

Abstract
The global widespread use of antimicrobials and accompanying increase in resistant bacterial strains is of major public health concern. Wastewater systems and wastewater treatment plants are considered a niche for antibiotic resistance genes (ARGs), with diverse microbial communities facilitating ARG transfer via mobile genetic element (MGE). In contrast to hospital sewage, wastewater from other health care facilities is still poorly investigated. At the instance of a nursing home located in south-west Germany, in the present study, shotgun metagenomics was used to investigate the impact on wastewater of samples collected up- and down-stream in different seasons. Microbial composition, ARGs and MGEs were analyzed using different annotation approaches with various databases, including Antibiotic Resistance Ontologies (ARO), integrons and plasmids. Our analysis identified seasonal differences in microbial communities and abundance of ARG and MGE between samples from different seasons. However, no obvious differences were detected between up- and downstream samples. The results suggest that, in contrast to hospitals, sewage from the nursing home does not have a major impact on ARG or MGE in wastewater, presumably due to much less intense antimicrobial usage. Possible limitations of metagenomic studies using high-throughput sequencing for detection of genes that seemingly confer antibiotic resistance are discussed.
Introduction
Antimicrobial consumption is constantly increasing, both in human and veterinary medicine, leading to a concomitant rise in antibiotic resistant bacteria [1]. It has been reported that antimicrobial resistance evolves in so called “genetic reactors” [2], e.g. farms, hospitals and long-term healthcare facilities (LTCF), which then act as a niche for bacterial exchange between individuals. The large quantities of antibiotics used in healthcare facilities exert a selection pressure on the microbial community. Furthermore, health care facilities emit the resistant bacteria that have evolved into the sewage system, where another potential recruitment pool for antibiotic resistant human pathogens can emerge [3,4]. Recent studies have reported on analyses of antibiotic resistant bacteria and resistance genes in sewage from hospitals [5–7], slaughterhouses and farms [8,9]. Despite the fact that resistant bacteria like methicillin-resistant Staphylococcus aureus, vancomycin-resistant enterococci and other multidrug-resistant enterics are endemic in the residents of LTCF [10–13], little is known about the release of resistant bacteria into the water cycle in sewage from LTCF. Indeed, in some countries the ageing population has prompted a considerable increase in the number of nursing homes. Thus, an investigation of corresponding wastewater systems is of interest.
In this study, a metagenomic approach was used to survey the taxonomic composition and content of resistance determinants in wastewater up- and downstream from a nursing home in south-west Germany. We identified differences in microbial communities in samples from different seasons but no obvious differences between the up- and downstream samples. Our results may suggest that the nursing home investigated has no marked influence on the wastewater system.
Materials and Methods
Ethics statement
The nursing home operator and the municipality responsible for the combined sewer system granted permission to analyze wastewater.
Water sampling
Sewage samples were collected from the combined sewer of a rural village (800 inhabitants) without any industry in south-west Germany (N: 48.40, E: 8.01). The water-samples were collected from two interconnected sites, one upstream (sample names: C1754, C1755), i.e. municipal sewage from the village, and the other 75 meters downstream, i.e. after wastewater discharge from a nursing home (280 residents) to the sewer system (sample names: C1756, C1757). According to the German Association for Water, Wastewater and Waste [14], the volume of wastewater calculated per day downstream of the nursing home is approximately 204 m3, of which roughly 25% originates from the nursing home.
The samples, each of which was taken every two hours over a 24-h period, were combined as “24-h composite samples”. To control for seasonal variations, samples were collected twice during the course of this study in November 2012 (November samples) and March 2013 (March samples).
DNA extraction and sequencing
The samples were stored and transported at + 4°C and processed immediately. Nine hundred ml were prepared for DNA extraction according to Lemarchand et al. [15]. Total DNA was extracted by use of the NucleoSpin-Soil Kit (Macherey-Nagel) and the concentrations were measured using a Qubit 2.0 Fluorometer and the Qubit dsDNA BR assay kit (Invitrogen). Additionally, gel electrophoresis was run to confirm the DNA integrity. The purified DNA was shipped to the ICMB (Institute of Clinical Molecular Biology, University of Kiel; Germany) for High-Throughput sequencing employing an Illumina-HighSeq sequencer (paired end sequencing; 101-bp reads; 6-bp Index sequence). The Illumina reads were preprocessed by means of the CASAVA-1.8.2. pipeline. Raw reads data have been deposited at the sequence read archive (SRA) of NCBI under Accession number SAMN02725022, SAMN02725023, SAMN02725024 and SAMN02725025.
Bioinformatics
For automated taxonomic and functional profiling, the sequences were submitted to MG-RAST (Meta Genome Rapid Annotation using Subsystem Technology, v3.2.2; website http://metagenomics.anl.gov; last access 16.06.2014) [16]. Subsequently, replicate sequences and reads with five or more ambiguous bases were eliminated.
Taxonomic analysis
The reads, clustered and identified as SSU-rDNA, were annotated against the M5RNA database in MG-RAST [17]. This database combined SSU-rDNA sequence databases RDP [18], Greengenes [19] and SILVA [20]. Taking a previous study [21], as reference, parameters in BLAT were adjusted to e-Value ≤ 10–5, identity ≥ 80% and alignment length of ≥ 50 bp.
Functional annotation
All reads were annotated against the SEED subsystem by means of BLAT [22] with the following parameters: e-Value ≤10–5, identity ≥ 60% and alignment length of ≥ 15 amino-acids (aa). The subsystems, i.e. clusters of functional roles, were created by curators to analyze annotations on differently detailed levels. In this study, we scrutinized at level 1 the subsystem “Virulence, Defense and Diseases” (“VDD”), which includes especially the level 2 subsystem “Resistance to antibiotics and toxic compounds” (RATC).
Assembly
CLC Genomic Workbench 6.5.1 was used to assemble the reads after removal of reads with more than 2 unknown bases. Assembly parameters were adjusted to a length fraction of ≥ 0.8, similarity to ≥ 0.95 and a minimum contig length of ≥ 250 bp [23].
Antibiotic resistance genes and mobile genetic elements
For subsequent analyses, reads were re-filtered using FASTX–Toolkit available at the Galaxy platform [24]. Reads with unknown nucleotides or having a score lower than minimum Illumina quality score (75% of all bases higher than 30) were removed [25]. The obtained reads and assembled contigs were blasted against antibiotic resistance genes and mobile genetic elements databases.
To generate a local BLAST database with antibiotic resistance genes and a controlled vocabulary of antibiotic resistance ontologies (ARO), the corresponding amino acid sequences, tagged specifically for antibiotic resistance, were downloaded from the Comprehensive Antibiotic Resistance Database (CARD) [26] (1,845 sequences—04.12.2013). This database was blasted using a BLASTx [27,28], implemented on the Galaxy-based framework Orion [29], against the unassembled reads and contigs. For extraction of the best hits the parameters were set according to Kristiansson et al. [3], e-Value ≤ 10–5, identity ≥ 90% and alignment length ≥ 25 aa. To check for signatures of mobile genetic elements, two additional databases were created. The first one was developed with 1,511 integron integrase gene sequences from the INTEGRALL database (kindly provided by Alexandra Moura) [30]. This was locally blasted using BLASTn [26, 27] against the unassembled and assembled reads (e-Value ≤10–5, identity ≥ 90% and alignment length of ≥ 50 bp) and the best hit in each alignment was selected [3]. For detection of reads and contigs associated with plasmids, the plasmid database available in NCBI (4,402 sequences—01.04.2014) was blasted against reads and contigs using BLASTn (cutoff e-Value ≤10–5; minimum similarity ≥ 95; alignment length above ≥ 90) and the best hit in each case was selected [31]. All local blast analyses were run on the Galaxy-based framework Orion.
Results
Sequencing and taxonomy
Illumina sequencing of the wastewater samples generated a total of 5.4 x 107 reads. The read number varied largely between the samples. Further details on QC-results and assembling are depicted in S1 Table. Two percent (± 0.3%) of the quality filtered reads from samples C1754, C1756, C1755 and C1757 contained SSU ribosomal RNA genes. They were assigned in MG-RAST to the M5RNA database in order to compare their domains. In all samples, 94.39% (± 1.78%) of the reads were assigned to the bacteria domain. The remaining reads were mapped to eukaryotes (2.37% ± 0.86%), archaea (0.18% ± 0.14%) and the virus domains (0.14% ± 0.08%) (Table 1). A high percentage of the reads from both November samples was assigned to the phylum Firmicutes (C1754: 77.43%; C1756: 71.85%) followed to a much lower extent by the phyla Actinobacteria (7.2 7%; 19.19%), Proteobacteria (9.55%; 4.48%), Bacteroidetes (1.60%; 0.69%) and Fusobacteria (0.24%; 0.08%) (Fig 1). In contrast, in both March samples (C1755, C1757), a considerably lower percentage of SSU rRNA-reads were assigned to Firmicutes (38.12%; 48.32%) and to a comparatively larger extent to Proteobacteria (38.71%; 32.83%). Remaining reads were classified to Actinobacteria (7.27%; 5.89%), Bacteroidetes (4.15%; 2.44%), and Fusobacteria (0.59%; 1.05%) (Fig 1). rRNA sequences, derived by unclassified bacteria and by SSU ribosomal RNA from other phyla together contained 12.16% and 9.47%, respectively, of the remaining rDNA reads. In contrast to the samples taken from both interconnected sampling sites on the same date, the taxonomic assignment showed high differences in phyla arrangement between the November and March samples.
Table 1. Alphabetically ordered functional gene categories in the wastewater samples (MG-RAST SEED Level 1 Distribution).
| Functional gene category | Percentage of assigned sequences | |||
|---|---|---|---|---|
| C1754 | C1756 | C1755 | C1757 | |
| Amino Acids and Derivatives | 8.35% | 8.14% | 8.94% | 8.87% |
| Carbohydrates | 11.74% | 12.21% | 10.01% | 10.37% |
| Cell Division and Cell Cycle | 1.73% | 1.78% | 1.51% | 1.55% |
| Cell Wall and Capsule | 3.54% | 3.54% | 3.43% | 3.51% |
| Clustering-based subsystems | 15.81% | 16.19% | 14.66% | 14.88% |
| Cofactors, Vitamins, Prosthetic Groups, Pigments | 5.90% | 5.82% | 6.70% | 6.49% |
| DNA Metabolism | 5.26% | 5.37% | 4.68% | 4.79% |
| Dormancy and Sporulation | 0.46% | 0.43% | 0.23% | 0.28% |
| Fatty Acids, Lipids, and Isoprenoids | 2.42% | 2.39% | 3.08% | 2.92% |
| Iron acquisition and metabolism | 0.66% | 0.60% | 0.65% | 0.66% |
| Membrane Transport | 3.09% | 3.25% | 3.36% | 3.35% |
| Metabolism of Aromatic Compounds | 0.97% | 0.89% | 1.95% | 1.76% |
| Miscellaneous | 7.78% | 8.11% | 7.86% | 7.82% |
| Motility and Chemotaxis | 0.64% | 0.59% | 1.04% | 0.94% |
| Nitrogen Metabolism | 1.22% | 1.14% | 1.85% | 1.73% |
| Nucleosides and Nucleotides | 3.44% | 3.59% | 3.05% | 3.16% |
| Phages, Prophages, Transposable elements, Plasmids | 2.29% | 1.86% | 1.79% | 1.73% |
| Phosphorus Metabolism | 0.72% | 0.76% | 0.78% | 0.79% |
| Photosynthesis | 0.05% | 0.04% | 0.08% | 0.08% |
| Potassium metabolism | 0.34% | 0.33% | 0.41% | 0.41% |
| Protein Metabolism | 9.04% | 8.99% | 7.93% | 8.22% |
| Regulation and Cell signaling | 1.30% | 1.32% | 1.52% | 1.51% |
| Respiration | 2.36% | 2.12% | 3.04% | 2.89% |
| RNA Metabolism | 4.59% | 4.46% | 4.14% | 4.24% |
| Secondary Metabolism | 0.32% | 0.36% | 0.31% | 0.32% |
| Stress Response | 2.10% | 2.03% | 2.62% | 2.55% |
| Sulfur Metabolism | 1.14% | 1.13% | 1.00% | 0.99% |
| Virulence, Disease and Defense | 2.73% | 2.56% | 3.38% | 3.20% |
Fig 1. Phylogenetic compositions of the bacteria domain in the samples.
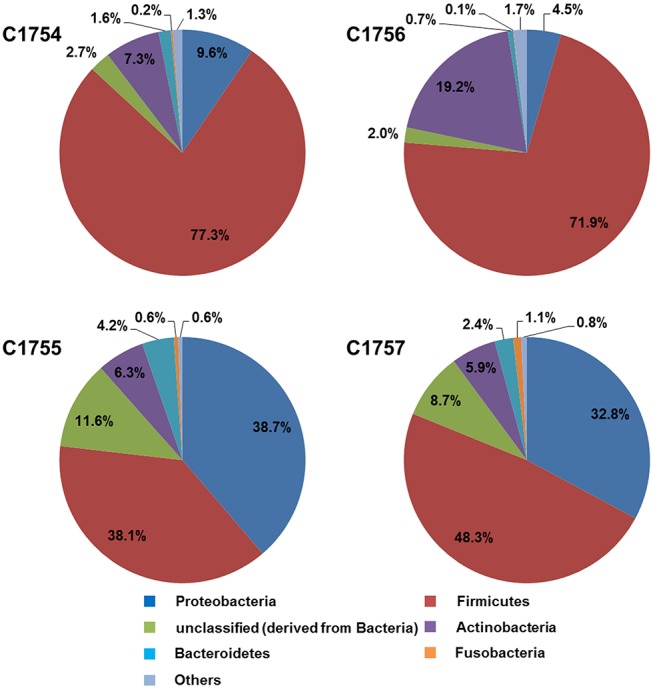
The samples were annotated using M5RNA database: C1754 and C1756: Upstream and downstream samples from November; C1755 and C1757: Upstream and downstream samples from March.
Functional analyses of wastewater sequences
After quality-filtering the reads by the MG-RAST pipeline, 30% to 49% of the remaining reads were annotated to level 1 subsystems (S1 Table). Based on SEED, the 12 most abundant subsystems were distributed similarly among all samples. Making up 41% to 45% of all aligned reads, the most frequent subsystems were “Clustering-based subsystems”, “Carbohydrates”, “Protein metabolism” and “Amino acids and derivatives” (Table 1). Between 2.5% and 3.3% of all level 1 reads were annotated to the subsystem “Virulence, disease and defense” (VDD) and scrutinized in depth. VDD consisted of seven level 2 clusters with the predominance of “Resistance to antibiotics and toxic compounds” (RATC). In all samples, around 68% (± 4%) of the VDD reads were assigned to the RATC subsystem (Table 2). This level 2 cluster contained genes assigned to resistance and adaption to antibiotics, metals and other environmental influences. A high percentage of assigned sequences belonged to five functional groups. The first group was attributed to “Resistance to fluoroquinolones” including DNA gyrase subunit A (gyrA), B (gyrB), Topoisomerase IV subunit A (parC) and B (parE) (C1754: 22.61%; C1756: 24.78%; C1755:13.48%; C1757:14.97%). The second was “Multidrug resistance efflux pumps”, such as members of the multi antimicrobial extrusion protein family and resistance-nodulation-cell division superfamily (14.67%; 12.00%; 13.18%; 13.70%). The third was “Copper homeostasis” (14.24%; 14.14%; 18.11%; 16.90%). The fourth was “BlaR1 family regulatory sensor-transducer disambiguation” mainly represented by Cu2+-exporting ATPase and rarely by regulatory sensor-transducer of the blaR1/mecR1 family (12.00%; 12.81%; 12.26%; 11.67%) and the fifth was “cobalt-zinc-cadmium resistance” (10.07%; 7.85%; 18.29%; 16.51%) (Table 2).
Table 2. Relative abundance of VDD and RATC genes (≥1%) in the samples.
| Subsystems | C1754 | C1756 | C1755 | C1757 |
|---|---|---|---|---|
| Level 1 VDD-hits (%) | 2.73 | 2.56 | 3.38 | 3.20 |
| Level 2 RATC within VDD (%) | 68.22 | 64.98 | 71.29 | 70.33 |
| Level 3 Categories within RATC (%) a) | ||||
| Resistance to fluoroquinolones | 22.61 | 24.78 | 13.48 | 14.97 |
| Multidrug Resistance Efflux Pumps | 14.67 | 12.00 | 13.18 | 13.70 |
| Copper homeostasis | 14.24 | 14.14 | 18.11 | 16.90 |
| BlaR1 Family Regulatory Sensor-transducer Disambiguation | 12.00 | 12.81 | 12.26 | 11.67 |
| Cobalt-zinc-cadmium resistance | 10.07 | 7.85 | 18.29 | 16.51 |
| Methicillin resistance in Staphylococci | 5.71 | 6.55 | 3.68 | 4.04 |
| Arsenic resistance | 3.56 | 3.29 | 2.89 | 3.17 |
| Multidrug efflux pump in Campylobacter jejuni (CmeABC operon) | 2.78 | 1.93 | 4.67 | 4.89 |
| Resistance to Vancomycin | 1.95 | 1.96 | 0.19 | 0.41 |
| Erythromycin resistance | 1.81 | 2.05 | 1.14 | 1.14 |
| Copper homeostasis: copper tolerance | 1.77 | 1.72 | 2.06 | 2.10 |
| Beta-lactamase | 1.46 | 1.76 | 1.91 | 1.98 |
| Cadmium resistance | 1.39 | 1.42 | 0.52 | 0.74 |
| Bile hydrolysis | 1.32 | 1.77 | 0.43 | 0.58 |
| The mdtABCD multidrug resistance cluster | 1.07 | 0.88 | 2.47 | 2.46 |
| Streptococcus pneumoniae Vancomycin Tolerance Locus | 0.82 | 1.42 | 0.19 | 0.20 |
| Mercury resistance operon | 0.97 | 1.27 | 1.89 | 1.73 |
| Mercuric reductase | 0.78 | 1.13 | 1.40 | 1.30 |
a)Legend: (RATC ≥1%).
Abundance and ARG diversity
After blasting by means of “BLASTX” against CARD, the best hits were normalized to the total number of quality filtered reads and later referred to as relative abundance. This abundance of reads was subsequently expressed in parts per million (ppm) according to Ma et al. [32]. Analyses of the four samples revealed reads of 526 ppm in sample C1754, 511 ppm in C1756, 639 ppm in C1755 and 710 ppm in C1757. Moreover, the genes were subsequently classified into resistance-related gene (RRG) clusters depending on their ARO numbers (Fig 2). In up- and downstream samples, we found 98, respectively 164 different RRGs corresponding to November samples, and 103, respectively 134 corresponding to March samples. Apparently, the inflows from nursing home sewage had no principal effect on RRG abundance in municipal wastewater. However, their diversity increased slightly and their composition changed. We classified the RRGs into groups with similar resistance spectrum (resistance against antimicrobial compounds from the same class). In all samples, rpoB genes prevailed, which, provided particular mutations have occurred, can confer resistance against antibiotics of the rifamycine group (Table 3).
Fig 2. Abundance and diversity of annotated reads.
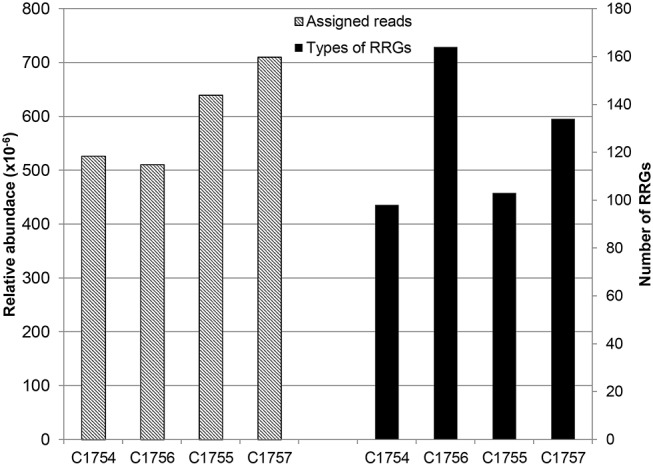
Abundance (left) and diversity (right) of reads annotated against CARD are depicted. The hits were normalized against the total number of reads in each sample after QC-filtering.
Table 3. Distribution of RRG-groups in the samples.
| RRG groups | C1754 [ppm] | C1756 [ppm] | C1755 [ppm] | C1757 [ppm] |
|---|---|---|---|---|
| Rifamycine | 203.2 | 155.9 | 243.4 | 245.1 |
| Multidrug | 70.3 | 86.1 | 87.0 | 136.3 |
| Quinolone | 58.8 | 45.6 | 102.5 | 102.7 |
| Tetracycline | 94.2 | 123.5 | 37.4 | 58 |
| Macrolides | 43.7 | 42.6 | 64.8 | 56.7 |
| Aminocoumarins | 27.7 | 25.3 | 50.6 | 45.1 |
| Beta-Lactam | 11.6 | 14.4 | 24.7 | 29.4 |
| Aminoglycosides | 7.6 | 8.7 | 17.6 | 19.3 |
| Sulfonamides | 1.5 | 3.3 | 2.1 | 6.8 |
| Chloramphenicol | 2.0 | 1.3 | 4.2 | 4.1 |
| Vancomycin | 5.7 | 3.3 | 1.3 | 2.7 |
| Others | 0.6 | 0.7 | 4.2 | 3.8 |
The number of hits against the CARD in each RRG-group is presented as hits per million reads (ppm).
For instance, in the November samples, the relative abundance of reads related to the rifamycine group decreased after the inflow. Moreover, March samples showed a smaller variation of reads tagged to the RRG group of multidrug resistance.
Notably, tetracycline related reads such as tetA, tetB(P), tetC, tetD, tetG. tetL, tetK, tetM, tetO, tetQ, tetS, tetT, tetW, tetX, tet32, tet39, tet43 and tet45 showed an increment of over 30% in both downstream samples. The other RRG-groups did not exhibit any remarkable changes.
The resistance genes with the highest frequency in the samples are presented in Fig 3. The RNA polymerase encoding rpoB gene, which can confer resistance against rifamycin in the presence of few particular mutations, was detected in all samples with an abundance of over 150 ppm (S2 Table). The resistance genes tetW and gyrA had a relative abundance higher than 50 ppm (S2 Table) in at least one sample from November and March. The following relevant RRGs [33] had a relative abundance higher than 10 ppm: aminoglycoside adenyltransferases, macrolide resistance genes ermB and macB, the multidrug gene acrB from the resistance-nodulation-division (RND) family. Furthermore, the quinolone resistance genes parC and both tetracycline genes tetO and tet32 were also detected with the same relative abundance. In the November samples, OXA gene family reads were present with a relative abundance lower than 10 ppm, in contrast to the March samples with a relative abundance higher than 10 ppm. This gene family belongs to β-lactam resistance gene ontologies among other detected determinants coding for β-lactamases (CTX-M, GES, AER CARB, PSE, R1, OMP, CphA, PC1, SHV, FEZ, TEM, AmpC and mecA) and was the one with the highest abundance in this group. Overall, sulfonamide resistance genes such as folP, sul1, sul2 and vanocmycin resistance related genes such as vanC, vanR, vanS, vanT, vanW vanXYC, vanY and vanZ were present below 5 ppm or at the detection limit (S2 Table).
Fig 3. Abundance of the 12 most frequent ARGs types with the highest blast hits number (Relative abundance in any sample ≥10 ppm).
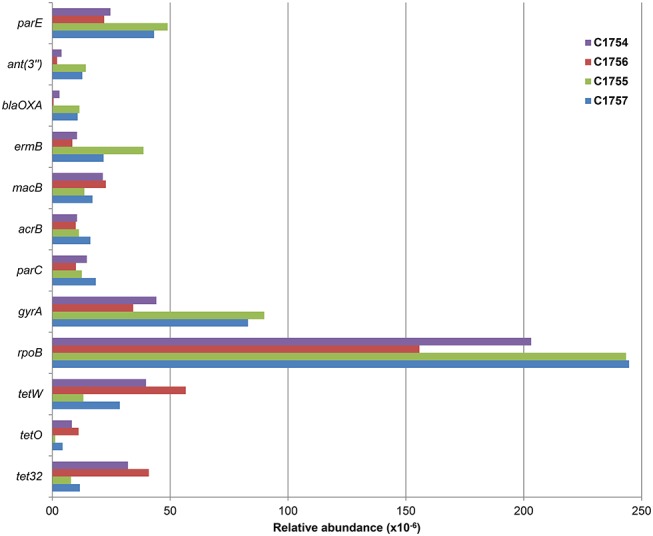
Reads assembled into contigs from C1754, C1756, C1755 and C1757 were blasted against CARD database with a total of 13, 193, 29 and 80 aligned contigs, respectively, and resulted in different types and numbers of RRG (8, 71, 19 and 80) (S2 Table).
Abundance of mobile genetic elements
Reads of seven to approximately 25 ppm could be assigned to integrase coding sequences of the INTEGRALL database (Fig 4). The November samples (C1754 and C1756) and the March samples (C1755 and C1757) were mapped against known integrase encoding genes with reads of 7, 8, 24 and 25 ppm, respectively. About 74% to 85% of the reads were assigned to class 1 integrase gene intI1. The remaining reads were associated to intI1delta, intI2, intI3, intIPac, groEL/intI1, intIA and intI ( S3 Table). Furthermore, assembled reads of all samples were simultaneously blasted against the integrall database. None of the 43,808 contigs from C1754 could be identified as encoding an integrase gene. Only 4 of 234,564 contigs (C1756), 4 of 23,420 contigs (C1755) and 7 of 74,589 (C1757) matched known integrase (S3 Table).
Fig 4. Relative abundance of integronase genes (A) and plasmids (B).
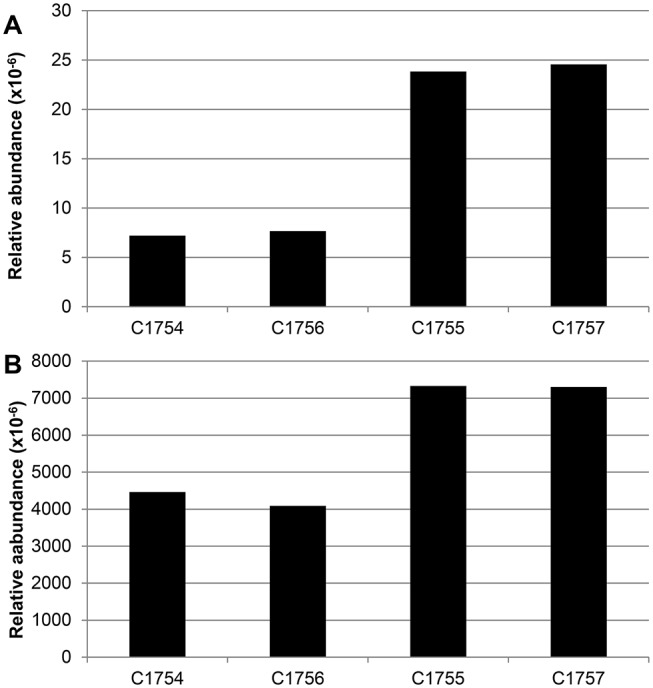
The hits were normalized against the total number of reads in each sample after QC-filtering.
Moreover, reads of 4460 and 4087 ppm from C1754 and C1756 respectively matched with plasmid sequences available in the NCBI RefSeq database. Of these, 31% were assigned to an unnamed plasmid of Eubacterium eligens ATCC 27750 (NC_012780.1). Interestingly, a higher number of reads from C1755 (7329 ppm) and C1757 (7303 ppm) were annotated (Fig 4), where 8.5% were assigned to the plasmid pKV29 of β-Proteobacterium Delftia sp. Strain KV29 (NC_019312.1). Notably, we did not find any dominating plasmids in either March samples (S4 Table).
Finally, after blasting of contigs against the plasmid database, we founded 210 contigs (C1754), 1440 contigs (C1756), 292 contigs (C1755) and 575 contigs (C1757) matching with 86, 366, 134 and 264 plasmids, respectively (S4 Table).
Discussion
In this study on the potential introduction of ARG into the water cycle by medical facilities other than hospitals, we investigated differences between the microbial community in up- and downstream samples of wastewater from a nursing home collected in two different seasons. We used the M5RNA database available on MG-RAST, which allows examination of microbial composition in metagenomic wastewater samples. In accordance with a recent study, which assumed 100 bp sequences as too short for accurate identification at sub-phyla levels [34], we only analyzed the community at the phylum level, and not in greater detail at the level of order or even genus. Our results did not show any differences at domain level when comparing samples from both sampling sites in the same season. However, comparison of the bacteria phyla distribution showed a seasonal difference between samples. The phylum Firmicutes dominated in the November samples. Firmicutes has been reported as the dominant phylum in wastewater samples with a high level of pollution and extreme conditions [35]. Firmicutes are frequently found alongside the phyla Bacteroidetes in human and animal feces [36–39]. Some studies describe them as being one of the dominating phyla in raw wastewater, while others describe low abundance [35,39–42]. Beside these phyla, it has been shown that Proteobacteria, belong to the microbial community of the sewer system and, dominate in sewage samples [35,42,43], although only found to represent c. 0.1% of the species in the human colon [37,42]. The low abundance of Proteobacteria and high number of feces-associated bacterial groups found in the November wastewater samples may indicate a domination of bacteria of fecal origin over other environmental bacteria. In contrast, the March sewage samples showed a different phyla distribution, with a similar number of reads linked to Firmicutes and to Proteobacteria in C1755 and an approximately comparable distribution in C1757. Our results indicate the presence of fecal bacteria independent of sampling upstream or downstream from the nursing home, yet to a different extent on both sampling dates. No extreme environmental conditions in the municipal sewage system were reported or investigated in further tests (data not shown). Due to the limited number of samples and without the possibility of statistical power tests our taxonomic analyses did not reveal an apparent alteration of the municipal sewage system community by the medical facility.
MG-RAST was used for functional gene annotations, especially those related to resistance against antibiotics and other compounds. Around 30% to 49% of the reads, which passed the MG-RAST quality control filter, were assigned to genes from the SEED level 1. These values were markedly higher than those reported or calculated on Illumina reads from other studies [25,34]. Our results were in accordance with a previous study by Wang et al. [25], where the number of annotated reads was closely related depending on the sample origin and the bacterial composition.
Regarding level 1 subsystem distribution, a large number of reads was assigned to the subsystem with basic prokaryotic processes such as carbohydrate metabolism. However, the samples did not show obvious differences at this level. Neither did the subsystem “Virulence, Disease and Defense” (VDD), which includes the level 2 system “Resistance to antibiotics and toxic compounds” (RATC), show any differences between the up- and downstream samples. Furthermore, in all four samples, the major number of reads in the subsystem VDD was assigned to the RATC system. For instance, resistance to fluoroquinolones, which proved to be one of the highest portions of RATC-assigned reads, has already been reported as dominating in the sludge of wastewater treatment plant [25]. The others categories were multidrug resistance efflux pumps, blaR1 family regulatory sensor-transducer disambiguation, methicillin resistance in staphylococci, multidrug efflux pump in Campylobacter jejuni (CmeABC operon), resistance to vancomycin, erythromycin, beta-lactamase and the mdtABCD multidrug resistance cluster. Overall, the samples were classified in the same range without any noteworthy differences in the number of reads. In addition to ARG, the RATC system includes heavy metal resistances against cobalt, zinc, copper and others. Different authors have already reported the traits resistance to antibiotics and heavy metals to be linked, due to their close association to mobile genetic elements and a possible co-selection with ARGs [44–46]. Based on this, heavy metal resistance genes could be added to the ARG index for better description of the antibiotics resistance potential [47]. Therefore, we included the reads associated with metal resistance in our analyses. At least 1% of reads in the RATC subsystem were assigned to copper [48,49], cobalt [44], zinc [50], cadmium [51] and arsenic resistance [52]. Like ARGs no apparent differences were detected between samples collected at the same time.
We used the strict and unambiguous resistance ontology of the CARD database to annotate genes possibly involved in contamination of wastewater with ARG. Up- and downstream samples contained ARG without salient differences between one another. These data may indicate that the ARG load from the nursing home did not have a detectable influence on the municipal wastewater. Notably, this result is not comparable to a hospital analysis, where wastewater can carry 27 times the number of antibiotic resistant bacteria than urban wastewater [5]. A conceivable reason could be the high use of antimicrobials in hospitals, whereas in Germany the antibiotic use point prevalence (defined daily doses per 1,000 residents per day) is relatively low in nursing homes [53]. Although the increase in ARG found between the up- and downstream samples was not considerable, slight differences were by all means detected between both sampling sites. Moreover, a large number of reads were assigned to genes involved in rifamycine resistance without any notable differences between samples. Rifamycine resistance genes, i.e. RNA polymerase subunit B encoding genes, are ubiquitous in many environments, and are often detected at the highest level [32]. Furthermore, no obvious differences were detected in genes causing resistance against antibiotics such as β-lactams, macrolides or sulfonamides found in samples taken up- and downstream. However, on both sampling dates there was an increase of tetracycline resistance genes and sequences encoding multidrug resistance systems downstream from the nursing home [54]. Both types of resistance are of medical importance, however the increase detected in the nursing home’s inflow was considered too small to allow to draw a reliable conclusion on the base of the limited sample size.
There are many differences in the output of metagenome annotation when using the MG-RAST platform or blast against CARD. Using CARD, the relative abundance of genes associated with resistance against tetracycline showed a similar abundance as genes associated with the quinolone RGG. In contrast, the subcategory “Resistance to fluoroquinolones” showed the highest relative abundance of all antibiotic resistance subcategories in MG-RAST; however, no resistance to tetracycline was reported in this RATC subcategory. These differences are primarily due to the organization of the databases. The SEED database focuses on the annotation of core metabolism rather than on categories such as resistance groups. Furthermore, not all related genes are summarized in one comprehensive category [16]. For instance, tetracycline resistance genes like tetO, are neither included in RATC-Category nor in the Level1 subsystem “VDD” as expected; rather they are located in sub-categories of the level 1 subsystem “Protein Metabolism”. Basically, the differences between the results are a consequence of the differences in the structure and design of the databases. Again, the dissimilarities in the used cutoffs and possible gene coverages render the results incomparable. MG-RAST provides a good overview of all the genes present in the metagenome. Special databases like CARD are nonetheless required especially for deep ARG analysis.
Mobile genetic elements (MGE) like integrons or plasmids play an important role in the transfer and spread of genes between bacteria, especially those related to antibiotic resistance [55]. In this study, the abundance of MGE was investigated to describe the potential of gene mobility in the metagenome samples, and to control for a possible spread of these elements by nursing home residents into the sewage [3,56]. The integrase genes, which were selected as unambiguous markers for integrons, showed nearly the same abundance at the interconnected sampling sites, albeit marked differences on both sampling dates, which might be in line with the different microbial composition in both sampling seasons. Additionally, IntI1 was the dominating integrase gene in all samples, confirming its higher prevalence in wastewater-associated environments [25,31].
Like integrase genes, plasmids were present to a similar extent at the interconnected sample sites, although the extent to which they were present differed in the two sampling seasons and the nursing home inflow showed no influence on the abundance of plasmids. The annotated plasmids were classified according to their host. The resulting taxonomy results revealed a notable relation between dominating phyla, based on the analysis of SSU reads and plasmids with high frequency. Comparison of the outcome of this plasmid BLAST with the taxonomy results revealed a relation between dominating phylum and the most frequent plasmid host. An unnamed plasmid of Eubacterium eligens [57], a species belonging to the phylum Firmicutes [58], was found in more than 30% of the total BLAST hits in the November samples. This result is in accordance with the high prevalence of Firmicutes SSU-reads in these samples. Yet, we did not detect similarly prevalent phyla in the March samples.
Besides providing new insights and opportunities, microbial metagenome studies are challenging and confined in their reproducibility [59]. However, the similar distribution of the various genetic traits found at both interconnected sampling sites argues for the reproducibility of the approach used in this work. Indeed, wastewater changes continuously, for which reason we collected “24-hour composite samples” in different seasons to avoid a bias due to differences in the microbial load of the wastewater at different times of day or seasonal variations. Despite these precautions, neither comparison of up- or downstream samples, nor samples taken from different seasons revealed major differences in ARG. Another limitation here is the read length of 100 bases, which, as mentioned above, might not be sufficient to provide a reliable annotation beyond taxon phylum [23]. Furthermore, housekeeping genes like the RNA polymerase subunit B encoding gene (rpoB) [60] confer resistance against antibiotics of the rifamycine group only, if mutations in a few particular codons are present [61,62]. Consequently, reads were assigned to resistance genes irrespective of whether the sequence contained a corresponding mutation or not. Similar restrictions are encountered with genes assigned to resistance against aminocumarines [63] and quinolones [64], which differ in only a few mutations from their susceptible types. Also, multidrug efflux systems, like the Resistance-Nodulation-Division Family, are usually present in the bacterial genome, and only overexpression of this pump, which cannot be detected without expression experiments, confers resistance against different antibiotics [65].
In an attempt to improve the sequence length and results, reads were assembled in contigs. Only about 23% to 51% of the reads were assembled in contigs larger than 250 bp. This leads to a loss of more than 49% of the sequence data. Previous studies reported similar drawbacks in assembling environmental sequences and explained it with the high bacterial diversity in environmental samples [43,66] (S1 Table). Additionally, the annotations of unassembled reads and contigs were not comparable because the information about the frequencies of reads was lost after assembly [16].
This study was conducted at the instance of a 280-resident nursing home to investigate the impact of ARG load in wastewater from a long-term health care facility on municipal sewage of an 800-inhabitant village. The results demonstrate that the microbial composition of the wastewater did not differ before or after the inflow from the nursing home. However, the study revealed seasonal changes. Different approaches in annotation to ARG and MGE also revealed mainly seasonal differences and only minute sampling site differences. In contrast to sewage from hospitals, which is well known to introduce ARG into the water cycle, similar pollution by a long term health care facility was not detected. Further studies with a higher number of interconnected samples should be performed for statistical proof.
Supporting Information
Details about the sequence quality control (QC) steps and the de novo assembling by use of the CLC Genomic Workbench 6.5.1 (Number of reads).
(DOCX)
Matched high-throughput sequencing reads/contigs of the samples C1754, C1755, C1756 and C1757 against CARD.
(DOCX)
Matched high-throughput sequencing reads/contigs of the samples C1754, C1755, C1756 and C1757 against the INTEGRALL database.
(DOCX)
Assigned high-throughput sequencing reads/contigs of the samples C1754, C1755, C1756 and C1757 against the RefSeq plasmid.
(DOCX)
Acknowledgments
The authors would like to thank Dr. A. Rehman, from the Christian-Albrechts-University, Kiel for helpful comments and Deborah Lawrie-Blum for assistance with preparation of the manuscript.
Data Availability
Raw reads data have been deposited at the sequence read archive (SRA) of NCBI under Accession numbers SAMN02725022, SAMN02725023, SAMN02725024 and SAMN02725025).
Funding Statement
This study was funded by the German Federal Ministry of Education and Research (02WRS1280A - J) as part of the research program “Risk Management of Emerging Compounds and Pathogens in the Water Cycle (RiSKWa)”.
References
- 1. Meyer E, Gastmeier P, Deja M, Schwab F. Antibiotic consumption and resistance: Data from Europe and Germany. Int J Med Microbiol. 2013;303: 388–395. 10.1016/j.ijmm.2013.04.004 [DOI] [PubMed] [Google Scholar]
- 2. Baquero F, Martínez J-L, Cantón R. Antibiotics and antibiotic resistance in water environments. Curr Opin Biotechnol. 2008;19: 260–265. 10.1016/j.copbio.2008.05.006 [DOI] [PubMed] [Google Scholar]
- 3. Kristiansson E, Fick J, Janzon A, Grabic R, Rutgersson C, Weijdegård B, et al. Pyrosequencing of Antibiotic-Contaminated River Sediments Reveals High Levels of Resistance and Gene Transfer Elements. PLoS ONE. 2011;6: e17038 10.1371/journal.pone.0017038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhang XX, Zhang T, Fang HH. Antibiotic resistance genes in water environment. Appl Microbiol Biotechnol. 2009;82: 397–414. 10.1007/s00253-008-1829-z [DOI] [PubMed] [Google Scholar]
- 5.Bréchet C, Plantin J, Sauget M, Thouverez M, Talon D, Cholley P, et al. Wastewater Treatment Plants Release Large Amounts of Extended-Spectrum β-Lactamase–Producing Escherichia coli Into the Environment. Clin Infect Dis. 2014; ciu190. 10.1093/cid/ciu190 [DOI] [PubMed]
- 6. Jakobsen L, Sandvang D, Hansen LH, Bagger-Skjot L, Westh H, Jorgensen C, et al. Characterisation, dissemination and persistence of gentamicin resistant Escherichia coli from a Danish university hospital to the waste water environment. Env Int. 2008;34: 108–115. [DOI] [PubMed] [Google Scholar]
- 7. Yang CM, Lin MF, Liao PC, Yeh HW, Chang BV, Tang TK, et al. Comparison of antimicrobial resistance patterns between clinical and sewage isolates in a regional hospital in Taiwan. Lett Appl Microbiol. 2009;48: 560–565. 10.1111/j.1472-765X.2009.02572.x [DOI] [PubMed] [Google Scholar]
- 8. Sabate M, Prats G, Moreno E, Balleste E, Blanch AR, Andreu A. Virulence and antimicrobial resistance profiles among Escherichia coli strains isolated from human and animal wastewater. Res Microbiol. 2008;159: 288–293. 10.1016/j.resmic.2008.02.001 [DOI] [PubMed] [Google Scholar]
- 9. Martins da Costa PM, Vaz-Pires PM, Bernardo FM. Antibiotic resistance of Enterococcus spp. isolated from wastewater and sludge of poultry slaughterhouses. J Env Sci Health B. 2006;41: 1393–1403. [DOI] [PubMed] [Google Scholar]
- 10. Manzur A, Gavalda L, Ruiz de Gopegui E, Mariscal D, Dominguez MA, Perez JL, et al. Prevalence of methicillin-resistant Staphylococcus aureus and factors associated with colonization among residents in community long-term-care facilities in Spain. Clin Microbiol Infect. 2008;14: 867–872. 10.1111/j.1469-0691.2008.02060.x [DOI] [PubMed] [Google Scholar]
- 11. Pop-Vicas A, Mitchell SL, Kandel R, Schreiber R, D’Agata EMC. Multidrug-Resistant Gram-Negative Bacteria in a Long-Term Care Facility: Prevalence and Risk Factors. J Am Geriatr Soc. 2008;56: 1276–1280. 10.1111/j.1532-5415.2008.01787.x [DOI] [PubMed] [Google Scholar]
- 12. Arvand M, Moser V, Pfeifer Y. Prevalence of extended-spectrum-β-lactamase-producing Escherichia coli and spread of the epidemic clonal lineage ST131 in nursing homes in Hesse, Germany. J Antimicrob Chemother. 2013;68: 2686–2688. 10.1093/jac/dkt226 [DOI] [PubMed] [Google Scholar]
- 13. Gruber I, Heudorf U, Werner G, Pfeifer Y, Imirzalioglu C, Ackermann H, et al. Multidrug-resistant bacteria in geriatric clinics, nursing homes, and ambulant care—Prevalence and risk factors. Int J Med Microbiol. 2013;303: 405–409. 10.1016/j.ijmm.2013.05.002 [DOI] [PubMed] [Google Scholar]
- 14. ATV. ATV—Working sheet A118: Hydraulic design and proof of sewerage networks 1st ed. Hennef, Germany: German Association for Water, Wastewater and Waste; 2006. [Google Scholar]
- 15. Lemarchand K, Berthiaume F, Maynard C, Harel J, Payment P, Bayardelle P, et al. Optimization of microbial DNA extraction and purification from raw wastewater samples for downstream pathogen detection by microarrays. J Microbiol Methods. 2005;63: 115–126. 10.1016/j.mimet.2005.02.021 [DOI] [PubMed] [Google Scholar]
- 16. Meyer F, Paarmann D, D’Souza M, Olson R, Glass EM, Kubal M, et al. The metagenomics RAST server—a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics. 2008;9: 386 10.1186/1471-2105-9-386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kent WJ. BLAT—The BLAST-Like Alignment Tool. Genome Res. 2002;12: 656–664. 10.1101/gr.229202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cole JR, Wang Q, Fish JA, Chai B, McGarrell DM, Sun Y, et al. Ribosomal Database Project: data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014;42: D633–642. 10.1093/nar/gkt1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, et al. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl Environ Microbiol. 2006;72: 5069–5072. 10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41: D590–D596. 10.1093/nar/gks1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bai Y, Liu R, Liang J, Qu J. Integrated Metagenomic and Physiochemical Analyses to Evaluate the Potential Role of Microbes in the Sand Filter of a Drinking Water Treatment System. PLoS ONE. 2013;8: e61011 10.1371/journal.pone.0061011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang H-Y, Cohoon M, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005;33: 5691–5702. 10.1093/nar/gki866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hewson I, Eggleston EM, Doherty M, Lee DY, Owens M, Shapleigh JP, et al. Metatranscriptomic Analyses of Plankton Communities Inhabiting Surface and Subpycnocline Waters of the Chesapeake Bay during Oxic-Anoxic-Oxic Transitions. Appl Environ Microbiol. 2014;80: 328–338. 10.1128/AEM.02680-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goecks J, Nekrutenko A, Taylor J, $author.lastName $author firstName. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11: R86 10.1186/gb-2010-11-8-r86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Z, Zhang X-X, Huang K, Miao Y, Shi P, Liu B, et al. Metagenomic profiling of antibiotic resistance genes and mobile genetic elements in a tannery wastewater treatment plant. PloS One. 2013;8: e76079 10.1371/journal.pone.0076079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McArthur AG, Waglechner N, Nizam F, Yan A, Azad MA, Baylay AJ, et al. The Comprehensive Antibiotic Resistance Database. Antimicrob Agents Chemother. 2013;57: 3348–3357. 10.1128/AAC.00419-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cock PJA, Grüning BA, Paszkiewicz K, Pritchard L. Galaxy tools and workflows for sequence analysis with applications in molecular plant pathology. PeerJ. 2013;1: e167 10.7717/peerj.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10: 421 10.1186/1471-2105-10-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cuccuru G, Orsini M, Pinna A, Sbardellati A, Soranzo N, Travaglione A, et al. Orione, a web-based framework for NGS analysis in microbiology. Bioinforma Oxf Engl. 2014; 10.1093/bioinformatics/btu135 [DOI] [PMC free article] [PubMed]
- 30. Moura A, Soares M, Pereira C, Leitão N, Henriques I, Correia A. INTEGRALL: a database and search engine for integrons, integrases and gene cassettes. Bioinformatics. 2009;25: 1096–1098. 10.1093/bioinformatics/btp105 [DOI] [PubMed] [Google Scholar]
- 31. Zhang T, Zhang X-X, Ye L. Plasmid Metagenome Reveals High Levels of Antibiotic Resistance Genes and Mobile Genetic Elements in Activated Sludge. PLoS ONE. 2011;6 10.1371/journal.pone.0026041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ma L, Li B, Zhang T. Abundant rifampin resistance genes and significant correlations of antibiotic resistance genes and plasmids in various environments revealed by metagenomic analysis. Appl Microbiol Biotechnol. 2014;98: 5195–5204. 10.1007/s00253-014-5511-3 [DOI] [PubMed] [Google Scholar]
- 33. Szczepanowski R, Linke B, Krahn I, Gartemann KH, Gutzkow T, Eichler W, et al. Detection of 140 clinically relevant antibiotic-resistance genes in the plasmid metagenome of wastewater treatment plant bacteria showing reduced susceptibility to selected antibiotics. Microbiology. 2009;155: 2306–2319. 10.1099/mic.0.028233-0 [DOI] [PubMed] [Google Scholar]
- 34. Chao Y, Ma L, Yang Y, Ju F, Zhang X-X, Wu W-M, et al. Metagenomic analysis reveals significant changes of microbial compositions and protective functions during drinking water treatment. Sci Rep. 2013;3 10.1038/srep03550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang ZH, Yang JQ, Zhang DJ, Zhou J, Zhang CD, Su XR, et al. Composition and structure of microbial communities associated with different domestic sewage outfalls. Genet Mol Res GMR. 2014;13: 7542–7552. 10.4238/2014.September.12.21 [DOI] [PubMed] [Google Scholar]
- 36. Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-Bacterial Mutualism in the Human Intestine. Science. 2005;307: 1915–1920. 10.1126/science.1104816 [DOI] [PubMed] [Google Scholar]
- 37. Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the Human Intestinal Microbial Flora. Science. 2005;308: 1635–1638. 10.1126/science.1110591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lamendella R, Domingo JWS, Ghosh S, Martinson J, Oerther DB. Comparative fecal metagenomics unveils unique functional capacity of the swine gut. BMC Microbiol. 2011;11: 103 10.1186/1471-2180-11-103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Singh KM, Jakhesara SJ, Koringa PG, Rank DN, Joshi CG. Metagenomic analysis of virulence-associated and antibiotic resistance genes of microbes in rumen of Indian buffalo (Bubalus bubalis). Gene. 2012;507: 146–151. 10.1016/j.gene.2012.07.037 [DOI] [PubMed] [Google Scholar]
- 40. Cai L, Ju F, Zhang T. Tracking human sewage microbiome in a municipal wastewater treatment plant. Appl Microbiol Biotechnol. 2014;98: 3317–3326. 10.1007/s00253-013-5402-z [DOI] [PubMed] [Google Scholar]
- 41. Shanks OC, Newton RJ, Kelty CA, Huse SM, Sogin ML, McLellan SL. Comparison of the Microbial Community Structures of Untreated Wastewaters from Different Geographic Locales. Appl Environ Microbiol. 2013;79: 2906–2913. 10.1128/AEM.03448-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. McLellan SL, Huse SM, Mueller-Spitz SR, Andreishcheva EN, Sogin ML. Diversity and population structure of sewage-derived microorganisms in wastewater treatment plant influent. Environ Microbiol. 2010;12: 378–392. 10.1111/j.1462-2920.2009.02075.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sanapareddy N, Hamp TJ, Gonzalez LC, Hilger HA, Fodor AA, Clinton SM. Molecular Diversity of a North Carolina Wastewater Treatment Plant as Revealed by Pyrosequencing. Appl Environ Microbiol. 2009;75: 1688–1696. 10.1128/AEM.01210-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Baker-Austin C, Wright MS, Stepanauskas R, McArthur JV. Co-selection of antibiotic and metal resistance. Trends Microbiol. 2006;14: 176–182. 10.1016/j.tim.2006.02.006 [DOI] [PubMed] [Google Scholar]
- 45. Jury KL, Vancov T, Stuetz RM, Khan SJ. Antibiotic resistance dissemination and sewage treatment plants In: Méndez-Vilas A, editor. Current research, technology and education topics in applied microbiology and microbial biotechnology. Badajoz, Spain: Formatex Research Center; 2010. pp. 509–519. Available: http://www.formatex.info/microbiology2/509-519.pdf [Google Scholar]
- 46. Wright GD. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat Rev Microbiol. 2007;5: 175–186. 10.1038/nrmicro1614 [DOI] [PubMed] [Google Scholar]
- 47. Port JA, Wallace JC, Griffith WC, Faustman EM. Metagenomic Profiling of Microbial Composition and Antibiotic Resistance Determinants in Puget Sound. PLoS ONE. 2012;7: e48000 10.1371/journal.pone.0048000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Finan TM, Weidner S, Wong K, Buhrmester J, Chain P, Vorhölter FJ, et al. The complete sequence of the 1,683-kb pSymB megaplasmid from the N2-fixing endosymbiont Sinorhizobium meliloti . Proc Natl Acad Sci. 2001;98: 9889–9894. 10.1073/pnas.161294698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Martins VV, Zanetti MOB, Pitondo-Silva A, Stehling EG. Aquatic environments polluted with antibiotics and heavy metals: a human health hazard. Environ Sci Pollut Res. 2014;21: 5873–5878. 10.1007/s11356-014-2509-4 [DOI] [PubMed] [Google Scholar]
- 50. Conejo MC, Garcia I, Martinez-Martinez L, Picabea L, Pascual A. Zinc Eluted from Siliconized Latex Urinary Catheters Decreases OprD Expression, Causing Carbapenem Resistance in Pseudomonas aeruginosa . Antimicrob Agents Chemother. 2003;47: 2313–2315. 10.1128/AAC.47.7.2313-2315.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. O’Driscoll J, Glynn F, Fitzgerald GF, van Sinderen D. Sequence Analysis of the Lactococcal Plasmid pNP40: a Mobile Replicon for Coping with Environmental Hazards. J Bacteriol. 2006;188: 6629–6639. 10.1128/JB.00672-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gilmour MW, Thomson NR, Sanders M, Parkhill J, Taylor DE. The complete nucleotide sequence of the resistance plasmid R478: defining the backbone components of incompatibility group H conjugative plasmids through comparative genomics. Plasmid. 2004;52: 182–202. 10.1016/j.plasmid.2004.06.006 [DOI] [PubMed] [Google Scholar]
- 53. McClean P, Hughes C, Tunney M, Goossens H, Jans B, European Surveillance of Antimicrobial Consumption (ESAC) Nursing Home Project Group. Antimicrobial prescribing in European nursing homes. J Antimicrob Chemother. 2011;66: 1609–1616. 10.1093/jac/dkr183 [DOI] [PubMed] [Google Scholar]
- 54. Martinez JL. Environmental pollution by antibiotics and by antibiotic resistance determinants. Env Pollut. 2009;157: 2893–2902. 10.1016/j.envpol.2009.05.051 [DOI] [PubMed] [Google Scholar]
- 55. Boerlin P, Reid-Smith RJ. Antimicrobial resistance: its emergence and transmission. Anim Health Res Rev Conf Res Work Anim Dis. 2008;9: 115–126. 10.1017/S146625230800159X [DOI] [PubMed] [Google Scholar]
- 56. Port JA, Cullen AC, Wallace JC, Smith MN, Faustman EM. Metagenomic Frameworks for Monitoring Antibiotic Resistance in Aquatic Environments. Environ Health Perspect. 2014;122: 222–228. 10.1289/ehp.1307009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mahowald MA, Rey FE, Seedorf H, Turnbaugh PJ, Fulton RS, Wollam A, et al. Characterizing a model human gut microbiota composed of members of its two dominant bacterial phyla. Proc Natl Acad Sci U S A. 2009;106: 5859–5864. 10.1073/pnas.0901529106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Holdeman LV, Moore WEC. New Genus, Coprococcus, Twelve New Species, and Emended Descriptions of Four Previously Described Species of Bacteria from Human Feces. Int J Syst Bacteriol. 1974;24: 260–277. 10.1099/00207713-24-2-260 [DOI] [Google Scholar]
- 59. Teeling H, Glöckner FO. Current opportunities and challenges in microbial metagenome analysis—a bioinformatic perspective. Brief Bioinform. 2012;13: 728–742. 10.1093/bib/bbs039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Case RJ, Boucher Y, Dahllöf I, Holmström C, Doolittle WF, Kjelleberg S. Use of 16S rRNA and rpoB Genes as Molecular Markers for Microbial Ecology Studies. Appl Environ Microbiol. 2007;73: 278–288. 10.1128/AEM.01177-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mokrousov I, Otten T, Vyshnevskiy B, Narvskaya O. Allele-Specific rpoB PCR Assays for Detection of Rifampin-Resistant Mycobacterium tuberculosis in Sputum Smears. Antimicrob Agents Chemother. 2003;47: 2231–2235. 10.1128/AAC.47.7.2231-2235.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Wichelhaus TA, Schafer V, Brade V, Boddinghaus B. Molecular Characterization of rpoB Mutations Conferring Cross-Resistance to Rifamycins on Methicillin-Resistant Staphylococcus aureus . Antimicrob Agents Chemother. 1999;43: 2813–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fujimoto-Nakamura M, Ito H, Oyamada Y, Nishino T, Yamagishi J. Accumulation of Mutations in both gyrB and parE Genes Is Associated with High-Level Resistance to Novobiocin in Staphylococcus aureus . Antimicrob Agents Chemother. 2005;49: 3810–3815. 10.1128/AAC.49.9.3810-3815.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yoshida H, Bogaki M, Nakamura M, Nakamura S. Quinolone resistance-determining region in the DNA gyrase gyrA gene of Escherichia coli . Antimicrob Agents Chemother. 1990;34: 1271–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nikaido H. Multidrug Resistance in Bacteria. Annu Rev Biochem. 2009;78: 119–146. 10.1146/annurev.biochem.78.082907.145923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rusch DB, Halpern AL, Sutton G, Heidelberg KB, Williamson S, Yooseph S, et al. The Sorcerer II Global Ocean Sampling Expedition: Northwest Atlantic through Eastern Tropical Pacific. PLoS Biol. 2007;5: e77 10.1371/journal.pbio.0050077 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Details about the sequence quality control (QC) steps and the de novo assembling by use of the CLC Genomic Workbench 6.5.1 (Number of reads).
(DOCX)
Matched high-throughput sequencing reads/contigs of the samples C1754, C1755, C1756 and C1757 against CARD.
(DOCX)
Matched high-throughput sequencing reads/contigs of the samples C1754, C1755, C1756 and C1757 against the INTEGRALL database.
(DOCX)
Assigned high-throughput sequencing reads/contigs of the samples C1754, C1755, C1756 and C1757 against the RefSeq plasmid.
(DOCX)
Data Availability Statement
Raw reads data have been deposited at the sequence read archive (SRA) of NCBI under Accession numbers SAMN02725022, SAMN02725023, SAMN02725024 and SAMN02725025).