

Abstract
Primitive neuroectodermal tumor (PNET) is a high grade malignant neoplasm of small round cell tumor family, commonly affecting children and young adults. Peripheral primitive neuroectodermal tumor (pPNET) is a predominately neural, nonepithelial malignancy seen outside the nervous system that can arise in any place throughout the body including the diverse tissues of the head and neck. The diagnosis of PNET is confounded by its clinical and histopathological similarity to Ewing’s sarcoma of the bone and has seldom been reported in the literature. The paucity of literature pertaining to the successful diagnosis and management of this lesion mandates its documentation and discussion. This article describes a case of an 11-year-old boy with an aggressive pPNET of the mandible. The clinical and radiographic presentations of this rare entity along with a detailed review on the current management modalities have been discussed.
Keywords: pPNET, Neuroectodermal tumor of mandible, Chemotherapy in pPNET
Introduction
Primitive neuroectodermal tumor (PNET) is a rare but extremely aggressive malignancy of the small round cell tumor family with histologic and immunologic evidence of neuroectodermal differentiation. PNET was primarily considered as a tumor localized to the central nervous system in the past. However, recognition of these tumors in the soft tissues of the trunk and the axial skeleton established the presence of peripheral primitive neuroectodermal tumor’s (pPNET’s) variants probably originating from undifferentiated mesenchyme. pPNET is considered a well differentiated version of the Small cell tumor family whereas Ewing’s sarcoma and the Askin’s tumor of the thorax are considered as the poorly differentiated variants of small round cell tumors (Ewings sarcoma family of tumors). Often the diagnosis of this lesion is confounded by its histopathologic similarity to Ewings sarcoma and rhabdomyosarcoma, as a result of which there is inadequate literature to establish definite treatment protocols for the management of the same. Hence it is of utmost importance to document and discuss their diagnostic morphologic, histologic presentation and the management protocol. The aim of this paper is to bring to light one such case of pediatric mandibular pPNET that had reported to our centre.
Case Report
An 11 year-old boy with Eastern Co-Operative Oncology Group (ECOG) Performance score of 1 reported to our unit with a rapidly enlarging painless mass of the left side of the mandible of 3 months duration. Examination revealed a solitary, smooth surfaced, firm, extensive swelling of 12 × 10 cm of the left mandible extending from the zygomatic arch upto the hyoid in the neck (Figs. 1, 2). Intra oral examination revealed a solid, erythematous smooth surfaced swelling extending from the canine anteriorly, obscuring the visualization of the posterior extent. Patient was unable to close his mouth due to the extensive swelling and the teeth in the region of the lesion were extracted prior to his presentation to us due to mobility. The swelling was associated with complaints of dysphagia with no signs of dyspnea or restriction in mouth opening.
Fig. 1.
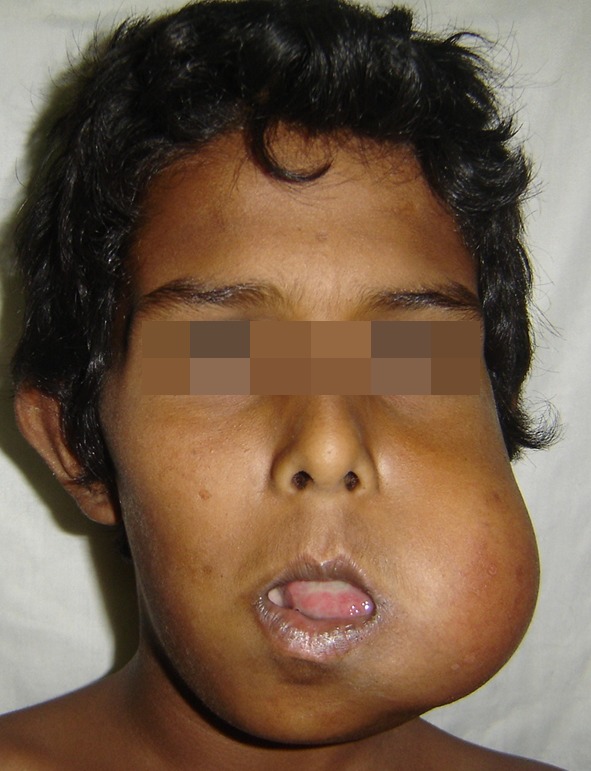
Pre operative frontal image of the patient
Fig. 2.
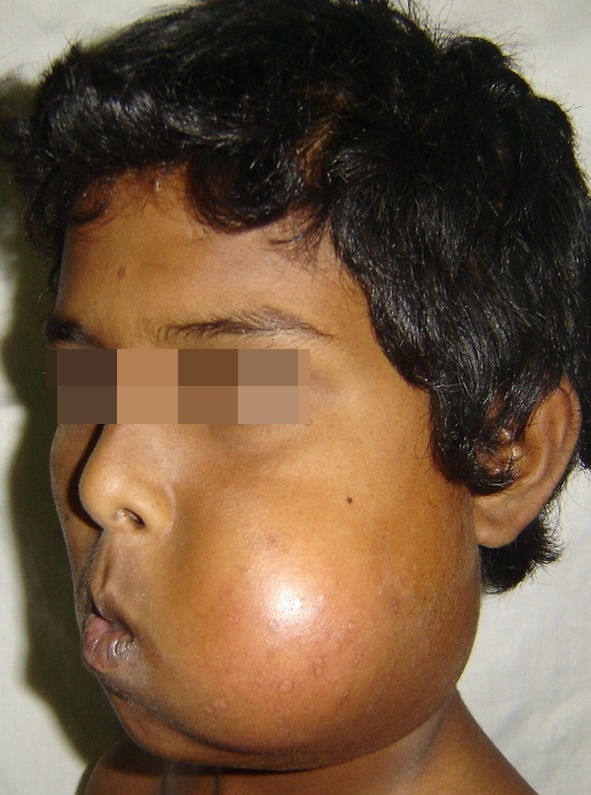
Pre operative lateral image showing the extensions of the lesion
A computed tomogram followed by an MRI (Fig. 3a–c) of the lesion were sought for, which revealed an expansile soft tissue mass with osteolysis of the left ramus, and the corpus of the mandible encroaching the retromolar trigone. Medially infiltration of the pterygoid muscles was present with displacement of the parapharyngeal fat. Distinct plane of dissection was evident between the lesion and the great vessels. A provisional diagnosis of sarcoma was arrived at and the patient was taken up for incision biopsy. Incision biopsy revealed it to be an angio-lipoma with sarcomatous changes. A metastatic work up was done including CT chest, ultrasound of the abdomen, long bone X-rays which were negative for metastasis. Surgery was considered the primary option for this patient after discussion at the multidisciplinary tumor board considering the report of incision biopsy, severe disability caused by the tumor and the possibility of pathological fracture (Fig. 4).
Fig. 3.

a Pre operative CT image. b Pre operative coronal CT image. c Pre operative MRI image
Fig. 4.
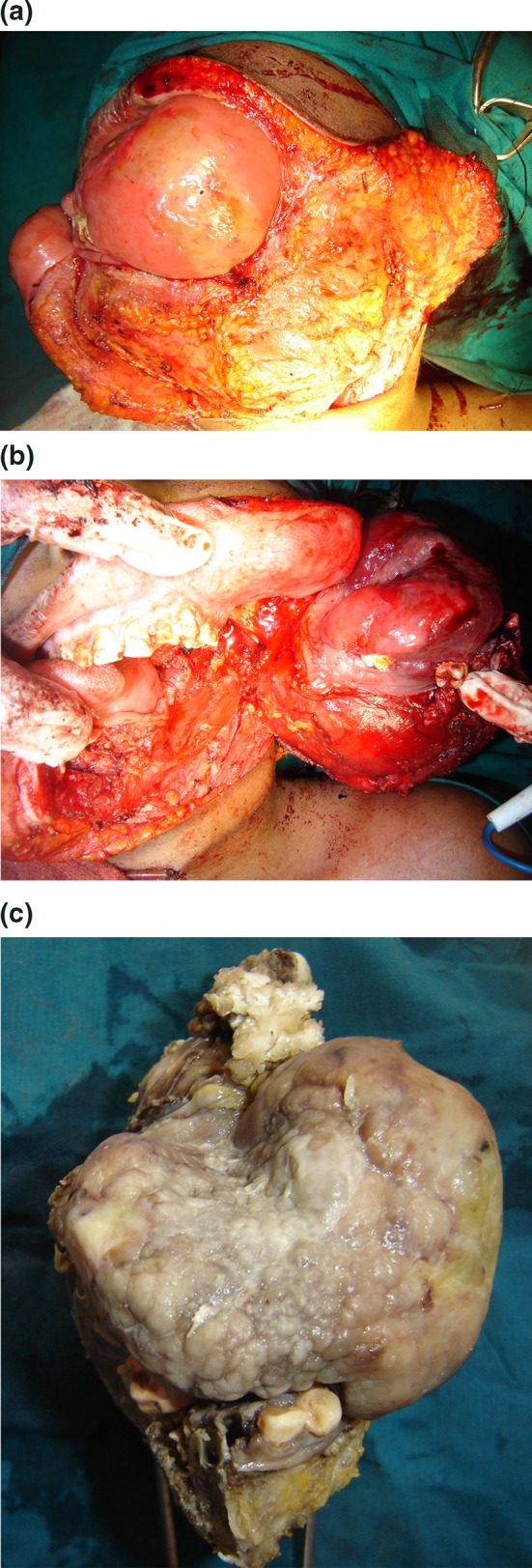
a Exposure of the lesion by elevating a cheek flap. b Tumor resection showing the extent of resection and the margins of clearance obtained. c The surgical specimen
The tumor was accessed through a commissurotomy incision continuing into the neck till the mastoid tip. Cheek flap was elevated in a sub cutaneous plane to provide adequate soft tissue clearance. Hemimandibulectomy was performed after the ligation of the maxillary artery at its exit from the parotid gland, with systematic dissection of the infra temporal fossa restricted lateral to the lateral pterygoid plate and spine of the sphenoid at the skull base to achieve 1 cm soft tissue clearance circumferentially. Reconstruction was performed with a regional flap (pectoralis major myocutaneous flap) (Fig. 5), and a definitive osseous reconstruction was deferred for a later stage.
Fig. 5.
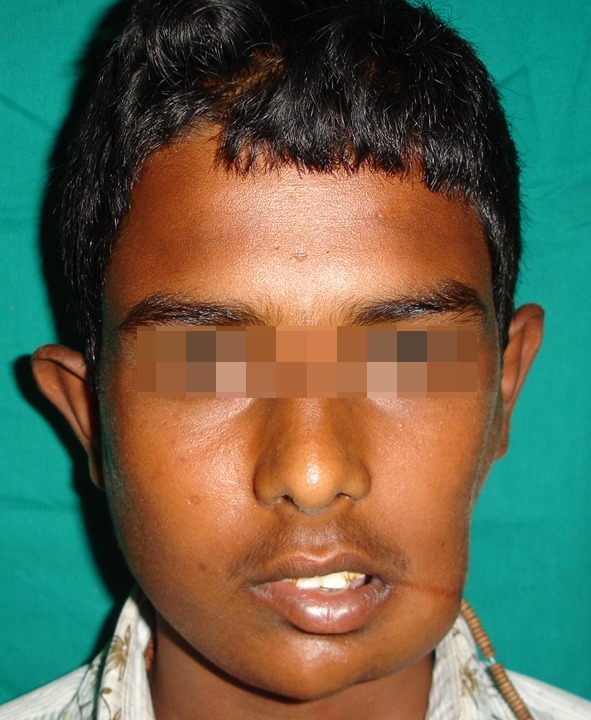
Post operative patient profile
Histologic examination of the specimen revealed a malignant neoplasm with diffuse group of small round cells arranged in a rosette pattern with scant cytoplasm and round vesicular nuclei with evident nucleoli. Mitotic figures were frequent, as were areas of necrosis and hemorrhage (Fig. 6a). Margins of the resection were free of tumor. Immunohistochemistry revealed the cells were positive for Vimentin and MIC 2 (Fig. 6b) and were immunonegative for cytokeratin and LCA confirming the diagnosis of pPNET. The patient was advised to undergo adjuvant chemotherapy. Two year follow-up revealed no signs of recurrence.
Fig. 6.
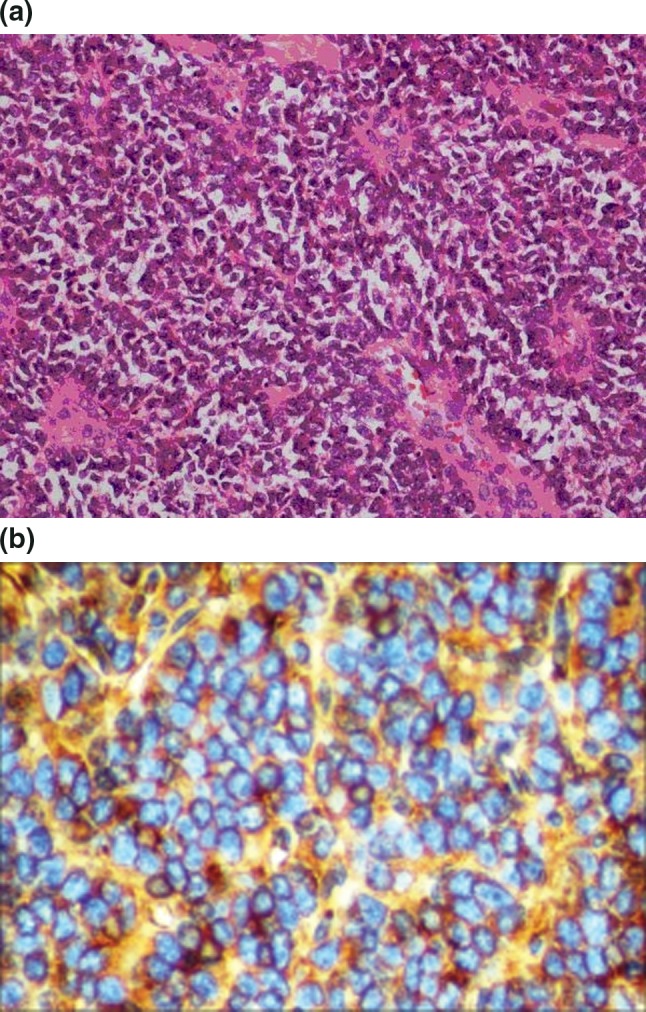
a Histopathological image. b IHC image showing MIC 2 positivity
Discussion
Primitive neuroectodermal tumor (PNET) is a rare aggressive variant of small round cell sarcomas that primarily originate from the neural crest cells. The tumors primarily arising from the neural crest cells can be classified based on the extent of differentiation of ectodermal and neural components into group A tumors that exhibit high ectodermal differentiation like the Pituitary tumors and the carcinoid tumors and group B tumors that show variable amount of neural differentiation as primitive neuroectodermal tumors [1].
Becker and Hinton [2] have further classified these primitive neuroectodermal tumors based on the amount of differentiation of the neural component into well differentiated neuronal tumor variants including ependymoblastoma, retinoblastoma, neuroblastoma and poorly differentiated neuronal variants into primitive neuroectodermal tumours, medulloblastoma. Initially PNETs were described to originate only from the components of the nervous system (CNS or the sympathetic nervous system) however the incidence of these tumors from the non neuronal soft tissues has enabled us to categorize these non neuronal soft tissue PNET’s into a separate category of peripheral PNET’s (pPNET’s). This brings forward the possibility of origin of these tumors from three cell sources including neural crest cells, primordial germ cells and the undifferentiated mesenchymal stem cells [3].
pPNET is a peripheral soft tissue sarcoma that clinically and histologically shows neuronal differentiation very similar to Ewing’s sarcoma. Cytogenetic evaluation of these tumors reveals t (11:22) translocation that results in the fusion of the amino terminus of the EWS (22q12) gene to the carboxyl terminus of the FLI1(11q24) gene. The resultant product is an aberrant transcription factor that is a potent transactivator of ‘c-myc’, a nuclear proto-oncogene C-myc, with another oncoprotein ‘max’ forms the ‘myc/max’ heterodimer along with over expression of other transcription factors that leads to an abnormal cell turnover leading to the tumor proliferation [4, 5].
Similar cytogenetic variations are seen to be associated with Ewings sarcoma. Apart from the cytogenetic studies, the immunohistochemical studies reveal the expression of neuronal markers by these tumors including CD99, Neuron Specific Enolase, neurofilament, HNK-1 (Leu-7), FLI-1, S-100 protein, and MB [5, 6]. p30/32MIC2 a cell surface glycoprotein encoded by MIC 2 gene on X and Y chromosomes is a specific marker for Ewing’s sarcoma and pPNETs that enables their differentiation from the other small round cell tumors [7, 8]. Both these tumors are believed to show neuroectodermal differentiation, albeit in different degree; Ewing’s sarcoma tends to be poorly differentiated, whereas PNET most often shows definite neuroectodermal differentiation. Although once viewed as distinct entities, Ewing’s sarcoma, Askin’s tumor, and PNET are now considered together as members of the Ewing family of tumors [9].
pPNET is a rare malignancy that comprises only around 1 % of all the sarcomas [10] with a 2 year survival rate of 65 % [11]. The poor prognosis is secondary to the high rate of metastasis of 14–50 % at the time of diagnosis of primary tumor [12]. Lungs, bone, and bone marrow are the common sites of metastasis and a through metastatic workup prior to the management of the primary is essential. This includes CT scan of the chest and abdomen, Bone scan, bone marrow aspiration/biopsy. Recent innovations like Proton MR spectroscopy aid in characterization of the tumor tissue especially in cases of tumor arising in the inaccessible sites. However metastatic work up may often be negative as pPNET’s (the primary tumor itself) are known to secrete angiostatin a chemical mediator that is known to suppress the angiogenesis that will prevent the homing-in of the metastatic tumor emboli and thus the metastatic disease may become evident only after the excision of the primary tumor. This probably makes addressing the micro circulatory metastasis essential in the form of pre operative chemotherapy to achieve complete cure [13].
National Comprehensive Cancer Network 2013 (NCCN) [14] recommends primary treatment in the form of multi agent chemotherapy along with local control therapy (surgery or definitive chemo-radiotherapy) followed by adjuvant therapy (chemotherapy or chemo- radiotherapy). This is similar to the protocol followed at MD Anderson Institute of Oncology (Table 1).
Table 1.
Flow chart showing the multidisciplinary treatment approach to pPNET management from MD Anderson Manual of Medical Oncology
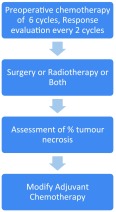
Primary treatment consists of multiagent chemotherapy along with appropriate growth factor support for 12–24 weeks followed by local control measures which include wide excision with 1.5 cm clearance circumferentially or definitive radiotherapy with chemotherapy or pre operative RT followed by wide excision. Chemotherapy is given as neo-adjuvant and Adjuvant therapy. The results of pooled analysis of INT-0091 and POG-9395 showed that the use of multiagent chemotherapy prior to surgery downstages the tumor in a majority of patients thereby increasing the probability of achieving complete resection with microscopically negative margins [15]. However the surgical margins are based on the pre-interventional imaging studies only (margins defined prior to the start of chemotherapy). Apart from this neo-adjuvant chemotherapy aids in assessing the percentage of tumor necrosis and in turn the tumor response to the chemotherapeutic which can aid in modifying the post surgical adjuvant chemotherapeutic settings. Microscopic metastasis that is not evident on the preoperative metastatic work up can be addressed by initiating neo-adjuvant chemotherapy.
Adjuvant chemotherapy with or without radiotherapy is recommended, regardless of surgical margins, following local control treatment.
The relative paucity of PNET cases is the primary cause for the lack of definitive treatment paradigms. Because of their rare occurrence, optimal therapy is challenging, particularly if they occur in the head and neck. However, chemotherapy and surgery remain the mainstay of treatment for these tumors with adjuvant radiotherapy in cases with microscopic or macroscopic positive margins [19, 20].
These tumors are extremely responsive to chemotherapeutic agents like Doxorubicin, Actinomycin D, Ifosfamide, Cyclophosphamide, Vincristine, Etoposide. The most commonly used combinations are Vincristine, Adriamycin (Doxorubicin), and cyclophosphamide (VAC); Ifosfamide and Etoposide (IE); and Vincristine, Adriamycin, and Ifosfamide (VAI). Multiple trials have shown that with these combinations of chemotherapy, survival rates >50 % can be achieved [16].
Though these tumors are considered radiosensitive, the higher incidence of the radiation induced sarcomas is of significant concern. The incidence of the radiation induced sarcomas varies with dosimetry, with a relatively lower incidence up to 45 Gy’s. An increase to 60 Gy has been associated with 20 % incidence of radiation sarcomas [17]. However Radiotherapy is used as salvage therapy in cases of incomplete excisions and close tumor margins. Age >10 years, radiation >50 Gy’s and high dose chemotherapy with hematopoietic rescue are poor prognostic factors [18]. The likelihood of complete tumor resection with a negative microscopic margin and consequent avoidance of external beam radiation and its potential complications is augmented with neoadjuvant chemotherapy and delayed resection [19, 20]. The follow-up of these patients is adequately essential. The protocol that is followed at our centre is discussed below (Table 2).
Table 2.
Follow-up protocol

Though evidence exists with respect to the application of neoadjuvant chemotherapy, multi institutional studies are necessary to formulate definite treatment paradigms for the management of these lesions.
Conflict of interest
None.
References
- 1.Mills S. Neuroectodermal neoplasms of the head and neck with emphasis on neuroendocrine carcinoma. Mod Pathol. 2002;15:264–278. doi: 10.1038/modpathol.3880522. [DOI] [PubMed] [Google Scholar]
- 2.Becker LE, Hinton D. Primitive neuroectodermal tumors of the central nervous system. Hum Pathol. 1983;14:538. doi: 10.1016/S0046-8177(83)80006-9. [DOI] [PubMed] [Google Scholar]
- 3.Dehner LP. Primitive neuroectodermal tumor and Ewing’s sarcoma. Am J Surg Pathol. 1993;17:1. doi: 10.1097/00000478-199301000-00001. [DOI] [PubMed] [Google Scholar]
- 4.May WA, Denny CT. Biology of EWS/FLI and related fusion genes in Ewing’s sarcoma and primitive neuroectodermal tumor. Curr Top Microbiol Immunol. 1997;220:143–150. [PubMed] [Google Scholar]
- 5.Ambros IM, Ambros P, Strehl S. MIC2 is a specific marker for Ewing’s sarcoma and peripheral primitive neuroectodermal tumor. Evidence for a common histogenesis of Ewing’s sarcoma and peripheral primitive neuroectodermal tumor from MIC2 expression and specific chromosome aberration. Cancer. 1991;67:1886–1893. doi: 10.1002/1097-0142(19910401)67:7<1886::AID-CNCR2820670712>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 6.Fellinger EJ, Garin-Chesa P, Triche TJ, et al. Immunohistochemical analysis of Ewing’s sarcoma cell surface antigen p30/32MIC2. Am J Pathol. 1991;139:317. [PMC free article] [PubMed] [Google Scholar]
- 7.Weidner N, Tjoe J. Immunohistochemical profile of monoclonal antibody O-13: antibody that recognizes glycoprotein p30/32MIC2 and is useful in diagnosing Ewing’s sarcoma and peripheral neuroepithelioma. Am J Surg Pathol. 1994;18:486. doi: 10.1097/00000478-199405000-00008. [DOI] [PubMed] [Google Scholar]
- 8.Shishikura A, Ushigome S, Shimoda T. Primitive neuroectodermal tumors of bone and soft tissue: histological subclassification and clinicopathologic correlations. Acta Pathol Jpn. 1993;43:176. doi: 10.1111/j.1440-1827.1993.tb01129.x. [DOI] [PubMed] [Google Scholar]
- 9.Alrawi SJ, Tan D, Sullivan M, Winston J, et al. Peripheral primitive neuroectodermal tumor of the mandible with cytogenetic and molecular biology aberrations. J Oral Maxillofac Surg. 2005;63:1216–1221. doi: 10.1016/j.joms.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 10.Kim MS, Kim B, Park CS, Song SY, Lee EJ, Park NH, et al. Radiological findings of peripheral primitive neuroectodermal tumor arising in the retroperitoneum. AJR. 2006;186:1125–1132. doi: 10.2214/AJR.04.1688. [DOI] [PubMed] [Google Scholar]
- 11.Jones JE, McGill T. Peripheral primitive neuroectodermal tumors of the head and neck. Arch Otolaryngol Head Neck Surg. 1995;121:1392. doi: 10.1001/archotol.1995.01890120050009. [DOI] [PubMed] [Google Scholar]
- 12.DeVita VT, Hellman S, Rosenberg S. Cancer: principles and practice. 4. Philadelphia, PA: Lippincott Williams and Wilkins; 1993. pp. 1778–1783. [Google Scholar]
- 13.Holmgren L, O’Reilly MS, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med. 1995;1:149–153. doi: 10.1038/nm0295-149. [DOI] [PubMed] [Google Scholar]
- 14.NCCN guidelines in Oncology (2013) Bone Cancer Version 1, pp 11–12
- 15.Shamberger RC, LaQuaglia MP, Gebhardt MC, et al. Ewing sarcoma/primitive neuroectodermal tumor of the chest wall: impact of initial versus delayed resection on tumor margins, survival, and use of radiation therapy. Ann Surg. 2003;238:563–567. doi: 10.1097/01.sla.0000089857.45191.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paulussen M, Ahrens S, Dunst J, et al. Localized Ewing tumor of bone: final results of the cooperative Ewing’s sarcoma study CESS 86. J Clin Oncol. 2001;19(6):1818–1829. doi: 10.1200/JCO.2001.19.6.1818. [DOI] [PubMed] [Google Scholar]
- 17.Kuttesch JF, Wexler LH, Marcus RB, Fairclough D, Weaver-McClure L, White M, et al. Second malignancies after Ewing sarcoma, radiation dose dependency of secondary sarcomas. J Clin Oncol. 1996;14:2818–2825. doi: 10.1200/JCO.1996.14.10.2818. [DOI] [PubMed] [Google Scholar]
- 18.Koscielniak E, Morgan M, Treuner J. Soft tissue sarcoma in children: prognosis and management. Drugs. 2002;1:21. doi: 10.2165/00128072-200204010-00003. [DOI] [PubMed] [Google Scholar]
- 19.Shamberger RC, et al. Ewing sarcoma/primitive neuroectodermal tumor of the chest wall. Impact of initial versus delayed resection on tumor margins, survival, and use of radiation therapy. Ann Surg. 2003;238:563–568. doi: 10.1097/01.sla.0000089857.45191.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunst J, Schuck A (2004) Indications for post-operative radiotherapy are unradical or marginal resections and poor histological response. Pediatr Blood Cancer 42(5):465–470. Role of radiotherapy in Ewing tumors [DOI] [PubMed]