

Abstract
Cerebral edema is a common complication following moderate and severe traumatic brain injury (TBI), and a significant risk factor for development of neuronal death and deterioration of neurological outcome. To this date, medical approaches that effectively alleviate cerebral edema and neuronal death after TBI are not available. Glucagon-like peptide-1 (GLP-1) has anti-inflammatory properties on cerebral endothelium and exerts neuroprotective effects. Here, we investigated the effects of GLP-1 on secondary injury after moderate and severe TBI. Male Sprague Dawley rats were subjected either to TBI by Controlled Cortical Impact (CCI) or sham surgery. After surgery, vehicle or a GLP-1 analogue, Liraglutide, were administered subcutaneously twice daily for two days. Treatment with Liraglutide (200 μg/kg) significantly reduced cerebral edema in pericontusional regions and improved sensorimotor function 48 hours after CCI. The integrity of the blood-brain barrier was markedly preserved in Liraglutide treated animals, as determined by cerebral extravasation of Evans blue conjugated albumin. Furthermore, Liraglutide reduced cortical tissue loss, but did not affect tissue loss and delayed neuronal death in the thalamus on day 7 post injury. Together, our data suggest that the GLP-1 pathway might be a promising target in the therapy of cerebral edema and cortical neuronal injury after moderate and severe TBI.
Introduction
Cerebral edema is a common life-threatening complication after traumatic brain injury (TBI) and represents a serious clinical problem due to the lack of specific treatments [1]. Cerebral edema promotes elevation of the intracranial pressure, and subsequently limits cerebral blood flow and tissue oxygenation leading to neuronal death and poor clinical prognosis [2]. The pathophysiological mechanisms behind the development of cerebral edema are multifaceted. It is generally agreed that both vasogenic and cytotoxic components contribute to edema in patients with moderate to severe TBI [3]. Vasogenic edema is caused by an inflammatory cascade with increased permeability of the blood-brain barrier (BBB) and movement of molecules and water to the brain interstitium [4].
Glucagon-like peptide-1 (GLP-1) is a gut-derived incretin hormone known for its effects on blood glucose homeostasis. GLP-1 interacts with a specific G-protein-coupled GLP-1 receptor (GLP-1R) that, besides being expressed on pancreatic cells, is also found on cerebral endothelial cells and various cells throughout the brain [5]. The half-life of endogenous GLP-1 is 1–2 minutes. Therefore, a long-acting GLP-1 analogue, Liraglutide, is used in the treatment of type 2 diabetes. Liraglutide binds selectively to the GLP-1R, and avoids being proteolyticly degraded by dipeptidyl peptidase-4 through binding to serum albumin [6]. Liraglutide has been reported to cross the BBB, and to have potent anti-inflammatory effects on cerebral endothelial cells and astrocytes [7–9]. Additionally, GLP-1R stimulation has been suggested to mediate neuroprotective effects in experimental stroke [10–13], Parkinson’s disease, and Alzheimer’s disease [14,15] and to promote neurogenesis [16].
The purpose of this study was to investigate whether treatment with Liraglutide improves neurological outcome in a rat model of moderate and severe TBI. We hypothesized that Liraglutide may attenuate BBB disruption and reduce neuronal loss after TBI.
Materials and Methods
Ethics Statement
All animal procedures were approved by the Malmö-Lund ethical committee (ethical permit number: M 19–12) and reported according to the ARRIVE guidelines. Adult male Sprague-Dawley rats (300–400 g, Charles River) were used. The rats were housed in standard laboratory cages (2 rats per cage) and in a reverse light-cycle. Behavioral tests were performed in the dark period when the rats were active. The rats had free access to food and water, and were kept in this environment for a minimum of 5 days before the experiments were performed.
Experimental design
Three separate experiments were conducted. A total of 79 rats were included in the experiments (for experimental outline see Fig. 1).
Fig 1. Experimental outline.
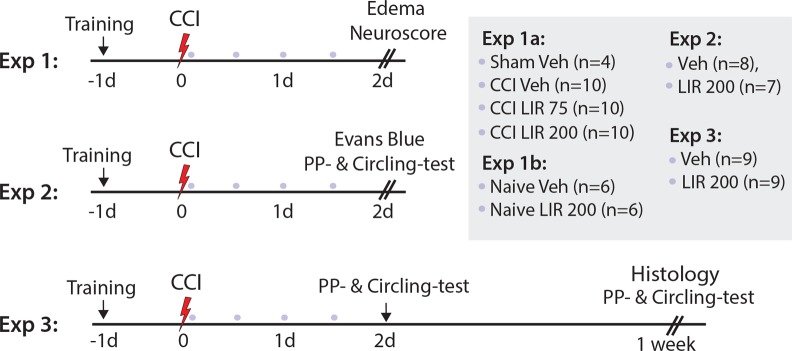
Flow diagram of the three experiments. The colored dots indicate treatment with a subcutaneous dose of Liraglutide 75 μg/kg (LIR75), Liraglutide 200 μg/kg (LIR200) or vehicle. Abbreviations: Controlled cortical impact (CCI), days (d), experiment (exp), Paw-placement test (PP-test).
For all experiments the first dose was given 10 min after Controlled Cortical Impact (CCI), and thereafter at 12, 24 and 36 hours after CCI. Investigators were blinded to injury status and pharmacological treatment of the rats.
Experiment 1.a: Thirty rats were subjected to CCI and randomized to subcutaneous treatment with either Liraglutide at a dose of 75 μg/kg (n = 10) or 200 μg/kg (n = 10), or vehicle (phosphate buffered saline, n = 10). Four rats were sham-operated. Neuroscore and brain edema were assessed 48 hours after CCI. Experiment 1.b: To assess the effect of Liraglutide on normal brain water content naive rats were injected with vehicle (n = 6) and Liraglutide 200 μg/kg (n = 6), and edema and Neuroscore was assessed 48 hours after CCI.
Experiment 2: The effect of vehicle (n = 8) and Liraglutide treatment (200 μg/kg; n = 7) twice daily on BBB integrity was tested by Evans blue extravasation 48 hours after CCI, together with circling test and paw-placement test.
Experiment 3: The effect of vehicle (n = 9) and Liraglutide 200 μg/kg (n = 9) on cortical lesion size and delayed thalamic neuronal death was determined 7 days after CCI. Results from the circling test and paw-placement test were recorded on day 2 and 7 post CCI.
Controlled cortical impact (CCI)
Experimental TBI is induced by CCI, a clinically relevant model of moderate and severe TBI [17]. The rats were anesthetized with sodium pentobarbital (60 mg/kg, intraperitoneal injection) and positioned in a stereotactic frame. This specific anesthetic regimen has minimal effects on cerebral metabolism and cerebral blood glucose concentration [18]. Following a midline scalp incision, a 5 mm craniotomy was performed centrally over the left parietal bone. The bone flap was removed and the dura was kept intact. A cortical impact of approximately 200 milliseconds (msec) was induced with a 5 mm pneumatically driven piston at a velocity of 4 m/s and a penetration depth of 3 mm as previously described [19,20]. Subsequently, the scalp incision was closed, and the rats were allowed to recover. Rectal temperature was recorded and maintained at 37±1°C by a heating pad throughout the surgical procedure and for a minimum of 3 hours post-injury until the rats were fully active. Sham-operated rats were exposed to the same surgical and peri-surgical procedures except the cortical impact.
Evaluation of neurological function
Composite Neuroscore
Sensorimotor function was evaluated by a composite Neuroscore adapted from Hunter et al. [21]. The composite Neuroscore consists of eight individual tests, each rated on an integral scale: general motility when placed on a platform (maximum 3 points), circling test (maximum 3), paw placement back to bench when slid over an edge (maximum 4), forepaw reaching (maximum 2), the rats ability to lift itself up when hanged by their forepaws on a horizontal bar (maximum 3), the ability to climb a 45° inclined plane without foot slips (maximum 3), contralateral reflex when lifted by the tail (maximum 1) and grip strength (maximum 2). A composite Neuroscore was calculated, giving a maximum score of 21 points. Rats were tested the day before injury to ensure that they were all able to obtain the maximum score in each subtest.
Circling test
This test assesses the function of front and hind limbs. It is recorded whether the rat turns towards the paretic side spontaneously or not, when lifted by its tail, and when pulled along the floor by its tail. For each sub-test, the rats got one point when showing symmetrical behavior [21,22].
Paw-placement test
The paw placement test was performed in order to assess tactile and proprioceptive sensory function [23,24]. The rats were placed on a tabletop and moved laterally towards the edge, until the limbs closest to the edge lost contact with the table surface. The deficit for each fore- and hindpaw was then evaluated as follows: When a paw was quickly moved back to the surface, the rat was assigned 1 point. If a paw was not moved back to the surface but inwards towards the side of the table edge and supinated, it scored 0.5 points. A persistent free-hanging limb was assigned 0 points. The score for each limb was combined to give a maximum score of 4 points. During the tests, care was taken that the head was straight at all times, and that the rats were not able to see their free-hanging limbs.
Brain Water Content
Brain water content was evaluated using the wet/dry-weight method, a reliable measure of posttraumatic brain edema [25]. Forty-eight hours after CCI, rats were anesthetized with 4% isoflurane (Baxter, Miami, FL) and decapitated. The brains were quickly removed from the cranium and cut into 6 mm coronal sections including the lesioned area. The tissue slab was quickly divided into ipsi- and contra-lateral cortex, hippocampus and thalamus. The samples for each region were placed on aluminum foil and weighed to obtain wet-weight. Samples were then dried at 90°C for 72 hours and reweighed for dry-weight. The percentage of water content was calculated using the following equation: ([wet weight—dry weight]/wet weight) x 100.
Blood-Brain Barrier integrity
The degree of BBB disruption was determined using the Evans blue dye extravasation as described previously [26]. Rats were anesthetized with pentobarbital as described above. Two percent Evans blue dissolved in phosphate-buffered saline (PBS) was injected intravenously (5 ml/kg). After one hour, the rats were perfused transcardially with a minimum of 400 ml saline through the left ventricle until colorless perfusion fluid was obtained from the right atrium. The brain was removed and sectioned into left and right hemispheres. The cerebellum was collected as an internal control. Each sample was then placed in 2 mL formamide and incubated at room temperature for 48 hours. The amount of Evans blue in the supernatant was measured by absorbance of the supernatant using a double beam spectrophotometer (U2800 Hitachi, Japan). A linear standard curve was made based on Evans blue external standards (0.47 to 37.5 ng/ml). The tissue was then dried at 95°C for 5 days. Based on the standard curve the amount of Evans blue was quantified as μg of Evans blue per mg dry weight of brain tissue.
Histochemical processing
Rats were anesthetized with isoflurane and transcardially perfused with PBS followed by 4% paraformaldehyde (PFA) in PBS. The brains were incubated over night in 4% PFA and thereafter transferred to a 25% sucrose solution for a minimum of 48 hours. The brains were cut into 40 μm coronal sections using a sliding microtome and one section was collected every 1 mm for measurement of lesion size.
Evaluation of neuronal tissue loss
The lesion size was assessed at day 7, at which point it is considered to be matured [27]. Free-floating sections were washed three times in PBS and quenched (3% H2O2 and 10% methanol) for 15 minutes. After blocking in 5% normal donkey serum and 0.25% Triton X-100 in PBS for 60 minutes, the sections were incubated in blocking solution containing a monoclonal mouse anti-NeuN antibody (1:1500, Millipore, Hampshire, UK) at 4°C overnight. After primary antibody incubation, the sections were rinsed and incubated with a biotinylated secondary donkey anti-mouse antibody (1:400, Vector Laboratories, USA) for 60 minutes in blocking solution. Visualization was done using a Vectorstain ABC kit (PK-6100, Vectorlab) and 3,3-diaminobenzidine/H2O2 (DabSafe, Saveen Werner, Sweden). Stained sections were mounted on glass. The slices were scanned, and the cortical, thalamic and hemispherical lesion volumes were determined using ImageJ by subtracting the area of healthy tissue on the contralateral side from that of the ipsilateral side for each section [28].
Evaluation of delayed neuronal death
Floating sections were stained with Fluoro-Jade C (FJC) in order to evaluate degenerating neurons [29]. Sections corresponding to 2.8 mm posterior of bregma were mounted and incubated with 1% NaOH in 80% alcohol for 5 min, 2 min in 70% alcohol, and 2 min in distilled water. The sections were then incubated in a 0.06% potassium permanganate solution for 10 min with agitation, later washed, and subsequently incubated in a solution of 0.0005% FJC (Millipore) in 0.1% acetic acid for 20 min. Finally, the slides were dried and coverslipped. Images were obtained on a LSM510 confocal microscope (Zeiss, Germany). A blinded researcher captured 5 images within the thalamic area guided by the DAPI-stain. For analysis ImageJ software with Point Picker plugin was used. Data are presented as an average of the 5 areas of interest.
Blood glucose
Blood glucose levels were measured with a Contour glucometer (Bayer, Germany). An average value of two consecutive measurements was recorded.
Statistical analysis
Analyses were performed using Prism Graph pad 6. All data are presented as mean ± SEM if nothing else is stated. P<0.05 was defined as statistically significant. Statistical differences between two groups were determined using students T-test. Neuroscore data were analyzed using a non-parametric Kruskal-Wallis test followed by Dunn’s test for multiple comparisons. Effects on subregional water content were tested using a two-way analysis of variance (ANOVA) test with treatment group and brain region as main variables, followed by Tukey’s post hoc test for multiple comparisons. Effects of treatment dose on total water content (experiment 1), and differences in temperature, blood glucose and body weight between treatment-groups, were analyzed with one-way ANOVA followed by Dunnet’s post-hoc test for each time point. Group sizes were decided before conducting any experiments based on previous investigations [8,26,30].
Results
Effect on physiological parameters
Overall, there were no effects of Liraglutide treatment on body temperature (Table 1). Mean blood glucose in sham animals ranged from 5.6–6.0 mmol/l throughout the experiment. At the 30 minute timepoint both treatment groups were slightly hyperglycemic, however, at this time point Liraglutide treated animals had significantly lower blood glucose compared to vehicle (Liraglutide: 6.6 ± 0.16 mmol/l, vehicle: 7.1 ± 0.17 mmol/l; p<0.05). At 12 hours, Liraglutide-treated animals had significant higher blood glucose than vehicle-treated animals (Liraglutide: 6.35 ± 0.28 mmol/l; vehicle: 5.3 ± 0.14 mmol/l; p<0.05), but did not differ significantly from sham-treated animals. Body weight % of baseline was significantly lower at all time points after CCI in Liraglutide-treated compared to vehicle-treated animals. However, at day 7, Liraglutide-treated animals had recovered their body weight to what it was before the injury (Table 1).
Table 1. Summary of blood glucose, core temperature and weight %.
| Core Temperature | ||||
|---|---|---|---|---|
| 12 hours | 24 hours | 26 hours | 48 hours | |
| Sham | 38.4 ± 0.19 | 37.5 ± 0.34 | 38.0 ± 0.00 | 38.1 ± 0.19 |
| Vehicle | 37.7 ± 0.21 | 38.3 ± 0.15 | 38.2 ± 0.13 | 38.4 ± 0.15 |
| LIR 75 | 37.7 ± 0.10 | 38.0 ± 0.17 | 38.1 ± 0.14 | 38.1 ± 0.07 |
| LIR 200 | 37.3 ± 0.19 | 37.6 ± 0.15* | 37.9 ± 0.08 | 38.1 ± 0.15 |
| Blood glucose | ||||
| 30 min | 12 hours | 24 hours | 48 hours | |
| Sham | - | 6.00 ± 0.08 | 5.38 ± 0.14 | 5.98 ± 0.30 |
| Vehicle | 7.1 ± 0.17 | 5.48 ± 0.20 | 5.16 ± 0.18 | 5.96 ± 0.29 |
| LIR 75 | - | 5.32 ± 0.14 | 4.82 ± 0.16 | 5.57 ± 0.18 |
| LIR 200 | 6.6 ± 0.16* | 6.35 ± 0.28* | 5.47 ± 0.18 | 5.33 ± 0.18 |
| Weight % of baseline | ||||
| 12 hours | 24 hours | 48 hours | 7 days | |
| Sham | 98.4 ± 0.82 | 97.1 ± 1.10 | 95.8 ± 1.32 | - |
| Vehicle | 96.3 ± 0.40 | 91.6 ± 0.36 | 94.4 ± 0.44 | 103 ± 0.87 |
| LIR 75 | 94.7 ± 0.34* | 94.3 ± 0.39* | 89.3 ± 0.55* | - |
| LIR 200 | 93.1 ± 0.43* | 90.5 ± 0.43* | 87.7 ± 0.43* | 96.1 ± 0.71* |
| Naive vehicle | 100 ± 0.49 | 100 ± 0.50 | 101 ± 0.80 | - |
| Naive LIR 200 | 93.7 ± 0.49* | 90.5 ± 0.61* | 91.1 ± 1.08* | |
* Indicates significant difference from vehicle.
Liraglutide treatment reduces CCI induced neurological deficits
At 48 hours post-CCI, injured vehicle treated animals demonstrated significant sensorimotor impairments when compared with sham-operated rats (p<0.001). Liraglutide treatment (200 μg/kg) significantly improved composite Neuroscore compared to vehicle treated rats (p<0.05, Fig. 2B).
Fig 2. Effect of Liraglutide on brain water content and Neuroscore after TBI.

A: Ipsilateral total water content of the 6 mm coronal sample area (Ipsi-Total) and corresponding contralateral region (Ctrl-Total) 48 hours following controlled cortical impact (CCI). Total water content increased significantly in vehicle treated animals compared to Sham. However, water content was mitigated significantly in animals treated with Liraglutide 200 μg/kg BID. B: Subregion water content of contralateral and ipsilateral areas 48 hours after CCI. Liraglutide 200 μg/kg BID significantly mitigated cerebral water content in the ipsilateral hippocampus and thalamus after CCI. C: Composite Neuroscore test was calculated through a battery of six sub-tests 48 hours after CCI, with a score of maximum 21 points. Values in A and B are means ± SEM. *:p<0.05, **:p<0.01. Values in C are presented as median and error bars indicate the 25th and 75th percentile. Abbreviations: LIR75—Liraglutide 75 μg/kg; n = 10, LIR200—Liraglutide 200 μg/kg; n = 10, vehicle n = 10 and Sham n = 4.
Liraglutide 75 μg/kg did not significantly improve Neuroscore after CCI. Individual scores for paw placement and circling tests in experiment 1 were significantly higher in the group treated with Liraglutide 200 μg/kg (data not shown). There was no effect of Liraglutide 200 μg/kg on Neuroscore when given to naive rats (Fig. 3B).
Fig 3. Effect of Liraglutide on normal brain water content and Neuroscore.

Water content in the uninjured naive brain was unaffected by Liraglutide 200 μg/kg 48 hours after CCI. Values are means ± SEM. B: There was no effect of Liraglutide on Neuroscore in naive animals. Values are median with 25th and 75th percentile.
In experiment 2 and 3, the circling test showed severe sensorimotor impairment in vehicle-treated rats 2 and 7 days after CCI (Fig. 4A and 4B). This impairment was significantly attenuated by Liraglutide (200 μg/kg) at day 2 (p<0.01) but not at day 7 after CCI. Likewise, Liraglutide-treated animals showed a significantly better improvement than vehicle-treated animals only at day 2 after CCI (p<0.05, Fig. 4C). Due to spontaneous recovery in paw placement both in vehicle and Liraglutide group no differences between the treatment groups were observed on day 7 after CCI (Fig. 4D). Together, these results demonstrate that administration of Liraglutide improves sensorimotor outcome 48 hours after CCI.
Fig 4. Paw placement and Circling test (experiment 2 and 3).

Performance of vehicle- and Liraglutide-treated rats in Circling test 2 days (A) and 7 days (B) after controlled cortical impact (CCI), and limb-placing ability for 2 days (C) and 7 days (D) after CCI. The normal score before injury is 2 for circling test and 4 for paw placement test. Values are presented as median and error bars indicate the 25th and 75th percentile, #:p<0.05.
Liraglutide treatment ameliorates cerebral edema
As shown in Fig. 2A, total water content of the brain tissue sample including all three regions studied was 77.6 ± 0.3% in sham-operated animals. In vehicle-treated rats subjected to CCI, water content increased to 81.1 ± 0.3%. Treatment with Liraglutide (200 μg/kg) led to a total water content of 80.19 ± 0.2%. This represents a significant mitigation of total water content elevation, i.e. edema, by 26% compared to vehicle treated rats (p<0.05). Similarly, subregion analysis of the hippocampus and thalamus representing the pericontusional areas revealed that Liraglutide significantly mitigated hippocampal edema by 39% (p<0.01, vehicle: 82.6 ± 0.3% vs. Liraglutide: 81.0 ± 0.6%; Sham: 78.4 ± 0.2%) and thalamic edema by 48% (p<0.01, vehicle: 78.9 ± 0.5% vs. Liraglutide: 77.5 ± 0.1%; Sham: 75.9 ± 0.1%) (Fig. 2C). There was no significant effect of Liraglutide on water content in the ipsilateral cerebral cortex region. The reduction of brain water content in animals treated with the lower Liraglutide dose (75 μg/kg) was insignificant. Liraglutide did not influence water content in normal brain tissue when given to naive animals (Fig. 3A).
Liraglutide treatment reduces BBB permeability
To further examine the effect of Liraglutide (200 μg/kg) on edema, we used Evans Blue as a marker of Albumin extravasation (Fig. 5B). The Evans blue content of the ipsilateral hemisphere of vehicle-treated animals (127 ± 11 μg EB/mg) was significant higher than the cerebellar control 48 hours after CCI (20.2 ± 1.9 μg EB/mg; p<0.0001). Liraglutide led to a significant reduction in Evans blue extravasation in the ipsilateral hemisphere (80.9 ±11 μg EB/mg; p<0.05) (Fig. 5A). The contralateral hemisphere had significantly less Evans blue in Liraglutide-treated animals (8.7 ± 1.2 μg EB/mg) compared to vehicle-treated animals (16.7 ± 2.7 μg EB/mg; p<0.05).
Fig 5. Evans blue exudation.

A: Analysis of Evans Blue extravasation (μg/mg of dry brain tissue) 48 hours after CCI in rats. Treatment with Liraglutide 200 μg/kg BID significantly reduced Evans Blue extravasation in both hemispheres. B: Illustration of coronal sections through the contusion center illustrates Evans blue distribution throughout the rat brain. Evans blue extravasation is particularly prominent in the pericontusional cortex, hippocampus and upper thalamus. Values are mean ± SEM. *:p<0.05.
Liraglutide reduced cortical lesion size but had no effect on delayed thalamic neuronal death
At day 7, a large lesion was apparent involving the cerebral cortex and subcortical structures including the corpus callosum, hippocampus and thalamus. The lesion was generally visible at +1.2 mm to -6.8 mm from bregma (Fig. 6A). Fig. 6B shows a significant reduction in cortical lesion size in Liraglutide-treated rats (41.3 ± 2.6 mm3) compared to vehicle treated animals (50.5 ± 3 mm3; p<0.05) at 7 days. There was no effect of Liraglutide on lesion size in the thalamus (Fig. 6C). Additionally, when estimating lesion size for the whole ipsilateral hemisphere, Liraglutide-induced reduction in lesion size was not significant compared to vehicle (data not shown). To assess degenerating neurons one week after CCI we performed FJC stainings of the sections collected at day 7 post injury. FJC positive (FJC+) cells were present in the ipsilateral thalamus. We found no difference in number of FJC+ cells between vehicle and Liraglutide-treated animals in any of the individual five thalamus ROI’s (Fig. 6C-E).
Fig 6. Effects on lesion volume and delayed neuronal death 7 days post-injury.

A: Illustration of representative lesions by NeuN stained coronal sections from rats treated either with vehicle or Liraglutide for two days. The successive coronal sections range from +2.2 to -6.8 mm from bregma. B: Calculated cortical lesion volume (mm3). C: Calculated lesion volume in the ipsilateral thalamus (mm3). D: NeuN stained coronal section -2.8 mm from bregma illustrating the 5 regions of interest (ROI) chosen for investigation of FJC+ cells within the thalamus. Each ROI represents a counting frame at 10X. E: Demonstration of counting frame with FJC+ cells 7 days after CCI. F: Degenerating neurons shown at 20X. G: The average number of FJC+ cells for the 5 ROI’s was unaffected by Liraglutide treatment for the first 2 days post injury. *:p<0.05. Values are means ± SEM.
Discussion
In this study we investigated the effect of post-TBI Liraglutide treatment on sensorimotor deficits, cerebral edema, BBB integrity, lesion volume, and delayed neuronal death.
Neurological function
TBI is associated with impairment of a wide range of neurobehavioral modalities including cognitive, emotional and sensorimotor function [31]. An emerging body of evidence indicates that treatment with Exendin-4 (Ex-4), a systemic GLP-1 agonist, abolishes cognitive deficits in rodent models of mild and moderate TBI [32–35]. In more detail, treatment with Ex-4 starting both 2 days before and 1 hour after mild concussive TBI improved memory function at day 7 and day 30 [33]. Additionally, in mild TBI, Tweedie et al. found that improvement in recognition memory by Ex-4 pretreatment was related to reduced changes in hippocampal genes related to Alzheimer’s disease [32]. Similar studies have been conducted in rats where Ex-4 administration 30 minutes after moderate fluid-percussion TBI improved memory function when assessed using a water maze test. In the above mentioned experiment, Ex-4 treatment was ended at a minimum of 2 days before any behavioral test to avoid confounding effects of Ex-4 on cognitive tests [32]. In the present study, the last dose was given 36 hours after TBI, that is, 12 hours before conducting any neurological test. However, as shown in Fig. 3B, this regime had no adverse effect on sensorimotor outcome in normal rats.
It has been reported that Liraglutide effectively improves sensorimotor function 24–72 hours post injury when, assessed by a variety of neuroscore tests, in different models of experimental stroke [8,13]. However, to our knowledge, the effect of GLP-1 receptor agonists on sensorimotor outcome after TBI has not previously been studied. Here we showed that Liraglutide treatment improved sensorimotor function 48 hours after TBI. However, this effect was attenuated on day 7, likely as a result of spontaneous functional recovery related to plasticity processes in the lesioned brain [36]. Cerebral edema peaks within the first two days after CCI, and is considered a primary causal factor of neuronal injury and sensorimotor deficits after moderate and severe TBI [25,37]. Therefore, it is likely that the observed positive effects of Liraglutide on neurological function 48 hours after CCI are to some extent related to anti-edema effects.
Edema and BBB
The levels of cerebral water content, and the regional differences in water content between cortical, hippocampal and thalamic areas that we report in this study, are in line with previous reports [26,38]. Liraglutide (200 μg/kg) BID significantly mitigated TBI induced water content increase in the hippocampus and thalamus by 39% and 48%, respectively. In contrast, Liraglutide did not significantly reduce edema in the cortical region. This might be caused by a local low Liraglutide delivery or different edema pathology in the contusion core, which has been shown to be markedly hypoperfused and sparsely vascularized [39]. In addition to cerebral edema, we found that Liraglutide markedly reduced Evans Blue extravasation in the ipsilateral and contralateral hemisphere. This indicates that GLP-R stimulation prevents endothelial barrier dysfunction, and suggests that the reduction in tissue water content, at least partially, is due to reduced vasogenic edema [26]. From Fig. 5B it can be discerned that Evans blue mainly is located pericontusionally in the contralateral hemisphere and to some extent in the contralateral cingulate cortex adjacent to the contusion. The increased Evans Blue extravasation in the uninjured contralateral hemisphere is likely due to the high magnitude of the injury.
Recruitment of neutrophils to the brain parenchyma plays a key role in edema formation [30,40–42]. Together with resident microglial cells, neutrophils mediate BBB disruption through release of glutamate, reactive oxygen species (ROS), and proteases such as matrix metallo-proteinases [43–45]. Glutamate and ROS also act in a toxic manner on parenchymal cells, thereby contributing to neuronal death and cytotoxic edema [4]. Several studies have pointed out an anti-inflammatory role of GLP-1 in acute brain injury, for instance Exendin-4 reduced IBA-1 positive microglia activation in experimental model of global ischemia and in stroked diabetic rats subjected to experimental stroke [10,11]. In addition, it has been shown that Liraglutide reduces cerebral edema formation in a mouse model of hemorrhagic stroke [8]. This effect was related to an anti-inflammatory effect on cerebral endothelial cells through reduced expression of ICAM-1 and subsequently a reduced recruitment of neutrophils to the brain parenchyma. Likewise in diabetic vascular disease, GLP-1 has anti-inflammatory effects on cardiovascular endothelial cells [46]. Liraglutide also reduced the expression of ROS and various adhesion molecules in cultures of human endothelial cells [47,48]. Thus, it is likely that the reduction in blood-brain barrier permeability and brain edema post-TBI demonstrated in the present study is related to the anti-inflammatory properties of Liraglutide.
Protection of neuronal tissue
It is evident that improvement in motor and cognitive functions in models of experimental TBI is associated with a decrease in macroscopic cortical tissue loss [49,50]. In concordance with this we found that systemic treatment with Liraglutide for two days after CCI reduced the cortical lesion volume of cortical neuronal injury by approximately 20%. This finding supports previous studies showing that Liraglutide reduces cryogenic brain injury at 2 days [51], and implantation of GLP-1 producing stem cells reduced hippocampal neuron loss after CCI [52]. Both Liraglutide and Ex-4 have demonstrated neuroprotective properties after experimental stroke [10,12,53]. These in vivo findings are supported by in vitro studies reporting that GLP-1 reduces oxidative stress in cultures of human neuroblastoma cells [54,55], and glutamate-induced excitotoxicity in neuronal cultures derived from rats [33,55]. Finally, preservation of cortical tissue by Liraglutide after CCI may indirectly be related to edema alleviation, and subsequently increased perfusion and oxygen tension in pericontusional areas [2,56].
The time course of neuronal death differs between different regions after cortical trauma. Focal cortical injury is associated with delayed death of thalamic neurons likely due to retrograde damage of cortical projections from the thalamus [57]. We analyzed Fluoro-Jade C positive (FJC+) neurons in the thalamus 7 days after CCI. There was no effect of Liraglutide when administered for 2 days after injury on FJC+ neurons. This can be due to the magnitude of the injury or the fact that the treatment was not continued beyond two days after the injury.
Glycemic control
Hyperglycemia occurs frequently after moderate and severe TBI [58,59], and is driven by a hypermetabolic sympathoadrenal stress response [60]. Several studies indicate that hyperglycemia in the early hours after severe TBI is associated with worse neurological outcome [58,61]. Hyperglycemia is believed to induce neurotoxicity through an increase of oxidative stress [62] and enhanced extracellular glutamate excitotoxicity [63]. In our study, both Liraglutide and vehicle treated animals were slightly hyperglycemic 30 minutes after the injury. However, Liraglutide treatment significantly reduced glucose levels at 30 minutes post injury.
Insulin is used worldwide for treatment of hyperglycemia in patients with severe TBI. However, several trials have reported that strict glucose control with insulin in patients with TBI is associated with variable degrees of hypoglycemia [64,65]. This is problematic since the injured brain is sensitive even to mild hypoglycemia [64]. On the other hand, it has been shown that GLP-1 enhances cerebral glucose transport and metabolism during hyperglycemia in humans [66], and that these effects are not present during hypoglycemia [67]. Furthermore GLP-1 reduces blood glucose mainly by stimulation of glucose dependent insulin synthesis, and suppression of glucagon secretion [5]. These effects of GLP-1 ceases when blood glucose reaches normal and hypoglycemic levels [68]. In accordance with this, treatment with Liraglutide did not cause hypoglycemia in our study. Therefore, our results support the hypothesis stated in a recent review by Greig et al., that GLP-1 might be considered as a safe therapy to diminish hyperglycemia after TBI [69].
Dose considerations
In humans Liraglutide has a half-life of around 13 hours, and is administered once daily in doses up to 1.8 mg. To investigate the effect of Liraglutide on secondary injury after TBI, we chose a dosing regime with two daily injections, starting immediately after injury, to compensate for a known shorter half-life of Liraglutide in rats (4 hours) compared to humans [70]. The lowest dose used in this study was 75 μg/kg BID, which is comparable with the currently used dose for antidiabetic treatment in humans [71]. The highest treatment dose used in this study (200 μg/kg BID), is somewhat higher than what is currently used for antidiabetic treatment. This dose, however, has been used previously in several rodent experiments [72,73]. In our experiments, both doses of Liraglutide resulted in a temporary reduction in body weight. This is a known side effect of Liraglutide [74], mediated by reduced appetite and gastric emptying [6]. Moreover, in a clinical trial by Astrup et al.[75] and recently in a phase III clinical trial [76], a high dose of 3 mg was effective at inducing weight loss and was well tolerated. Thus, even though 200 μg/kg BID currently has less translational value, it is likely that higher doses will be in clinical use for other conditions within the near future.
Conclusions
Treatment with Liraglutide reduced cerebral edema and cortical neuronal tissue loss, and improved BBB integrity and sensorimotor outcome after experimental TBI. These novel findings extend prior research with GLP-1 agonists in TBI and experimental stroke. Further experiments are needed to delineate the exact mechanisms by which Liraglutide decreases cerebral edema and cortical tissue loss after TBI. Furthermore, it will be important to elucidate if longer treatment periods will provide better neuroprotective effect and functional outcome after CCI, and whether a later treatment onset is efficacious.
Prevention of cerebral edema and neuronal injury remains an important challenge in TBI patients. This study indicates that GLP-1R stimulation is a promising target in terms of sensorimotor sequelae after TBI. Given that several GLP-1 agonists, including Liraglutide, are already approved for clinical use and are well tolerated, further exploration of these effects is warranted.
Acknowledgments
The authors want to thank Peter Bentzer for kindly sharing the wet/dry-weight method, Brian DellaValle for productive discussions, Lasse Maretty for statistical advice and Kerstin Beirup for technical assistance.
Data Availability
All relevant data are within the paper.
Funding Statement
The authors received no specific funding for this work.
References
- 1. Xiong Y, Mahmood A, Chopp M. Emerging treatments for traumatic brain injury. Expert Opin Emerg Drugs. 2009;14: 67–84. 10.1517/14728210902769601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Narotam PK, Morrison JF, Nathoo N. Brain tissue oxygen monitoring in traumatic brain injury and major trauma: outcome analysis of a brain tissue oxygen-directed therapy. J Neurosurg. 2009;111: 672–82. 10.3171/2009.4.JNS081150 [DOI] [PubMed] [Google Scholar]
- 3. Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience. 2004;129: 1021–1029. [DOI] [PubMed] [Google Scholar]
- 4. Chodobski A, Zink BJ, Szmydynger-Chodobska J. Blood-brain barrier pathophysiology in traumatic brain injury. Transl Stroke Res. 2011;2: 492–516. 10.1007/s12975-011-0125-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Holst JJ, Burcelin R, Nathanson E. Neuroprotective properties of GLP-1: theoretical and practical applications. Curr Med Res Opin. 2011;27: 547–58. 10.1185/03007995.2010.549466 [DOI] [PubMed] [Google Scholar]
- 6. Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;87: 1409–39. [DOI] [PubMed] [Google Scholar]
- 7. Hunter K, Hölscher C. Drugs developed to treat diabetes, liraglutide and lixisenatide, cross the blood brain barrier and enhance neurogenesis. BMC neurosci. BioMed Central Ltd; 2012;13: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hou J, Manaenko A, Hakon J, Hansen-Schwartz J, Tang J, Zhang JH. Liraglutide, a long-acting GLP-1 mimetic, and its metabolite attenuate inflammation after intracerebral hemorrhage. J cereb Blood Flow Metab. Nature Publishing Group; 2012; 1–10. 10.1038/jcbfm.2012.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Iwai T, Ito S, Tanimitsu K, Udagawa S, Oka J-I. Glucagon-like peptide-1 inhibits LPS-induced IL-1beta production in cultured rat astrocytes. Neurosci Res. 2006;55: 352–60. [DOI] [PubMed] [Google Scholar]
- 10. Darsalia V, Mansouri S, Ortsäter H, Olverling A, Nozadze N, Kappe C, et al. Glucagon-like peptide-1 receptor activation reduces ischaemic brain damage following stroke in Type 2 diabetic rats. Clin Sci (Lond). 2012;122: 473–83. 10.1042/CS20110374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee CH, Yan B, Yoo K-Y, Choi JH, Kwon S-H, Her S, et al. Ischemia-induced changes in glucagon-like peptide-1 receptor and neuroprotective effect of its agonist, exendin-4, in experimental transient cerebral ischemia. J Neurosci Res. 2011;89: 1103–13. 10.1002/jnr.22596 [DOI] [PubMed] [Google Scholar]
- 12. Briyal S, Gulati K, Gulati A. Repeated administration of exendin-4 reduces focal cerebral ischemia-induced infarction in rats. Brain res. Elsevier B.V.; 2012;1427: 23–34. 10.1016/j.brainres.2011.10.026 [DOI] [PubMed] [Google Scholar]
- 13. Sato K, Kameda M, Yasuhara T, Agari T, Baba T, Wang F, et al. Neuroprotective effects of liraglutide for stroke model of rats. Int J Mol Sci. 2013;14: 21513–24. 10.3390/ijms141121513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li Y, Perry T, Kindy MS, Harvey BK, Tweedie D, Holloway HW, et al. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc Natl Acad Sci U S A. 2009;106: 1285–90. 10.1073/pnas.0806720106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McClean PL, Hölscher C. Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease. Neuropharmacology. Elsevier; 2014;76 Pt A: 57–67. 10.1016/j.neuropharm.2013.08.005 [DOI] [PubMed] [Google Scholar]
- 16. Bertilsson G, Patrone C, Zachrisson O, Andersson A, Dannaeus K, Heidrich J, et al. Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson’s disease. J Neurosci Res. 2008;86: 326–38. [DOI] [PubMed] [Google Scholar]
- 17. Albert-Weissenberger C, Sirén A-L. Experimental traumatic brain injury. Exp Transl Stroke Med. 2010;2: 16 10.1186/2040-7378-2-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schwarzkopf TM, Horn T, Lang D, Klein J. Blood gases and energy metabolites in mouse blood before and after cerebral ischemia: the effects of anesthetics. Exp Biol Med. 2013;238: 84–9. 10.1258/ebm.2012.012261 [DOI] [PubMed] [Google Scholar]
- 19. Dixon CE, Clifton GL, Lighthall JW, Yaghmai a a, Hayes RL. A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods. 1991;39: 253–62. [DOI] [PubMed] [Google Scholar]
- 20. Hamm RJ, Dixon CE, Gbadebo DM, Singha a K, Jenkins LW, Lyeth BG, et al. Cognitive deficits following traumatic brain injury produced by controlled cortical impact. J Neurotrauma. 1992;9: 11–20. [DOI] [PubMed] [Google Scholar]
- 21. Hunter AJ, Hatcher J, Virley D, Nelson P, Irving E, Hadingham SJ, et al. Functional assessments in mice and rats after focal stroke. Neuropharmacology. 2000;39: 806–16. [DOI] [PubMed] [Google Scholar]
- 22. Yrjänheikki J, Koistinaho J, Kettunen M, Kauppinen R a, Appel K, Hüll M, et al. Long-term protective effect of atorvastatin in permanent focal cerebral ischemia. Brain Res. 2005;1052: 174–9. [DOI] [PubMed] [Google Scholar]
- 23. De Ryck M, Van Reempts J, Duytschaever H, Van Deuren B, Clincke G. Neocortical localization of tactile/proprioceptive limb placing reactions in the rat. Brain Res. 1992;573: 44–60. [DOI] [PubMed] [Google Scholar]
- 24. Madinier A, Quattromani MJ, Sjölund C, Ruscher K, Wieloch T. Enriched housing enhances recovery of limb placement ability and reduces aggrecan-containing perineuronal nets in the rat somatosensory cortex after experimental stroke. PLoS One. 2014;9: e93121 10.1371/journal.pone.0093121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Başkaya MK, Doğan A, Temiz C, Dempsey RJ. Application of 2,3,5-triphenyltetrazolium chloride staining to evaluate injury volume after controlled cortical impact brain injury: role of brain edema in evolution of injury volume. J Neurotrauma. 2000;17: 93–9. [DOI] [PubMed] [Google Scholar]
- 26. Fukui S, Fazzina G, Amorini AM, Dunbar JG, Marmarou A. Differential effects of atrial natriuretic peptide on the brain water and sodium after experimental cortical contusion in the rat. J cereb Blood Flow Metab. 2003;23: 1212–8. [DOI] [PubMed] [Google Scholar]
- 27. Hall ED, Bryant YD, Cho W, Sullivan PG. Evolution of post-traumatic neurodegeneration after controlled cortical impact traumatic brain injury in mice and rats as assessed by the de Olmos silver and fluorojade staining methods. J Neurotrauma. 2008;25: 235–47. 10.1089/neu.2007.0383 [DOI] [PubMed] [Google Scholar]
- 28. Swanson RA, Morton MT, Tsao-Wu G, Savalos RA, Davidson C, Sharp FR. A semiautomated method for measuring brain infarct volume. J Cereb Blood Flow Metab. 1990;10: 290–293. [DOI] [PubMed] [Google Scholar]
- 29. Zhang Y-B, Li S-X, Chen X-P, Yang L, Zhang Y-G, Liu R, et al. Autophagy is activated and might protect neurons from degeneration after traumatic brain injury. Neurosci Bull. 2008;24: 143–9. 10.1007/s12264-008-1108-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kenne E, Erlandsson A, Lindbom L, Hillered L, Clausen F. Neutrophil depletion reduces edema formation and tissue loss following traumatic brain injury in mice. J Neuroinflammation. BioMed Central Ltd; 2012;9: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Draper K, Ponsford J. Long-term outcome following traumatic brain injury: a comparison of subjective reports by those injured and their relatives. Neuropsychol Rehabil. 2009;19: 645–61. 10.1080/17405620802613935 [DOI] [PubMed] [Google Scholar]
- 32. Tweedie D, Rachmany L, Rubovitch V, Lehrmann E, Zhang Y, Becker KG, et al. Exendin-4, a glucagon-like peptide-1 receptor agonist prevents mTBI-induced changes in hippocampus gene expression and memory deficits in mice. Exp Neurol. Elsevier Inc.; 2013;239: 170–82. 10.1016/j.expneurol.2012.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rachmany L, Tweedie D, Li Y, Rubovitch V, Holloway HW, Miller J, et al. Exendin-4 induced glucagon-like peptide-1 receptor activation reverses behavioral impairments of mild traumatic brain injury in mice. Age. 2012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tweedie D, Rachmany L, Rubovitch V, Zhang Y, Becker KG, Perez E, et al. Changes in mouse cognition and hippocampal gene expression observed in a mild physical- and blast-traumatic brain injury. Neurobiol Dis. Elsevier Inc.; 2013;54: 1–11. 10.1016/j.nbd.2013.02.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eakin K, Li Y, Chiang Y-H, Hoffer BJ, Rosenheim H, Greig NH, et al. Exendin-4 ameliorates traumatic brain injury-induced cognitive impairment in rats. PLoS One. 2013;8: e82016 10.1371/journal.pone.0082016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nishibe M, Barbay S, Guggenmos D, Nudo RJ. Reorganization of motor cortex after controlled cortical impact in rats and implications for functional recovery. J Neurotrauma. 2010;27: 2221–32. 10.1089/neu.2010.1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Feickert HJ, Drommer S, Heyer R. Severe head injury in children: impact of risk factors on outcome. J Trauma. 1999;47: 33–8. [DOI] [PubMed] [Google Scholar]
- 38. Sztriha L, Joó F, Szerdahelyi P. Time-course of changes in water, sodium, potassium and calcium contents of various brain regions in rats after systemic kainic acid administration. Acta Neuropathol. 1986;70: 169–76. [DOI] [PubMed] [Google Scholar]
- 39. Forbes ML, Hendrich KS, Kochanek PM, Williams DS, Schiding JK, Wisniewski SR, et al. Assessment of cerebral blood flow and CO2 reactivity after controlled cortical impact by perfusion magnetic resonance imaging using arterial spin-labeling in rats. J cereb Blood Flow Metab. 1997;17: 865–74. [DOI] [PubMed] [Google Scholar]
- 40. Knoblach SM, Faden AI. Administration of either anti-intercellular adhesion molecule-1 or a nonspecific control antibody improves recovery after traumatic brain injury in the rat. J Neurotrauma. 2002;19: 1039–50. [DOI] [PubMed] [Google Scholar]
- 41. Whalen MJ, Carlos TM, Dixon CE, Robichaud P, Clark RS, Marion DW, et al. Reduced brain edema after traumatic brain injury in mice deficient in P-selectin and intercellular adhesion molecule-1. J Leukoc Biol. 2000;67: 160–8. [DOI] [PubMed] [Google Scholar]
- 42. Schoettle RJ, Kochanek PM, Magargee MJ, Uhl MW, Nemoto EM. Early polymorphonuclear leukocyte accumulation correlates with the development of posttraumatic cerebral edema in rats. J Neurotrauma. 1990;7: 207–17. [DOI] [PubMed] [Google Scholar]
- 43. Collard CD, Park K a, Montalto MC, Alapati S, Buras J a, Stahl GL, et al. Neutrophil-derived glutamate regulates vascular endothelial barrier function. J Biol Chem. 2002;277: 14801–11. [DOI] [PubMed] [Google Scholar]
- 44. Fischer S, Wiesnet M, Renz D, Schaper W. H2O2 induces paracellular permeability of porcine brain-derived microvascular endothelial cells by activation of the p44/42 MAP kinase pathway. Eur J Cell Biol. 2005;84: 687–97. [DOI] [PubMed] [Google Scholar]
- 45. Shigemori Y, Katayama Y, Mori T, Maeda T, Kawamata T. Matrix metalloproteinase-9 is associated with blood-brain barrier opening and brain edema formation after cortical contusion in rats. Acta Neurochir Suppl. 2006;96: 130–3. [DOI] [PubMed] [Google Scholar]
- 46. Ceriello A, Novials A, Ortega E, Canivell S, La Sala L, Pujadas G, et al. Glucagon-Like Peptide 1 Reduces Endothelial Dysfunction, Inflammation, and Oxidative Stress Induced by Both Hyperglycemia and Hypoglycemia in Type 1 Diabetes. Diabetes Care. 2013; 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Shiraki A, Oyama J, Komoda H, Asaka M, Komatsu A, Sakuma M, et al. The glucagon-like peptide 1 analog liraglutide reduces TNF-α-induced oxidative stress and inflammation in endothelial cells. Atherosclerosis. Elsevier Ireland Ltd; 2012;221: 375–82. [DOI] [PubMed] [Google Scholar]
- 48. Krasner NM, Ido Y, Ruderman NB, Cacicedo JM. Glucagon-like peptide-1 (GLP-1) analog liraglutide inhibits endothelial cell inflammation through a calcium and AMPK dependent mechanism. PLoS One. 2014;9: e97554 10.1371/journal.pone.0097554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang G, Jiang X, Pu H, Zhang W, An C, Hu X, et al. Scriptaid, a novel histone deacetylase inhibitor, protects against traumatic brain injury via modulation of PTEN and AKT pathway: scriptaid protects against TBI via AKT. Neurotherapeutics. 2013;10: 124–42. 10.1007/s13311-012-0157-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Shein NA, Grigoriadis N, Alexandrovich AG, Simeonidou C, Lourbopoulos A, Polyzoidou E, et al. Histone deacetylase inhibitor ITF2357 is neuroprotective, improves functional recovery, and induces glial apoptosis following experimental traumatic brain injury. FASEB J. 2009;23: 4266–75. 10.1096/fj.09-134700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dellavalle B, Hempel C, Johansen FF, Anders J, Kurtzhals L. GLP-1 improves neuropathology after murine cold lesion brain trauma. Ann Clin Transl Neurol. 2014;1: 721–732. 10.1002/acn3.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Heile AMB, Wallrapp C, Klinge PM, Samii A, Kassem M, Silverberg G, et al. Cerebral transplantation of encapsulated mesenchymal stem cells improves cellular pathology after experimental traumatic brain injury. Neurosci Lett. 2009;463: 176–81. 10.1016/j.neulet.2009.07.071 [DOI] [PubMed] [Google Scholar]
- 53. Teramoto S, Miyamoto N, Yatomi K, Tanaka Y, Oishi H, Arai H, et al. Exendin-4, a glucagon-like peptide-1 receptor agonist, provides neuroprotection in mice transient focal cerebral ischemia. J cereb Blood Flow Metab. Nature Publishing Group; 2011;31: 1696–705. 10.1038/jcbfm.2011.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li Y, Tweedie D, Mattson MP, Holloway HW, Greig NH. Enhancing the GLP-1 receptor signaling pathway leads to proliferation and neuroprotection in human neuroblastoma cells. J Neurochem. 2010;113: 1621–31. 10.1111/j.1471-4159.2010.06731.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Perry T, Haughey NJ, Mattson MP, Egan JM, Greig NH. Protection and reversal of excitotoxic neuronal damage by glucagon-like peptide-1 and exendin-4. J Pharmacol Exp Ther. 2002;302: 881–8. [DOI] [PubMed] [Google Scholar]
- 56. Kochanek PM, Marion DW, Zhang W, Schiding JK, White M, Palmer AM, et al. Severe controlled cortical impact in rats: assessment of cerebral edema, blood flow, and contusion volume. J Neurotrauma. 1995;12: 1015–25. [DOI] [PubMed] [Google Scholar]
- 57. Conti AC, Raghupathi R, Trojanowski JQ, McIntosh TK. Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post-traumatic period. J Neurosci. 1998;18: 5663–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jeremitsky E, Omert LA, Dunham CM, Wilberger J, Rodriguez A. The impact of hyperglycemia on patients with severe brain injury. J Trauma. 2005;58: 47–50. [DOI] [PubMed] [Google Scholar]
- 59. Eakins J. Blood glucose control in the trauma patient. J Diabetes Sci Technol. 2009;3: 1373–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. McCowen KC, Malhotra A, Bistrian BR. Stress-induced hyperglycemia. Crit Care Clin. 2001;17: 107–24. [DOI] [PubMed] [Google Scholar]
- 61. Laird AM, Miller PR, Kilgo PD, Meredith JW, Chang MC. Relationship of Early Hyperglycemia to Mortality in Trauma Patients. J Trauma. 2004;56: 1058–1062. [DOI] [PubMed] [Google Scholar]
- 62. Tomlinson DR, Gardiner NJ. Glucose neurotoxicity. Nat Rev Neurosci. 2008;9: 36–45. [DOI] [PubMed] [Google Scholar]
- 63. Li P, Shuaib A, Miyashita H, He Q-P, Siesjo BK, Warner DS. Hyperglycemia Enhances Extracellular Glutamate Accumulation in Rats Subjected to Forebrain Ischemia Editorial Comment. Stroke. 2000;31: 183–192. [DOI] [PubMed] [Google Scholar]
- 64. Vespa P, Boonyaputthikul R, McArthur DL, Miller C, Etchepare M, Bergsneider M, et al. Intensive insulin therapy reduces microdialysis glucose values without altering glucose utilization or improving the lactate/pyruvate ratio after traumatic brain injury. Crit Care Med. 2006;34: 850–6. [DOI] [PubMed] [Google Scholar]
- 65. Godoy DA, Di Napoli M, Rabinstein AA. Treating hyperglycemia in neurocritical patients: benefits and perils. Neurocrit Care. 2010;13: 425–38. 10.1007/s12028-010-9404-8 [DOI] [PubMed] [Google Scholar]
- 66. Gejl M, Egefjord L, Lerche S, Vang K, Bibby BM, Holst JJ, et al. Glucagon-like peptide-1 decreases intracerebral glucose content by activating hexokinase and changing glucose clearance during hyperglycemia. J cereb Blood Flow Metab. 2012;32: 2146–52. 10.1038/jcbfm.2012.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gejl M, Lerche S, Egefjord L, Brock B, Møller N, Vang K, et al. Glucagon-like peptide-1 (GLP-1) raises blood-brain glucose transfer capacity and hexokinase activity in human brain. Front Neuroenergetics. 2013;5: 2 10.3389/fnene.2013.00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nauck MA, Heimesaat MM, Behle K, Holst JJ, Nauck MS, Ritzel R, et al. Effects of glucagon-like peptide 1 on counterregulatory hormone responses, cognitive functions, and insulin secretion during hyperinsulinemic, stepped hypoglycemic clamp experiments in healthy volunteers. J Clin Endocrinol Metab. 2002;87: 1239–46. [DOI] [PubMed] [Google Scholar]
- 69. Greig NH, Tweedie D, Rachmany L, Li Y, Rubovitch V, Schreiber S, et al. Incretin mimetics as pharmacologic tools to elucidate and as a new drug strategy to treat traumatic brain injury. Alzheimers Dement. Elsevier; 2014;10: S62–75. 10.1016/j.jalz.2013.12.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sturis J, Gotfredsen CF, Rømer J, Rolin B, Ribel U, Brand CL, et al. GLP-1 derivative liraglutide in rats with beta-cell deficiencies: influence of metabolic state on beta-cell mass dynamics. Br J Pharmacol. 2003;140: 123–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Services USDoHaH, Administration FaD. Guidance for Industry Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. [Internet]. 2005.
- 72. Raun K, von Voss P, Gotfredsen CF, Golozoubova V, Rolin B, Knudsen LB. Liraglutide, a long-acting glucagon-like peptide-1 analog, reduces body weight and food intake in obese candy-fed rats, whereas a dipeptidyl peptidase-IV inhibitor, vildagliptin, does not. Diabetes. 2007;56: 8–15. [DOI] [PubMed] [Google Scholar]
- 73. Secher A, Jelsing J, Baquero AF, Hecksher-Sørensen J, Cowley MA, Dalbøge LS, et al. The arcuate nucleus mediates GLP-1 receptor agonist liraglutide-dependent weight loss. J Clin Invest. 2014;124: 4473–88. 10.1172/JCI75276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Cummings BP, Stanhope KL, Graham JL, Baskin DG, Griffen SC, Nilsson C, et al. Chronic administration of the glucagon-like peptide-1 analog, liraglutide, delays the onset of diabetes and lowers triglycerides in UCD-T2DM rats. Diabetes. 2010;59: 2653–61. 10.2337/db09-1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Astrup A, Rössner S, Van Gaal L, Rissanen A, Niskanen L, Al Hakim M, et al. Effects of liraglutide in the treatment of obesity: a randomised, double-blind, placebo-controlled study. Lancet. Elsevier Ltd; 2009;374: 1606–16. [DOI] [PubMed] [Google Scholar]
- 76. DeFronzo R, Bergenstal R, Bode B, Kushner R, Lewin A, Skjøth T, et al. Effects of Liraglutide 3.0 Mg and 1.8 Mg on Body Weight and Cardiometabolic Risk Factors in Overweight and Obese Adults with Type 2 Diabetes Mellitus (T2DM): The Scale Diabetes Randomized, Double-Blind, Placebo-Controlled, 56-Week Trial. Int Congr Endocrinol Endocr Soc Chicago, IL. 2014; [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.