list. In a Joint GWAS Analysis, the list of genes obtained by translating the Joint GWAS SNP list into genes using a genomic map (e.g., HG18).
Target GWAS SNP list. In a Joint GWAS Analysis, the list of SNPs obtained by taking the top Nsnp SNPs from the Target GWAS, where Nsnp is the size of the Joint GWAS SNP list.
Target GWAS gene list. In a Joint GWAS Analysis, the list of genes obtained by translating enough of the top SNPs from the Target GWAS SNP list into genes, in order to obtain Ng unique genes, where Ng is the size of the Joint GWAS gene list.
NHGRI disease SNP list. A list of SNPs associated with the Target Disease in any GWAS listed in the NHGRI GWAS catalog conducted on the Target Disease.
NHGRI disease gene list. A list of genes associated with the Target Disease in any GWAS listed in the NHGRI GWAS catalog conducted on the Target Disease.
NHGRI disease pathway clusters. A list of pathway clusters identified as enriched by the DAVID pathway aggregator tool, upon the input of the NHGRI Disease gene list.
M. In a Joint GWAS Analysis, the number of SNPs maximizing the enrichment p-value for common SNPs between the Cross GWAS and Target GWAS.
Nsnp. The size of the Joint GWAS SNP list.
Ng. The size of the Joint GWAS gene list.
Common SNPs. In a Joint GWAS Analysis, the SNPs occurring in both the top M SNPs of the Target GWAS and the Cross GWAS, for any value of M.
Covered. The genes (or the number of genes) from either the Joint GWAS gene list or Target GWAS gene list that occur in an enriched pathway of that list, and that occur in an NHGRI Disease pathway cluster.
Left-over gene list. List of genes obtained from the Joint GWAS gene list that did not cover any NHGRI Disease pathway clusters.
Appendix B. Supplementary data
Figure S1: Venn Diagram of Joint GWAS Analysis for SNP-level comparison.
Table S1: False-positive rates for Joint GWAS gene list and Target GWAS gene list identification of NHGRI genes.
Table S2: Differential gene identification for Joint GWAS gene list and Target GWAS gene list identification of NHGRI genes.
Table S3: False-positive rates for Joint GWAS gene list and Target GWAS gene list identification of NHGRI gene functional categories.
Table S4: Differential gene identification by gene functional cluster overlap for Joint GWAS gene list and Target GWAS gene list identification of NHGRI genes.
Tables S5: Pathway comparison of Joint GWAS Analysis to Combined Target-Cross Diseases.
Tables S6: Pathways identified as novel associations to Target Diseases.
Table S7: General Characteristics of Null GWAS Joint GWAS Analysis vs. VEGF GWAS Joint GWAS Analysis.
Table S8: Gene Level and Gene-Functional Level results for Null GWAS Joint GWAS Analysis vs. VEGF GWAS Joint GWAS Analysis.
Table S9: Pathway Cluster Level results for Null GWAS Joint GWAS Analysis vs. VEGF GWAS Joint GWAS Analysis.
Tables S10 through S30: NHGRI Disease pathway cluster coverage for Joint GWAS Analysis of each pair of WTCCC diseases.
References
- 1.Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A. An integrated map of genetic variation from 1092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barabasi A.L., Gulbahce N., Loscalzo J. Network medicine: a network-based approach to human disease. Nat. Rev. Genet. 2011;12:56–68. doi: 10.1038/nrg2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barrenas F., Chavali S., Alves A.C., Coin L., Jarvelin M.R., Jornsten R., Langston M.A., Ramasamy A., Rogers G., Wang H. Highly interconnected genes in disease-specific networks are enriched for disease-associated polymorphisms. Genome Biol. 2012;13:R46. doi: 10.1186/gb-2012-13-6-r46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barrenas F., Chavali S., Holme P., Mobini R., Benson M. Network properties of complex human disease genes identified through genome-wide association studies. PLoS One. 2009;4:e8090. doi: 10.1371/journal.pone.0008090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chavali S., Barrenas F., Kanduri K., Benson M. Network properties of human disease genes with pleiotropic effects. BMC Syst. Biol. 2010;4:78. doi: 10.1186/1752-0509-4-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cotsapas C., Voight B.F., Rossin E., Lage K., Neale B.M., Wallace C., Abecasis G.R., Barrett J.C., Behrens T., Cho J. Pervasive sharing of genetic effects in autoimmune disease. PLoS Genet. 2011;7:e1002254. doi: 10.1371/journal.pgen.1002254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crowley J.J., Sullivan P.F., McLeod H.L. Pharmacogenomic genome-wide association studies: lessons learned thus far. Pharmacogenomics. 2009;10:161–163. doi: 10.2217/14622416.10.2.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elbers C.C., van Eijk K.R., Franke L., Mulder F., van der Schouw Y.T., Wijmenga C., Onland-Moret N.C. Using genome-wide pathway analysis to unravel the etiology of complex diseases. Genet. Epidemiol. 2009;33:419–431. doi: 10.1002/gepi.20395. [DOI] [PubMed] [Google Scholar]
- 10.Evangelou E., Ioannidis J.P. Meta-analysis methods for genome-wide association studies and beyond. Nat. Rev. Genet. 2013;14:379–389. doi: 10.1038/nrg3472. [DOI] [PubMed] [Google Scholar]
- 11.Fanale D., Amodeo V., Corsini L.R., Rizzo S., Bazan V., Russo A. Breast cancer genome-wide association studies: there is strength in numbers. Oncogene. 2012;31:2121–2128. doi: 10.1038/onc.2011.408. [DOI] [PubMed] [Google Scholar]
- 12.Fehringer G., Liu G., Briollais L., Brennan P., Amos C.I., Spitz M.R., Bickeboller H., Wichmann H.E., Risch A., Hung R.J. Comparison of pathway analysis approaches using lung cancer GWAS data sets. PLoS One. 2012;7:e31816. doi: 10.1371/journal.pone.0031816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gottesman O., Drill E., Lotay V., Bottinger E., Peter I. Can genetic pleiotropy replicate common clinical constellations of cardiovascular disease and risk? PLoS One. 2012;7:e46419. doi: 10.1371/journal.pone.0046419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gudmundsson J., Sulem P., Steinthorsdottir V., Bergthorsson J.T., Thorleifsson G., Manolescu A., Rafnar T., Gudbjartsson D., Agnarsson B.A., Baker A. Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat. Genet. 2007;39:977–983. doi: 10.1038/ng2062. [DOI] [PubMed] [Google Scholar]
- 15.Hemani G., Yang J., Vinkhuyzen A., Powell J.E., Willemsen G., Hottenga J.J., Abdellaoui A., Mangino M., Valdes A.M., Medland S.E. Inference of the genetic architecture underlying BMI and height with the use of 20,240 sibling pairs. Am. J. Hum. Genet. 2013;93:865–875. doi: 10.1016/j.ajhg.2013.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hindorff L.A., Sethupathy P., Junkins H.A., Ramos E.M., Mehta J.P., Collins F.S., Manolio T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. U. S. A. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang da W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang da W., Sherman B.T., Tan Q., Collins J.R., Alvord W.G., Roayaei J., Stephens R., Baseler M.W., Lane H.C., Lempicki R.A. The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8:R183. doi: 10.1186/gb-2007-8-9-r183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang da W., Sherman B.T., Tan Q., Kir J., Liu D., Bryant D., Guo Y., Stephens R., Baseler M.W., Lane H.C. DAVID bioinformatics resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007;35:W169–W175. doi: 10.1093/nar/gkm415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang W., Wang P., Liu Z., Zhang L. Identifying disease associations via genome-wide association studies. BMC Bioinform. 2009;10(Suppl. 1):S68. doi: 10.1186/1471-2105-10-S1-S68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee Y., Li J., Gamazon E., Chen J.L., Tikhomirov A., Cox N.J., Lussier Y.A. AMIA Summits Transl Sci Proc. 2010. Biomolecular Systems of Disease Buried Across Multiple GWAS Unveiled by Information Theory and Ontology; pp. 31–35. [PMC free article] [PubMed] [Google Scholar]
- 22.Lettre G., Jackson A.U., Gieger C., Schumacher F.R., Berndt S.I., Sanna S., Eyheramendy S., Voight B.F., Butler J.L., Guiducci C. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat. Genet. 2008;40:584–591. doi: 10.1038/ng.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyer L.R., Zweig A.S., Hinrichs A.S., Karolchik D., Kuhn R.M., Wong M., Sloan C.A., Rosenbloom K.R., Roe G., Rhead B. The UCSC Genome Browser database: extensions and updates 2013. Nucleic Acids Res. 2013;41:D64–D69. doi: 10.1093/nar/gks1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mootha V.K., Lindgren C.M., Eriksson K.F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstrale M., Laurila E. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 25.Park S.H., Lee J.Y., Kim S. A methodology for multivariate phenotype-based genome-wide association studies to mine pleiotropic genes. BMC Syst. Biol. 2011;5(Suppl. 2):S13. doi: 10.1186/1752-0509-5-S2-S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Puzyrev V.P., Freidin M.B. Genetic view on the phenomenon of combined diseases in man. Acta Nat. 2009;1:52–57. [PMC free article] [PubMed] [Google Scholar]
- 28.Ramanan V.K., Shen L., Moore J.H., Saykin A.J. Pathway analysis of genomic data: concepts, methods, and prospects for future development. Trends Genet. 2012;28:323–332. doi: 10.1016/j.tig.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramos P.S., Criswell L.A., Moser K.L., Comeau M.E., Williams A.H., Pajewski N.M., Chung S.A., Graham R.R., Zidovetzki R., Kelly J.A. A comprehensive analysis of shared loci between systemic lupus erythematosus (SLE) and sixteen autoimmune diseases reveals limited genetic overlap. PLoS Genet. 2011;7:e1002406. doi: 10.1371/journal.pgen.1002406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rzhetsky A., Wajngurt D., Park N., Zheng T. Probing genetic overlap among complex human phenotypes. Proc. Natl. Acad. Sci. U. S. A. 2007;104:11694–11699. doi: 10.1073/pnas.0704820104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shriner D. Moving toward system genetics through multiple trait analysis in genome-wide association studies. Front. Genet. 2012;3:1. doi: 10.3389/fgene.2012.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sirota M., Schaub M.A., Batzoglou S., Robinson W.H., Butte A.J. Autoimmune disease classification by inverse association with SNP alleles. PLoS Genet. 2009;5:e1000792. doi: 10.1371/journal.pgen.1000792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sivakumaran S., Agakov F., Theodoratou E., Prendergast J.G., Zgaga L., Manolio T., Rudan I., McKeigue P., Wilson J.F., Campbell H. Abundant pleiotropy in human complex diseases and traits. Am. J. Hum. Genet. 2011;89:607–618. doi: 10.1016/j.ajhg.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Soldatos T.G., O'Donoghue S.I., Satagopam V.P., Jensen L.J., Brown N.P., Barbosa-Silva A., Schneider R. Martini: using literature keywords to compare gene sets. Nucleic Acids Res. 2010;38:26–38. doi: 10.1093/nar/gkp876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Solovieff N., Cotsapas C., Lee P.H., Purcell S.M., Smoller J.W. Pleiotropy in complex traits: challenges and strategies. Nat. Rev. Genet. 2013;14:483–495. doi: 10.1038/nrg3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stranger B.E., Stahl E.A., Raj T. Progress and promise of genome-wide association studies for human complex trait genetics. Genetics. 2011;187:367–383. doi: 10.1534/genetics.110.120907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Torkamani A., Topol E.J., Schork N.J. Pathway analysis of seven common diseases assessed by genome-wide association. Genomics. 2008;92:265–272. doi: 10.1016/j.ygeno.2008.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trzaskowski M., Davis O.S., DeFries J.C., Yang J., Visscher P.M., Plomin R. DNA evidence for strong genome-wide pleiotropy of cognitive and learning abilities. Behav. Genet. 2013;43:267–273. doi: 10.1007/s10519-013-9594-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vinkhuyzen A.A., Wray N.R., Yang J., Goddard M.E., Visscher P.M. Estimation and partition of heritability in human populations using whole-genome analysis methods. Annu. Rev. Genet. 2013;47:75–95. doi: 10.1146/annurev-genet-111212-133258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang K., Li M., Hakonarson H. Analysing biological pathways in genome-wide association studies. Nat. Rev. Genet. 2010;11:843–854. doi: 10.1038/nrg2884. [DOI] [PubMed] [Google Scholar]
- 42.Willer C.J., Speliotes E.K., Loos R.J., Li S., Lindgren C.M., Heid I.M., Berndt S.I., Elliott A.L., Jackson A.U., Lamina C. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat. Genet. 2009;41:25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang J., Benyamin B., McEvoy B.P., Gordon S., Henders A.K., Nyholt D.R., Madden P.A., Heath A.C., Martin N.G., Montgomery G.W. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang J., Lee S.H., Goddard M.E., Visscher P.M. GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang X., Bentink S., Scheid S., Spang R. Similarities of ordered gene lists. J. Bioinforma. Comput. Biol. 2006;4:693–708. doi: 10.1142/s0219720006002120. [DOI] [PubMed] [Google Scholar]
- 46.Zeggini E., Ioannidis J.P. Meta-analysis in genome-wide association studies. Pharmacogenomics. 2009;10:191–201. doi: 10.2217/14622416.10.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Venn Diagram of Joint GWAS Analysis for SNP-level comparison.
Table S1: False-positive rates for Joint GWAS gene list and Target GWAS gene list identification of NHGRI genes.
Table S2: Differential gene identification for Joint GWAS gene list and Target GWAS gene list identification of NHGRI genes.
Table S3: False-positive rates for Joint GWAS gene list and Target GWAS gene list identification of NHGRI gene functional categories.
Table S4: Differential gene identification by gene functional cluster overlap for Joint GWAS gene list and Target GWAS gene list identification of NHGRI genes.
Tables S5: Pathway comparison of Joint GWAS Analysis to Combined Target-Cross Diseases.
Tables S6: Pathways identified as novel associations to Target Diseases.
Table S7: General Characteristics of Null GWAS Joint GWAS Analysis vs. VEGF GWAS Joint GWAS Analysis.
Table S8: Gene Level and Gene-Functional Level results for Null GWAS Joint GWAS Analysis vs. VEGF GWAS Joint GWAS Analysis.
Table S9: Pathway Cluster Level results for Null GWAS Joint GWAS Analysis vs. VEGF GWAS Joint GWAS Analysis.
Tables S10 through S30: NHGRI Disease pathway cluster coverage for Joint GWAS Analysis of each pair of WTCCC diseases.