

Abstract
A better understanding of drug resistance mechanisms is required to improve outcomes in patients with pancreatic cancer. Here we characterized patterns of sensitivity and resistance to three conventional chemotherapeutic agents with divergent mechanisms of action (gemcitabine, 5-fluorouracil, and cisplatin) in pancreatic cancer cells. Four (L3.6pl, BxPC-3, CFPAC-1, SU86.86) were sensitive and five (PANC-1, Hs766T, AsPC-1, MIAPaCa-2, Mpanc96) were resistant to all 3 agents based on GI50 (50% growth inhibition). Gene expression profiling and unsupervised hierarchical clustering revealed that the sensitive and resistant cells formed two distinct groups and differed in expression of specific genes including several features of “epithelial-mesenchymal transition” (EMT). Interestingly, an inverse correlation between E-cadherin and its transcriptional suppressor, Zeb-1 was observed in the gene expression data and was confirmed by real time PCR. Independent validation experiment using 5 new pancreatic cancer cell lines confirmed that an inverse correlation between E-cadherin and Zeb-1 correlated closely with resistance to gemcitabine, 5-fluorouracil, and cisplatin. Silencing Zeb-1 in the mesenchymal lines not only increased the expression of E-cadherin but also other epithelial markers such as EVA1 and MAL2 and restored drug sensitivity. Importantly, immunohistochemical analysis of E-cadherin and Zeb-1 in primary tumors confirmed that expression of the two proteins was mutually exclusive (p=0.012). Therefore, our results suggest that Zeb-1 and other regulators of EMT may maintain drug resistance in human pancreatic cancer cells, and therapeutic strategies to inhibit Zeb-1 and reverse EMT should be evaluated.
Keywords: Zeb-1, E-cadherin, EMT, Gene expression profiling
Introduction
Pancreatic cancer is the fourth leading cause of cancer death in the United States (ACS 2007). Due to a lack of early detection methods and an absence of effective biomarkers, patients are usually diagnosed at a late stage with a less than 5 % 5-year survival rate. Unfortunately, there remains no effective therapy available to treat this aggressive tumor. Although most chemotherapy regimens utilize gemcitabine as the clinical standard of care for pancreatic cancer, patients generally have limited response to this therapy. Combination therapy and targeted therapies have also been overall quite disappointing. Thus, a better global understanding of the molecular mechanisms underlying drug resistance in pancreatic cancer may lead to the development of more effective therapeutic interventions.
Gene expression profiling has been used to identify biomarkers and therapeutic targets in pancreatic cancer (1, 2). Also, identification of genes related to chemo-sensitivity in pancreatic cancer has been performed in an attempt to improve the efficacy of pancreatic cancer therapy, and a number of different biomarkers, including S100A4, S100P, BNIP3, Cox-2, and periostin, have been advanced as therapeutic targets (3–7). However, even though there is an impression that pancreatic cancers tend to be cross-resistant to a large variety of cancer therapies, a mechanistic understanding of drug resistance has not been obtained.
Epithelial to mesenchymal transition (EMT) is a developmental process that appears to play an important role in tumor progression and metastasis in diverse solid tumors, including pancreatic cancer (8, 9). The EMT phenotype is characterized by the loss of cell-to-cell adhesion with the disintegration of tight, adherens and gap junctions and a phenotypic change from an “epithelial” morphology to a motile, fibroblast-like morphology (10, 11). The hallmark of EMT is loss of the epithelial homotypic adhesion molecule E-cadherin and gain of mesenchymal markers such as vimentin and/or fibronectin. Suppression of E-cadherin expression in normal cells and cancers is mediated by a number of genetic factors including mutation or promoter methylation of CDH1, or direct promoter repression mediated by Snail, Slug, Twist, Zeb-1and Sip1 (10–13), all of which interact with E-box elements located within the proximal region of the E-cadherin promoter (10). A number of clinical studies have demonstrated that increased expression of E-cadherin is associated with improved survival in several tumor types(14, 15), and there is some evidence that siRNA-mediated silencing of these transcriptional suppressors can increase cellular sensitivity to genotoxic stress (16).
Here, we characterized the effects of 3 conventional cancer chemotherapeutic agents on cell death in a panel of human pancreatic cancer cell lines to determine whether cells that were resistant to gemcitabine were also resistant to agents that act via independent mechanisms. We then used global gene expression profiling and unsupervised hierarchical clustering to demonstrate that the drug-resistant cells contained a number of features consistent with EMT. Reversal of EMT via silencing of Zeb-1 not only restored the expression of typical epithelial marker genes but also increased cellular sensitivity to therapeutic reagents. Our data suggest that Zeb-1 and other regulators of EMT maintain drug resistance in human pancreatic cancer cells, and therapeutic strategies to inhibit Zeb-1 and reverse EMT should be evaluated.
Materials and Methods
Cell Lines and Reagents
Ten pancreatic cancer cell lines were used for the generation of transcriptome data. Seven pancreatic cancer cell lines were obtained from American Type Tissue Collection (Manassas, VA) including AsPC-1, MIAPaCa-2, PANC-1, BxPC-3, CFPAC-1, Hs766T and SU86.86. MPanc96 and human pancreatic ductal epithelial (HPDE) cells were obtained from Dr Timothy J. Eberlein (St Louis, MO) and Dr. M. Tsao (Ontario Cancer Institute, Toronto, ON, Canada), respectively. L3.6pl cells were derived from COLO357 that had undergone metastasis from the pancreas to the liver of nude mice (17). For the validation experiments, 5 cell lines (Suit2, SW1990, Capan-1, T3M4 and Colo357) which were generously provided by Drs. Eric Collisson, Joe Gray, and Martin McMahon (University of California and Lawrence Livermore Laboratory, San Francisco) were used. All of the cell lines were genotyped by DNA fingerprinting (PowerPlex 16, Promega, Inc., Madison, WI). Gemcitabine was purchased from Eli Lilly and Co. (Indianapolis, IN). 5-fluorouracil (5FU), and cisplatin were purchased from Sigma (St. Louis, MO), respectively.
MTS Assay
Cells (3000/well) were plated in 96-well plate. After 24 hrs, cells were treated with increasing concentrations of the chemotherapy agents. 20μl of MTS solution (Promega, G358B, Madison, WI) was added to each well and the cells incubated at 37°C with 5% CO2 for 1hour. Absorbance at 490 nm was then measured with a Micro-plate reader (MRX, Danatech Laboratory, Houston, TX).
PI-FACS Analysis
Standard propidium iodide (PI) staining by the hypotonic lysis method was used for apoptosis studies. Apoptosis was induced in 106 cells by gemcitabine treatment. After 48/72hours, the cells were trypsinized, washed once with cold PBS, then incubated for 30 minutes in 500μL of hypotonic solution (0.1% sodium citrate, 0.1% Triton X-100, 100μg/mL RNase, and 50μg/mL PI), and analyzed by flow cytometry (Beckman Coulter Inc, Fullerton, CA).
RNA Isolation, Microarray Platform and Microarray Experiments
All transcriptome data were generated from duplicates of the cell lines. Cells were plated and total RNA was isolated independently using Trizol reagent (Molecular lab), followed by clean up with RNeasy® Mini kit (Qiagen, Valencia, CA). RNA was used for the synthesis of biotin labeled cRNA which was prepared using the Illumina RNA amplification kit (Ambion, Inc, Austin, TX) then cRNA can be hybridized to Illumina Human-6v2 (Illumina, Inc., San Diego, CA) chip. After being washed, the slides were scanned with Bead station 500X (Illumina, Inc.) and the signal intensities were quantified with Bead Studio (Illumina, Inc.). Quantile normalization was used to normalize the data. Microarray data is available on Gene Expression Omnibus with accession number GSE15550.
Data Processing
BRB ArrayTools version 3.6 developed by National Cancer Institute (18) was used to analyze the data. To select genes that are differentially expressed between the two different sub-groups (sensitive and resistant), a class comparison tool within BRB ArrayTools was used. The values were averaged over replicates of samples. This software uses a two-sample t-test to calculate the significance of the observations (i.e., P < 0.001). To see global gene expression patterns, each genes values, adjusted to be a mean of zero, were used for unsupervised hierarchical clustering with Cluster and TreeView (19). The Pearson correlation coefficient was used to analyze the correlation between gene expression and chemosensitivity. Growth-inhibitory IC50 (GI50) values were calculated using GraphPad software (San Diego CA).
Pathway Analysis
Functional and pathway analysis was performed using The Ingenuity™ Pathway Analysis software. This software contains a database for identifying networks and pathways of interest in genomic data.
Real-time PCR Analysis
Real-time PCR technology (StepOne™; Applied Biosystems, Foster City, CA) was used in conjunction with Assays-on-Demand (Applied Biosystems). The comparative CT method was used to determine relative gene expression levels for each target gene and Cyclophilin A served as an internal control to normalize for the amount of amplifiable RNA in each reaction.
Silencing of Zeb-1 Expression by siRNA
Dharmacon SMART Pool® control and Zeb-1 siRNA were used (Dharmacon, Lafayette, CO) with Oligofectamine according to the manufacturer's protocol (Invitrogen Corp, Carlsbad, CA). Cells were incubated with the siRNA complex for 48 hours, then treated with chemotherapeutic reagents and harvested for mRNA and protein expression changes (assayed via real-time RT-PCR and western blot) at 48 hrs. Cell death was measured by PI-FACS analysis.
Western Blotting
Total cellular protein extract was isolated from harvested cells, protein concentration determined, and western blotting carried out as described previously (20). The antibodies used were anti-Zeb-1 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), anti-E-cadherin (Zymed Laboratories Inc., San Francisco, CA) and anti-actin (Sigma chemical Co, St. Louis, MO). Same antibodies were used for immunoistochemistry.
Immunohistochemistry
Cells grown in chambered slides were washed with cold PBS and fixed with iced acetone for 5 mins at room temperature (RT). Cells were washed with PBS and blocked with blocking solution (horse serum: goat serum =1:4) for 30 mins. After cells were incubated with anti-E-cadherin/Zeb-1/Vimentin (Santa Cruz Biotechnology) overnight at 4°C, cells were washed with PBS and incubated with FITC conjugated secondary antibody (1:200, Santa Cruz) for 1 hour. Cells were washed with PBS and mounted with Prolong Gold anti-faded reagent with DAPI (Invitrogen).
For the human samples, unstained pancreatic tissue microarray (TMA) slides were deparaffinized with xylene and rehydrated with ethanol. Antigen retrieval was carried out by repetitive boiling and cooling cycles for a total of 15 min in antigen unmasking solution (Vector Laboratories). Endogenous peroxidase activity was blocked with 6% hydrogen peroxide in methanol, and nonspecific binding sites were blocked with normal donkey serum. Primary antibody diluted (1:50) in 2% BSA/0.2% Triton in PBS was added, and samples were incubated overnight at 4°C, after which biotinylated secondary antibody was added and incubated for 30 min followed by Vectastain Elite ABC reagent (Vector Laboratories) and incubation for an additional 30 min. Finally, slides were developed with 3, 3'-diaminobenzidine, counterstained with hematoxylin, dehydrated with ethanol, and fixed with xylene and mounted. The staining results were evaluated by a pathologist to determine the intensity and the percentage of positive tumor cells. The staining for zeb-1 was categorized as positive (nuclear staining in ≥ 10% of tumor cells) or negative (no nuclear staining or nuclear staining in < 10% of tumor cells). The staining for E-cadherin was categorized as 0 (negative or less than 10% moderate to strong membranous staining in tumor cells), 1 (≥10%, but less than 50% moderate to strong membranous staining) or 2 (≥ 50% moderate to strong membranous staining). The correlation between zeb-1 and E-cadherin was analyzed by Fisher's exact tests using Statistical Package for Social Sciences software for Windows (Version 12.0, SPSS Inc., Chicago, IL). We used a two-sided significance level of 0.05.
Results
Characterization of Pancreatic Cancer Cells Based on Multi-drug Sensitivity
We examined the native sensitivity of one immortalized “normal” human pancreatic epithelial line (HPDE) and 9 commonly available pancreatic cell lines to gemcitabine in vitro by PI-FACS analysis. All of the lines were genotyped by DNA fingerprinting using a commercial kit. Five of the lines (HPDE, L3.6pl, BxPC3, CFPAC, SU86.86) were sensitive and five (PANC-1, Hs766T, AsPC-1, MIAPaCa-2, MPanc96) were resistant based on the level of apoptosis (sub-G0/G1 population) stimulated with 10μM gemcitabine (Fig.1A). To test the multi-drug sensitivity of those cell lines, we performed cell viability assays using 2 additional chemotherapeutic agents, cisplatin and 5-FU. Interestingly, all of the gemcitabine-sensitive cancer cell lines were substantially more sensitive to 5-FU and cisplatin as compared to the gemcitabine-resistant lines (Fig.1B & C). Log10(GI50) values of a panel of cell lines and cluster analysis using log10(GI50) values were plotted (Fig.1D). In the sensitive cells log10(GI50) values were below the mean log10(GI50) across the panel, whereas resistant cell lines had log10(GI50) values above the mean.
Figure 1.

Effect of chemotherapeutic drugs on pancreatic cancer cells. A: Proapoptotic effects of gemcitabine on pancreatic cancer cells. 1.0×106 of pancreatic cancer cells were seeded in culture plates, and incubated with and without 10 μM gemcitabine. After 72 hours, apoptotic cells were measured by FACS analysis. Data shown are mean ± SEM from three experiments. B&C: Effects of various chemotherapeutic drugs on pancreatic cancer cell viability. Cells were treated with increasing concentrations of gemcitabine, 5-FU, and cisplatin (1–100,000 nM). After 72 hours, viable cells were quantified using the MTS reagent. Data shown are mean ± SEM from three experiments. D: log10 (GI50) values and cluster analysis of using log10 (GI50) values were plotted. If log10 (GI50) of the cell lines were below the mean of log10 (GI50) across the panel of cells, the cell lines were defined as sensitive, whereas resistant cell lines had log10 (GI50) values above the mean.
Identification of Global Gene Expression Patterns of Pancreatic Cancer Cells
Our results suggested that drug sensitivity and resistance might be controlled by broad-based mechanism(s). In an attempt to elucidate these mechanisms, we performed global gene expression profiling on the 9 pancreatic cancer cell lines and the normal HPDE cells using duplicate mRNA isolates obtained at different times. Unsupervised hierarchical clustering using whole genome expression patterns in the array demonstrated two distinct subgroups that clearly separated the drug-sensitive from the drug-resistant cell lines (Fig.2A). Clustering of the 6127 genes that were differentially expressed in at least two arrays with at least a two-fold change demonstrated the same expression pattern as the whole genome (Supp.Fig.1). This unsupervised clustering analysis revealed two subgroups that correlated with the multi-drug sensitivity data identified in the proliferation and apoptosis assays in Fig.1 (Fig.2A).
Figure 2.

Differentially expressed genes between sensitive and resistant cells. A: Unsupervised hierarchical clustering using whole genome expression patterns in the array (average linkage clustering). Cell lines are separated as two distinct subgroups that correlated with drug sensitivity (Blue box: sensitive cells, Red box: resistant cells). B: EMT related genes with significant differences in expression between sensitive and resistant cells (p<0.001). Highly expressed genes are in red and lower expressed genes are in green. C: The differential expression of MAL2, S100A4 and EVA1 were confirmed by real-time PCR. Mean ± SEM from triplicate samples.
Differential Expression of Cellular Adhesion and Motility Molecules
To identify genes which may play a role in drug sensitivity/resistance, the differentially expressed genes between sensitive and resistant cell lines were extracted using the class comparison tool in BRB Array Tools (p<0.001). We identified several interesting networks/pathways that were statistically significantly enriched, including those involved in cellular movement, cellular development, molecular transport and cancer using Ingenuity™ pathway analysis (Supp.Table.1). The 115 genes that were used for the pathway analyses are listed in Supp.Table.2 (p<0.001). However, one of the most obvious patterns that emerged from these analyses involved genes that had been previously implicated in EMT, including genes required for cell polarity (MAL2, and RABGEF1), adherens junction formation (EVA1 and PTPRM) and cell motility (S100A4, GNA11, EFEMP1, WDR44 and PKD1) (Fig 2B). All genes in Fig.2B had statistical significance at the cut-off of p<0.001 except EVA1 (p=0.0025). The differential expression of MAL2, S100A4 and EVA1 were confirmed by real-time PCR (Fig.2C), and p-values and fold changes of the genes are summarized in Table.1A. In addition to t-test, to determine the relationship between drug sensitivity and gene expression, Pearson correlation analysis was performed using GI50 values (in Fig.1D) and expression of genes in Fig.2.B (Supp.Table.3). Not all the genes' expression was directly correlated with GI50 though the significant patterns observed in class comparison analysis (t-test). We speculate that the EMT phenotype is roughly bimodal in the cells, which may explain why gene expression and GI50 are not be linearly correlated.
1.
Table..Adhesion and motility molecules with significant difference in expression between sensitive and resistant cells
| PTPRM | WDR44 | GNA11 | EVA1 | EFEMP1 | MAL2 | S100A4 | PKD1 | |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| p-value | 0.00035 | 0.00066 | 0.00076 | 0.00256 | 2.1e–06 | 7.9e–06 | 4.75e–05 | 0.00067 |
| Fold changes (S/R)* | 3.98 | −1.64 | 1.55 | 4.95 | 13.33 | 40.21 | −35.16 | −2.12 |
S/R: Sensitive cells/Resistant cells
Cellular Morphology and the Expression of EMT Markers
We subsequently performed experiments to assess the role of EMT in drug resistance in more detail. Simple light microscopic analysis of the cell lines confirmed that the drug-sensitive cells were uniform in shape and grew in tightly adherent “sheets” of cells in monolayer culture, whereas the drug-resistant cells were more irregular in shape and did not form close attachments in culture (data not shown), indicating that the drug-resistant cells displayed a more “mesenchymal” phenotype. To further confirm our observation, we assessed the levels of E-cadherin (epithelial marker, p=0.0084), Zeb-1 (a transcriptional suppressor of E-cadherin, p=0.0085) and vimentin (mesenchymal markers, p=0.12) in the gene expression data. We observed a statistically significant inverse correlation (r=−0.797, p=0.006) between E-cadherin and Zeb-1 (Fig.3A) that was associated with the EMT phenotype. We confirmed these expression data by real-time RT-PCR using the same RNA from the array experiment (Fig.3A). We then analyzed the protein levels of epithelial marker E-cadherin and its transcriptional suppressor Zeb-1, and also the mesenchymal marker vimentin in 3 sensitive and 3 resistant cells by immunofluorescence staining (Fig.3B–D). The staining patterns clearly confirmed the gene expression and real-time RT-PCR data, in that all of the epithelial cell lines expressed E-cadherin with no expression of Zeb-1 and vimentin, whereas all the resistant cell lines highly expressed Zeb-1 with no expression of E-cadherin and vimentin, with the exception of Hs766T, which does not express vimentin. To test the hypothesis that mesenchymal cells would show more migratory properties, we performed migration assays. As we expected, in general, the epithelial cells were less migratory than the mesenchymal cells (Supp.Fig.4).
Figure 3.

Expression of EMT markers in pancreatic cancer cells. A: Expression patterns of E-cadherin, Zeb-1 and Vimentin in the array data were generated via heat map and confirmed by quantitative real-time PCR. An inverse correlation between E-cadherin and Zeb-1 was observed across the cell lines. Mean ± SEM from triplicate samples. B- D: Immunofluorescence localization of E-cadherin, Zeb-1 and Vimentin confirms the association of epithelial and mesenchymal phenotype in drug sensitivity and resistance.
Effects of Silencing Zeb-1 on EMT-related Gene Expression and Drug Sensitivity
The gene expression and real time PCR data suggested to us that Zeb-1 might negatively regulate E-cadherin expression and possibly drug sensitivity. To test this hypothesis, we knocked down Zeb-1 using siRNA in Panc-1, and assessed the effects on EMT-related gene expression. Zeb-1-specific siRNA significantly reduced expression of Zeb-1 and that this was associated with a significant induction of E-cadherin mRNA and protein expression (Fig.4A–B). Furthermore, Zeb-1silencing up-regulated expression of EVA1 and MAL2, two other epithelial markers that were expressed at low levels in the mesenchymal cells, demonstrating that all 3 “epithelial” genes are directly or indirectly regulated by this transcriptional repressor (Fig.4B). Then we measured the effects of Zeb-1 silencing on drug-induced cell death in several of the drug-resistant cell lines (Panc-1, MIAPaCa-2 and Hs766T). Significant increases of apoptotic cell death were measured in Zeb-1 silencing cells after gemcitabine, 5 FU and cisplatin treatment in Panc-1 and MIAPaCa-2 while moderate effects was observed in Hs766T demonstrating that Zeb-1 does play a direct role in drug resistance in pancreatic cancer cells (Fig.5C).
Figure 4.

Effects of silencing Zeb-1 on EMT-related gene expression and drug sensitivity. A: Cells were transiently transfected with Zeb-1 specific siRNA or non-specific control for 48 h, and the expression of Zeb-1 and E-cadherin protein were measured in Panc-1 cells. B. The mRNA expression of Zeb-1, E-cadherin, EVA1 and MAL2 were measured after Zeb-1 silencing. Zeb-1-specific siRNA significantly reduced the expression of Zeb-1 and this was associated with a significant induction of E-cadherin, EVA1 and MAL2 mRNA levels. Mean ± SEM from triplicate samples. C: Effects of Zeb-1 silencing on drug sensitivity in the resistant cells. Zeb-1 specific siRNA or non-specific control transfected Panc-1, MIAPaCa-2 and Hs766T cells were incubated with 10 μM gemcitabine, 5FU and cisplatin for up to 72 hrs, then apoptotic cells were measured by PI-FACS analysis. Data converted into percentage of apoptosis over the control.
Figure 5.

A: The expression of E-cadherin and Zeb-1 in primary patient tumors. a and b: E-cadherin positive cancer cells are negative for Zeb-1. c and d: Zeb-1 positive cells are negative for E-cadherin. B: The inverse correlation between E-cadherin and Zeb-1 expression in independent validation experiment. C: Apoptotic effects of gemcitabine, cisplatin and 5-FU in a validation set of 5 new cell lines measured by PI-FACS analysis.
Validation of the Inverse Correlation of E-cadherin and Zeb-1 Expression in Primary Patient Tissues and 5 independent pancreatic cancer cell lines
Qualitative analyses of E-cadherin and Zeb-1 expression in a pancreatic patient TMA revealed an inverse correlation between Zeb-1 and E-cadherin expression (p=0.012). E-cadherin was identified primarily in the cell junctions as an adherent molecule and Zeb-1 was found primarily in the nucleus as a transcriptional factor (Fig. 5A).
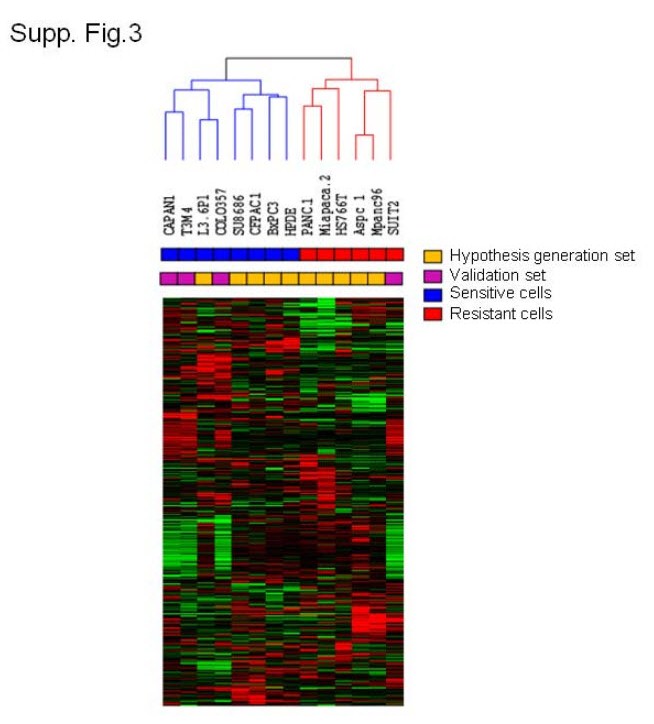
To further evaluate the relationship between the inverse correlation between E-cadherin and Zeb-1 and drug sensitivity, we performed an independent validation experiment using 5 new pancreatic cancer cell lines. Two cell lines (Suit2, SW1990) expressed high-levels of Zeb-1 and low-levels expression of E-cadherin, whereas the other three cell lines (Capan-1, T3M4, COLO357) showed high expression of E-cadherin with low expression of Zeb-1 (Fig.5B). As anticipated, the epithelial cells (Capan-1, T3M4, COLO357) were relatively sensitive to gemcitabine, cisplatin and 5-FU and opposite trend was observed in mesenchymal cells (Suit2, SW1990) (Fig.5C). Expression profiling of four out of five cell lines (T3M4, COLO357, Capan-1, Suit2) further confirmed our hypothesis showing both of groups clustered along with sensitive or resistant sub-types in Fig 2A (Suppl. Fig.6). COLO357, the parental cell line for L3.6pl, expressed high levels of E-cadherin and low levels of Zeb-1 and was highly sensitive to drug, demonstrating that the phenotype is stable even after multiple in vitro and in vivo passaging of the cells.
Discussion
In this study, we identified a molecular mechanism of multi-drug resistance in pancreatic cancer cells. We evaluated the sensitivity of a panel of pancreatic cancer cells to three chemotherapeutic agents and identified a correlation between the patterns of drug sensitivity/resistance and global gene expression. Importantly, Zeb-1-mediated EMT appears to be a major mediator of this drug resistance in pancreatic cancer cell lines.
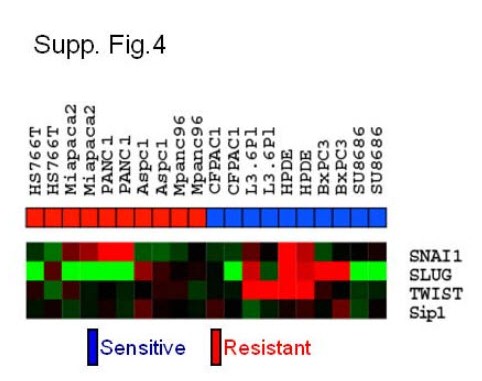
Our global gene expression patterns demonstrate that there are shared drug sensitivity and resistance mechanisms in pancreatic cancer (Fig.2A). Morphologic changes identified by light microscopy led us to examine the expressional correlation of E-cadherin and its transcriptional regulators in our transcriptome analysis. The loss of E-cadherin expression mediated by transcriptional suppression has been associated with a poor clinical outcome in several types of cancers (14, 21). The importance of particular transcriptional repressors in maintaining the EMT phenotype may vary depending on the tissue type (14, 15, 21–24). However, our pharmacogenomics approach suggests that the Zeb-1 plays a dominant role in the pancreatic cancer cell lines we studied here and that Snail, Slug, Twist and Sip1 do not correlate significantly with drug resistance (supp. Fig.2 and Fig.3). We confirmed that the inverse correlation between E-cadherin and Zeb-1 was also present in primary patient tumor samples (Fig.5A). Whether or not EMT correlates with gemcitabine resistance in patients will require further investigation.
A recent study using siRNA to silence Zeb-1 showed that Zeb-1 can suppress the transcription of multiple genes involved in determining epithelial polarity including the cadherin families, components of tight- and gap- junctions, demonstrating the ubiquitous role of the EMT phenotype genes (25). Similarly, we have shown that increased E-cadherin expression after silencing of Zeb-1 was accompanied by increased drug sensitivity (Fig.4C). Importantly, EMT also correlates with resistance to the EGFR inhibitor erlotinib (26), a biological agent that is currently being combined with gemcitabine in pancreatic cancer patients (27), and our data demonstrate that Zeb-1 silencing also enhances sensitivity to erlotinib in Panc-1 cells (A Kwan, unpublished data). Zeb-1 represses E-cadherin expression by recruiting histone deacetylases (HDACs) to E-box elements within the E-cadherin promoter. Thus, recent studies have demonstrated that clinically relevant HDAC inhibitors such as suberoylanilide hydroxamic acid (SAHA) can restore E-cadherin expression and sensitivity to gemcitabine and other agents (28, 29). We have also found that SAHA causes up-regulation of E-cadherin and down-regulation of Zeb-1 and restores gemcitabine and gefitinib sensitivity in several of the mesenchymal lines characterized in this study (K. Fournier, A. Kwan, manuscripts in preparation). Thus, it may be possible to use HDAC inhibitors to reverse the EMT phenotype and restore drug sensitivity to pancreatic cancers and other solid tumors.
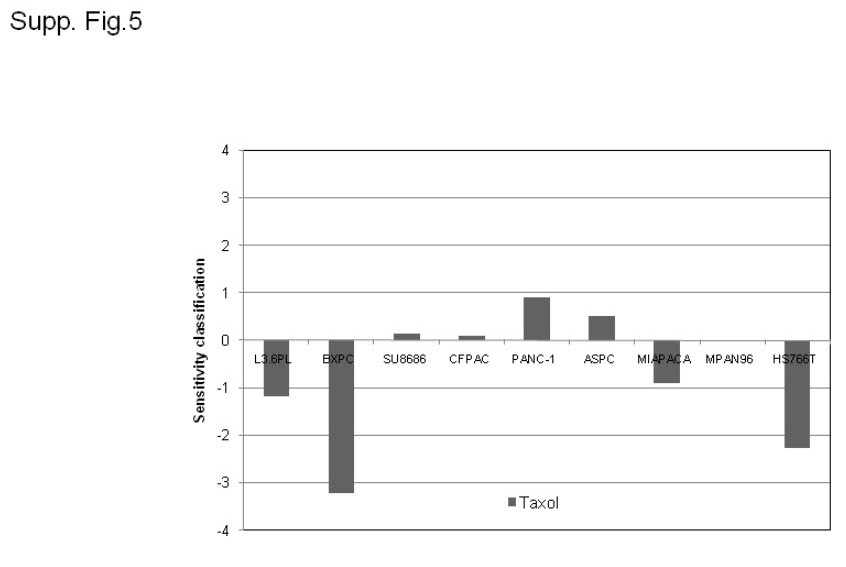
All of the drugs used in this study are DNA damaging agents. Among commonly used conventional cancer chemotherapeutic agents, taxanes are unique because they do not appear to act by inducing DNA damage but rather target microtubules leading to mitotic arrest and apoptosis. For reasons that are not entirely clear, taxanes have not been used for pancreatic cancer treatment. We tested the effects of paclitaxel in our panel of cell lines to determine whether EMT correlated with taxol resistance. Interestingly, paclitaxel resistance did not correlate well with EMT (data not shown). For example, the GI50s of paclitaxel in the Hs766T and MIA.PaCa-2 cells were lower than the mean GI50 across the panel (Supp. Fig.5). It has been reported that paclitaxel sensitivity does not correlate with sensitivity to cisplatin and oxaliplatin in primary human pancreatic cancer cells (30). In future studies we plan to explore the molecular basis for the observed heterogeneity in taxane responsiveness in our cells. It is possible that taxanes might have activity in pancreatic cancers that are resistant to the agents that are currently being used in the treatment of the disease.
Gemcitabine efficacy is limited due to the rapid development of resistance in patients with pancreatic cancer. There are multiple known factors responsible for this resistance including up-regulation of PI3K/AKT, S100A4, HMGA, and the ATP binding cassette (ABC) transporter genes (3, 31, 32). With respect to the latter, 48 distinct transporters within seven different subfamilies have been identified. They function as trans-membrane proteins that transport lipid, cholesterol, and drug metabolites out of the cell. It has been shown that several of these ABC transporters are associated with gemcitabine resistance (32, 33). Interestingly, we did not detect any relationship between the expression of ABC transporters and drug sensitivity within our panel of cells (suppl. Fig.3). S100A4, one of the calcium binding protein family of S100's has been linked to migratory and invasive properties in a number of other solid tumors including prostate cancer, breast cancer, and gastric cancer (34–36). We also confirmed that the resistant cells with high expression of S100A4 have greater migratory properties than the sensitive cells which have a low expression of S100A4 (Supp Fig.4). Knock-down of S100A4 also has been reported to cause an increase in E-cadherin expression and an increased latency of tumor formation (34). Conversely, increased expression of S100A4 and decreased expression of E-cadherin have been shown to correlate with a poor prognosis in pancreatic cancer patients (36). Evaluation of patients samples has demonstrated hypo-methylation of S100A4, causing downregulation of E-cadherin in the majority of pancreatic cancers. This demonstrates its role as a potential prognostic marker and possible therapeutic target (37, 38). Our study confirms the inverse relationship between S100A4 and E-cadherin as well. However, the mechanism of regulation of S100A4 and E-cadherin is not clear, and further investigation of this mechanism will be required.
The loss of cellular polarity and homotypic adhesion are major components of EMT. Our transcriptome analyses revealed various EMT related genes such as PKD, MAL2, and EVA1 that appear to be associated with drug sensitivity in pancreatic cancer cells. Transforming growth factor (TGF)-β is a known activator of the EMT program via the major signaling pathways Smad, raf, notch and PKD (39–41). The blockage of PKD expression by siRNA or a specific inhibitor attenuated TGF-β induced EMT (41). Likewise, EVA1 and MAL2 are expressed on many types of epithelial cells (42–44). The functions of EVA1 and MAL2 in human cancer are not well characterized, but their mRNA expression levels correlated with sensitivity to EGFR inhibitors and the epithelial phenotype (26, 45). Interestingly, knock-down of Zeb-1 expression restored the expression of MAL2 (46). Our data also identify an inverse correlation between EVA1/MAL2 and Zeb-1 expression in pancreatic cancer cells (Fig. 5B) again suggesting an important role of Zeb-1 in regulating the EMT program.
In this study, the measurement of basal gene expression levels enabled us to identify Zeb-1-mediated EMT as a multi-drug resistance mechanism in pancreatic cancer. Zeb-1 silencing can partially restore the drug sensitivity in resistant cells implying other signaling pathways might be responsible for drug resistance mechanism. Because of the limited number of cell lines used in our experiments, there may be many other important biological factors that contribute to drug resistance in pancreatic cancer. Furthermore, pancreatic cancer is exemplified by the presence of an extensive tumor-associated stroma, and recent studies have confirmed that stromal cells also contribute to drug resistance (47). We are currently expanding our panel of cell lines and models to include conventional and primary xenografts directly address these issues. Overall, we remain confident that by using gene expression profiling as a starting point to identify the biological properties associated with drug sensitivity or resistance, we will ultimately be able to better match tumors to effective therapies and improve disease control in patients.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgement
We thank Dr. Eric Collisson for providing pancreatic cancer cells.
REFERENCES
- 1.Logsdon CD, Simeone DM, Binkley C, et al. Molecular profiling of pancreatic adenocarcinoma and chronic pancreatitis identifies multiple genes differentially regulated in pancreatic cancer. Cancer Res. 2003;63:2649–57. [PubMed] [Google Scholar]
- 2.Lowe AW, Olsen M, Hao Y, et al. Gene expression patterns in pancreatic tumors, cells and tissues. PLoS ONE. 2007;2:e323. doi: 10.1371/journal.pone.0000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mahon PC, Baril P, Bhakta V, et al. S100A4 contributes to the suppression of BNIP3 expression, chemoresistance, and inhibition of apoptosis in pancreatic cancer. Cancer Res. 2007;67:6786–95. doi: 10.1158/0008-5472.CAN-07-0440. [DOI] [PubMed] [Google Scholar]
- 4.Eibl G, Takata Y, Boros LG, et al. Growth stimulation of COX-2-negative pancreatic cancer by a selective COX-2 inhibitor. Cancer Res. 2005;65:982–90. [PubMed] [Google Scholar]
- 5.Baril P, Gangeswaran R, Mahon PC, et al. Periostin promotes invasiveness and resistance of pancreatic cancer cells to hypoxia-induced cell death: role of the beta4 integrin and the PI3k pathway. Oncogene. 2007;26:2082–94. doi: 10.1038/sj.onc.1210009. [DOI] [PubMed] [Google Scholar]
- 6.Akada M, Crnogorac-Jurcevic T, Lattimore S, et al. Intrinsic chemoresistance to gemcitabine is associated with decreased expression of BNIP3 in pancreatic cancer. Clin Cancer Res. 2005;11:3094–101. doi: 10.1158/1078-0432.CCR-04-1785. [DOI] [PubMed] [Google Scholar]
- 7.Arumugam T, Simeone DM, Van Golen K. Logsdon CD. S100P promotes pancreatic cancer growth, survival, and invasion. Clin Cancer Res. 2005;11:5356–64. doi: 10.1158/1078-0432.CCR-05-0092. [DOI] [PubMed] [Google Scholar]
- 8.Nakajima S, Doi R, Toyoda E, et al. N-cadherin expression and epithelial-mesenchymal transition in pancreatic carcinoma. Clin Cancer Res. 2004;10:4125–33. doi: 10.1158/1078-0432.CCR-0578-03. [DOI] [PubMed] [Google Scholar]
- 9.Ellenrieder V, Hendler SF, Ruhland C, Boeck W, Adler G, Gress TM. TGF-beta-induced invasiveness of pancreatic cancer cells is mediated by matrix metalloproteinase-2 and the urokinase plasminogen activator system. Int J Cancer. 2001;93:204–11. doi: 10.1002/ijc.1330. [DOI] [PubMed] [Google Scholar]
- 10.Peinado H, Olmeda D, Cano A. Snail. Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–28. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 11.Kang Y, Massague J. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell. 2004;118:277–9. doi: 10.1016/j.cell.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 12.Vandewalle C, Comijn J, De Craene B, et al. SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic acids Res. 2005;33:6566–78. doi: 10.1093/nar/gki965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lombaerts M, van Wezel T, Philippo K, et al. E-cadherin transcriptional downregulation by promoter methylation but not mutation is related to epithelial-to-mesenchymal transition in breast cancer cell lines. Br J cancer. 2006;94:661–71. doi: 10.1038/sj.bjc.6602996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pena C, Garcia JM, Silva J, et al. E-cadherin and vitamin D receptor regulation by SNAIL and ZEB1 in colon cancer: clinicopathological correlations. Hum Mol Genet. 2005;14:3361–70. doi: 10.1093/hmg/ddi366. [DOI] [PubMed] [Google Scholar]
- 15.Uchikado Y, Natsugoe S, Okumura H, et al. Slug Expression in the E-cadherin preserved tumors is related to prognosis in patients with esophageal squamous cell carcinoma. Clin Cancer Res. 2005;11:1174–80. [PubMed] [Google Scholar]
- 16.Kajita M, McClinic KN, Wade PA. Aberrant expression of the transcription factors snail and slug alters the response to genotoxic stress. Mol Cell Biol. 2004;24:7559–66. doi: 10.1128/MCB.24.17.7559-7566.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bruns CJ, Harbison MT, Kuniyasu H, Eue I, Fidler IJ. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia. 1999;1:50–62. doi: 10.1038/sj.neo.7900005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wright GW, Simon RM. A random variance model for detection of differential gene expression in small microarray experiments. Bioinformatics. 2003;19:2448–55. doi: 10.1093/bioinformatics/btg345. [DOI] [PubMed] [Google Scholar]
- 19.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A. 1998;95:14863–8. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi W, Gerner EW, Ramdas L, et al. Combination of 5-fluorouracil and N1,N11-diethylnorspermine markedly activates spermidine/spermine N1-acetyltransferase expression, depletes polyamines, and synergistically induces apoptosis in colon carcinoma cells. J Biol Chem. 2005;280:3295–304. doi: 10.1074/jbc.M409930200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dohadwala M, Yang SC, Luo J, et al. Cyclooxygenase-2-dependent regulation of E-cadherin: prostaglandin E(2) induces transcriptional repressors ZEB1 and snail in non-small cell lung cancer. Cancer Res. 2006;66:5338–45. doi: 10.1158/0008-5472.CAN-05-3635. [DOI] [PubMed] [Google Scholar]
- 22.Elloul S, Elstrand MB, Nesland JM, et al. Snail, Slug, and Smad-interacting protein 1 as novel parameters of disease aggressiveness in metastatic ovarian and breast carcinoma. Cancer. 2005;103:1631–43. doi: 10.1002/cncr.20946. [DOI] [PubMed] [Google Scholar]
- 23.Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–39. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 24.Haddad Y, Choi W, McConkey DJ. Delta-crystallin enhancer binding factor 1 controls the epithelial to mesenchymal transition phenotype and resistance to the epidermal growth factor receptor inhibitor erlotinib in human head and neck squamous cell carcinoma lines. Clin Cancer Res. 2009;15:532–42. doi: 10.1158/1078-0432.CCR-08-1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aigner K, Dampier B, Descovich L, et al. The transcription factor ZEB1 (deltaEF1) promotes tumour cell dedifferentiation by repressing master regulators of epithelial polarity. Oncogene. 2007;26:6979–88. doi: 10.1038/sj.onc.1210508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yauch RL, Januario T, Eberhard DA, et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res. 2005;11:8686–98. doi: 10.1158/1078-0432.CCR-05-1492. [DOI] [PubMed] [Google Scholar]
- 27.Morgan MA, Parsels LA, Kollar LE, Normolle DP, Maybaum J, Lawrence TS. The combination of epidermal growth factor receptor inhibitors with gemcitabine and radiation in pancreatic cancer. Clin Cancer Res. 2008;14:5142–9. doi: 10.1158/1078-0432.CCR-07-4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumagai T, Wakimoto N, Yin D, et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (Vorinostat, SAHA) profoundly inhibits the growth of human pancreatic cancer cells. Int J Cancer. 2007;121:656–65. doi: 10.1002/ijc.22558. [DOI] [PubMed] [Google Scholar]
- 29.Witta SE, Gemmill RM, Hirsch FR, et al. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006;66:944–50. doi: 10.1158/0008-5472.CAN-05-1988. [DOI] [PubMed] [Google Scholar]
- 30.Michalski CW, Erkan M, Sauliunaite D, et al. Ex vivo chemosensitivity testing and gene expression profiling predict response towards adjuvant gemcitabine treatment in pancreatic cancer. Br J cancer. 2008;99:760–7. doi: 10.1038/sj.bjc.6604528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Asano T, Yao Y, Zhu J, Li D, Abbruzzese JL, Reddy SA. The PI 3-kinase/Akt signaling pathway is activated due to aberrant Pten expression and targets transcription factors NF-kappaB and c-Myc in pancreatic cancer cells. Oncogene. 2004;23:8571–80. doi: 10.1038/sj.onc.1207902. [DOI] [PubMed] [Google Scholar]
- 32.Szakacs G, Annereau JP, Lababidi S, et al. Predicting drug sensitivity and resistance: profiling ABC transporter genes in cancer cells. Cancer Cell. 2004;6:129–37. doi: 10.1016/j.ccr.2004.06.026. [DOI] [PubMed] [Google Scholar]
- 33.Oguri T, Achiwa H, Sato S, et al. The determinants of sensitivity and acquired resistance to gemcitabine differ in non-small cell lung cancer: a role of ABCC5 in gemcitabine sensitivity. Mol Cancer Ther. 2006;5:1800–6. doi: 10.1158/1535-7163.MCT-06-0025. [DOI] [PubMed] [Google Scholar]
- 34.Saleem M, Kweon MH, Johnson JJ, et al. S100A4 accelerates tumorigenesis and invasion of human prostate cancer through the transcriptional regulation of matrix metalloproteinase 9. Proc Natl Acad Sci U S A. 2006;103:14825–30. doi: 10.1073/pnas.0606747103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rudland PS, Platt-Higgins A, Renshaw C, et al. Prognostic significance of the metastasis-inducing protein S100A4 (p9Ka) in human breast cancer. Cancer Res 2000. 60:1595–603. [PubMed] [Google Scholar]
- 36.Yonemura Y, Endou Y, Kimura K, et al. Inverse expression of S100A4 and E-cadherin is associated with metastatic potential in gastric cancer. Clin Cancer Res. 2000;6:4234–42. [PubMed] [Google Scholar]
- 37.Sato N, Maitra A, Fukushima N, et al. Frequent hypomethylation of multiple genes overexpressed in pancreatic ductal adenocarcinoma. Cancer Res. 2003;63:4158–66. [PubMed] [Google Scholar]
- 38.Oida Y, Yamazaki H, Tobita K, et al. Increased S100A4 expression combined with decreased E-cadherin expression predicts a poor outcome of patients with pancreatic cancer. Onco Rep. 2006;16:457–63. [PubMed] [Google Scholar]
- 39.Janda E, Nevolo M, Lehmann K, Downward J, Beug H, Grieco M. Raf plus TGFbeta-dependent EMT is initiated by endocytosis and lysosomal degradation of E-cadherin. Oncogene. 2006;25:7117–30. doi: 10.1038/sj.onc.1209701. [DOI] [PubMed] [Google Scholar]
- 40.Zavadil J, Cermak L, Soto-Nieves N, Bottinger EP. Integration of TGF-beta/Smad and Jagged1/Notch signalling in epithelial-to-mesenchymal transition. The EMBO J. 2004;23:1155–65. doi: 10.1038/sj.emboj.7600069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang Y, Pan X, Lei W, Wang J, Song J. Transforming growth factor-beta1 induces epithelial-to-mesenchymal transition and apoptosis via a cell cycle-dependent mechanism. Oncogene. 2006;25:7235–44. doi: 10.1038/sj.onc.1209712. [DOI] [PubMed] [Google Scholar]
- 42.Marazuela M, Acevedo A, Garcia-Lopez MA, Adrados M, de Marco MC, Alonso MA. Expression of MAL2, an integral protein component of the machinery for basolateral-to-apical transcytosis, in human epithelia. J Histochem Cytochem. 2004;52:243–52. doi: 10.1177/002215540405200212. [DOI] [PubMed] [Google Scholar]
- 43.de Marco MC, Martin-Belmonte F, Kremer L, et al. MAL2, a novel raft protein of the MAL family, is an essential component of the machinery for transcytosis in hepatoma HepG2 cells. J Cell Biol. 2002;159:37–44. doi: 10.1083/jcb.200206033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guttinger M, Sutti F, Panigada M, et al. Epithelial V-like antigen (EVA), a novel member of the immunoglobulin superfamily, expressed in embryonic epithelia with a potential role as homotypic adhesion molecule in thymus histogenesis. J Cell Biol. 1998;141:1061–71. doi: 10.1083/jcb.141.4.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Frederick BA, Helfrich BA, Coldren CD, et al. Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Mol Cancer Ther. 2007;6:1683–91. doi: 10.1158/1535-7163.MCT-07-0138. [DOI] [PubMed] [Google Scholar]
- 46.Spaderna S, Schmalhofer O, Wahlbuhl M, et al. The transcriptional repressor ZEB1 promotes metastasis and loss of cell polarity in cancer. Cancer Res. 2008;68:537–44. doi: 10.1158/0008-5472.CAN-07-5682. [DOI] [PubMed] [Google Scholar]
- 47.Hwang RF, Moore T, Arumugam T, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918–26. doi: 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.