

Abstract
Trypanosoma cruzi, the aetiological agent of Chagas disease, invades nucleated mammalian cells including macrophages. In this study, we investigated the crosstalk between T. cruzi-induced immune activation of reactive oxygen species (ROS) and pro-inflammatory responses, and their role in myocardial pathology. Splenocytes of infected mice (C3H/HeN) responded to Tc-antigenic stimulus by more than a two-fold increase in NADPH oxidase (NOX) activity, ROS generation, cytokine production (IFN-γ > IL-4 > TNFα > IL1-β ≈ IL6), and predominant expansion of CD4+ and CD8+ T cells. Inhibition of NOX, but not of myeloperoxidase and xanthine oxidase, controlled the ROS (>98%) and cytokine (70–89%) release by Tc-stimulated splenocytes of infected mice. Treatment of infected mice with apocynin (NOX inhibitor) in drinking water resulted in a 50–90% decline in endogenous NOX/ROS and cytokine levels, and splenic phagocytes’ proliferation. The splenic percentage of T cells was maintained, though more than a 40% decline in splenic index (spleen weight/body weight) indicated decreased T-cell proliferation in apocynin-treated/infected mice. The blood and tissue parasite burden were significantly increased in apocynin-treated/infected mice, yet acute myocarditis, ie inflammatory infiltrate consisting of macrophages, neutrophils, and CD8+ T cells, and tissue oxidative adducts (eg 8-isoprostanes, 3-nitrotyrosine, and 4-hydroxynonenal) were diminished in apocynin-treated/infected mice. Consequently, hypertrophy (increased cardiomyocytes’ size and β-MHC, BNP, and ANP mRNA levels) and fibrosis (increased collagen, glycosaminoglycans, and lipid contents) of the heart during the chronic phase were controlled in apocynin-treated mice. We conclude that NOX/ROS is a critical regulator of the splenic response (phagocytes, T cells, and cytokines) to T. cruzi infection, and bystander effects of heart-infiltrating phagocytes and CD8+ T cells resulting in cardiac remodelling in chagasic mice.
Keywords: Chagas disease, Trypanosoma cruzi, reactive oxygen species, NADPH oxidase, apocynin, inflammation
Introduction
Chagas disease is caused by the protozoan Trypanosoma cruzi [1]. During the acute phase of infection, parasites can be found in the circulating blood, and the host may develop fever or swelling around the site of inoculation and, rarely, severe inflammation in the heart muscle or brain. A majority of patients (>95%) recover from acute parasitaemia and enter a prolonged clinically asymptomatic phase, sometimes exhibiting a remnant of mild myocardial changes with scattered microscopic foci of fibrosis and lymphocytic infiltration. Several years later, ~30% of the infected individuals develop chronic cardiac disease with progressive cardiomegaly, arrhythmia, thromboembolic events, and heart failure [2,3], suggested to be associated with the persistent presence of inflammatory infiltrate [4], oxidative stress-induced cell and tissue damage [3], and fibrosis in the heart [5].
NADPH oxidase (NOX) activation and ROS production, termed the ‘oxidative burst’ of activated phagocytic cells, might play an important role in the control of T. cruzi [4,6]. Of the seven known homologues of NOX, Nox2 is the primary source of ROS in activated phagocytes. The inducible Nox1, predominantly expressed in vascular smooth muscle cells [7], and Nox4, constitutively expressed in blood vessels, produce low levels of ROS under basal conditions [8] and may also contribute to the redox state in the heart during T. cruzi infection. NOX isozymes are multi-subunit complexes and utilize NAD(P)H as an electron donor to reduce O2 to superoxide (O2•−), which is then dismutated into other oxidants (eg H2O2, •OH) [9]. In prototypic Nox2, plasma membrane-associated proteins gp91phox and p22phox make up the flavocytochrome-b558 complex that is the major component responsible for enzyme stability and activity. Phosphorylation of cytosolic factors (p47phox, p67phox, and p40phox) and small Rho GTPases in response to exogenous or endogenous stimuli initiates their translocation to the cell membrane and NOX activation [10,11]. Activated phagocytes, a key defence response to the invading pathogens [12], exert cytotoxic effects via ROS-mediated modification of DNA, proteins, and lipids. Alternatively, O2•− may combine with nitric oxide (•NO) to produce peroxynitrite and peroxynitrous acid, which have been shown to kill T. cruzi [13,14]. Studies in models of atherosclerosis and other forms of peripheral vascular disease have implicated NOX/ROS as obligatory mediators of inflammation induced by growth factors and cytokines [10]. The specific role of NOX/ROS in triggering myocardial inflammatory processes during Chagas disease is not known.
In this study, we investigated the role of NOX/ROS in the activation of cytokines and infiltration of inflammatory infiltrate in the heart, and in perpetuating chagasic myocarditis. For this, mice were infected with T. cruzi and treated with NOX inhibitor (apocynin) in drinking water. Our data suggest that NOX/ROS play an important role in splenic activation of inflammatory cells and cytokine production, and contribute to myocarditis and chronic fibrosis in Chagas disease.
Materials and methods
Additional details may be found in the Supporting information, Supplementary materials and methods.
Parasites and mice
T. cruzi trypomastigotes (SylvioX10/4 strain) were propagated in C2C12 cells [15]. All reagents for tissue culture were purchased from Gibco-Invitrogen (Carlsbad, CA, USA). C3H/HeN male mice (6–8 weeks old; Harlan Labs, Houston, TX, USA) were infected with T. cruzi (10 000 per mouse, intra-peritoneally) and treated with 1.5 mM apocynin in drinking water. Apocynin is a naturally occurring methoxy-substituted catechol, experimentally used as an inhibitor of NADPH oxidase [16]. Mice were sacrificed at days 25 (acute phase) and 150 (chronic phase) post-infection, and sera/plasma and tissue samples were stored at 4 °C or −80 °C. Animal experiments were performed according to the National Institutes of Health Guide for the Care and Use of Experimental Animals.
Cell activation and proliferation
T. cruzi antigenic lysate (TcL) was prepared by subjecting parasites (1 × 109 per ml PBS, 50% amastigotes–50% trypomastigotes) to five to six freeze–thaw cycles followed by sonication on ice for 30 min. Freshly harvested spleens were crushed to prepare a single cell suspension and depleted of red blood cells by hypotonic lysis. Splenocytes were seeded in 24-well plates (106 per ml RPMI/10% FBS) and incubated with concanavalin A (ConA, 5 μg/ml) or TcL (25 μg/ml) at 37 °C and 5% CO2. In some experiments, cells were incubated in the presence of 100 μM allopurinol (Allo), 50 μM salicyl hydroxamate (SHX) or 600 μM apocynin (Apo), which are specific inhibitors of xanthine oxidase (XOD), myeloperoxidase (MPO), and Nox2, respectively, or with 7.5 μM N-acetylcysteine (NAC; an ROS scavenger) or 20 μM FCCP (a mitochondrial uncoupler). Cells and supernatants were stored at −80 °C.
Flow cytometry
Splenocytes (106 per 100 μl) were incubated for 30 min with FITC-conjugated anti-CD68 (macrophages), Cy3-conjugated anti-Ly-6G/Gr-1 (neutrophils) (from BD-Pharmingen, San Jose, CA, USA), FITC-conjugated anti-CD8, or PE-conjugated anti-CD4 (eBioscience, San Diego, CA, USA) antibodies (1 : 50 dilution). Following incubation, cells were fixed with 2% paraformaldehyde and flow cytometry was performed on a FACScan apparatus. Flow data were analysed using BD Cell Quest software. Cells stained with isotype-matched IgGs were used as negative controls.
Tissue homogenates
Spleen or heart tissue (50 mg) was washed with ice-cold Tris-buffered saline and homogenized in lysis buffer (tissue : buffer ratio, 1 : 10, w/v) [17]. Homogenates were centrifuged at 3000 g at 4 °C for 10 min and the supernatants were stored at −80 °C.
Biochemical assays
Splenic and heart homogenates were used for in-gel detection of NOX activity [18]. The extracellular matrix (ECM) components [collagen and glycosaminoglycans (GAGs)] and metalloproteinases in heart homogenates were assessed using the methods described in the Supporting information, Supplementary materials and methods.
ROS measurements
Intracellular ROS levels were determined using CM-H2DCF-DA (Ex498 nm/Em598 nm) and Amplex® Red/horseradish peroxidase (Ex563 nm/Em587 nm) fluorescent probes (Invitrogen, Carlsbad, CA, USA) [19]. For in situ ROS detection, 10 μm cryostat sections were equilibrated in Kreb’s buffer and incubated in the dark for 30 min with 5 μM dihydroethidium (DHE). DHE, when oxidized, intercalates within the cell’s DNA, staining its nucleus a bright fluorescent red (Ex518 nm/Em605 nm). Fluorescence was detected on an Olympus BX-15 microscope and images were captured by using a mounted digital camera.
Cytokine levels
A Bio-Plex Multiplex Cytokine Assay (Bio-Rad, Hercules, CA, USA) was employed to profile the concentration of nine murine cytokines. Briefly, plates were coated with cytokine-specific antibodies conjugated with different colour-coded beads and then sequentially incubated with splenic supernatants, biotinylated cytokine-specific detection antibodies, and streptavidin–phycoerythrin conjugate. Fluorescence was recorded using a SpectraMax® M5 microplate reader, and cytokine concentrations were calculated with Bio-Plex Manager software by using a standard curve derived from recombinant cytokines (2–32 000 pg/ml). In some experiments, IL-6, IL-4, IFN-γ, and TNF-α levels in the culture supernatants were measured using optEIA™ ELISA kits (Pharmingen, San Diego, CA, USA), according to the manufacturer’s specifications.
Histology and immunohistochemistry
Left ventricular (LV) tissue sections were fixed in formalin and embedded in paraffin. Tissue sections (5 μm) were stained with haematoxylin and eosin (H&E) and Masson’s Trichrome [20] to examine the inflammatory infiltrate/myocarditis and collagen deposition, respectively. Tissue sections (5 μm) were subjected to immunostaining with antibody against 4-hydroxynonenal (4-HNE; 1 : 500; Alpha-Diagnostics, San Antonio, TX, USA) and gp91phox (1 : 100; Santa Cruz Biotech, Santa Cruz, CA, USA) to visualize oxidative adducts and NOX activation, respectively, and with antibody against CD68, Ly-6G/Gr-1, Ly-3.2/CD8b.2, and CD4/L3T4 (1 : 100, BD-Pharmingen) to visualize immune cell populations (Supporting information, Supplementary materials and methods).
Western blotting
Tissue homogenates (5 μg) were resolved on denaturing 10% acrylamide gels and proteins transferred to PVDF membranes. Membranes were probed with specific antibodies against gp91phox (1 : 1000; Santa Cruz Biotech, Santa Cruz, CA, USA), 8-isoprostane (1 : 2000; Oxford-Biomed, Rochester Hills, MI, USA), 3-nitrotyrosine (3-NT; 1 : 2000; Cell Signaling, Danvers, MA, USA) and angiotensin II receptor (AT1; 1 : 500; Millipore, Billerica, MA, USA), and signal was developed by a chemiluminescence detection system (GE-Healthcare, Piscataway, NJ, USA) [21].
Real-time RT-PCR
Total RNA from tissue samples (50 mg) was extracted using Trizol reagent (Invitrogen, Carlsbad, CA, USA) and reverse transcribed. First-strand cDNA was used as a template in a real-time PCR on an iCycler thermal cycler (Bio-Rad) with SYBR Green Supermix (Bio-Rad, Hercules, CA, USA), and specific oligonucleotides were used for hypertrophy (β-MHC, ANP, and BNP) and fibrosis (collagen I, collagen III) markers [22,23]. The threshold cycle (Ct) values for each target mRNA were normalized to GAPDH mRNA, and the relative expression level of each target gene was calculated with the formula n-fold change = , where ΔCt represents Ct (infected) − Ct (control) [24,25].
Tissue parasite burden
Total DNA isolated from blood and cardiac and skeletal muscle was used as a template for the amplification of T. cruzi-specific 18SrDNA sequence by traditional and real-time PCR [26].
Data analysis
Data (mean ± SD) were derived from at least triplicate observations per sample (n ≥ 6 animals per group). Normally distributed data were analysed by Student’s t-test. The level of significance is presented by *,#p < 0.01, **,##p < 0.001 (*normal versus infected; # infected/untreated versus apocynin-treated/infected).
Results
NADPH oxidase is a major source of ROS in infected mice
We performed in-gel catalytic staining to assess NOX activation by T. cruzi. Splenocytes of acutely infected mice, in vitro-stimulated with TcL, exhibited a more than two-fold increase in NOX activity (Figure 1A). Apocynin treatment normalized the T. cruzi-induced NOX activity in TcL-stimulated splenocytes, thus validating the reaction specificity (Figure 1A).
Figure 1.
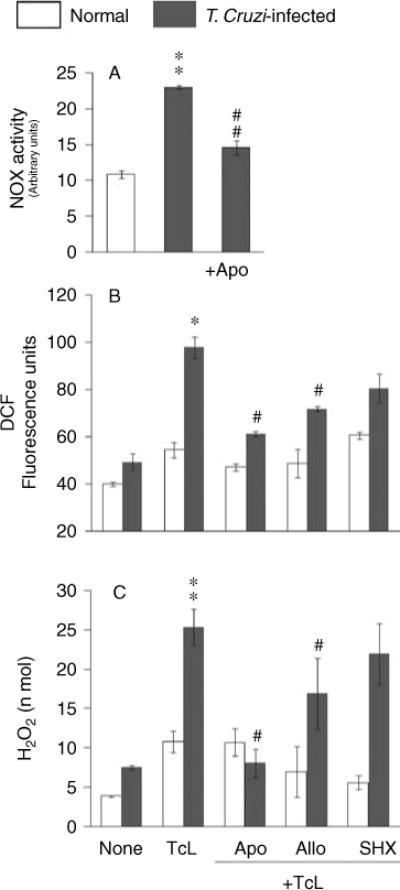
(A) Splenic NOX activity is increased in response to T. cruzi infection. C3H/HeN mice were infected with T. cruzi (10 000 parasites per mouse) and harvested during the acute infection phase (25 dpi). Splenocytes were in vitro-stimulated with TcL (± apocynin) for 48 h, and in-gel catalytic staining was performed to measure NOX activity. Gels were incubated in the presence of allopurinol, to inhibit xanthine oxidase interference, and apocynin, to validate the specificity of the reaction. Densitometric units of NOX activity are shown. (B, C) NOX-dependent ROS are elicited by splenocytes of infected mice. Splenocytes from normal and acutely infected mice were in vitro-incubated with TcL (as above). At 48 h post-incubation, cells were stained with H2DCFDA (which detects intracellular ROS) (B) or supernatants were utilized to examine the H2O2 levels by the Amplex® Red/HRP method (C). In some experiments, splenocytes were incubated in the presence of inhibitors of NADPH oxidase (apocynin, Apo), xanthine oxidase (allopurinol, Allo), and myeloperoxidase (salicyl hydroxamate, SHX), and ROS levels were measured. Data (mean±SD) are representative of three independent experiments (*,#±p < 0.01; **,##p < 0.001; *normal versus infected; # infected/treated versus infected/untreated).
Next, we employed H2DCFDA and Amplex® Red fluorescent probes to investigate whether NOX produces ROS as a host defence against T. cruzi. Splenocytes from acutely infected mice, in vitro-stimulated with TcL for 48 h, exhibited a 79% and two-fold increase in DCF fluorescence and H2O2 levels, respectively (Figures 1B and 1C, **p<0.001). In vitro treatment of splenocytes of infected mice with apocynin resulted in a 70–80% decline in ROS production, while allopurinol and SHX resulted in a 30–40% and 20–35% decline in Tc-stimulated ROS production, respectively (Figures 1B and 1C, #p < 0.01). FCCP (mitochondrial uncoupler) had no significant effect on ROS release in splenocytes of infected mice (data not shown). These results demonstrated that splenocytes of infected mice respond to T. cruzi (and TcL) by activation of NOX-dependent ROS release.
NOX/ROS signal TcL-stimulated cytokine response in splenocytes from infected mice
ROS may signal cytokine gene expression and contribute to the host’s defense against T. cruzi. To investigate this possibility, splenocytes of normal and acutely infected mice were in vitro-stimulated with TcL, and culture supernatants were analysed using a Bio-Plex Multiplex Cytokine Assay. These data showed a distinct increase in IL-1β, IL-6, TNF-α, IFN-γ, and IL-4 release (two-, four-, five-, 158-, and four-fold, respectively; p < 0.01) in TcL-stimulated splenocytes of infected mice compared with normal controls. The extent of cytokine release was significantly inhibited in the presence of apocynin (70–100%; all ##p < 0.001), moderately decreased by allopurinol (30–50%; #p < 0.01), and not altered by SHX (MPO inhibitor) (Figures 2A–2E). Incubation with NAC (ROS scavenger) blocked the cytokine release by more than 80%, while ConA elicited cytokine release in all groups (data not shown). These results suggested that NOX/ROS contribute to Tc antigen-specific activation and production of cytokines by the splenocytes of infected mice.
Figure 2.
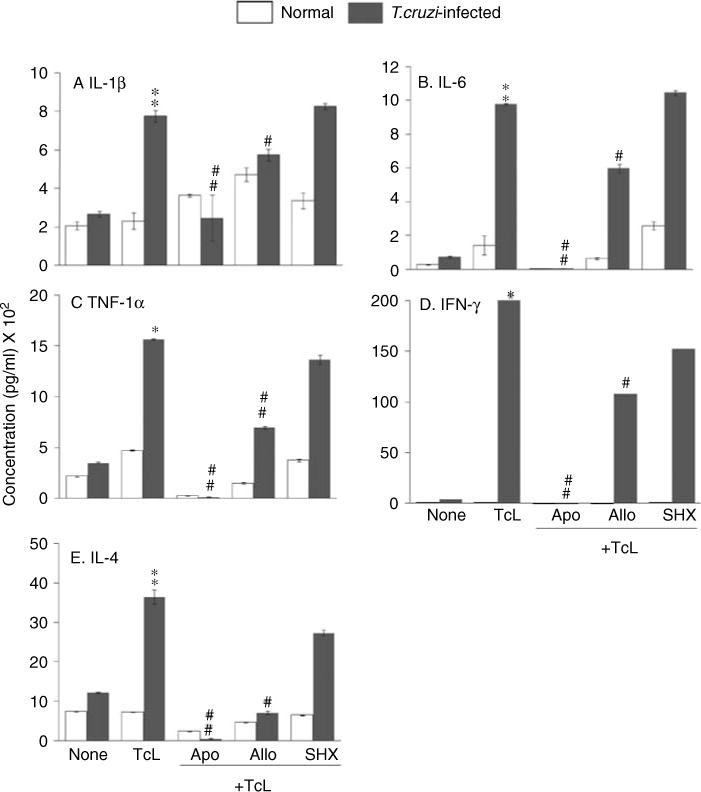
Splenocytes of T. cruzi-infected mice, in vitro-stimulated with Tc antigenic lysate (TcL), produce cytokines in a NOX/ROS-dependent manner. Splenocytes from normal and acutely infected mice were in vitro-stimulated as in Figure 1. The expression levels of IL-1β (A), IL-6 (B), TNF-α (C), IFN-γ (D), and IL-4 (E) cytokines in cell-free supernatants, measured using a Bio-Plex Multiplex Cytokine Assay, are shown (n = 6 mice per group; **,##p < 0.001).
NOX/ROS inhibition by apocynin decreased the T. cruzi-elicited splenic response in vivo
To investigate if NOX/ROS signal cytokine production in vivo, we treated the infected mice with apocynin in drinking water. In-gel catalytic staining of splenocyte homogenates showed that the in vivo NOX activity was increased by three-fold in acutely infected mice and was largely controlled in apocynin-treated/infected mice (Figure 3A). Likewise, splenic ROS levels were increased by three-fold in acutely infected mice (**p < 0.001) and controlled by 75% in apocynin-treated/infected mice (Figure 3B, #p < 0.01). Furthermore, apocynin treatment resulted in decreased endogenous production of splenic cytokines in acutely infected mice. The splenic levels of IL-6, IFN-γ, and IL-4, determined by ELISA, were increased by seven-, three-, and eight-fold, respectively, in acutely infected mice and decreased by 55%, 95%, and 91%, respectively, in apocynin-treated/infected mice (##p < 0.001, Figures 3C, 3E, and 3F). It is important to note that in vivo depletion of NOX activity by apocynin did not alter the splenocytes’ ability to produce cytokines. This was evident by the observation that spleen cells from apocynin-treated/infected mice responded to in vitro incubation with TcL (Figures 3G–3J) or ConA (Supporting information, Supplementary Figure 1) by a robust increase in cytokines, similar to that noted in TcL-stimulated splenocytes of untreated/infected mice. When apocynin was added during in vitro incubation with TcL, the release of cytokines by splenic cultures of infected mice was reduced to the levels detected in normal controls (Figures 3K–3N).
Figure 3.
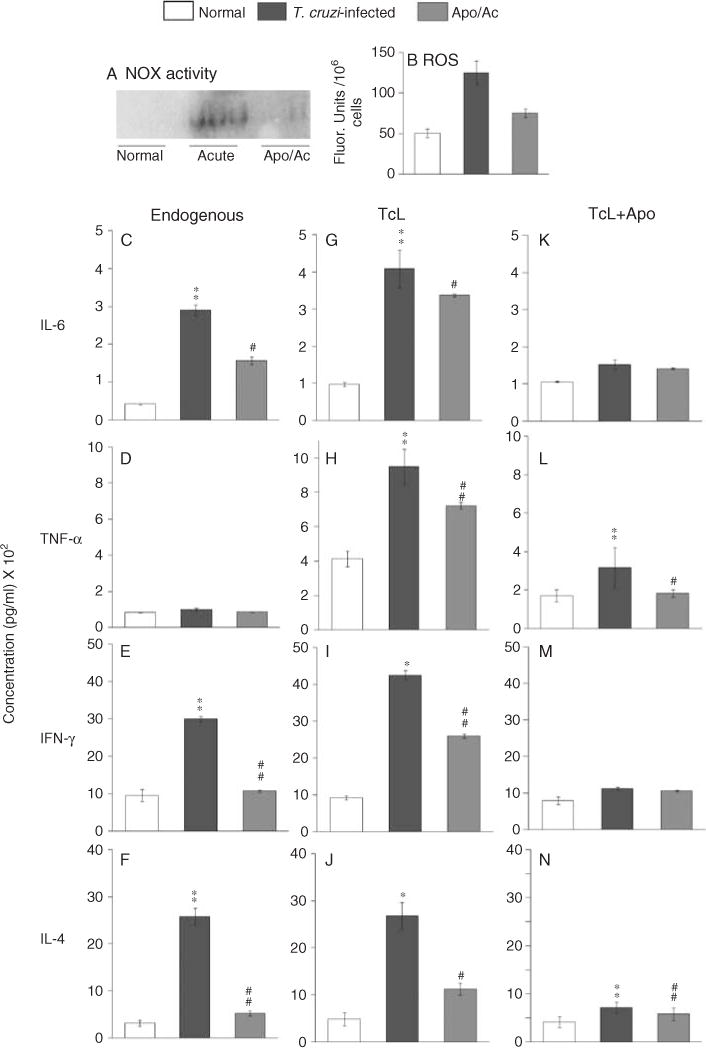
Apocynin treatment was beneficial in controlling splenic NOX/ROS and the cytokine response in acutely infected mice. Mice were infected with T. cruzi and given 1.5 mM apocynin in their drinking water. Mice were harvested at day 25 post-infection. (A) In-gel catalytic staining of endogenous NOX activity in splenocyte homogenates. (B) Fluorometric evaluation of splenic ROS levels using the H2DCFDA probe. (C–N) Splenocytes from infected/untreated and infected/apocynin-treated mice were incubated without antigenic lysate (C–F) or stimulated with TcL (G–N) in the presence (K–N) or absence of apocynin (G–J). The levels of IL-6, TNF-α, IFN-γ, and IL-4 in cell-free supernatants were measured by an ELISA (n = 6 mice/group; **,##p < 0.001).
We noted a more than two-fold increase in spleen weight and splenic index (spleen weight/body weight), indicative of immune cell proliferation (Figure 4), and therefore examined the splenic immune cell population by flow cytometry. The mean percentages of CD68+ macrophages (6 ± 0.5), Ly-6G/Gr-1+ neutrophils (7 ± 0.6), CD4+ T cells (18 ± 1.5), and CD8+ T cells (25 ± 1.2) indicated a predominant expansion of T cells in the spleens of infected mice (Table 1). Upon apocynin treatment, the increase in splenic weight and splenic index was controlled by 45% in infected mice (Figure 4). The mean percentages of splenic T-cell subsets were not altered, while those of CD68+ macrophages (3 ± 0.4) and Ly-6G/Gr-1+ neutrophils (4 ± 0.2) were decreased by 50% and 40%, respectively, in apocynin-treated/infected mice. These data suggested that NOX/ROS directly signal macrophage and neutrophil proliferation and have an effect on T-cell proliferation in infected mice.
Figure 4.
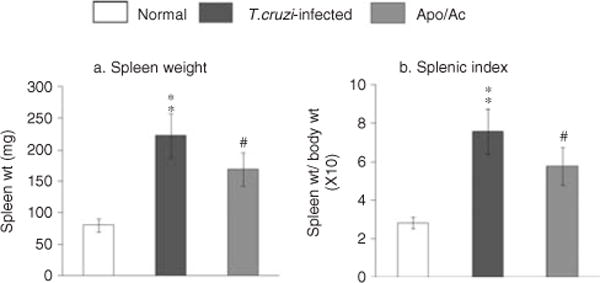
NOX/ROS enhanced immune cell proliferation in infected mice. The increase in (a) spleen weight and (b) splenic index in acutely infected mice was diminished by apocynin treatment.
Table 1.
Mean percentage of cell phenotype in the spleen and myocardium of T. cruzi-infected mice (± apocynin)
| Mice | Inflammation score | Macrophages | Neutrophils | CD8+ T cells | CD4+ T cells |
|---|---|---|---|---|---|
| Splenic cell population (percentage) | |||||
| Normal | 1 ± 0.2 | 1 ± 0.4 | 0 | 0 | |
| Acutely infected | 6 ± 0.5 | 7 ± 0.6 | 25 ± 1.2 | 18 ± 1.5 | |
| Apocynin-treated/infected | 3 ± 0.4 | 4 ± 0.2 | 25 ± 2.2 | 19 ± 1.1 | |
| Cardiac infiltrate composition (No of cells/mf) | |||||
| Normal | 1 ± 0.64 | 3 ± 0.5 | 2 ± 0.5 | 0 | 0 |
| Acutely infected | 3 ± 0.79 | 32 ± 2.5 | 13 ± 3.0 | 10 ± 3.0 | 6 ± 0.5 |
| Apocynin-treated/infected | 1 ± 0.67 | 8 ± 2.0 | 8 ± 2.5 | 8 ± 1.0 | 6 ± 2.0 |
C3H/HeN mice were infected with T. cruzi and treated with apocynin in drinking water. Mice were harvested at 25 days post-infection (acute phase). Splenic single cell suspension was stained with fluorescence-tagged specific antibodies, and flow cytometry was performed to obtain the mean percentage of macrophages, neutrophils, CD8+ T cells and CD4+ T cells. Hematoxylin and Eosin-stained heart tissue sections were scored for myocarditis by a semi-quantitative analysis of the presence of inflammatory cells as 0, absent/none; 1, focal or mild myocarditis with ≤1 focus; 2, moderate with ≥2 inflammatory foci; 3, extensive with generalized coalescing of inflammatory foci; and 4, severe with diffused inflammation. Immunohistochemistry was performed to evaluate the average number of positively stained cell subsets (macrophages, neutrophils, and CD4+ and CD8+ T cells) per microscopic field (>10 mf/tissue). Data are presented as mean ± SD (n = 6 per group).
NOX/ROS effects on myocardial infiltration of inflammatory cells in infected mice
We investigated whether inhibition of NOX activity by apocynin alters myocardial response. Histological studies showed that the intense inflammatory infiltrate in the heart of acutely infected mice (Figure 5b, score 4) was significantly decreased by apocynin treatment (Figure 5c, score 1). The inflammatory infiltrate in the infected myocardium consisted predominantly of macrophages (CD68+), neutrophils (Ly-6G+), and CD8+ T cells (Figures 5e, 5h, and 5k, and Table 1). In agreement with the splenic data, the numbers of macrophages and neutrophils were significantly decreased in apocynin-treated/infected mice (Figures 5f and 5i and Table 1). The decline in cardiac infiltration of macrophages was associated with a similar level of decline in NOX activity. Immunostaining of gp91phox, a haem-binding subunit that is an indicator of NOX activity, was significantly increased in the myocardium of infected mice and controlled in apocynin-treated/infected mice (Figures 5q and 5r). These results suggested that NOX-producing phagocytes constitute the major inflammatory infiltrate, and apocynin-mediated inhibition of NOX activity resulted in decreased proliferation (Figure 4) and myocardial infiltration of phagocytes in infected myocardium.
Figure 5.
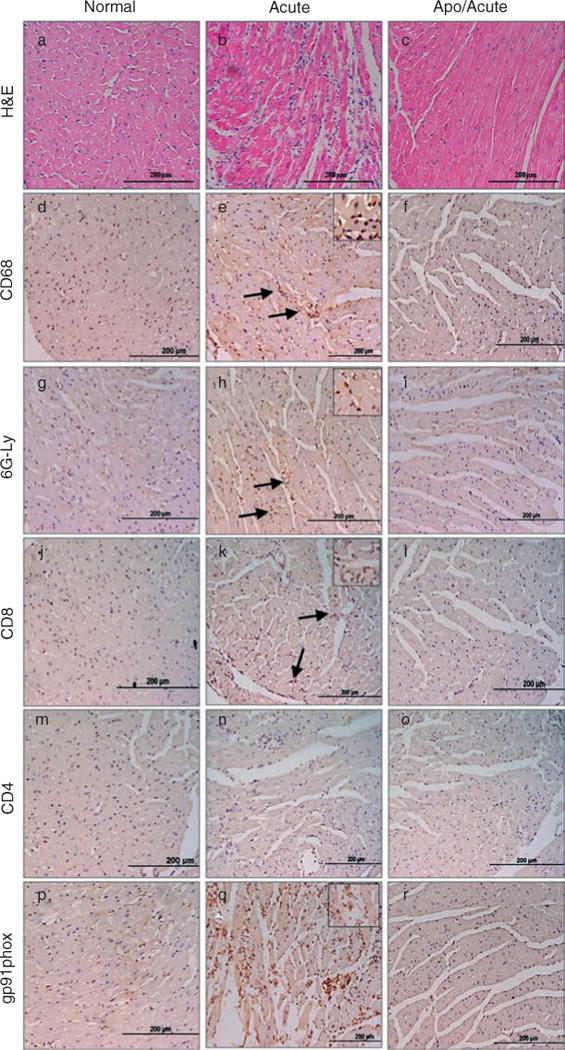
Myocardial inflammatory infiltrate is decreased in apocynin-treated/infected mice. Paraffin-embedded left ventricular (LV) heart sections (5 μm) were examined by H&E staining (blue: nuclear; pink: muscle/cytoplasm) (a–c) or subjected to immunostaining with antibodies against CD68 (macrophages, d–f), Ly-6G/GR-1 (neutrophils, g–i), Ly-3.2/CD8b.2 (CD8+ T cells, j–l), L3T4/CD4 (CD4+ T cells, m–o), and gp91phox (NOX activity, p–r). Colour (brown) was developed using HRP-conjugated secondary antibody. Tissue sections from normal (a, d, g, j, m, p), acutely infected (b, e, h, k, n, o), and apocynin-treated/infected (c, f, i, l, o, r) mice are shown. Arrows and insets in panels e, h, k, and q indicate the abundant brown stain in inflammatory cells and positively stained cells, respectively. The results shown are representative of three different experiments (3–4 mice per group), and were utilized to score the tissues as described in the Materials and methods section. Scale bar = 200 μm.
NOX is required for acute parasite control in mice
We first performed a semi-quantitative PCR to examine the cardiac parasite burden. These data showed a significant level of Tc18SrDNA signal in the myocardium of acutely infected mice that was not altered by apocynin treatment (Figures 6A and 6B). We then employed a sensitive real-time PCR to obtain quantitative evaluation of the parasite burden. These data showed a two- to three-fold increase in circulatory (blood), myocardial, and skeletal muscle levels of parasite burden in apocynin-treated/infected mice compared with that detected in infected/untreated mice (Figure 6C, #p < 0.01). These results suggested that NOX-induced responses (oxidative burst, cytokine production, phagocytic cells) are required for parasite control in infected mice.
Figure 6.
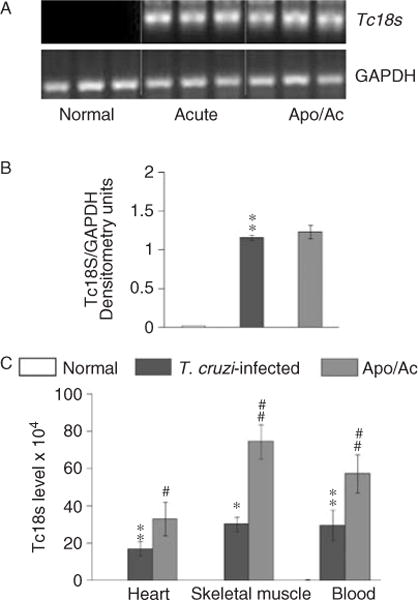
Quantitation of parasite burden. Total DNA was isolated from the myocardium, skeletal muscle, and blood of acutely infected mice (± apocynin), and used as a template for T. cruzi 18SrDNA amplification by PCR (A) and real-time PCR (C). Densitometric quantification of Tc18SrDNA PCR-amplicons, normalized to mGAPDH, is shown (B).
NOX/ROS cause oxidative stress in infected myocardium
ROS, when produced in excess, can contribute to oxidative stress. DHE, when oxidized, binds DNA and exhibits bright red fluorescence. In situ staining with DHE showed that the myocardial fluorescence was significantly increased in acutely infected mice (Figure 7A). Consequently, myocardial 8-isoprostane and 3-NT levels (oxidative stress markers) were significantly increased in acutely infected mice (Figure 7B, a–c; **p < 0.001). When treated with apocynin, the enhanced myocardial level of ethidium fluorescence, and 8-isoprostane and 3-NT adducts were decreased by more than 50% in infected mice (Figures 7A and 7B). 4-Hydroxynonenal (4-HNE) is a major lipid peroxidation product formed during oxidative stress. Immunostaining showed an eight-fold increase in antibody detection of the 4-HNE-stained area in acutely infected heart compared with normal controls (17% versus 1.9% of total area; Figure 7C, b, d) that was significantly controlled in apocynin-treated/infected mice (Figure 7C, c, d). Together, these results suggested that NOX/ROS cause bystander oxidative damage in the heart, and apocynin inhibition of NOX prevented, at least partially, the oxidative adducts in the myocardium of acutely infected mice.
Figure 7.
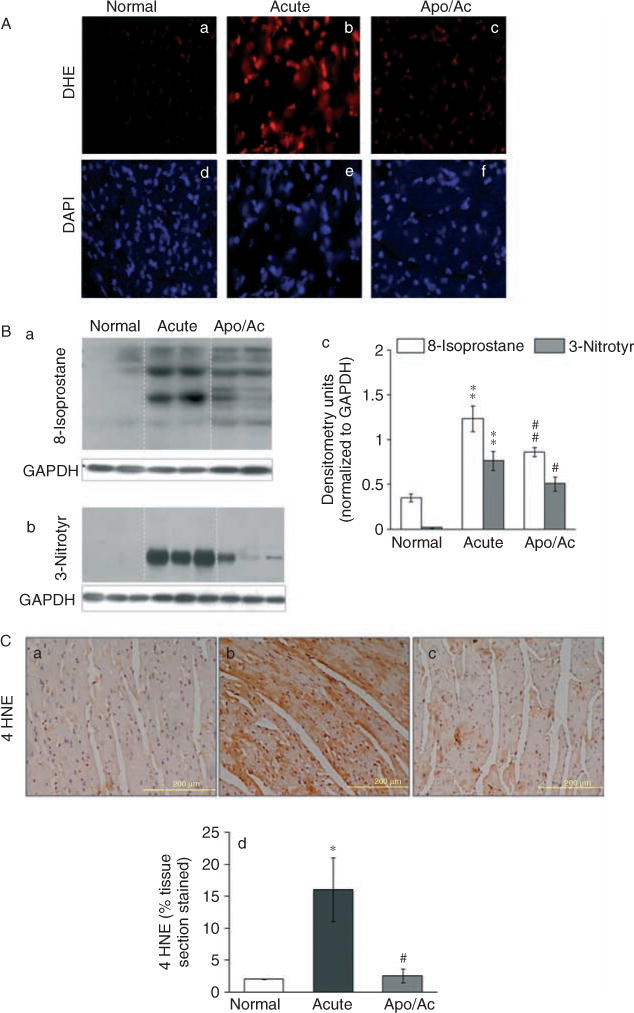
NOX/ROS contribute to myocardial oxidative stress in acutely infected mice. (A) Frozen heart sections from normal (a, d), acutely infected (b, e), and apocynin-treated/infected (c, f) mice were loaded with DHE (detects O2•−), and its oxidation resulting in nuclear bright fluorescent red staining (a–c) was determined by fluorescence microscopy. Tissue sections were counter-stained with DAPI (blue fluorescence, d–f). (B) Heart homogenates were subjected to western blotting using monoclonal antibodies against 8-isoprostane (a) and 3-nitrotyrosine (b) to detect lipid peroxidation products and nitrosative adducts, respectively (control: anti-GAPDH antibody), and signal was quantitated by densitometry (c). (C) Immunohistochemical staining of paraffin-embedded heart sections with anti-4-HNE antibody detected 4-hydroxynonenal adducts (brown colour) (a–c). Scale bar = 200 μm. The mean percentage of the 4-HNE-stained tissue section is shown (d).
Apocynin treatment was beneficial in controlling the cardiac remodelling responses in chagasic mice
ROS and inflammatory mediators have been suggested to promote the development of interstitial and perivascular fibrosis, as well as myocardial hypertrophy [27–30]. We observed no change in heart size or heart/body ratio (wt/wt) in acutely infected mice (Figure 8B). The chronically infected mice exhibited a 50% and 43% increase in heart weight (range 121–182 mg; *p < 0.01) and heart/body (wt/wt) ratio (range 4.2–6.0; *p < 0.01), respectively (Figures 8A and 8B). Histological staining with Masson’s Trichrome and Oil Red O of the heart tissue sections detected no significant evidence of collagen and lipid deposition, respectively, in acutely infected myocardium (data not shown). In comparison, chronically infected mice exhibited a high degree of myocardial remodelling (Figures 8C and 8D, and Supporting information, Supplementary Figure 2). We noted up to a 60% increase in cardiomyocytes’ size (Figure 8C, b, j) that was associated with a four-fold, ten-fold, and 16-fold increase in the mRNA levels for hypertrophy markers β-MHC, ANP, and BNP, respectively, in chronically infected murine hearts (Figure 8D, a–c; *p < 0.01). Likewise, myocardial collagen contents (Figure 8C, e, k) and lipid deposition (score 3 ± 0.92; Figure 8C, h), specifically around the vasculature, ± were markedly increased in chronically infected mice. A significant increase in fibrosis in chronically infected heart was evidenced by a six-fold, 38-fold, and twofold increase in the hydroxyproline amino acid content (a major component of collagens; Supporting information, Supplementary Figure 2A), the mRNA level of type I collagen (Supporting information, Supplementary Figure 2B), and matrix metalloproteinase 9 activity (MMP-9; a type IV collagenase; Supporting information, Supplementary Figure 2C, a, b), respectively. We also noted a four-fold increase in glycosaminoglycans (GAGs; ECM carbohydrate polymers) in chronically infected murine hearts (Supporting information, Supplementary Figure 2D). In comparison, the apocynin-treated/chronically infected mice exhibited heart size and heart weight similar to those noted in control/uninfected mice (Figures 8A and 8B). Subsequently, cardiomyocytes’ size was normalized to the control level (Figure 8C, c, j), and apocynin-treated/chronically infected mice exhibited a distinct decline in enhanced β-MHC, ANP, and BNP mRNA levels (55%, 95%, and 47%, respectively; #p < 0.01) compared with that noted in the myocardium of untreated/chronically infected mice (Figure 8D). Likewise, apocynin-treated/chronically infected mice exhibited a significant control of myocardial fibrosis, evidenced by the detection of moderate to no Masson’s Trichrome (Figure 8C, f, k) and Oil Red O (score 0; Figure 8C, i) staining, and a 77%, 89%, 66%, and 72% decline in the enhanced levels of hydroxyproline, collagen I mRNA, MMP-9 activity, and GAG content, respectively, compared with that noted in the myocardium of untreated/chronically infected mice (Supporting information, Supplementary Figure 2). The cardiac renin–angiotensin system (RAS) has been implicated in inducing myocardial fibrosis [31]. Angiotensin II mediates most of its relevant biological effects via angiotensin II type 1 (AT1) receptor activation [32]. Our data showed a more than two-fold increase in the AT1 receptor (37 kD) level in chronic myocardium (**p < 0.001) which was partially controlled (46% decline, #p < 0.01) in apocynin-treated/chronic mice (Figure 8E). Together, these results, along with those presented in Figures 5 and 7, suggested that acute control of NOX/ROS and inflammatory reactions was beneficial in arresting the chronic evolution of hypertrophic and fibrotic responses in chagasic mice.
Figure 8.
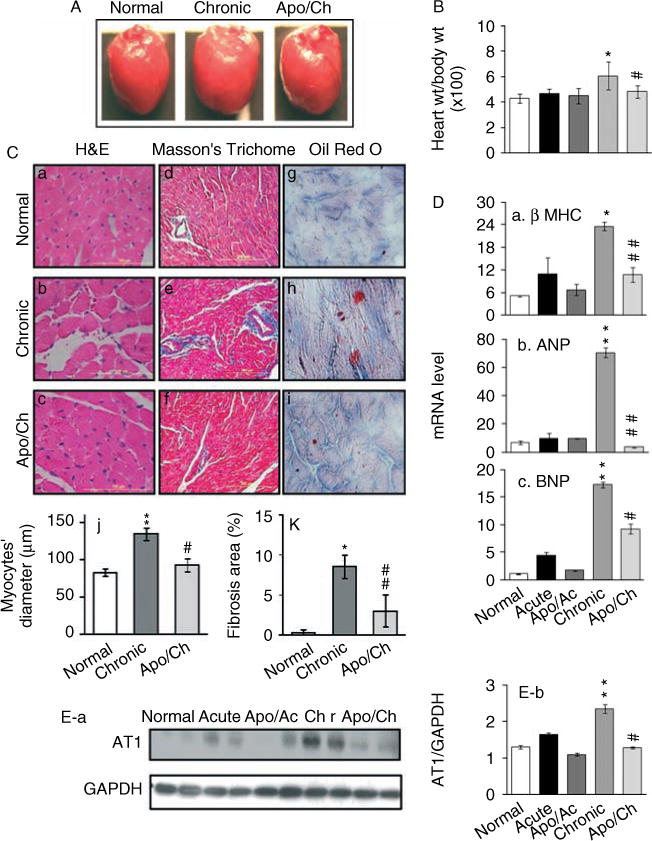
Acute control of NOX/ROS activation by apocynin was beneficial in limiting chronic myocardial hypertrophy in chagasic mice. (A, B) Representative photographs of the whole heart (A) and heart/body ratios (wt/wt) (B) of normal mice, and mice acutely (Ac) or chronically (Ch) infected by T. cruzi [± apocynin (apo) treatment] are shown. (C) Representative micrographs of heart (LV) tissue sections stained with H&E (a–c), Masson’s Trichome (d–f), and Oil Red O (g–i) showed an increase in cardiomyocyte size, interstitial collagen content (blue), and intramyocyte lipid deposition (red tint), respectively, in chagasic mice [scale bar 100 μm (a–c); 200 μm (d–i)]. The cardiomyocyte size (c), and collagen (f) and lipid (i) deposition were decreased in apocynin-treated/chronically = infected mice. Quantitative scoring of cardiomyocytes’ transnuclear diameter from H&E-stained tissue sections (j) and fibrosis, ie the percentage of Masson’s Trichome stained collagen area (k), is shown. (D) Myocardial levels of mRNAs for β-MHC (a), atrial natriuretic peptide (ANP) (b), and brain natriuretic peptide (BNP) (c) were determined by real-time RT-PCR. Data (mean ± SD) represent values (n = 6 mice per group). (E) Heart homogenates were subjected to western blotting using anti-AT1 receptor antibody (a) (control: anti-GAPDH antibody), and signal was quantitated by densitometry (b). n = 6 mice per group (**,##p < 0.001).
Discussion
In this study, we provide evidence that phagocytic oxidative burst (NOX-induced ROS production) in response to T. cruzi infection plays an essential role in immune cell activation and inflammatory cytokine production. We also demonstrate that diminishing the NOX/ROS responses is beneficial in controlling acute myocarditis characterized by extensive oxidative injuries and infiltration of inflammatory infiltrate, and subsequently limited the cardiac remodelling in chronically infected chagasic mice.
NADPH oxidase produces ROS as a major product and the prototypic Nox2 was first identified in phagocytes. Upon exposure to micro-organisms or other stimuli, NOX is activated and catalyses one electron reduction of O2, resulting in a rapid increase in ROS production that serves as the first line of host defence against microbes. Early studies have reported cytochemical detection of NOX at the plasma membrane of peritoneal mouse macrophages exposed to T. cruzi [4]. We have extended these observations in in vivo infection models. Our data showed that mice respond to T. cruzi infection by splenic NOX activation and ROS production. How NOX is activated in response to T. cruzi is not known. A robust response of the splenocytes of infected mice to Tc antigenic lysate (Figure 1) suggests that parasite factors (and not active invasion) are sufficient to activate NOX and ROS generation. Indeed, T. cruzi-derived components are recognized by toll-like receptors (GPI anchor and Tc52: TLR2, GIPL: TLR4, and TcDNA: TLR9) and NOD, implicated in NOX activation [33–35], thus allowing us to propose that these parasite components, individually or in synergy, contribute to NOX activation and ROS generation. Future studies will be required to identify TLR- and NOD-signalling mechanisms that initiate the translocation of cytosolic components (p47phox, p67phox, and G-protein) and NOX assembly in response to T. cruzi. Irrespective of the mode of activation, NOX/ROS played an important role in the control of acute T. cruzi infection, as evidenced by an increase in parasite burden in mice when NOX activity was inhibited (Figure 6). This notion is supported by recent observations demonstrating the importance of NOX in eliciting macrophage-derived peroxynitrite-dependent cytotoxicity against T. cruzi [14].
Splenomegaly in rodents and humans infected by T. cruzi is well documented [36]. Others have shown that T. cruzi promotes expression of TNF-α and other cytokines in macrophages [4,37,38]. The innate response is followed by B and T-lymphocyte activation, and cytokine (eg IFN-γ, IL-4, and IL-10) synthesis and secretion by activated T lymphocytes in infected mice [39–41]. Our observations of an increase in splenic index, predominant proliferation of CD8+ and CD4+ T cells, and a mixed (type 1/type 2) cytokine response to T. cruzi in mice are in alignment with the published reports. However, to the best of our knowledge, this is the first report demonstrating that splenic activation of NOX/ROS is required for immune cell proliferation and cytokine expression upon T. cruzi infection. The significance of NOX/ROS was demonstrated by the fact that apocynin treatment resulted in a more than 45% decline in the T. cruzi-induced increase in splenic index and immune cell proliferation in infected mice (Figure 4). Likewise, NOX inhibition resulted in a 55–95% decline in inflammatory mediators in the spleens of apocynin-treated/infected mice in vivo (Figure 3), as well as when splenocytes of infected mice were in vitro-stimulated in the presence of apocynin (Figure 2). Further studies will be required to determine whether NOX/ROS signal nuclear transport and activation of transcription factors (eg NF-κB and AP-1) [42,43] and promote cytokine gene expression, or whether NOX/ROS elicit immune cell proliferation and thereby indirectly alter the cytokine profile in infected mice. However, our data provide clues to these mechanisms. The finding of no change in the percentage of T-cell subsets despite a 45% decline in splenic index in apocynin-treated/infected mice suggests that NOX/ROS indirectly, or in synergy with cytokines, affect the T-cell proliferation in infected mice. In contrast, the observation of a direct correlation in the percentage decline in spleen weight and splenic percentage of macrophages and neutrophils in apocynin-treated/infected mice suggests an autocrine mechanism of NOX/ROS activation and proliferation of phagocytic cells in response to T. cruzi. Overall, we surmise that NOX/ROS are a critical regulator of phagocyte activation and proliferation, T-cell proliferation, and cytokine response during T. cruzi infection.
Cellular injury occurs as a result of the oxidation of proteins, DNA, and lipids [44,45]. Our published reports have shown an unvarying high degree of oxidative/nitrosative damage in the myocardium of T. cruzi-infected mice evidenced by high levels of malonyl aldehydes, protein carbonyls, 3-NT, and glutathione disulphide contents [21,46]. A considerable decline in the myocardial levels of isoprostane, 3-NT, and 4-HNE adducts in apocynin-treated/infected mice (Figure 7) provides the first evidence that NOX-dependent ROS contribute to acute oxidative stress in T. cruzi-infected mice.
Myocarditis, characterized by the presence of inflammatory infiltrate, and enhanced hypertrophy and fibrosis [47], was advanced in infected mice. The acutely infected mice exhibited extensive infiltration of CD68+ phagocytes and CD8+ T cells in the heart (Figure 5). In the chronic stage, the persistence of inflammatory infiltrate in the heart was associated with extensive cardiac remodelling evidenced by an increased size of cardiomyocytes, enhanced mRNA levels of hypertrophy markers (β-MHC, ANP, and BNP), extensive deposition of ECM components (collagen and GAGs), and increased MMP-9 (collagenase) activity (Figure 8 and Supporting information, Supplementary Figure 2). Several observations allow us to propose that activation of NOX/ROS was detrimental and contributed to myocarditis in infected mice. First we noted up-regulation of apocynin-sensitive gp91phox expression and NOX activity in the myocardium of acutely infected mice. Apocynin treatment resulted in a noticeable decline in gp91phox expression, and consequently, infiltration of inflammatory infiltrate and oxidative adducts were diminished in acutely infected myocardium (Figures 7 and 8). Furthermore, we noted beneficial effects of apocynin in preserving cardiac structure (Figure 8 and Supporting information, Supplementary Figure 2) that occurred despite an increase in parasite burden (Figure 6). Apocynin-treated mice exhibited a substantial decline in myocardial hypertrophic gene expression, collagen and GAG contents, intramyocyte lipid deposition, and angiotensin II and MMP-9 levels during the chronic disease phase (Figure 8 and Supporting information, Supplementary Figure 2). CD8+ T cells are known to exert cytotoxic effects in chagasic myocardium [48]. Our data allow us to surmise that heart-infiltrating NOX/ROS-producing phagocytes also exert bystander effects and contribute to cardiac remodelling and fibrosis in Chagas disease. We propose that phagocytes affect the myocardium due to the cytotoxicity of NOX/ROS and cytokines/chemokines that they produce, or by enhancing the infiltration of cytotoxic CD8+ T cells, which is to be investigated in future studies.
In summary, we have shown that T. cruzi-induced NOX activation and oxidative burst play an important role in inflammatory cell proliferation and cytokine production in infected mice. A delicate balance between the beneficial and detrimental effects of the immune response is required to obtain an ideal outcome of inflammation with efficient pathogen control and tissue repair. In mice infected by T. cruzi, consequences of the infiltration of NOX/ROS-producing phagoyctes and CD8+ T cells persisted, with structural alteration and remodelling of the heart. Apocynin treatment decreased the host’s defence against acute parasitaemia, due to decreased activation of inflammatory and oxidative processes; however, it was beneficial in arresting progressive myocarditis in infected mice. Further studies will provide insights into the mechanisms by which NOX/ROS signalling could be regulated to maintain anti-parasite inflammatory responses, and unravel novel therapeutic strategies aimed at limiting the inflammatory and oxidative damage in chagasic myocardium.
Supplementary Material
Figure S1. Apocynin treatment did not alter the ability of splenocytes to produce cytokines.
Figure S2. Apocynin treatment controlled fibrosis in chagasic mice.
Acknowledgments
This work was supported by a grant from the National Institutes of Health/National Institute of Allergy and Infectious Diseases (RO1AI054578) to NJG.
Abbreviations
- Allo
allopurinol
- Apo
apocynin
- ConA
concanavalin A
- DAB
diaminobenzidine tetrahydrochloride
- DAPI
4′, 6-diamidino-2-phenylindole-dihydrochloride
- DHE
dihydroethidium
- dpi
days post-infection
- 4-HNE
4-hydroxynonenal
- HRP
horseradish peroxidase
- IFN-γ
interferon gamma
- IL-1β
interleukin 1 beta
- MPO
myeloperoxidase
- NAC
N-acetylcysteine
- NBT
nitroblue tetrazolium salt
- NOX
NAD(P)H oxidase
- 3-NT
3-nitrotyrosine
- ROS
reactive oxygen species
- SHX
salicyl hydroxamate
- TcL
T. cruzi antigenic lysate
- T. cruzi
Trypanosoma cruzi
- TNF-α
tumour necrosis factor alpha
- XOD
xanthine oxidase
Footnotes
No conflicts of interest were declared.
Author contribution statement
All reagents and resources were provided by NJG, and all experiments and data analysis were performed by MD. MD and NJG designed the experiments and wrote the manuscript together.
SUPPORTING INFORMATION ON THE INTERNET
The following supporting information may be found in the online version of this article.
References
- 1.Higuchi Mde L, Benvenuti LA, Martins Reis M, et al. Pathophysiology of the heart in Chagas’ disease: current status and new developments. Cardiovasc Res. 2003;60:96–107. doi: 10.1016/s0008-6363(03)00361-4. [DOI] [PubMed] [Google Scholar]
- 2.Tostes S, Jr, Bertulucci Rocha-Rodrigues D, de Araujo Pereira G, et al. Myocardiocyte apoptosis in heart failure in chronic Chagas’ disease. Int J Cardiol. 2005;99:233–237. doi: 10.1016/j.ijcard.2004.01.026. [DOI] [PubMed] [Google Scholar]
- 3.Zacks MA, Wen J-J, Vyatkina G, et al. An overview of chagasic cardiomyopathy: pathogenic importance of oxidative stress. Ann Acad Bras Cienc. 2005;77:695–715. doi: 10.1590/s0001-37652005000400009. [DOI] [PubMed] [Google Scholar]
- 4.Cardoni RL, Antunez MI, Morales C, et al. Release of reactive oxygen species by phagocytic cells in response to live parasites in mice infected with Trypanosoma cruzi. Am J Trop Med Hyg. 1997;56:329–334. doi: 10.4269/ajtmh.1997.56.329. [DOI] [PubMed] [Google Scholar]
- 5.Leon JS, Wang K, Engman DM. Captopril ameliorates myocarditis in acute experimental Chagas disease. Circulation. 2003;107:2264–2269. doi: 10.1161/01.CIR.0000062690.79456.D0. [DOI] [PubMed] [Google Scholar]
- 6.Piacenza L, Alvarez MN, Peluffo G, et al. Fighting the oxidative assault: the Trypanosoma cruzi journey to infection. Curr Opin Microbiol. 2009;12:415–421. doi: 10.1016/j.mib.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 7.Weintraub NL. Nox response to injury. Arterioscler Thromb Vasc Biol. 2002;22:4–5. [PubMed] [Google Scholar]
- 8.Wang HD, Johns DG, Xu S, et al. Role of superoxide anion in regulating pressor and vascular hypertrophic response to angiotensin II. Am J Physiol Heart Circ Physiol. 2002;282:H1697–H1702. doi: 10.1152/ajpheart.00914.2001. [DOI] [PubMed] [Google Scholar]
- 9.Griendling KK. Novel NAD(P)H oxidases in the cardiovascular system. Heart. 2004;90:491–493. doi: 10.1136/hrt.2003.029397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 11.Bataller R, Schwabe RF, Choi YH, et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest. 2003;112:1383–1394. doi: 10.1172/JCI18212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Babior BM, Lambeth JD, Nauseef W. The neutrophil NADPH oxidase. Arch Biochem Biophys. 2002;397:342–344. doi: 10.1006/abbi.2001.2642. [DOI] [PubMed] [Google Scholar]
- 13.Piacenza L, Peluffo G, Alvarez MN, et al. Peroxiredoxins play a major role in protecting Trypanosoma cruzi against macrophage-and endogenously-derived peroxynitrite. Biochem J. 2008;410:359–368. doi: 10.1042/BJ20071138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alvarez MN, Peluffo G, Piacenza L, et al. Intraphagosomal peroxynitrite as a macrophage-derived cytotoxin against internalized Trypanosoma cruzi: consequences for oxidative killing and role of microbial peroxiredoxins in infectivity. J Biol Chem. 2011;286:6627–6640. doi: 10.1074/jbc.M110.167247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Plata F, Garcia Pons F, Eisen H. Antigenic polymorphism of Trypanosoma cruzi: clonal analysis of trypomastigote surface antigens. Eur J Immunol. 1984;14:392–399. doi: 10.1002/eji.1830140503. [DOI] [PubMed] [Google Scholar]
- 16.Paterniti I, Galuppo M, Mazzon E, et al. Protective effects of apocynin, an inhibitor of NADPH oxidase activity, in splanchnic artery occlusion and reperfusion. J Leukoc Biol. 88:993–1003. doi: 10.1189/jlb.0610322. [DOI] [PubMed] [Google Scholar]
- 17.Wen JJ, Vyatkina G, Garg N. Oxidative damage during chagasic cardiomyopathy development: role of mitochondrial oxidant release and inefficient antioxidant defense. Free Radic Biol Med. 2004;37:1821–1833. doi: 10.1016/j.freeradbiomed.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 18.Lopez-Huertas E, Corpas FJ, Sandalio LM, et al. Characterization of membrane polypeptides from pea leaf peroxisomes involved in superoxide radical generation. Biochem J. 1999;337:531–536. [PMC free article] [PubMed] [Google Scholar]
- 19.Wen JJ, Garg NJ. Mitochondrial generation of reactive oxygen species is enhanced at the Q(o) site of the complex III in the myocardium of Trypanosoma cruzi-infected mice: beneficial effects of an antioxidant. J Bioenerg Biomembr. 2008;40:587–598. doi: 10.1007/s10863-008-9184-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garg N, Popov VL, Papaconstantinou J. Profiling gene transcription reveals a deficiency of mitochondrial oxidative phosphorylation in Trypanosoma cruzi-infected murine hearts: implications in chagasic myocarditis development. Biochim Biophys Acta. 2003;1638:106–120. doi: 10.1016/s0925-4439(03)00060-7. [DOI] [PubMed] [Google Scholar]
- 21.Dhiman M, Nakayasu ES, Madaiah YH, et al. Enhanced nitrosative stress during Trypanosoma cruzi infection causes nitrotyrosine modification of host proteins: implications in Chagas’ disease. Am J Pathol. 2008;173:728–740. doi: 10.2353/ajpath.2008.080047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Byrne JA, Grieve DJ, Bendall JK, et al. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ Res. 2003;93:802–805. doi: 10.1161/01.RES.0000099504.30207.F5. [DOI] [PubMed] [Google Scholar]
- 23.Kang BY, Khan JA, Ryu S, et al. Curcumin reduces angiotensin II-mediated cardiomyocyte growth via LOX-1 inhibition. J Cardiovasc Pharmacol. 2010;55:176–183. doi: 10.1097/FJC.0b013e3181ca4ba1. [DOI] [PubMed] [Google Scholar]
- 24.Bhatia V, Sinha M, Luxon B, et al. Utility of Trypanosoma cruzi sequence database for the identification of potential vaccine candidates: in silico and in vitro screening. Infect Immun. 2004;72:6245–6254. doi: 10.1128/IAI.72.11.6245-6254.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garg NJ, Gerstner A, Bhatia V, et al. Gene expression analysis in mitochondria from chagasic mice: alterations in specific metabolic pathways. Biochem J. 2004;381:743–52. doi: 10.1042/BJ20040356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gupta S, Garg NJ. Prophylactic efficacy of TcVac2 against Trypanosoma cruzi in mice. PLoS Negl Trop Dis. 2010;4:e797. doi: 10.1371/journal.pntd.0000797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakamura K, Fushimi K, Kouchi H, et al. Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-alpha and angiotensin II. Circulation. 1998;98:794–799. doi: 10.1161/01.cir.98.8.794. [DOI] [PubMed] [Google Scholar]
- 28.Pimentel DR, Amin JK, Xiao L, et al. Reactive oxygen species mediate amplitude-dependent hypertrophic and apoptotic responses to mechanical stretch in cardiac myocytes. Circ Res. 2001;89:453–460. doi: 10.1161/hh1701.096615. [DOI] [PubMed] [Google Scholar]
- 29.Murdoch CE, Zhang M, Cave AC, et al. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc Res. 2006;71:208–215. doi: 10.1016/j.cardiores.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 30.Cave AC, Brewer AC, Narayanapanicker A, et al. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal. 2006;8:691–728. doi: 10.1089/ars.2006.8.691. [DOI] [PubMed] [Google Scholar]
- 31.Fielitz J, Hein S, Mitrovic V, et al. Activation of the cardiac renin–angiotensin system and increased myocardial collagen expression in human aortic valve disease. J Am Coll Cardiol. 2001;37:1443–1449. doi: 10.1016/s0735-1097(01)01170-6. [DOI] [PubMed] [Google Scholar]
- 32.Wassmann S, Nickenig G. Pathophysiological regulation of the AT1-receptor and implications for vascular disease. J Hypertens Suppl. 2006;24:S15–S21. doi: 10.1097/01.hjh.0000220402.53869.72. [DOI] [PubMed] [Google Scholar]
- 33.Silva GK, Gutierrez FR, Guedes PM, et al. Cutting edge: nucleotide-binding oligomerization domain 1-dependent responses account for murine resistance against Trypanosoma cruzi infection. J Immunol. 2010;184:1148–1152. doi: 10.4049/jimmunol.0902254. [DOI] [PubMed] [Google Scholar]
- 34.Lipinski S, Till A, Sina C, et al. DUOX2-derived reactive oxygen species are effectors of NOD2-mediated antibacterial responses. J Cell Sci. 2009;122:3522–3530. doi: 10.1242/jcs.050690. [DOI] [PubMed] [Google Scholar]
- 35.Kayama H, Takeda K. The innate immune response to Trypanosoma cruzi infection. Microbes Infect. 2010;12:511–517. doi: 10.1016/j.micinf.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 36.de Meis J, Morrot A, Farias-de-Oliveira DA, et al. Differential regional immune response in Chagas disease. PLoS Negl Trop Dis. 2009;3:e417. doi: 10.1371/journal.pntd.0000417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Munoz-Fernandez MA, Fernandez MA, Fresno M. Activation of human macrophages for the killing of intracellular Trypanosoma cruzi by TNF-alpha and IFN-gamma through a nitric oxide-dependent mechanism. Immunol Lett. 1992;33:35–40. doi: 10.1016/0165-2478(92)90090-b. [DOI] [PubMed] [Google Scholar]
- 38.Melo RC, Fabrino DL, D’Avila H, et al. Production of hydrogen peroxide by peripheral blood monocytes and specific macrophages during experimental infection with Trypanosoma cruzi in vivo. Cell Biol Int. 2003;27:853–861. doi: 10.1016/s1065-6995(03)00173-2. [DOI] [PubMed] [Google Scholar]
- 39.Cuervo H, Pineda MA, Aoki MP, et al. Inducible nitric oxide synthase and arginase expression in heart tissue during acute Trypanosoma cruzi infection in mice: arginase I is expressed in infiltrating CD68+ macrophages. J Infect Dis. 2008;197:1772–1782. doi: 10.1086/529527. [DOI] [PubMed] [Google Scholar]
- 40.Silva EM, Guillermo LV, Ribeiro-Gomes FL, et al. Caspase-8 activity prevents type 2 cytokine responses and is required for protective T cell-mediated immunity against Trypanosoma cruzi infection. J Immunol. 2005;174:6314–6321. doi: 10.4049/jimmunol.174.10.6314. [DOI] [PubMed] [Google Scholar]
- 41.Miyahira Y. Trypanosoma cruzi infection from the view of CD8+ T cell immunity—an infection model for developing T cell vaccine. Parasitol Int. 2008;57:38–48. doi: 10.1016/j.parint.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 42.Cave A, Grieve D, Johar S, et al. NADPH oxidase-derived reactive oxygen species in cardiac pathophysiology. Philos Trans R Soc London Ser B Biol Sci. 2005;360:2327–2334. doi: 10.1098/rstb.2005.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang H, Petkova SB, Cohen AW, et al. Activation of transcription factors AP-1 and NF-kappa B in murine chagasic myocarditis. Infect Immun. 2003;71:2859–2867. doi: 10.1128/IAI.71.5.2859-2867.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Butterfield DA, Koppal T, Howard B, et al. Structural and functional changes in proteins induced by free radical-mediated oxidative stress and protective action of the antioxidants N-tert-butyl-alpha-phenylnitrone and vitamin E. Ann N Y Acad Sci. 1998;854:448–462. doi: 10.1111/j.1749-6632.1998.tb09924.x. [DOI] [PubMed] [Google Scholar]
- 45.Marnett LJ. Oxyradicals and DNA damage. Carcinogenesis. 2000;21:361–370. doi: 10.1093/carcin/21.3.361. [DOI] [PubMed] [Google Scholar]
- 46.Wen JJ, Dhiman M, Whorton EB, et al. Tissue-specific oxidative imbalance and mitochondrial dysfunction during Trypanosoma cruzi infection in mice. Microbes Infect. 2008;10:1201–1209. doi: 10.1016/j.micinf.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Godsel LM, Leon JS, Engman DM. Angiotensin converting enzyme inhibitors and angiotensin II receptor antagonists in experimental myocarditis. Curr Pharm Des. 2003;9:723–735. doi: 10.2174/1381612033455440. [DOI] [PubMed] [Google Scholar]
- 48.Reis DD, Jones EM, Tostes S, Jr, et al. Characterization of inflammatory infiltrates in chronic chagasic myocardial lesions: presence of tumor necrosis factor-alpha+ cells and dominance of granzyme A+, CD8+ lymphocytes. Am J Trop Med Hyg. 1993;48:637–644. doi: 10.4269/ajtmh.1993.48.637. [DOI] [PubMed] [Google Scholar]
- 49.Moore RE, Navab M, Millar JS, et al. Increased atherosclerosis in mice lacking apolipoprotein A-I attributable to both impaired reverse cholesterol transport and increased inflammation. Circ Res. 2005;97:763–771. doi: 10.1161/01.RES.0000185320.82962.F7. [DOI] [PubMed] [Google Scholar]
- 50.Wen JJ, Gupta S, Guan Z, et al. Phenyl-alpha-tert-butyl-nitrone and benzonidazole treatment controlled the mitochondrial oxidative stress and evolution of cardiomyopathy in chronic chagasic rats. J Am Coll Cardiol. 2010;55:2499–2508. doi: 10.1016/j.jacc.2010.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bartlett EJ, Lenzo JC, Sivamoorthy S, et al. Type I IFN-beta gene therapy suppresses cardiac CD8+ T-cell infiltration during autoimmune myocarditis. Immunol Cell Biol. 2004;82:119–126. doi: 10.1046/j.0818-9641.2004.01234.x. [DOI] [PubMed] [Google Scholar]
- 52.Liu YY, Cai WF, Yang HZ, et al. Bacillus Calmette-Guerin and TLR4 agonist prevent cardiovascular hypertrophy and fibrosis by regulating immune microenvironment. J Immunol. 2008;180:7349–7357. doi: 10.4049/jimmunol.180.11.7349. [DOI] [PubMed] [Google Scholar]
- 53.Hoemann CD. Molecular and biochemical assays of cartilage components. Methods Mol Med. 2004;101:127–156. doi: 10.1385/1-59259-821-8:127. [DOI] [PubMed] [Google Scholar]
- 54.Altieri P, Brunelli C, Garibaldi S, et al. Metalloproteinases 2 and 9 are increased in plasma of patients with heart failure. Eur J Clin Invest. 2003;33:648–656. doi: 10.1046/j.1365-2362.2003.01187.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Apocynin treatment did not alter the ability of splenocytes to produce cytokines.
Figure S2. Apocynin treatment controlled fibrosis in chagasic mice.