

Abstract
Gestational hypoxia inhibits steroid hormone–induced upregulation of Ca2+-activated K+ (KCa) channel activities in uterine arteries. We tested the hypothesis that increased reactive oxygen species play an important role in hypoxia-mediated inhibition of KCa channel activities. Uterine arteries were isolated from nonpregnant (nonpregnant uterine artery) and near-term (≈142–145 day) pregnant (pregnant uterine artery) sheep maintained at either sea level or high altitude (3820 m, Pao2: 60 mm Hg) for 110 days. In pregnant uterine arteries, hypoxia significantly decreased large conductance channel opener NS1619- and small conductance channel opener NS309-induced relaxations, which were partially restored by reactive oxygen species inhibitor N-acetylcysteine (NAC). NAC significantly increased large conductance KCa but not small conductance KCa current densities in uterine arterial smooth muscle cells in pregnant animals acclimatized to high altitude. The NAC-sensitive component of small conductance KCa–induced relaxations was diminished in endothelium-denuded arteries. In nonpregnant uterine arteries, NS1619- and NS309-induced relaxations were diminished compared with those in pregnant uterine arteries. Treatment of nonpregnant uterine arteries with 17β-estradiol and progesterone for 48 hours increased small conductance KCa type 3 protein abundance and NS1619- and NS309-induced relaxations, which were inhibited by hypoxia. This hypoxia-mediated inhibition was reversed by NAC. Consistently, steroid hormone treatment had no significant effects on large conductance KCa current density in nonpregnant uterine arteries of hypoxic animals in the absence of NAC but significantly increased it in the presence of NAC. These results suggest an important role of hypoxia-mediated reactive oxygen species in negatively regulating steroid hormone–mediated upregulation of KCa channel activity and adaptation of uterine vascular reactivity in pregnancy, which may contribute to the increased incidence of preeclampsia and fetal intrauterine growth restriction associated with gestational hypoxia.
Keywords: anoxia, oxidative stress, uterine artery
Ca2+-activated K+ (KCa) channels are key regulators of vascular tone.1,2 Based on their conductance, KCa channels are divided into large conductance (BKCa), intermediate conductance (IKCa), and small conductance (SKCa) channels. BKCa channels are expressed in vascular smooth muscle, and SKCa channels are thought to be expressed predominantly in endothelial cells.2 Hyperpolarization produced by the activation of SKCa in endothelial cells may be transmitted to vascular smooth muscle cells via the myoendothelial gap junction.3 Our recent studies demonstrated that both BKCa and SKCa, but not IKCa channels, contributed to the regulation of uterine vascular function.4–6 Uterine blood flow increases dramatically during pregnancy to optimize the supply of oxygen and nutrition for the fetal development because of both vascular remodeling/growth and a decrease in uterine vascular tone.7 Steroid hormones (estrogen and progesterone) play an important role in the hemodynamic adaption to pregnancy, in part, by upregulating KCa channel function,4 leading to decreased uterine vascular tone. However, this adaptive change is severely compromised by gestational hypoxia, leading to increased incidence of preeclampsia and fetal intrauterine growth restriction.8–11 This regulation of the uterine circulation involves increased vascular tone because of impaired KCa channel function.5,6 However, the mechanism underlying the impairment of KCa channel function remains poorly understood.
Reactive oxygen species (ROS) in the cardiovascular system primarily include superoxide, hydrogen peroxide, and hydroxyl radical. Increased levels of ROS during exposure to hypoxia have been demonstrated in several arterial beds including the uterine arteries from pregnant sheep12,13; and oxidative stress has been implicated in the pathogenesis of various cardiovascular disorders.14,15 Activities of KCa channels in vascular smooth muscle cells are subject to modulation by ROS.16,17 However, the role of increased ROS in regulating KCa channel function and uterine vascular tone in response to hypoxia remains unknown. The present studies investigated whether the hypoxia-mediated heightened ROS altered KCa channel activities and their mediated relaxations of uterine arteries in pregnancy. In addition, we also investigated the effects of steroid hormones on KCa channel activities and their mediated uterine vasorelaxations and determined whether the effect of steroid hormones was changed by hypoxia-enhanced ROS.
Materials and Methods
Tissue Preparation and Treatment
Uterine arteries were harvested from both nonpregnant and near-term (≈142–145 days of gestation) pregnant sheep maintained at sea level (≈300 m) or exposed to high-altitude (3801 m) hypoxia (Pao2: 60 mm Hg) for 110 days (from 30 days of gestation for pregnant animals).18 Animals were anesthetized with intravenous injection of propofol (2 mg/kg) followed by intubation, and anesthesia was maintained on 1.5% to 3.0% isoflurane balanced in O2 throughout the surgery. An incision was made in the abdomen and the uterus exposed. The uterine arteries were isolated and removed without stretching and placed into a Krebs solution containing (in mmol/L) 130.0 NaCl, 10.0 HEPES, 6.0 glucose, 4.0 KCl, 4.0 NaHCO3, 1.8 CaCl2, 1.2 MgSO4, 1.18 KH2PO4, and 0.025 EDTA (pH 7.4) in room air with Po2 of ≈100 mm Hg. To determine the ex vivo hypoxic effect, uterine arteries were treated under normoxic or hypoxic conditions for 48 hours. Given an ≈50% decrease in arterial Po2 observed in high-altitude hypoxic sheep,5,18,19 uterine arteries were incubated in a given culture dish with 5 mL of phenol red–free DMEM with 1% charcoal-stripped fetal bovine serum, 100 U/mL penicillin, 100 μg/mL streptomycin and treated at 37°C in humidified incubators for 48 hours with oxygen levels at either 21% O2 for normoxic or 10.5% O2 for hypoxic conditions as described previously.5,18,19 In some tissues, the endothelium was removed by gentle rotation of arterial rings on an approximately sized blunt hypodermic needle, and the validity of endothelium removal was confirmed by the absence of endothelial nitric oxide synthase in immunohistochemical staining as shown previously.6 Endothelium removal had no significant effect on KCl- or norepinephrine-induced contractions. Each experimental group had 4 to 6 animals. All procedures and protocols were approved by the Institutional Animal Care and Use Committee and followed the guidelines by the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Contraction Studies
The fourth generation branches of main uterine arteries were separated from surrounding tissues and cut into 2-mm ring segments. In uterine arteries of pregnant sheep, isometric tension was measured in the Krebs solution in a tissue bath at 37°C as described previously.17–20 Briefly, each ring segment was equilibrated for 60 minutes and then gradually stretched to the optimal resting tension, as determined by the tension that developed in response to 3× 120 mmol/L KCl, each added at a stretch level. In the studies of arterial contractility, vessels are commonly stimulated with repeated (normally 3×) maximal concentrations of KCl to stabilize the vascular reactivity. Without the KCl stimulation, norepinephrine-induced contractions and subsequent NS1619 compound–mediated relaxations were unstable and were not comparable between vessels. After stable responses to KCl were obtained, arteries were treated with 0 or 1 mmol/L N-acetylcysteine (NAC) for 20 minutes. Tissues were then precontracted with submaximal concentrations of norepinephrine that produced ≈70% to 80% of the maximal contraction, followed by additions of NS1619 or NS309 in a cumulative manner.
In uterine arteries of nonpregnant sheep, tissues were treated with 17β-estradiol (0.3 nmol/L, Sigma) and progesterone (P4, 100 nmol/L, Sigma) for 48 hours in the absence or presence of 1 mmol/L NAC. For hormonal treatments, arteries from nonpregnant sheep were incubated under normoxia of 21% O2 for tissues from normoxic animal or hypoxia of 10.5% O2 for tissues from hypoxic animals in the absence or presence of 17β-estradiol and P4 as reported previously.4,5,18–20 The concentrations of 17β-estradiol and P4 were chosen based on the previous study of their concentration-dependent effects on uterine arteries,20 and they are physiologically relevant as observed in ovine pregnancy.21 Our previous studies have shown that the hormonal treatment regulates KCa channel activity and pressure-dependent myogenic tone in the uterine artery.4,5,18–20 After the treatments, NS1619- or NS309-induced relaxations were determined as described above.
Western Immunoblotting
Our previous study demonstrated that pregnancy increased protein abundance of type 2 (SK2) and type 3 (SK3) SKCa channels in ovine uterine arteries.6 In the present study, the effect of steroid hormones on SK2 and SK3 expression was measured in uterine arteries of nonpregnant sheep. Briefly, fourth order uterine arteries were treated with 17β-estradiol (0.3 nmol/L) and progesterone (P4, 100 nmol/L) or vehicle control for 48 hours. Tissues from each animal provided enough protein for Western blotting. After the treatments, tissues were homogenized in a lysis buffer followed by centrifugation at 4°C for 10 minutes at 10 000g, and supernatants were collected. Samples with equal proteins were loaded onto 7.5% polyacrylamide gel with 0.1% sodium dodecyl sulfate and were separated by electrophoresis at 100 V for 2 hours. Proteins were then transferred onto nitrocellulose membranes. After blocking nonspecific binding sites by dry milk, membranes were incubated with primary antibodies against SK2 channel (Alomone Ltd, Jerusalem, Israel) and SK3 channel (Santa Cruz Biotechnology, Santa Cruz, CA). After washing, membranes were incubated with secondary horseradish peroxidase–conjugated antibodies. Proteins were visualized with enhanced chemiluminescence reagents, and blots were exposed to Hyperfilm. Results were quantified with the Kodak electrophoresis documentation and analysis system and Kodak ID image analysis software.
Measurement of KCa Channel Current
Arterial smooth muscle cells were enzymatically dissociated from resistance-sized uterine arteries, and whole-cell K+ currents were recorded using an EPC 10 patch-clamp amplifier with Patchmaster software (HEKA, Lambrecht/Pfalz, Germany) at room temperature as previously described.4–6 Briefly, cell suspension drops were placed in a recording chamber, and adherent cells were continuously superfused with HEPES-buffered physiological salt solution containing (in mmol/L) 140.0 NaCl, 5.0 KCl, 1.8 CaCl2, 1.2 MgCl2, 10.0 HEPES, and 10.0 glucose (pH 7.4). Only relaxed and spindle-shaped myocytes were used for recording. Micropipettes were pulled from borosilicate glass and had resistances of 2 to 5 mol/LΩ when filled with the pipette solution containing (in mmol/L) 140.0 KCl, 1.0 MgCl2, 5.0 Na2ATP, 5.0 EGTA, 10.0 HEPES (pH 7.2). CaCl2 was added to bring free Ca2+ concentrations to 200 nmol/L as determined using WinMAXC software (Chris Patton, Stanford University). Cells were held at −50 mV, and whole-cell K+ currents were evoked by voltage steps from −60 mV to +80 mV by stepwise 10-mV depolarizing pulses (350-ms duration, 10-second intervals) in the absence and presence of 1 mmol/L BKCa channel blocker tetraethylammonium4,5 or 1 μmol/L SKCa channel blocker apamin.6 The K+ currents were normalized to cell capacitance and were expressed as picoampere per picofarad (pA/pF).
Data Analysis
Concentration–response curves were analyzed by computer-assisted nonlinear regression to fit the data using GraphPad Prism (GraphPad Software, San Diego, CA). Results were expressed as means±SEM obtained from the number of experimental animals given. Differences were evaluated for statistical significance (P<0.05) by ANOVA or t test where appropriate.
Results
NAC Inhibited the Hypoxic Effect in Suppressing KCa Channel–Mediated Relaxations of Uterine Arteries in Pregnant Sheep
In normoxic animals, both BKCa channel opener NS1619 and SKCa channel opener NS309 induced concentration-dependent relaxations of uterine arteries from pregnant sheep, which were not significantly affected by NAC (Figure 1A and 1B). In pregnant animals acclimatized to high-altitude hypoxia, both NS1619- and NS309-induced relaxations were significantly attenuated (Figure 1C and 1D). Compared with the relaxations seen in normoxic animals, chronic hypoxia caused a significant decrease in the maximal response of NS1619 from 39.3±3.8% to 13.7±3.8% (P<0.05) and NS309 from 58.0±11.2% to 17.7±3.3% (P<0.05). As shown in Figure 1C and 1D, treatment of the vessels with NAC inhibited the hypoxic effect and significantly increased the maximal response of NS1619-induced relaxations from 13.7±3.8% to 46.2±9.5% (P<0.05) and NS306-induced relaxations from 17.7±3.3% to 34.9±4.6% (P<0.05). In the presence of NAC, there was no significant difference in NS1619-induced relaxations between normoxic and hypoxic animals (43.9±5.0% versus 46.2±9.5%; P>0.05). Unlike NS1619, NAC partially blocked the effect of hypoxia on NS309-mediated relaxations.
Figure 1.
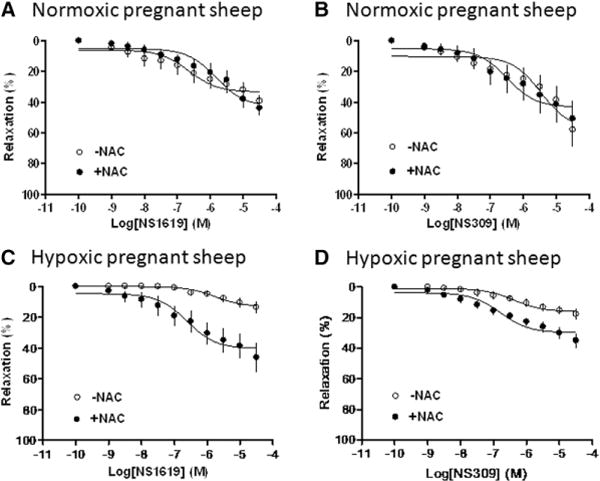
Effect of N-acetylcysteine (NAC) on Ca2+-activated K+ channel–induced relaxations of uterine arteries from pregnant sheep. Uterine arteries were isolated from normoxic (A and B) and hypoxic (C and D) pregnant sheep. Concentration–response curves were determined in NS1619-induced relaxations and NS309-induced relaxations in the absence or presence of NAC (1 mmol/L). The x axis indicates cumulative increases of the agonists in approximately one-half log increments. Data are means±SEM from 4 to 5 animals in each group. The Emax values are presented in the text.
NAC Increased BKCa Channel Activities in Uterine Arteries of Hypoxic Animals
To determine the role of ROS in the hypoxia-mediated effect on KCa channel activities in uterine arteries, whole-cell K+ and KCa currents were determined in the absence or presence of NAC in smooth muscle cells freshly isolated from uterine arteries of normoxic and hypoxic pregnant animals. Consistent with the previous finding,5 chronic hypoxia significantly decreased both whole-cell K+ currents (from 52.8±1.7 to 42.4±0.8 pA/pF at +80 mV; P<0.05) and BKCa current density (from 29.1±2.0 to 18.5±1.4 pA/pF; P<0.05). NAC was without effect on whole-cell K+ currents (50.8±2.0 versus 52.8±1.7 pA/pF; P>0.05), BKCa current density (27.8±1.8 versus 29.1±2.0 pA/pF; P>0.05), or SKCa current density (10.8±1.1 versus 12.5±2.2 pA/pF; P>0.05) in myocytes of normoxic animals. However, NAC significantly increased whole-cell K+ currents from 42.4±0.8 to 50.4±2.1 pA/pF (P<0.05; Figure 2A) and BKCa currents from 18.5±1.4 to 26.6±2.4 pA/pF (P<0.05; Figure 2B) in uterine arterial myocytes of hypoxic animals. Consistent with the previous finding,6 no apamin-sensitive SKCa current was detected in uterine arterial myocytes of hypoxic animals, and NAC treatment failed to upregulate SKCa currents (Figure S1 in the online-only Data Supplement).
Figure 2.
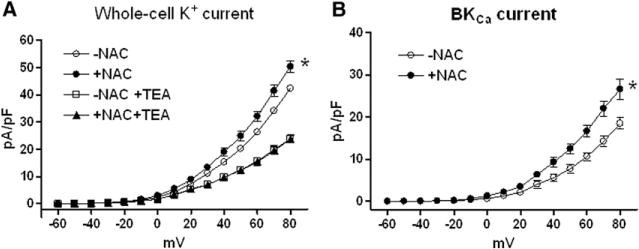
Effect of N-acetylcysteine (NAC) on large conductance Ca2+-activated K+ (BKCa) channel current density in myocytes of uterine arteries from hypoxic pregnant sheep. A, Whole-cell K+ currents were recorded in the absence or presence of NAC (1 mmol/L) and tetraethylammonium (TEA, 1 mmol/L). B, BKCa currents were determined as the difference between the whole-cell K+ current in the absence of TEA and that in the presence of TEA. The K+ currents were normalized to cell capacitance and were expressed as picoampere per picofarad (pA/pF; y axis), as a function of stepwise 10-mV depolarizing pulses (x axis). Data are means±SEM of cells from 5 animals of each group (*P<0.05; +NAC vs −NAC).
Endothelium Mediated the NAC-Sensitive Component of SKCa-Induced Relaxations of Uterine Arteries in Pregnant Sheep
The finding that NAC increased NS309-induced relaxations of uterine arteries from hypoxic animals but had no effect on SKCa channel current density in uterine arterial smooth muscle cells is intriguing and suggests an endothelium-dependent mechanism in the NAC-sensitive component of SKCa-mediated relaxation under hypoxic conditions. To test this hypothesis, intact and endothelium-denuded uterine arteries of pregnant sheep were treated ex vivo under normoxic (21% O2) or hypoxic (10.5% O2) conditions for 48 hours. After the treatment, NS309-induced relaxations were determined in the absence or presence of NAC pretreatment for 20 minutes. Our previous study showed that endothelium removal caused a modest 20% decrease in the maximal response of NS309-induced relaxations in normoxic vessels.6 As shown in Figure 3A, NAC had no effect on NS309-mediated relaxations in the normoxic condition. Hypoxia significantly decreased the maximal response of NS309-induced relaxations (22.7±2.6% versus 56.0±2.6%; P<0.05), and NAC significantly increased NS306-induced relaxations from 22.7±2.6% to 44.8±1.9% (P<0.05) in endothelium intact vessels (Figure 3B). In contrast, NAC showed no effect on NS306-induced relaxations in endothelium-denuded uterine arteries (Figure 3C).
Figure 3.
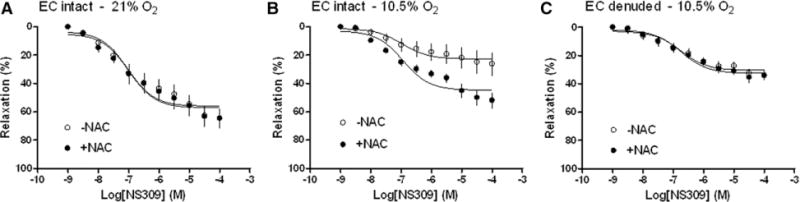
Effect of endothelium on N-acetylcysteine (NAC)–mediated response in NS309-induced relaxations. Uterine arteries were isolated from pregnant sheep. Endothelium (EC) intact and denuded vessels were treated ex vivo under 21% O2 (A) or 10.5% O2 (B and C) for 48 hours. Concentration–response curves were determined in NS309-induced relaxations in the absence or presence of NAC (1 mmol/L). The x axis indicates cumulative increases of the agonist in approximately one-half log increments. Data are means±SEM from 5 animals in each group. The Emax values are presented in the text.
Steroid Hormones Enhanced KCa Channel–Mediated Relaxations of Uterine Arteries From Nonpregnant Sheep
Compared with uterine arteries of pregnant animals (Figure 1A and 1B), both NS1619- and NS309-induced relaxations were significantly attenuated in uterine arteries of nonpregnant animals (Figure 4). Our recent study demonstrated that steroid hormone treatment upregulated BKCa channel expression and current density in uterine arteries.4 In the present study, we further examined the functional significance of steroid hormone–mediated effect on KCa channel–induced relaxations of uterine arteries. As shown in Figure 4A, the hormonal treatment significantly enhanced the potency (pD2, −log EC50; 5.4±0.3 versus 7.4±0.4; P<0.05) and had a tendency to increase the maximal response (12.6±2.1% versus 18.8±2.4%; P=0.08) of NS1619-induced relaxations. Both the potency (5.6±0.2 versus 6.4±0.2; P<0.05) and the maximal response (10.4±1.0% versus 24.2±2.1%; P<0.05) of NS309-induced relaxations were significantly increased by the hormonal treatment (Figure 4B). Consistent with the increased NS309-induced relaxations, the hormonal treatment significantly upregulated the protein abundance of SK3 but not SK2 channels in uterine arteries (Figure 5).
Figure 4.
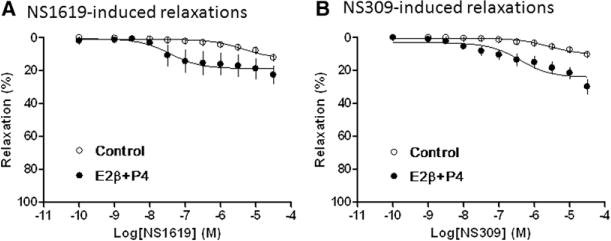
Effect of hormonal treatment on Ca2+-activated K+ channel–induced relaxations of uterine arteries from normoxic nonpregnant sheep. Uterine arteries were isolated from normoxic nonpregnant sheep and treated ex vivo with 17β-estradiol (E2β; 0.3 nmol/L) plus progesterone (P4; 100 nmol/L) under 21% O2 for 48 hours. Concentration–response curves were determined in NS1619-induced relaxations (A) and NS309-induced relaxations (B) in uterine arteries treated without (control) or with steroid hormones (E2β+P4). The x axis indicates cumulative increases of the agonists in approximately one-half log increments. Data are means±SEM from 4 to 6 animals in each group. The Emax and pD2 values are presented in the text.
Figure 5.
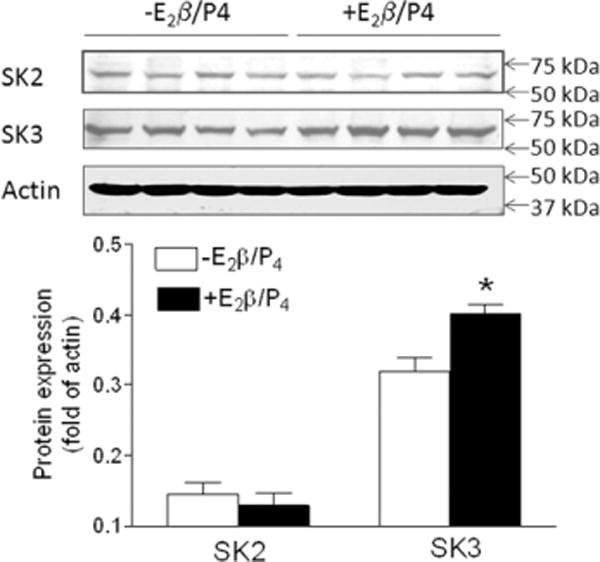
Effect of steroid hormones on small conductance calcium–activated K+ channel (SK)2 and SK3 protein abundance in uterine arteries from normoxic nonpregnant sheep. Uterine arteries were isolated from normoxic nonpregnant sheep and treated ex vivo with 17β-estradiol (E2β; 0.3 nmol/L) plus progesterone (P4; 100 nmol/L) under 21% O2 for 48 hours. Protein abundance of SK2 and SK3 was determined by Western blot analyses. Data are means±SEM of tissues from 4 animals of each group (*P<0.05; +E2β/P4 vs −E2β/P4).
NAC Rescued the Effect of Steroid Hormones on KCa Channel–Induced Relaxations of Uterine Arteries in Hypoxic Nonpregnant Sheep
Steroid hormones increased KCa channel–mediated relaxations of uterine arteries in normoxic nonpregnant animals (Figure 4). However, the hormonal treatment had no effect on either NS1619-induced relaxations (Emax: 11.2±1.0% versus 12.9±1.1%; P>0.05; Figure 6A) or NS309-induced relaxations (Emax: 11.0±0.5% versus 10.3±1.4%; P>0.05; Figure 6B) of uterine arteries in hypoxic nonpregnant sheep. Inhibition of ROS by NAC rescued the effect of steroid hormones on uterine arteries of hypoxic animals, and both NS1619- and NS309-induced relaxations were significantly increased by the hormonal treatment in the presence of NAC (Figure 6).
Figure 6.
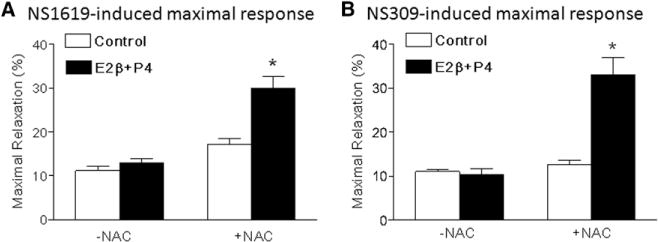
Effect of N-acetylcysteine (NAC) on steroid hormone–mediated response in KCa channel–induced relaxations of uterine arteries from hypoxic nonpregnant sheep. Uterine arteries were isolated from hypoxic nonpregnant sheep and treated ex vivo with 17β-estradiol (E2β; 0.3 nmol/L) plus progesterone (P4; 100 nmol/L) under 10.5% O2 for 48 hours in the absence and presence of NAC (1 mmol/L). NS1619-and NS309-induced relaxations were determined after the treatment. A, NS1619-induced maximal response. B, NS309-induced maximal response. Data are means±SEM from 4 to 6 animals in each group (*P<0.05; E2β+P4 vs control).
NAC Restored the Effect of Steroid Hormones on Upregulation of BKCa Channel Activities in Uterine Arteries of Hypoxic Nonpregnant Sheep
We further examined KCa channel activities in uterine arteries from hypoxic nonpregnant sheep treated with steroid hormones in the presence of NAC for 48 hours. Our recent study demonstrated that chronic hypoxia abolished steroid hormone–induced upregulation of BKCa channel activity in uterine arteries of nonpregnant sheep.5 The present study showed that in the presence of NAC, the hormonal treatment of uterine arteries from nonpregnant animals acclimatized to long-term high-altitude hypoxia was able to increase whole-cell K+ densities (Figure 7A) and BKCa current densities significantly (Figure 7B), similar to those seen in normoxic animals in the previous study.5 In contrast, whole-cell K+ currents in uterine arteries treated with NAC were not sensitive to SKCa blocker apamin, and enhanced whole-cell K+ currents by the hormonal treatment in the presence of NAC were not inhibited by apamin (Figure S2).
Figure 7.
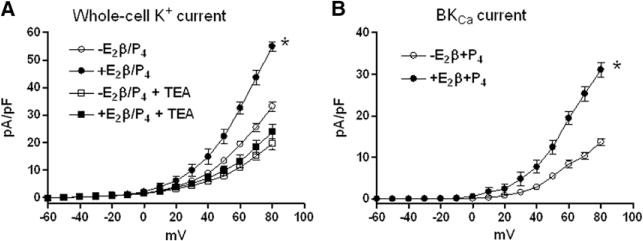
Effect of N-acetylcysteine (NAC) on steroid hormone–mediated response in large conductance Ca2+-activated K+ (BKCa) channel current density in uterine arteries from hypoxic nonpregnant sheep. Uterine arteries were isolated from hypoxic nonpregnant sheep and treated ex vivo with 17β-estradiol (E2β; 0.3 nmol/L) plus progesterone (P4; 100 nmol/L) under 10.5% O2 for 48 hours in the presence of NAC (1 mmol/L). After the treatment, whole-cell K+ currents were recorded in the absence or presence of tetraethylammonium (TEA, 1 mmol/L) in myocytes isolated from uterine arteries. The K+ currents were normalized to cell capacitance and were expressed as picoampere per picofarad (pA/Pf; y axis), as a function of stepwise 10-mV depolarizing pulses (x axis). A, Whole-cell K+ currents. B, BKCa currents were determined as the difference between the whole-cell K+ current in the absence of TEA and that in the presence of TEA. Data are means±SEM of cells from 5 animals of each group (*P<0.05; +E2β/P4 vs −E2β/P4).
Discussion
In the present study, we have demonstrated that the function of both BKCa and SKCa channels in uterine arteries of pregnant animals is suppressed by heightened oxidative stress during chronic hypoxia. Our results suggest that chronic hypoxia–induced oxidative stress exerts its adverse effect on KCa channel–mediated relaxations of uterine arteries through suppressing steroid hormone–induced upregulation of KCa channel activities. These findings provide strong evidence that heightened ROS is a common mechanism to impair BKCa and SKCa channel function in uterine arteries and contributes to the dysfunction of uterine circulation caused by chronic hypoxia during gestation.
Consistent with our previous studies,6 the present finding that both NS1619- and NS309-induced relaxations of uterine arteries were significantly attenuated by chronic hypoxia in pregnant animals, further suggesting that chronic hypoxia downregulates both BKCa and SKCa channel activities. However, the molecular mechanisms underlying chronic hypoxia–mediated downregulation of BKCa channel– and SKCa channel–mediated uterine arterial relaxation remain incompletely understood. The present finding that treatment with NAC, an antioxidant to scavenge free radicals,22 significantly enhanced both NS1619- and NS309-induced relaxations in hypoxic but not normoxic animal suggests that hypoxia-mediated increase in ROS may be one of the key mechanisms in attenuating both BKCa channel– and SKCa channel–mediated relaxations of uterine arteries during gestation. ROS play an important role in pathogenesis of cardiovascular dysfunctions.14,15 Increased ROS have been shown to inhibit BKCa channel activity.16,23–25 We recently demonstrated that gestational hypoxia increased ROS production in uterine arteries.17 Thus, heightened ROS likely attribute to the suppressed function of BKCa and SKCa channels in uterine arteries from pregnant sheep by chronic hypoxia. In the present study, our finding that NAC partially reversed the impairment of BKCa channel activity suggests that ROS may directly alter BKCa channel activity, resulting in the regulation of BKCa-mediated uterine vascular contractility in response to hypoxia exposure. These observations are consistent with previous findings that the nicotinamide adenine dinucleotide phosphate oxidase inhibitor apocynin partially reversed the suppression of BKCa channel function by chronic hypoxia.17 Other studies in insulin resistant rats showed that impaired BKCa channel–mediated relaxations of cerebral arteries were restored by superoxide dismutase plus catalase that catalyze the breakdown of free radicals.26
A novel and interesting finding in the present study is that heightened ROS regulate SKCa channel–mediated uterine vascular relaxations but without alteration of SKCa channel activity. Our data indicated that hypoxia-mediated impairment of SKCa channel–mediated relaxations of uterine arteries were alleviated by NAC. However, NAC treatment failed to restore SKCa channel current density in vascular smooth muscle cells of uterine arteries. It is likely that the NAC enhanced SKCa-mediated relaxations of uterine arteries via an endothelium-dependent mechanism. SKCa channels are present in both vascular smooth muscle cells and endothelial cells of uterine arteries.6 The finding that the effect of NAC on NS309-mediated relaxations was lost in endothelium-denuded uterine arteries under the hypoxic condition suggests an endothelium-dependent mechanism in the NAC-sensitive component of SKCa-mediated relaxations, possibly by releasing vasodilators (eg, nitric oxide or prostacyclin) and transmitting hyperpolarization into vascular smooth muscle through myoendothelial gap junctions.3 Given the finding that the increased blood pressure in the rat model with reduced uterine perfusion pressure was alleviated by NAC,27 the upregulation of KCa function by NAC may have therapeutic implications.
Whereas 48 hours of estrogen plus progesterone treatment is a short duration compared with the long duration of estrogen and progesterone being exposed to the blood vessels in vivo during gestation, our previous studies demonstrated that this ex vivo hormonal treatment increased BKCa channel activity and decreased uterine arterial myogenic tone.4,5,18–20 We have demonstrated that BKCa channel function is directly regulated by estrogen and progesterone through a genomic effect.4 Similarly, 17β-estradiol and progesterone also selectively upregulated the expression of SKCa type 3 channels in uterine arteries. This finding mimicked enhanced SKCa channel activity seen in pregnant uterine arteries form normoxic animals.6 The lack of hormonal effect on SK2 channels is intriguing, but the mechanisms remain unknown at present. Both estrogen receptors α (ER-α) and β (ER-β) are present in the uterine artery.18,28 Estrogen has been shown to control SKCa channel expression in human myometrial cells via the specificity protein family of transcription factors.29 Although this significant, albeit small, change in SK3 channel expression may contribute to the channel function, the finding of the acute effect of NAC in the present study suggests that chronic hypoxia (either in vivo or ex vivo for 48 hours)–mediated ROS persist and predominately affect KCa channel activities in the uterine arteries.
In contrast to the upregulation of KCa channel activities and KCa channel–mediated relaxations in uterine arteries of normoxic animals by estrogen and progesterone, the effect of steroid hormones on the regulation of KCa channel activities and their function of inducing relaxations was diminished in hypoxic animals. These findings suggest that hypoxia-mediated downregulation of KCa channel activities and function may be regulated through steroid hormone–mediated signaling. These observations are not surprising because ER-α receptor is downregulated during gestational hypoxia via increased methylation of the receptor gene.18,19 Thus, it is possible that downregulation of ER-α may play a role in reduced KCa channel activities.
Of interest, the present study demonstrates that the hormonal treatment in the presence of NAC restored KCa channel–mediated relaxations of uterine arteries from hypoxic animals, suggesting that increased oxidative stress causing by chronic hypoxia diminishes the effect of steroid hormones to regulate of KCa channel activities. ROS has been shown to induce post-translational modifications of ER-α, leading to ER-α downregulation in human breast cancer cells.30 Scavenging free radicals by NAC removed inhibitory effects of ROS, allowing steroid hormones to upregulate KCa channel function. Correspondingly, BKCa channel activity in uterine arterial smooth muscle cells was also restored by NAC with the hormonal treatment. Although cotreatment of uterine arteries from hypoxic animals with steroid hormones and NAC failed to upregulate SKCa channel activity in vascular smooth muscle cells, it remains possible to upregulate SKCa channel function in endothelial cells, which in turn results in enhanced SKCa channel–mediated relaxations via the interaction of endothelial and smooth muscle cells of uterine arteries as aforementioned. It has been shown that NAC supplement improves human coronary and peripheral endothelium-dependent vasodilation.31
In conclusion, the results suggest that heightened ROS production by chronic hypoxia attenuates steroid hormone–mediated signaling, which leads to downregulation of KCa channel activities and results in decreased relaxations of uterine arteries during gestation. The attenuation of KCa channel–mediated relaxations may contribute to enhanced uterine vascular tone and increased incidence of preeclampsia and fetal intrauterine growth restriction associated with maternal hypoxia.
Perspectives
The present study demonstrates a novel mechanism that hypoxia-mediated heightened ROS attenuates KCa channel activities and their mediated uterine vascular relaxation in pregnancy. Given that KCa channels play an important role in regulating vascular tone and thus blood flow and pressure, dysregulation of KCa channel function in the uterine artery may contribute significantly to maladaptation of uterine vascular hemodynamics in pregnancy, leading to an increased risk of preeclampsia. Reductions in uteroplacental blood flow and chronic uteroplacental ischemia in a variety of animal models lead to a hypertensive state that closely resembles preeclampsia in women. The present findings provide a mechanistic understanding of dysfunction of KCa channels in uterine vascular adaptation to pregnancy in chronic hypoxia and may suggest new insights of therapeutic strategies by enhancing the KCa channel activities in vascular smooth muscle, which may be beneficial for pregnant women with preeclampsia.
Supplementary Material
Novelty and Significance.
What Is New?
Chronic hypoxia during gestation attenuates both large conductance Ca2+-activated K+ (KCa) and small conductance KCa channel mediated relaxations of uterine arteries.
Increased reactive oxygen species production contributes to hypoxia-induced attenuation of KCa channel function in uterine arteries during pregnancy.
Gestational hypoxia inhibits steroid hormonal regulation of KCa channel activities and their mediated uterine vascular relaxations via reactive oxygen species signaling.
What Is Relevant?
Chronic hypoxia during pregnancy is one of the most common factors in increasing uterine vascular tone and decreasing uterine blood flow.
KCa channels play an important role in maintaining vascular tone. Chronic hypoxia–mediated maladaptation of KCa channel function may be a key mechanism in aberrant uteroplacental circulation and fetal growth restriction.
Summary
The present study demonstrates a novel role of heighted oxidative stress on sex steroid hormone–mediated KCa channel function in uterine arterial smooth muscle in pregnancy acclimated to high-altitude hypoxia. Understanding of the interaction of reactive oxygen species and steroid hormones in the regulation of KCa channel function will help improve our understanding of pathophysiological mechanisms underlying maladaptation of the uteroplacental circulation and pregnancy complications including preeclampsia and fetal growth restriction associated with chronic hypoxia during gestation.
Acknowledgments
Sources of Funding: This work was supported by National Institutes of Health grants HL110125 (to L. Zhang), HL089012 (to L. Zhang), HD031226 (to L. Zhang), and DA032510 (to D. Xiao) and by the regents of the University of California Tobacco-Related Disease Research Program grant 22XT-0022 (to D. Xiao).
Footnotes
Disclosures
None.
The online-only Data Supplement is available with this article at http://hyper.ahajournals.org/lookup/suppl/doi:10.1161/HYPERTENSIONAHA.114.03555/-/DC1.
Hypertension is available at http://hyper.ahajournals.org
References
- 1.Ledoux J, Werner ME, Brayden JE, Nelson MT. Calcium-activated potassium channels and the regulation of vascular tone. Physiology (Bethesda) 2006;21:69–78. doi: 10.1152/physiol.00040.2005. [DOI] [PubMed] [Google Scholar]
- 2.Hu XQ, Zhang L. Function and regulation of large conductance Ca(2+)-activated K+ channel in vascular smooth muscle cells. Drug Discov Today. 2012;17:974–987. doi: 10.1016/j.drudis.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Félétou M. Calcium-activated potassium channels and endothelial dysfunction: therapeutic options? Br J Pharmacol. 2009;156:545–562. doi: 10.1111/j.1476-5381.2009.00052.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu XQ, Xiao D, Zhu R, Huang X, Yang S, Wilson S, Zhang L. Pregnancy upregulates large-conductance Ca(2+)-activated K(+) channel activity and attenuates myogenic tone in uterine arteries. Hypertension. 2011;58:1132–1139. doi: 10.1161/HYPERTENSIONAHA.111.179952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hu XQ, Xiao D, Zhu R, Huang X, Yang S, Wilson SM, Zhang L. Chronic hypoxia suppresses pregnancy-induced upregulation of large-conductance Ca2+-activated K+ channel activity in uterine arteries. Hypertension. 2012;60:214–222. doi: 10.1161/HYPERTENSIONAHA.112.196097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu R, Hu XQ, Xiao D, Yang S, Wilson SM, Longo LD, Zhang L. Chronic hypoxia inhibits pregnancy-induced upregulation of SKCa channel expression and function in uterine arteries. Hypertension. 2013;62:367–374. doi: 10.1161/HYPERTENSIONAHA.113.01236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenfeld CR. Mechanisms regulating angiotensin II responsiveness by the uteroplacental circulation. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1025–R1040. doi: 10.1152/ajpregu.2001.281.4.R1025. [DOI] [PubMed] [Google Scholar]
- 8.Zamudio S, Palmer SK, Dahms TE, Berman JC, Young DA, Moore LG. Alterations in uteroplacental blood flow precede hypertension in preeclampsia at high altitude. J Appl Physiol (1985) 1995;79:15–22. doi: 10.1152/jappl.1995.79.1.15. [DOI] [PubMed] [Google Scholar]
- 9.Zamudio S, Palmer SK, Droma T, Stamm E, Coffin C, Moore LG. Effect of altitude on uterine artery blood flow during normal pregnancy. J Appl Physiol (1985) 1995;79:7–14. doi: 10.1152/jappl.1995.79.1.7. [DOI] [PubMed] [Google Scholar]
- 10.Keyes LE, Armaza JF, Niermeyer S, Vargas E, Young DA, Moore LG. Intrauterine growth restriction, preeclampsia, and intrauterine mortality at high altitude in Bolivia. Pediatr Res. 2003;54:20–25. doi: 10.1203/01.PDR.0000069846.64389.DC. [DOI] [PubMed] [Google Scholar]
- 11.Julian CG, Galan HL, Wilson MJ, Desilva W, Cioffi-Ragan D, Schwartz J, Moore LG. Lower uterine artery blood flow and higher endothelin relative to nitric oxide metabolite levels are associated with reductions in birth weight at high altitude. Am J Physiol Regul Integr Comp Physiol. 2008;295:R906–R915. doi: 10.1152/ajpregu.00164.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marshall C, Mamary AJ, Verhoeven AJ, Marshall BE. Pulmonary artery NADPH-oxidase is activated in hypoxic pulmonary vasoconstriction. Am J Respir Cell Mol Biol. 1996;15:633–644. doi: 10.1165/ajrcmb.15.5.8918370. [DOI] [PubMed] [Google Scholar]
- 13.Rathore R, Zheng YM, Niu CF, Liu QH, Korde A, Ho YS, Wang YX. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic Biol Med. 2008;45:1223–1231. doi: 10.1016/j.freeradbiomed.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolin MS, Ahmad M, Gupte SA. Oxidant and redox signaling in vascular oxygen sensing mechanisms: basic concepts, current controversies, and potential importance of cytosolic NADPH. Am J Physiol Lung Cell Mol Physiol. 2005;289:L159–L173. doi: 10.1152/ajplung.00060.2005. [DOI] [PubMed] [Google Scholar]
- 15.Schnabel R, Blankenberg S. Oxidative stress in cardiovascular disease: successful translation from bench to bedside? Circulation. 2007;116:1338–1340. doi: 10.1161/CIRCULATIONAHA.107.728394. [DOI] [PubMed] [Google Scholar]
- 16.Brakemeier S, Eichler I, Knorr A, Fassheber T, Köhler R, Hoyer J. Modulation of Ca2+-activated K+ channel in renal artery endothelium in situ by nitric oxide and reactive oxygen species. Kidney Int. 2003;64:199–207. doi: 10.1046/j.1523-1755.2003.00051.x. [DOI] [PubMed] [Google Scholar]
- 17.Xiao D, Hu XQ, Huang X, Zhou J, Wilson SM, Yang S, Zhang L. Chronic hypoxia during gestation enhances uterine arterial myogenic tone via heightened oxidative stress. PLoS One. 2013;8:e73731. doi: 10.1371/journal.pone.0073731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang K, Xiao D, Huang X, Xue Z, Yang S, Longo LD, Zhang L. Chronic hypoxia inhibits sex steroid hormone-mediated attenuation of ovine uterine arterial myogenic tone in pregnancy. Hypertension. 2010;56:750–757. doi: 10.1161/HYPERTENSIONAHA.110.155812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dasgupta C, Chen M, Zhang H, Yang S, Zhang L. Chronic hypoxia during gestation causes epigenetic repression of the estrogen receptor-α gene in ovine uterine arteries via heightened promoter methylation. Hypertension. 2012;60:697–704. doi: 10.1161/HYPERTENSIONAHA.112.198242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiao D, Huang X, Yang S, Zhang L. Direct chronic effect of steroid hormones in attenuating uterine arterial myogenic tone: role of protein kinase c/extracellular signal-regulated kinase ½. Hypertension. 2009;54:352–358. doi: 10.1161/HYPERTENSIONAHA.109.130781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Catchpole HR. Hormonal mechanisms in pregnancy and parturition. In: Cole HH, Cupps PT, editors. Reproduction in Domestic Animals. 3. New York, NY: Academic Press; 1977. [Google Scholar]
- 22.Sun SY. N-acetylcysteine, reactive oxygen species and beyond. Cancer Biol Ther. 2010;9:109–110. doi: 10.4161/cbt.9.2.10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Y, Terata K, Chai Q, Li H, Kleinman LH, Gutterman DD. Peroxynitrite inhibits Ca2+-activated K+ channel activity in smooth muscle of human coronary arterioles. Circ Res. 2002;91:1070–1076. doi: 10.1161/01.res.0000046003.14031.98. [DOI] [PubMed] [Google Scholar]
- 24.Soto MA, González C, Lissi E, Vergara C, Latorre R. Ca(2+)-activated K+ channel inhibition by reactive oxygen species. Am J Physiol Cell Physiol. 2002;282:C461–C471. doi: 10.1152/ajpcell.00167.2001. [DOI] [PubMed] [Google Scholar]
- 25.Tang XD, Garcia ML, Heinemann SH, Hoshi T. Reactive oxygen species impair Slo1 BK channel function by altering cysteine-mediated calcium sensing. Nat Struct Mol Biol. 2004;11:171–178. doi: 10.1038/nsmb725. [DOI] [PubMed] [Google Scholar]
- 26.Erdös B, Simandle SA, Snipes JA, Miller AW, Busija DW. Potassium channel dysfunction in cerebral arteries of insulin-resistant rats is mediated by reactive oxygen species. Stroke. 2004;35:964–969. doi: 10.1161/01.STR.0000119753.05670.F1. [DOI] [PubMed] [Google Scholar]
- 27.Chang EY, Barbosa E, Paintlia MK, Singh A, Singh I. The use of N-acetylcysteine for the prevention of hypertension in the reduced uterine perfusion pressure model for preeclampsia in Sprague-Dawley rats. Am J Obstet Gynecol. 2005;193(3 Pt 2):952–956. doi: 10.1016/j.ajog.2005.05.083. [DOI] [PubMed] [Google Scholar]
- 28.Byers MJ, Zangl A, Phernetton TM, Lopez G, Chen DB, Magness RR. Endothelial vasodilator production by ovine uterine and systemic arteries: ovarian steroid and pregnancy control of ER-α and ER-β levels. J Physiol. 2005;565:85–99. doi: 10.1113/jphysiol.2005.085753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pierce SL, England SK. SK3 channel expression during pregnancy is regulated through estrogen and Sp factor-mediated transcriptional control of the KCNN3 gene. Am J Physiol Endocrinol Metab. 2010;299:E640–E646. doi: 10.1152/ajpendo.00063.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weitsman GE, Weebadda W, Ung K, Murphy LC. Reactive oxygen species induce phosphorylation of serine 118 and 167 on estrogen receptor alpha. Breast Cancer Res Treat. 2009;118:269–279. doi: 10.1007/s10549-008-0221-0. [DOI] [PubMed] [Google Scholar]
- 31.Andrews NP, Prasad A, Quyyumi AA. N-acetylcysteine improves coronary and peripheral vascular function. J Am Coll Cardiol. 2001;37:117–123. doi: 10.1016/s0735-1097(00)01093-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.