

Abstract
Adoptive transfer of tumor-specific T cells has shown some success for treating metastatic melanoma. We evaluated a novel strategy to improve adoptive therapy by administering both T cells and oncolytic myxoma virus to mice with syngeneic B16.SIY melanoma brain tumors. Adoptive transfer of activated CD8+ 2C T cells that recognize SIY peptide doubled survival time, but SIY-negative tumors recurred. Myxoma virus killed B16.SIY cells in vitro, and intratumoral injection of virus led to selective and transient infection of the tumor. Virus treatment recruited innate immune cells to the tumor and induced IFNβ production in the brain, resulting in limited oncolytic effects in vivo. To counter this, we evaluated the safety and efficacy of co-administering 2C T cells, myxoma virus, and either rapamycin or neutralizing antibodies against IFNβ. Mice that received either triple combination therapy survived significantly longer with no apparent side effects, but eventually relapsed. Importantly, rapamycin treatment did not impair T cell-mediated tumor destruction, supporting the feasibility of combining adoptive immunotherapy and rapamycin-enhanced virotherapy. Myxoma virus may be a useful vector for transient delivery of therapeutic genes to a tumor to enhance T cell responses.
Keywords: Brain tumor, Melanoma, T cells, Myxoma virus, Rapamycin
Introduction
Patients with malignant cutaneous melanoma that develop brain metastases have very poor prognoses [1]. Adoptive T cell therapy is currently the most successful treatment for metastatic melanoma [2]. Approximately 50% of patients show an objective response (partial or complete) after lymphodepletion with chemotherapy and subsequent infusion of ex vivo activated, autologous tumor-infiltrating T cells [3]. Importantly, T cell responses against metastases have been observed at various sites including the brain [3, 4].
T cell-based immunotherapies for cancer are promising, but can result in tumor escape due to a variety of mechanisms such as immune editing of tumors [5–9], including brain tumors [10, 11]. Recurrent tumors are comprised of antigen-loss variant (ALV) cancer cells that can evade antigen-specific T cell killing. One potential strategy to prevent tumor recurrence is to combine multiple treatments, such as T cell immunotherapy and oncolytic virotherapy, with complementary anti-tumor mechanisms [12–14].
Concurrent delivery of an oncolytic virus could complement and enhance adoptive T cell therapy for cancer in several ways: tumor-specific viral infection may lead to oncolysis and release of tumor antigens to stimulate a T cell response, recruit additional immune cells, induce cytokine production, and in the case of engineered recombinant viruses, express therapeutic genes (e.g., antigens or cytokines) that could promote clearance of ALV cancer cells by immune cells.
Many human and murine cancer cells are permissive to infection by myxoma virus (MYXV) [15–22], a replication-competent poxvirus pathogenic only in European rabbits [23, 24]. Productive replication of MYXV in cancer cells results in cell death. With regard to use in the brain, MYXV has demonstrated safety and potent efficacy against orthotopic glioma xenografts in nude mice [16] and more limited, yet significant oncolytic effects against syngeneic gliomas in immunocompetent rats [22].
In the present studies, we assessed the feasibility and efficacy of combining adoptive T cell therapy and myxoma virotherapy to treat syngeneic B16.SIY mouse melanoma brain tumors. The ability of adoptively transferred CD8+ 2C T cells to target established, aggressively growing B16.SIY brain tumors was evaluated using C57BL/6 RAG1−/− host mice that lack endogenous T and B cells, imitating the lymphodepletion protocol used in adoptive therapy clinical trials. The ability of MYXV to infect, express a virally delivered gene, and kill B16.SIY cells was evaluated in vitro and in vivo. The brain cytokine and innate immune cell responses to intratumoral injection of MYXV were also characterized. Finally, the novel treatment combination of T cell adoptive therapy and myxoma virotherapy for brain tumors was assessed, as well as the benefits of co-administering either neutralizing antibodies against interferon-β (IFNβ) or rapamycin. Neutralizing antibodies were used to minimize the well-known anti-viral effects of IFNβ. Rapamycin and similar compounds improve the oncolytic effects of several viruses in vivo including MYXV [17, 21, 22, 25–30] via inhibition of both Type I interferon production and macrophage recruitment [22], but the potential immunosuppressive effects of rapamycin treatment on concurrent T cell immunotherapy were unknown.
Materials and methods
Mice
C57BL/6 and C57BL/6 RAG1−/− mice originally purchased from The Jackson Laboratory (Bar Harbor, Maine) are maintained as colonies at the University of Illinois. 2C T cell receptor (TCR) transgenic mice on the C57BL/6 background are maintained as a heterozygous colony and screened for expression of the 2C TCR on Thy1.2+ peripheral blood cells with 1B2 clonotypic antibody by flow cytometry. Mice were 2–6 months of age at the time of experiments. All procedures were approved by the Institutional Animal Care and Use Committee at the University of Illinois at Urbana-Champaign.
Cancer cell lines
B16-F10 mouse melanoma cells (H-2b) were purchased from American Type Culture Collection (Manassas, VA). B16.SIY cancer cells (gift from Dr. Thomas Gajewski, University of Chicago, IL) were derived by retroviral transduction of B16-F10 cells. B16.SIY cells express a fusion protein of the Kb-binding peptide SIYRYYGL (SIY) and enhanced green fluorescent protein (GFP) as previously described [31]. CD8+ 2C T cells isolated from 2C TCR mice recognize SIY peptide in the context of MHC I Kb. Cancer cells were grown in Dulbecco’s Modified Eagle Medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS), l-glutamine, penicillin, and streptomycin.
Intracranial cancer cell infusions
Cancer cells were trypsinized, washed twice with Hanks Balanced Salt Solution (HBSS; Mediatech, Inc., Manassas, VA), and stereotaxically infused into the brains of mice anesthetized with isoflurane (Aerrane, Baxter, Deerfield, IL). 2 × 103 B16.SIY cells in 300 nL HBSS were infused into ventral striatum (AP 0.5 mm, Lat 2.5 mm, DV −4 mm). In some experiments, mice received bilateral infusions. Mice were euthanized at 75% of baseline body weight or signs of neurological impairment, in accordance with IACUC guidelines.
Preparation of activated 2C T cells for adoptive transfer
Lymphocytes from spleens and lymph nodes of 2C TCR mice were prepared by mechanical tissue dissociation through nylon mesh followed by ammonium chloride buffer lysis of erythrocytes. Mixed lymphocytes were incubated at 37°C, 5% CO2 for 48 h with 3 μM SIY peptide and 5% rat ConA supe to activate and expand effector 2C T cells. Cells were then collected using PBS/EDTA buffer and washed with HBSS. Mice were briefly restrained under a heat lamp, and approximately 1 × 107 cells in 200 μL HBSS were injected into the tail vein. Control mice were injected with 200 μL HBSS. 2C T cell adoptive transfers were given on day six or seven in survival experiments.
Histological analysis of 2C T cell response
Mice with 7-day-old B16.SIY brain tumors were treated with 2C T cell adoptive transfers and killed 2 or 7 days later, or followed for survival and euthanized upon symptoms of tumor relapse. Brains and cervical lymph nodes were collected, snap-frozen in Tissue-Tek OCT medium (Ted Pella, Inc., Redding, CA), and sectioned. 1B2 monoclonal antibody specific for the 2C TCR was purified from the hybridoma and biotinylated in our laboratory. Unfixed 10-micron tissue sections were blocked with Superblock (Pierce, Rockford, IL), stained with 1B2 antibody overnight, then amplified with the TSA Biotin System (Perkin Elmer, Boston, MA), and detected with streptavidin-Alexa 594 (Invitrogen Molecular Probes, Eugene, OR). Tissue was counterstained with 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI, Invitrogen Molecular Probes) and analyzed by fluorescence microscopy.
Flow cytometry experiments
Brain tumors were excised and gently minced to dissociate cells. Explanted cells were cultured for 4–7 days in supplemented DMEM to enrich for live melanin-positive cells, trypsinized, and washed twice with PBS/0.5% BSA before analysis of GFP expression on a BD FACS Canto instrument (BD Biosciences, San Jose, CA). MYXV-Red-infected B16.SIY cells were trypsinized, washed with PBS, fixed with 1% MeOH-free paraformaldehyde in PBS, and analyzed for tdTomato red expression on a BD LSR II instrument (BD Biosciences) using FCS Express software (De Novo Software, Los Angeles, CA).
Viruses
The Lausanne strain of MYXV and RK13 rabbit kidney cells were a gift from Dr. Richard Moyer (University of Florida, Gainesville, FL). RK13 cells were maintained in minimum essential medium (MEM-C; Mediatech, Manassas, VA) with Earle’s salts supplemented with 2 mM glutamine, 50 U/mL penicillin G, 50 μg/mL streptomycin, 1 mM sodium pyruvate, and 0.1 mM nonessential amino acids. MYXV-Red expresses tdTomato red fluorescent protein under the control of a synthetic vaccinia virus early/late promoter and was originally designated vMyx-tdTr [32]. Virus replicated in RK13 cells was sucrose pad-purified prior to use in mouse experiments. UV-inactivated virus was prepared by exposing MYXV-Red to a G30T8 germicidal UV light producing irradiation at 254 nm for 24 h.
Electron microscopy
B16.SIY cells infected with 10 infectious MYXV particles per cell [multiplicity of infection (MOI) of 10] were collected 24 h p.i. for transmission electron microscopy (TEM). Infected cell pellets were suspended in Karnovsky’s fixative and shipped to the University of Florida Interdisciplinary Center for Biotechnology Research for processing and imaging with a Hitachi H-7000 with Olympus Soft-Imaging Systems MegaView III digital camera.
Viral growth curves
Growth kinetics of MYXV-Red in cancer cells were determined by inoculating confluent cultures at MOI 0.1 in serum-free medium at 37°C in 5% CO2 for 1 h. The inoculum was then removed, and cells were washed with PBS three times before replacing with fresh medium. Cells were harvested at 0, 12, 24, and 48 h post-inoculation (p.i.). Pellets were resuspended in MEM-C without serum and stored at −20°C. Titers were determined by replicating the virus in RK13 cells as described below. Cells were infected similarly using different MOIs for in vitro cell viability assays and for the in vivo pre-infection survival experiment.
Titration of infectious virions
Cell or tissue samples were homogenized, frozen then thawed three times, and sonicated. A series of tenfold dilutions were made and added to confluent RK13 cells. After 1 h of incubation at 37°C in 5% CO2, a 1:1 mixture of 1% SeaKem high gelling temperature agarose and 2X MEM-C with 20% serum was added to the infected cells. Plates were incubated at 37°C in 5% CO2 for 5–7 days until viral foci developed. A minimum of three separate series of tenfold dilutions were counted to calculate the average number of viral focus forming units per mL (ffu/mL; virus titer) in each sample.
Cell viability assays
CellTiter-Blue Viability Assay (Promega, Madison, WI) was used according to kit instructions. Viable cells convert a redox dye (resazurin) into a fluorescent product (resorufin) detectable at 590 nm. B16.SIY cells growing in 96-well plates were mock-infected or infected at MOI 1 or 10 with wild-type MYXV. Blue reagent was added 1.5 h prior to fluorescence readings. Images of cytopathic effect of virus infection were acquired using a Leica DMI 4000 B microscope and ImagePro Express software (Media Cybernetics, Bethesda, MD).
Intratumoral injection of MYXV and characterization of infection in vivo
Wild-type and RAG1−/− mice with 4- to 7-day-established B16.SIY brain tumors were anesthetized and stereotaxically injected intratumorally (i.t.) with approximately 5x106 ffu of MYXV-Red or UV-inactivated MYXV-Red in 0.7-μL total volume. Mice were euthanized 1, 2, 3, 5, 6, 7, or 8 days post-injection for analysis of tdTomato red expression (n = 2–3 per day). Brains were immersion fixed in 10% formalin to preserve tdTomato red protein expression. Ten micron thick sections were counterstained with DAPI and analyzed with an Olympus fluorescence microscope.
Immunostaining and histopathological analyses
Wild-type and RAG1−/− mice with five-day-established B16.SIY brain tumors were injected i.t. with MYXV-Red, UV-MYXV-Red or PBS (n = 4 per group). Mice were killed 48 h later, and brains were either immersion fixed in 10% formalin for paraffin-embedding/processing for hematoxylin and eosin staining, or snap-frozen in OCT medium for cryosectioning and immunostaining (n = 2 per group). Ten-μm cryosections were fixed in cold 95% ethanol and blocked with Superblock. Sections were incubated with purified anti-mouse CD11b, anti-mouse 4D11, or anti-mouse GR-1 (all antibodies from BD Pharmingen, San Jose, CA) overnight, washed with PBS + 0.1% Tween-20, and incubated with biotinylated rabbit anti-rat antibody (Vector, Burlingame, CA) with 5% normal rabbit serum (Vector) for 2 h. Slides were washed and incubated with streptavidin-Alexa 594 and DAPI. Control slides omitting each primary antibody were negative for Alexa594.
Cytokine ELISAs
MYXV-Red (7.6 × 106 ffu in 1 μL), a sucrose pad-purified preparation of uninfected, lysed RK13 cells (mock), or PBS was injected i.t. into RAG1−/− mice with B16.SIY brain tumors on day 8. Tumors and adjacent brain tissue were excised 24 h later, weighed, and pooled (2 per sample). Tissues were homogenized in 2-mL ice-cold PBS + 0.1% Igepal detergent. Supernatants were concentrated and tested in triplicate. Mouse TNFα Ready-Set-Go ELISA kits (eBiosciences, San Diego, CA) and mouse IFNα and IFNβ ELISA kits (PBL Biomedical Labs, Piscataway, NJ) were used according to manufacturer instructions.
Treatment with Interferon β neutralizing antibodies
Mice with 5-day-established B16.SIY tumors received i.t. MYXV-Red (5 × 106 ffu in 0.7 μL) injections and both i.t. and i.p. anti-IFNβ injections. Rabbit polyclonal antibodies against mouse IFNβ (PBL Interferon Source) were diluted in sterile saline and injected into mice i.t. as a single dose (12.5 ng in 0.5 μL along with 0.7 μL virus) and i.p. at 100 μg/kg body weight diluted in 100 μL sterile PBS once daily for 4 days starting on day five.
Rapamycin experiments
For in vitro experiments, B16.SIY cells were plated at 2 × 105 per well in 24-well plates. After 24 h, medium was replaced with fresh DMEM containing 50 ng/mL TNFα (R&D Systems, Minneapolis, MN), 100 U/mL IFNβ (R&D Systems), and 20 mM rapamycin (LC Laboratories, Woburn, MA) dissolved in dimethyl sulfoxide (DMSO) for 6 h. Cells were then washed and inoculated with MYXV-Red at MOI 1 in serum-free medium for 1 h. After 1 h, fresh medium (+ 10% FCS) was added, and the plates were incubated for 3 days at 37°C, 5% CO2. Cells were collected with trypsin/EDTA, washed with PBS, and fixed in 1% paraformaldehyde in PBS before analysis of tdTomato red expression on a BD LSRII cytometer.
For survival experiments, rapamycin was dissolved in DMSO to make a 50-mg/mL stock and frozen at −80°C. RAG1−/− mice with 5-day-old B16.SIY brain tumors were injected i.t. with MYXV-Red (approximately 7.6 × 106 ffu in 1 μL) or PBS. On day six, some mice were injected with approximately 2 × 107 preactivated 2C T cells i.v. Rapamycin treatment (3 mg/kg i.p.) was started on day 5. Mice received a total of six injections given every other day. Control mice received PBS.
Data analysis
GraphPad Prism software was used for all statistical analyses: cell viability data by two-way ANOVA with Bonferroni post-test, survival curves by Log-rank test, cytokine ELISAs and rapamycin flow data by one-way ANOVA and unpaired t test. Significance was considered P < 0.05.
Results
Adoptive T cell therapy increases survival duration of tumor-bearing mice
Wild-type C57BL/6 mice and C57BL/6 RAG1−/− mice with established syngeneic B16.SIY brain tumors were injected with activated 2C T cells on day 7 and followed for survival (Fig. 1a). All mice treated with 2C T cells lived more than twice as long as untreated mice (P < 0.05), and the survival durations of RAG1−/− and wild-type mice were similar. However, adoptive therapy alone was insufficient to cure any of the mice.
Fig. 1.

a Survival of C57BL/6 and RAG1−/− mice with B16.SIY brain tumors injected with activated 2C T cells on day 7. Control mice (dashed lines) died within 2 weeks (median survival: RAG1−/− 14 days, C57BL/6 13 days) while mice treated with 2C T cells (solid lines) survived more than twice as long (RAG1−/− 35 days, C57BL/6 30 days). C57BL/6 mice are represented in gray, RAG1−/− mice in black. n = 3 per group. *P < 0.05 vs. respective control group without 2C treatment. b Immunostaining of 2C T cells in brain tumors 2 and 7 days after transfer. 2C T cells are labeled in left images with an idiotype specific antibody and Alexa-594 (red); middle images show DAPI counterstain (blue), right images are merged. Scale bar 200 μm. Images are representative of three mice. c Low or absent SIY/GFP expression by cancer cells cultured from brain tumors that recurred following adoptive T cell therapy (n = 4). GFP-negative B16-F10 cells (filled black curve) and freshly cultured B16.SIY cells (red curve) were included as negative and positive controls, respectively
T cells infiltrate and destroy tumors, but tumors recur and consist of ALV cells
C57BL/6 mice with established B16.SIY brain tumors were treated with activated 2C T cells on day seven and euthanized 2 or 7 days after transfer, or when mice required euthanasia from tumor symptoms. Immunostaining revealed low numbers of 2C T cells in the tumors 2 days after transfer, but a massive infiltration at day 7 (Fig. 1b) when tumors were necrotic and lacked clear margins. 2C T cells were absent from the brain at the point of relapse when tumors filled most of the hemisphere (data not shown). However, 2C T cells were still present in cervical lymph nodes, although in much smaller numbers than at 2 and 7 days after transfer (data not shown).
To test the hypothesis that late-stage tumors recurred from SIY-negative cancer cells no longer recognized by 2C T cells, tumors were collected and the cells analyzed for SIY-peptide expression using GFP as a marker for the SIY-GFP fusion protein (Fig. 1c). Tumor explant cells were compared to fresh B16.SIY cells (high GFP) and parental B16-F10 cells (GFP-negative). All cancer cells (melanin-positive) recovered from the brain tumors had very low or absent expression of SIY/GFP.
B16.SIY melanoma cancer cells are permissive to infection by MYXV-Red
To attempt a strategy that might facilitate elimination of antigen-loss variant cells, we took advantage of the finding that parental B16-F10 cells are permissive to MYXV infection [21]. Two methods confirmed that recombinant MYXV expressing tdTomato red protein (MYXV-Red) can also productively infect and replicate in B16.SIY cells. First, all virion stages were observed in infected B16.SIY cells indicating complete viral morphogenesis (Fig. 2a). Second, MYXV-Red exhibited similar growth kinetics in B16.SIY cells as in permissive B16-F10 and RK13 cells (Fig. 2b).
Fig. 2.

a Stages of MYXV-Red morphogenesis in infected B16.SIY cells. Cells were analyzed by TEM 24 h post-inoculation at MOI 10. Immature viral crescents (CR) form in cytoplasmic virus factories (VF). Crescents grow into immature virions (IV) with nucleoid (Nu) that condense to form mature virus (MV). b Multi-step growth kinetics of MYXV-Red in RK13, B16-F10, and B16.SIY cells. HPI = hrs post-inoculation at MOI 0.1. Error bars show mean ± SEM. Data are averaged from three independent experiments. c In vitro B16.SIY cell viability following infection with MYXV. Cells were mock-infected or infected at MOI 1 or 10, and metabolic activity was assessed over 72 h p.i. Viable cells are fluorescent. Data are representative of two independent experiments. Error bars show mean ± SEM, n = 3. *P < 0.05. d In vitro cytopathic effect of MYXV-Red infection (MOI 1) on confluent B16.SIY cells 48 h p.i. Panels one and three were imaged using phase; two and four used a Texas red filter
MYXV infection of cancer cells in vitro results in oncolysis
The metabolic activity of infected B16.SIY cells was measured to assess the oncolytic effects of MYXV (Fig. 2c). Mock-infected cells multiplied over 72 h with a corresponding increase in total metabolic activity. At a multiplicity of infection (MOI) of ten, viability was significantly decreased by 24 h post-inoculation (p.i.), and at a MOI of one, viability was significantly decreased by 48 h p.i. At 72 h p.i., the viability at both MOIs was low, but some cancer cells did survive. Cytopathic effects of MYXV-Red infection on B16.SIY cell monolayers were also evident (Fig. 2d). At a MOI of one, robust expression of tdTomato red protein was visible by 24 h p.i. and significant numbers of cells detached from the wells and died by 48 h p.i.
Intratumoral injection of MYXV-Red results in safe, tumor-specific infection
MYXV-Red was injected into B16.SIY brain tumors growing in both C57BL/6 and C57BL/6 RAG1−/− mice to assess safety and characterize the duration and spread of infection in vivo. Intratumoral (i.t.) injection of virus resulted in robust expression of tdTomato red protein in the tumors, but not in surrounding normal brain by 24 h p.i. (Fig. 3). Some cancer cells expressing tdTomato red protein could be found up to eight days post-injection, but expression peaked at 48–72 h p.i. (data not shown). Expression of tdTomato was not observed in tumors injected with UV-inactivated MYXV-Red or in contralateral tumors following i.t. injection of mice with bilateral tumors (data not shown). In all experiments, mice maintained body weight and behaved normally following injection of live or UV-inactivated virus into the brain. Peripheral tissues were collected one, three, or 5 days p.i. and homogenates were inoculated onto RK13 cells to assess spread of infectious virus outside the brain. Live virus was not detected in blood, spleen, lymph nodes, liver, kidney, lung, heart, intestine, or reproductive tract of any mice (n = 2 or 3 per group).
Fig. 3.
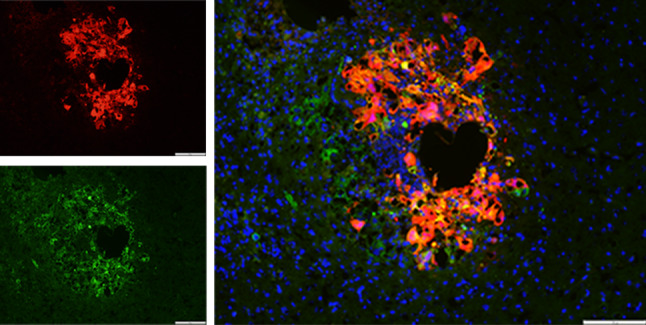
Tumor-selective infection and viral gene expression following intratumoral injection of MYXV-Red into B16.SIY brain tumors. tdTomato Red protein (left top image) is expressed in GFP+ cancer cells (left bottom image), but not in normal surrounding brain tissue 24 h following injection of virus into a four-day-old tumor. Right image shows the merged red, green and DAPI counterstained (blue) images. Scale bar 200 μm
Pre-incubation of cancer cells with virus improves survival of mice
We evaluated whether MXYV-Red could improve the survival of C57BL/6 RAG1−/− mice infused in the brain with B16.SIY cells that were briefly pre-incubated with MYXV-Red (MOI 1 for 1 h). Pre-incubated (100%) or mock-infected cells, or a 1:1 mixture of both (50%), were washed and injected into mice. Mice injected with either 50 or 100% pre-incubated cells lived slightly longer than mice injected with mock-infected cells, but there were no long-term survivors (18 and 20 vs. 15 days, respectively, P < 0.05, data not shown). This finding, combined with the reduction of tdTomato red expression by 8 days p.i., suggests that anti-viral mechanisms exist in vivo which hinder the replication and spread of virus.
Strong cellular immune response to MYXV-Red may promote clearance of virus
The striking difference in the oncolytic ability of MYXV in vitro and the modest survival advantage conferred in vivo may involve rapid immune clearance or inactivation of the virus. To characterize the innate immune response to the virus, wild-type and RAG1−/− mice with B16.SIY brain tumors were injected i.t. with MYXV-Red, UV-inactivated MYXV-Red or PBS. Brains were collected after 48 h, and sections were stained with hematoxylin and eosin (Fig. 4a). Foci of necrosis containing neutrophils and macrophages were present in tumors injected with MYXV-Red and UV-MYXV-Red, but not PBS, and were most concentrated at the site of virus injection (Fig. 4 and data not shown). There were no apparent differences between wild-type and RAG1−/− mice.
Fig. 4.

a H&E staining of B16.SIY brain tumors injected intratumorally with live or dead virus or PBS. Degenerative neutrophils are found in UV-MYXV-Red and MYXV-Red-injected brain tumors (right) but are absent from PBS-injected tumors (left) 48 h p.i. Wild-type and RAG1−/− hosts had similar responses. UV-MYXV-Red and MYXV-Red-injected tumors were indistinguishable, so a representative MYXV-Red image is shown. Scale bar 200 μm. b Innate immune response to intratumoral injection of live or dead virus or PBS into 5-day-established B16.SIY brain tumors in RAG1−/− mice. Brain tissue collected 48 h p.i. was stained for CD11b+ macrophages/microglia, GR-1+ neutrophils, and Ly49G2+ NK cells. Virus injection sites can be identified at the right side of the top right and middle right images where CD11b+ and GR-1+ cell infiltrates are most concentrated. UV-MYXV-Red and MYXV-Red-injected tumors were indistinguishable, so representative MYXV-Red images are shown (right column). Primary Abs detected by Alexa594 (red), DAPI stain (blue). Scale bar 200 μm
Immunostaining revealed CD11b+ cells located at the rim of all tumors including those injected with PBS (Fig. 4b). Most CD11b+ macrophages/microglia and GR-1+ neutrophils were present within tumors injected with live or UV-inactivated virus, but not PBS, especially at the virus injection site. Low numbers of Ly49G2+ NK cells were found in tumors injected with live or UV-inactivated virus, but not in PBS-injected tumors.
Brain cytokine response to MYXV-Red may restrict spread of virus in vivo
Cytokines produced by normal brain and immune cells may abort viral replication and prevent robust tumor infection. MYXV-Red, a purified preparation of uninfected lysed RK13 cells (mock), or PBS was injected i.t. into established B16.SIY brain tumors growing in C57BL/6 RAG1−/− mice. TNFα, IFNβ, and IFNα levels in brain homogenates were assessed 24 h p.i. (Fig. 5a). We observed significant increases in both IFNβ and TNFα, but not IFNα, in response to virus. The cytokine response was virus-specific because the mock preparation elicited a response similar to PBS. The cytokine response to MYXV-Red was also similar whether or not a tumor was present (data not shown).
Fig. 5.

a In vivo cytokine response to intratumoral injection of MYXV-Red. RAG1−/− mice with 8-day-old B16.SIY brain tumors were injected with MYX-Red, a purified preparation of uninfected, lysed RK13 cells (mock), or PBS. Supernatants of homogenized tumors and adjacent brain tissue were assessed for TNFα, IFNβ, and IFNα by ELISA 24 h p.i. Gray lines indicate baseline response defined as cytokine release following i.t. injection of PBS. Error bars show mean ± SEM, n = 3. *P < 0.05. Data from two separate experiments were combined. Two brains were pooled per sample; samples normalized by tissue weight. b Survival of tumor-bearing mice treated with MYXV-Red, 2C T cell adoptive therapy and neutralizing antibodies against IFNβ. RAG1−/− mice with established B16.SIY brain tumors received i.t. injections of MYXV-Red and anti-IFNβ Abs on day 5, activated 2C T cell transfers on day 6, and daily i.p. injections of anti-IFNβ Abs for 4 days starting at virus treatment. Mice that received the triple combination treatment lived significantly longer than mice that received only MYXV-Red and 2C T cells, *P < 0.05. Data were pooled from two separate experiments
Treatment with neutralizing antibodies (Abs) against IFNβ is safe and improves survival of MYXV-Red-treated mice when combined with adoptive T cell therapy
We evaluated the safety and efficacy of treating tumor-bearing mice with virus and neutralizing Abs against IFNβ. C57BL/6 RAG1−/− mice with established B16.SIY brain tumors were injected i.t. with MYXV-Red and polyclonal neutralizing Abs against IFNβ on day 5. Mice were given i.v. injections of activated 2C T cells on day 6 and received daily intraperitoneal (i.p.) injections of anti-IFNβ Abs for 4 days after virus treatment. At 4 days p.i., strong expression of tdTomato Red protein was observed and remained limited to the tumor (data not shown). Mice that received the triple combination treatment (Fig. 5b) lived three times longer than untreated mice (median survival 39 vs. 13 days, respectively), and significantly longer than mice receiving only MYXV-Red and 2C T cells (39 vs. 32 days, P < 0.05).
Rapamycin treatment partially reverses IFNβ suppression of MYXV-Red in vitro and improves survival of tumor-bearing mice
We evaluated the effect of rapamycin on cytokine suppression of MYXV-Red infection of B16.SIY cells in vitro (Fig. 6a). Cells were pre-treated with rapamycin, TNFα and/or IFNβ for 6 h prior to infection with MYXV-Red (MOI 1) and analyzed for tdTomato red expression at 72 h p.i. IFNβ pre-treatment significantly inhibited the expression of tdTomato red for at least 3 days p.i., while TNFα had no effect, alone or in combination with IFNβ (Fig. 6a and data not shown). When cells were pretreated with both rapamycin and IFNβ, tdTomato red expression was partially rescued.
Fig. 6.

a In vitro effect of rapamycin on cytokine suppression of MYXV-Red gene expression in cancer cells. B16.SIY cells were pre-incubated with IFNβ, TNFα, and rapamycin for 6 h prior to inoculation with MYXV-Red at MOI 1. Cells were fixed and analyzed for tdTomato red expression by flow at 72 h p.i. Error bars show mean ± SEM, n = 3. P < 0.05. Data are representative of two independent experiments. b Survival of RAG1−/− mice with B16.SIY brain tumors treated with intratumoral injections of MYXV-Red on day 5 and i.p. rapamycin injections every other day for six total treatments. Median survival 13.5 days PBS, 14 days MYXV-Red, 15.5 days PBS + rapa, 21.5 days MYXV-Red + rapa. *P < 0.05 MYXV-Red + rapa compared to all other groups. c Survival of RAG1−/− mice with B16.SIY brain tumors that received triple combination treatment of i.t. MYXV-Red on day 5, rapamycin (six i.p. treatments every other day starting day 5), and adoptive 2C T cell transfers on day 6. Median survival PBS + 2C = 31 days, MYXV-Red + 2C = 32 days, PBS + rapa + 2C = 31 days, MYXV-Red + rapa + 2C = 36.5 days. *P < 0.05, MYXV-Red + rapa + 2C vs. PBS + 2C. Data were pooled from three separate experiments
To test whether rapamycin could improve the survival of C57BL/6 RAG1−/− mice treated with MYXV-Red, virus was injected into B16.SIY brain tumors on day five and mice received i.p. rapamycin injections every other day for six total treatments. Mice that received both MYXV-Red and rapamycin lived significantly longer (P < 0.05) than mice receiving either single treatment (Fig. 6b). The efficacy of a triple combination treatment of MYXV-Red, rapamycin, and adoptive T cell transfer was uncertain due to the potential cytotoxicity of rapamycin on T cells. Tumor-bearing mice received i.t. MYXV-Red injections on day 5, 2C T cell adoptive transfers on day 6, and rapamycin every other day for six treatments total. Mice receiving the triple combination treatment lived significantly longer (P < 0.05) than mice receiving only T cells (Fig. 6c), and the combination of virus and rapamycin was required for the survival benefit.
Discussion
T cells are exceedingly proficient at antigen-specific killing, but successful treatment of cancer requires elimination of both antigen-expressing and ALV cancer cells. We demonstrate the feasibility of combining adoptive T cell therapy with concurrent administration of an oncolytic virus. Tumor infection by MYXV could complement adoptive therapy by at least three potential mechanisms. First, MYXV could directly kill melanoma cells (including ALV cells) without harming normal brain parenchymal cells. Second, tumor infection could lead to local production of anti-tumor cytokines (e.g. TNFα) and recruitment of immune cells (macrophages, NK cells) to aid in tumor clearance. Third, cancer cells killed by MYXV could be a source of tumor peptides to be cross-presented by tumor stromal cells to further enhance a cytotoxic T cell response [33, 34].
In vitro 2C T cell killing of B16.SIY melanoma cells is surprisingly inefficient, presumably due to low MHC I levels and PD-L1 expression [31]. Impressively, activated 2C T cells almost completely eliminated B16.SIY brain tumors in vivo. However, tumors arising from ALV cancer cells were the cause of death in all treated mice. IFNγ produced by activated T cells may have upregulated class I MHC or co-stimulatory molecules on the tumor cells resulting in the observed initial killing. Homeostatic proliferation of the transferred cells [35–37] was probably not critical because RAG1−/− mice did not live significantly longer than wild-type mice.
We observed productive infection, spread, and oncolysis by MYXV in vitro, but more limited oncolytic effects in vivo in a syngeneic brain tumor model. Tumor-specific viral expression of tdTomato red protein peaked at 48–72 h p.i., and only a few fluorescent cells were observed at 1 week. Tumor cells near the virus injection site were more likely to express tdTomato red. Macrophages, microglia, and neutrophils may have participated indirectly in viral clearance via cytokine production. We observed similar infectivity and innate immune cell recruitment in wild-type and RAG1−/− mice, indicating a minimal role of the adaptive immune response in viral clearance.
Several lines of evidence support the hypothesis that local cytokines impaired the spread of infection. Only a small number of infectious virions were recovered from most brain tumors weeks after injection (data not shown), and virus was not detected in any peripheral tissues. Exogenous IFNβ induced an anti-viral state in B16.SIY melanoma cells in vitro, consistent with its role as the major restrictive cytokine of MYXV replication in primary mouse cells [38]. IFNβ and TNFα produce a synergistic anti-viral state in human cells [39], but we did not observe an effect of TNFα on mouse melanoma cells. IFNβ production by MYXV-Red-infected B16.SIY cells in vitro was not detectable (data not shown), so we attribute the limitation in oncolytic effects in vivo to IFNβ produced by surrounding brain or immune cells. A small but significant survival benefit was achieved by the addition of anti-IFNβ antibodies to treatment with MYXV-Red and T cells. Furthermore, intracranial infusion of neutralizing antibodies against IFNβ was not associated with infection of normal brain cells. The role of Type I interferon in anti-cancer immune responses is complex [40, 41], but cancers with defective Type I interferon signaling [9, 42–44] may be ideal targets for MYXV therapy, eliminating the need for IFN neutralization.
Rapamycin activates Akt through an mTORC1-dependent pathway, increasing MYXV tropism for some nonpermissive cancer cells [45]. Rapamycin can also inhibit Type I interferon production [22, 29]. Rapamycin had no effect on MYXV-Red infection of B16.SIY cells in vitro, but significantly increased the percentage of tdTomato red positive cells following IFNβ treatment. Treatment of tumor-bearing mice with MYXV-Red and rapamycin together improved survival by a few days, although the single largest benefit in this aggressive tumor model was adoptive T cell therapy.
Rapamycin is best known for its immunosuppressive effects and can prevent rejection of organ transplants in part by inhibiting T cell proliferation and inducing anergy. It is now appreciated that rapamycin can also have immunostimulatory effects on CD8+ T cells, and treatment of mice with rapamycin promotes the generation and function of memory CD8+ T cells [46]. It is possible that rapamycin could improve vaccination strategies for both infection and cancer. In our model, we show that multiple days of rapamycin treatment does not restrict the survival benefit conferred by the adoptively transferred T cells. Furthermore, rapamycin treatment improved the overall survival of mice treated with both MYXV-Red and T cells, supporting the feasibility of combining immunotherapy, virotherapy, and rapamycin treatment. Myxoma virus could be engineered to transiently express tumor antigens or cytokines in cancer cells to further enhance antigen-specific T cell responses.
Acknowledgments
We thank the anonymous donor that funded this research, Thomas Gajewski and Maciej Lesniak for the B16.SIY cells, and the Flow Cytometry Facility at University of Illinois at Urbana–Champaign.
References
- 1.Sampson JH, Carter JH, Jr, Friedman AH, Seigler HF. Demographics, prognosis, and therapy in 702 patients with brain metastases from malignant melanoma. J Neurosurg. 1998;88(1):11–20. doi: 10.3171/jns.1998.88.1.0011. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg SA, Dudley ME. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Curr Opin Immunol. 2009;21(2):233–240. doi: 10.1016/j.coi.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, Robbins PF, Huang J, Citrin DE, Leitman SF, Wunderlich J, Restifo NP, Thomasian A, Downey SG, Smith FO, Klapper J, Morton K, Laurencot C, White DE, Rosenberg SA. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26(32):5233–5239. doi: 10.1200/JCO.2008.16.5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hong JJ, Rosenberg SA, Dudley ME, Yang JC, White DE, Butman JA, Sherry RM (2010) Successful Treatment of Melanoma Brain Metastases With Adoptive Cell Therapy. Clin Cancer Res. doi:10.1158/1078-0432.CCR-10-1507 [DOI] [PMC free article] [PubMed]
- 5.Jager E, Ringhoffer M, Karbach J, Arand M, Oesch F, Knuth A. Inverse relationship of melanocyte differentiation antigen expression in melanoma tissues and CD8 + cytotoxic-T-cell responses: evidence for immunoselection of antigen-loss variants in vivo. Int J Cancer. 1996;66(4):470–476. doi: 10.1002/(SICI)1097-0215(19960516)66:4<470::AID-IJC10>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 6.Riker A, Cormier J, Panelli M, Kammula U, Wang E, Abati A, Fetsch P, Lee KH, Steinberg S, Rosenberg S, Marincola F. Immune selection after antigen-specific immunotherapy of melanoma. Surgery. 1999;126(2):112–120. doi: 10.1016/S0039-6060(99)70143-1. [DOI] [PubMed] [Google Scholar]
- 7.Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, Greenberg PD. Adoptive T cell therapy using antigen-specific CD8 + T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci USA. 2002;99(25):16168–16173. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldberger O, Volovitz I, Machlenkin A, Vadai E, Tzehoval E, Eisenbach L. Exuberated numbers of tumor-specific T cells result in tumor escape. Cancer Res. 2008;68(9):3450–3457. doi: 10.1158/0008-5472.CAN-07-5006. [DOI] [PubMed] [Google Scholar]
- 9.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6(11):836–848. doi: 10.1038/nri1961. [DOI] [PubMed] [Google Scholar]
- 10.Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Bigner DD. Tumor-specific immunotherapy targeting the EGFRvIII mutation in patients with malignant glioma. Semin Immunol. 2008;20(5):267–275. doi: 10.1016/j.smim.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas DL, Kim M, Bowerman NA, Narayanan S, Kranz DM, Schreiber H, Roy EJ. Recurrence of intracranial tumors following adoptive T cell therapy can be prevented by direct and indirect killing aided by high levels of tumor antigen cross-presented on stromal cells. J Immunol. 2009;183(3):1828–1837. doi: 10.4049/jimmunol.0802322. [DOI] [PubMed] [Google Scholar]
- 12.Diaz RM, Galivo F, Kottke T, Wongthida P, Qiao J, Thompson J, Valdes M, Barber G, Vile RG. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007;67(6):2840–2848. doi: 10.1158/0008-5472.CAN-06-3974. [DOI] [PubMed] [Google Scholar]
- 13.Qiao J, Wang H, Kottke T, Diaz RM, Willmon C, Hudacek A, Thompson J, Parato K, Bell J, Naik J, Chester J, Selby P, Harrington K, Melcher A, Vile RG. Loading of oncolytic vesicular stomatitis virus onto antigen-specific T cells enhances the efficacy of adoptive T-cell therapy of tumors. Gene Ther. 2008;15(8):604–616. doi: 10.1038/sj.gt.3303098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang YQ, Tsai YC, Monie A, Wu TC, Hung CF. Enhancing the therapeutic effect against ovarian cancer through a combination of viral oncolysis and antigen-specific immunotherapy. Mol Ther. 2010;18(4):692–699. doi: 10.1038/mt.2009.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sypula WFJ, Ma Y, Bell J, McFadden G. Myxoma virus tropism in human tumor cells. Gene Ther Mol Biol. 2004;8:103–114. [Google Scholar]
- 16.Lun X, Yang W, Alain T, Shi ZQ, Muzik H, Barrett JW, McFadden G, Bell J, Hamilton MG, Senger DL, Forsyth PA. Myxoma virus is a novel oncolytic virus with significant antitumor activity against experimental human gliomas. Cancer Res. 2005;65(21):9982–9990. doi: 10.1158/0008-5472.CAN-05-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lun XQ, Zhou H, Alain T, Sun B, Wang L, Barrett JW, Stanford MM, McFadden G, Bell J, Senger DL, Forsyth PA. Targeting human medulloblastoma: oncolytic virotherapy with myxoma virus is enhanced by rapamycin. Cancer Res. 2007;67(18):8818–8827. doi: 10.1158/0008-5472.CAN-07-1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woo Y, Kelly KJ, Stanford MM, Galanis C, Chun YS, Fong Y, McFadden G. Myxoma virus is oncolytic for human pancreatic adenocarcinoma cells. Ann Surg Oncol. 2008;15(8):2329–2335. doi: 10.1245/s10434-008-9924-z. [DOI] [PubMed] [Google Scholar]
- 19.Wu Y, Lun X, Zhou H, Wang L, Sun B, Bell JC, Barrett JW, McFadden G, Biegel JA, Senger DL, Forsyth PA. Oncolytic efficacy of recombinant vesicular stomatitis virus and myxoma virus in experimental models of rhabdoid tumors. Clin Cancer Res. 2008;14(4):1218–1227. doi: 10.1158/1078-0432.CCR-07-1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim M, Madlambayan GJ, Rahman MM, Smallwood SE, Meacham AM, Hosaka K, Scott EW, Cogle CR, McFadden G. Myxoma virus targets primary human leukemic stem and progenitor cells while sparing normal hematopoietic stem and progenitor cells. Leukemia. 2009;23(12):2313–2317. doi: 10.1038/leu.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stanford MM, Shaban M, Barrett JW, Werden SJ, Gilbert PA, Bondy-Denomy J, Mackenzie L, Graham KC, Chambers AF, McFadden G. Myxoma virus oncolysis of primary and metastatic B16F10 mouse tumors in vivo. Mol Ther. 2008;16(1):52–59. doi: 10.1038/sj.mt.6300348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lun X, Alain T, Zemp FJ, Zhou H, Rahman MM, Hamilton MG, McFadden G, Bell J, Senger DL, Forsyth PA. Myxoma virus virotherapy for glioma in immunocompetent animal models: optimizing administration routes and synergy with rapamycin. Cancer Res. 2010;70(2):598–608. doi: 10.1158/0008-5472.CAN-09-1510. [DOI] [PubMed] [Google Scholar]
- 23.McFadden G. Poxvirus tropism. Nat Rev Microbiol. 2005;3(3):201–213. doi: 10.1038/nrmicro1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jackson EW, Dorn CR, Saito JK, McKercher DG. Absence of serological evidence of myxoma virus infection in humans exposed during an outbreak of myxomatosis. Nature. 1966;211(5046):313–314. doi: 10.1038/211313a0. [DOI] [PubMed] [Google Scholar]
- 25.Homicsko K, Lukashev A, Iggo RD. RAD001 (everolimus) improves the efficacy of replicating adenoviruses that target colon cancer. Cancer Res. 2005;65(15):6882–6890. doi: 10.1158/0008-5472.CAN-05-0309. [DOI] [PubMed] [Google Scholar]
- 26.Alonso MM, Gomez-Manzano C, Jiang H, Bekele NB, Piao Y, Yung WK, Alemany R, Fueyo J. Combination of the oncolytic adenovirus ICOVIR-5 with chemotherapy provides enhanced anti-glioma effect in vivo. Cancer Gene Ther. 2007;14(8):756–761. doi: 10.1038/sj.cgt.7701067. [DOI] [PubMed] [Google Scholar]
- 27.Alonso MM, Jiang H, Yokoyama T, Xu J, Bekele NB, Lang FF, Kondo S, Gomez-Manzano C, Fueyo J. Delta-24-RGD in combination with RAD001 induces enhanced anti-glioma effect via autophagic cell death. Mol Ther. 2008;16(3):487–493. doi: 10.1038/sj.mt.6300400. [DOI] [PubMed] [Google Scholar]
- 28.Lun XQ, Jang JH, Tang N, Deng H, Head R, Bell JC, Stojdl DF, Nutt CL, Senger DL, Forsyth PA, McCart JA. Efficacy of systemically administered oncolytic vaccinia virotherapy for malignant gliomas is enhanced by combination therapy with rapamycin or cyclophosphamide. Clin Cancer Res. 2009;15(8):2777–2788. doi: 10.1158/1078-0432.CCR-08-2342. [DOI] [PubMed] [Google Scholar]
- 29.Alain T, Lun X, Martineau Y, Sean P, Pulendran B, Petroulakis E, Zemp FJ, Lemay CG, Roy D, Bell JC, Thomas G, Kozma SC, Forsyth PA, Costa-Mattioli M, Sonenberg N. Vesicular stomatitis virus oncolysis is potentiated by impairing mTORC1-dependent type I IFN production. Proc Natl Acad Sci USA. 2010;107(4):1576–1581. doi: 10.1073/pnas.0912344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lun X, Chan J, Zhou H, Sun B, Kelly JJ, Stechishin OO, Bell JC, Parato K, Hu K, Vaillant D, Wang J, Liu TC, Breitbach C, Kirn D, Senger DL, Forsyth PA. Efficacy and safety/toxicity study of recombinant vaccinia virus JX-594 in two immunocompetent animal models of glioma. Mol Ther. 2010;18(11):1927–1936. doi: 10.1038/mt.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, Gajewski TF. PD-L1/B7H–1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8 + T cells. Cancer Res. 2004;64(3):1140–1145. doi: 10.1158/0008-5472.CAN-03-3259. [DOI] [PubMed] [Google Scholar]
- 32.Liu J, Wennier S, Reinhard M, Roy E, MacNeill A, McFadden G. Myxoma virus expressing interleukin-15 fails to cause lethal myxomatosis in European rabbits. J Virol. 2009;83(11):5933–5938. doi: 10.1128/JVI.00204-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang B, Bowerman NA, Salama JK, Schmidt H, Spiotto MT, Schietinger A, Yu P, Fu YX, Weichselbaum RR, Rowley DA, Kranz DM, Schreiber H. Induced sensitization of tumor stroma leads to eradication of established cancer by T cells. J Exp Med. 2007;204(1):49–55. doi: 10.1084/jem.20062056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calzascia T, Di Berardino-Besson W, Wilmotte R, Masson F, de Tribolet N, Dietrich PY, Walker PR. Cutting edge: cross-presentation as a mechanism for efficient recruitment of tumor-specific CTL to the brain. J Immunol. 2003;171(5):2187–2191. doi: 10.4049/jimmunol.171.5.2187. [DOI] [PubMed] [Google Scholar]
- 35.Kline J, Brown IE, Zha YY, Blank C, Strickler J, Wouters H, Zhang L, Gajewski TF. Homeostatic proliferation plus regulatory T-cell depletion promotes potent rejection of B16 melanoma. Clin Cancer Res. 2008;14(10):3156–3167. doi: 10.1158/1078-0432.CCR-07-4696. [DOI] [PubMed] [Google Scholar]
- 36.Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity. 2008;29(6):848–862. doi: 10.1016/j.immuni.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 37.Ge Q, Hu H, Eisen HN, Chen J. Different contributions of thymopoiesis and homeostasis-driven proliferation to the reconstitution of naive and memory T cell compartments. Proc Natl Acad Sci USA. 2002;99(5):2989–2994. doi: 10.1073/pnas.052714099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang F, Ma Y, Barrett JW, Gao X, Loh J, Barton E, Virgin HW, McFadden G. Disruption of Erk-dependent type I interferon induction breaks the myxoma virus species barrier. Nat Immunol. 2004;5(12):1266–1274. doi: 10.1038/ni1132. [DOI] [PubMed] [Google Scholar]
- 39.Bartee E, Mohamed MR, Lopez MC, Baker HV, McFadden G. The addition of tumor necrosis factor plus beta interferon induces a novel synergistic antiviral state against poxviruses in primary human fibroblasts. J Virol. 2009;83(2):498–511. doi: 10.1128/JVI.01376-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fujita M, Scheurer ME, Decker SA, McDonald HA, Kohanbash G, Kastenhuber ER, Kato H, Bondy ML, Ohlfest JR, Okada H. Role of type 1 IFNs in antiglioma immunosurveillance–using mouse studies to guide examination of novel prognostic markers in humans. Clin Cancer Res. 2010;16(13):3409–3419. doi: 10.1158/1078-0432.CCR-10-0644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Willmon CL, Saloura V, Fridlender ZG, Wongthida P, Diaz RM, Thompson J, Kottke T, Federspiel M, Barber G, Albelda SM, Vile RG. Expression of IFN-beta enhances both efficacy and safety of oncolytic vesicular stomatitis virus for therapy of mesothelioma. Cancer Res. 2009;69(19):7713–7720. doi: 10.1158/0008-5472.CAN-09-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu B, Grander D, Sangfelt O, Einhorn S. Primary leukemia cells resistant to alpha-interferon in vitro are defective in the activation of the DNA-binding factor interferon-stimulated gene factor 3. Blood. 1994;84(6):1942–1949. [PubMed] [Google Scholar]
- 43.Wong LH, Krauer KG, Hatzinisiriou I, Estcourt MJ, Hersey P, Tam ND, Edmondson S, Devenish RJ, Ralph SJ. Interferon-resistant human melanoma cells are deficient in ISGF3 components, STAT1, STAT2, and p48-ISGF3gamma. J Biol Chem. 1997;272(45):28779–28785. doi: 10.1074/jbc.272.45.28779. [DOI] [PubMed] [Google Scholar]
- 44.Dunn GP, Sheehan KC, Old LJ, Schreiber RD. IFN unresponsiveness in LNCaP cells due to the lack of JAK1 gene expression. Cancer Res. 2005;65(8):3447–3453. doi: 10.1158/0008-5472.CAN-04-4316. [DOI] [PubMed] [Google Scholar]
- 45.Stanford MM, Barrett JW, Nazarian SH, Werden S, McFadden G. Oncolytic virotherapy synergism with signaling inhibitors: Rapamycin increases myxoma virus tropism for human tumor cells. J Virol. 2007;81(3):1251–1260. doi: 10.1128/JVI.01408-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460(7251):108–112. doi: 10.1038/nature08155. [DOI] [PMC free article] [PubMed] [Google Scholar]