

Abstract
A 62-year-old woman was admitted for nephrotic syndrome and lung tumor. A renal biopsy showed membranous features of the glomeruli. Immunofluorescence studies revealed granular IgG4-κ deposits along with the glomerular basement membrane. Electron microscopy revealed granular electron-dense deposits. Further study denied multiple myeloma. Light microscopy of the resected lung tumor revealed IgG4-related lung disease with no malignancy. Steroid therapy induced a remission of the nephrotic syndrome, with no recurrence of the lung tumor. We consider that this is the first case of a proliferative glomerulonephritis with monoclonal IgG deposits of IgG4 subclass, and a rare concurrence with IgG4-related disease.
Keywords: IgG4-related disease, nephrotic syndrome, proliferative glomerulonephritis with monoclonal IgG deposits
Background
Proliferative glomerulonephritis with monoclonal IgG deposits (PGNMID) is a rare entity. It is characterized by mesangial/endocapillary proliferative, membranoproliferative or membranous features on light microscopy, granular immunoglobulin deposits restricted to a single IgG subclass and a single light chain by immunofluorescence microscopy, and immune-complex type deposits by electron microscopy [1, 2]. To date, reports of PGNMID consist of monoclonal IgG deposition of IgG1, IgG2 and IgG3 heavy chains, but none for monoclonal IgG4 deposits [1–7].
On the other hand, IgG4-related disease is a newly recognized disease, characterized by IgG4-positive plasma cell infiltration, storiform fibrosis, and it is often accompanied with elevated serum IgG4 concentrations. Target organ varies such as pancreas, salivary glands, kidneys, lymph nodes and lungs [8]. Today its notion is widely known and clinical reports in number are increasing. However there has been no report of IgG4-related disease complicated with PGNMID.
Herein, we report the first case of PGNMID with monoclonal deposits of IgG4-κ. The patient had a right lung tumor, and partial resection of the tumor revealed fibrotic changes in the lung interstitium with heavy infiltration of IgG4-positive plasma cells, without malignant change. We also discuss the relation between PGNMID and IgG4-related disease.
Case report
A 62-year-old woman was admitted for suspected pneumonia and nephrotic syndrome in June 2012. She had suffered from cough and sputum from 2 weeks prior to admission. On admission, her blood pressure was 111/58 mmHg, temperature 36.5°C and pulse 63 bpm. A physical examination revealed pale conjunctiva and slight bilateral pretibial edema. No abnormal sign was observed in the lungs, heart or abdomen.
Urinalysis showed heavy proteinuria (4.3 g/day) with hematuria (5–10/HPF). White blood cell count was 8300/μL with normal distribution, hemoglobin 9.9 g/dL, hematocrit 29.1% and a platelet count of 230 000/μL. Serum total protein was 5.8 g/dL, albumin 2.5 g/dL, blood urea nitrogen 23.8 mg/dL, creatinine 1.56 mg/dL, alanine aminotransferase 23 U/L, aspartate aminotransferase 15 U/L, lactate dehydrogenase 172 U/L and total cholesterol 138 mg/dL. Serum IgG was 1255 mg/dL, IgG1 746 mg/dL, IgG2 446 mg/dL, IgG3 86.1 mg/dL and IgG4 35.9 mg/dL. IgA was 250 mg/dL, IgM 128 mg/dL, C3 86 mg/dL and C4 32 mg/dL. Anti-nuclear antibodies were 40-fold with speckled pattern. Both MPO- and PR3-antineutrophil cytoplasmic antibodies were negative. Chest X-ray revealed a right lower lobe infiltrate.
A renal biopsy was performed. Light microscopy showed 13 global scleroses, 15 cellular and 2 fibrotic crescents within 38 glomeruli, with bubbling and spike appearance of the glomerular basement membrane (GBM) in the functioning glomeruli (Figures 1 and 2). In the tubulointerstitium, severe lymphocyte infiltration, mild tubular atrophy and interstitial fibrosis were observed (Figure 1). Immunofluorescence studies showed granular staining 3+ for γ heavy chain, 2+ for κ-light chain and trace staining for C3, along with the GBM, but no significant staining for λ-light chain or C1q. Immunofluorescence staining for γ-heavy chain subclasses showed a dominant 3+ granular staining for IgG4, trace staining for IgG2, negative for IgG1 or IgG3 (Figure 3). Immunofluorescence staining for antibodies to M-type phospholipase A2 receptor (PLA2R) (Sigma-Aldrich, USA) was performed. The specimen had negative glomerular staining for anti-PLA2R. Immunohistochemistry study revealed IgG1 dominant and minor IgG4 staining of the infiltrate plasma cells in the tubulointerstitium, and normal ratio of κ/λ light chain positive plasma cells. Electron microscopy revealed granular electron-dense deposits without organized structures in the subepithelial space and in the mesangial area (Figure 4). Bone marrow aspiration was performed because of the presence of monoclonal immunoglobulin deposition in the glomeruli. The smears were normocellular with no morphological abnormality, and 1.8% of the cells were plasma cells. Serum M-protein and urine Bence-Jones protein were not detected by immunofixation electrophoresis. The serum free κ/λ light chain ratio was 1.06, which is within the normal range. By the above results, multiple myeloma or underlying plasma cell dyscrasia was denied.
Fig. 1.

Light microscopy reveals crescentic glomerulonephritis and lymphocyte infiltration in the tubulointerstitium, and interstitial fibrosis. Periodic acid-Schiff staining (×100).
Fig. 2.

Light microscopy shows bubbling and spike appearances of the glomerular base membrane in a functioning glomeruli. Periodic acid-methnamine-silver stain (×400 and magnified).
Fig. 3.

Immunofluorescence microscopy for IgG heavy chain, κ light chain, λ light chain, C3, IgG heavy chain subclasses of IgG1, IgG2, IgG3 and IgG4. It displays granular staining of IgG heavy chain dominant for IgG4 and κ light chain along glomerular capillary walls, but no significant staining for others including λ light chain (×400).
Fig. 4.

Electron microscopy reveals granular electron-dense deposits without organized structures in the subepithelial space and in the mesangial area. The bar indicates 2 μm.
Computed tomography of the chest revealed a tumor-like mass of 2.3 cm in diameter in the base of the right lung. Partial resection of the lung was performed by video-assisted thracoscopic surgery. Microscopic examination of the section revealed fibrotic changes of the lung interstitium, proliferation of lymphocytes and plasma cells, with no malignant changes. Immunohistochemical study showed positive IgG4 staining in half of the proliferated plasma cells (Figure 5). Light chain restriction was not detected by κ and λ light chain immunostains. No abnormal expression of CD56 or cyclin D1 was observed.
Fig. 5.
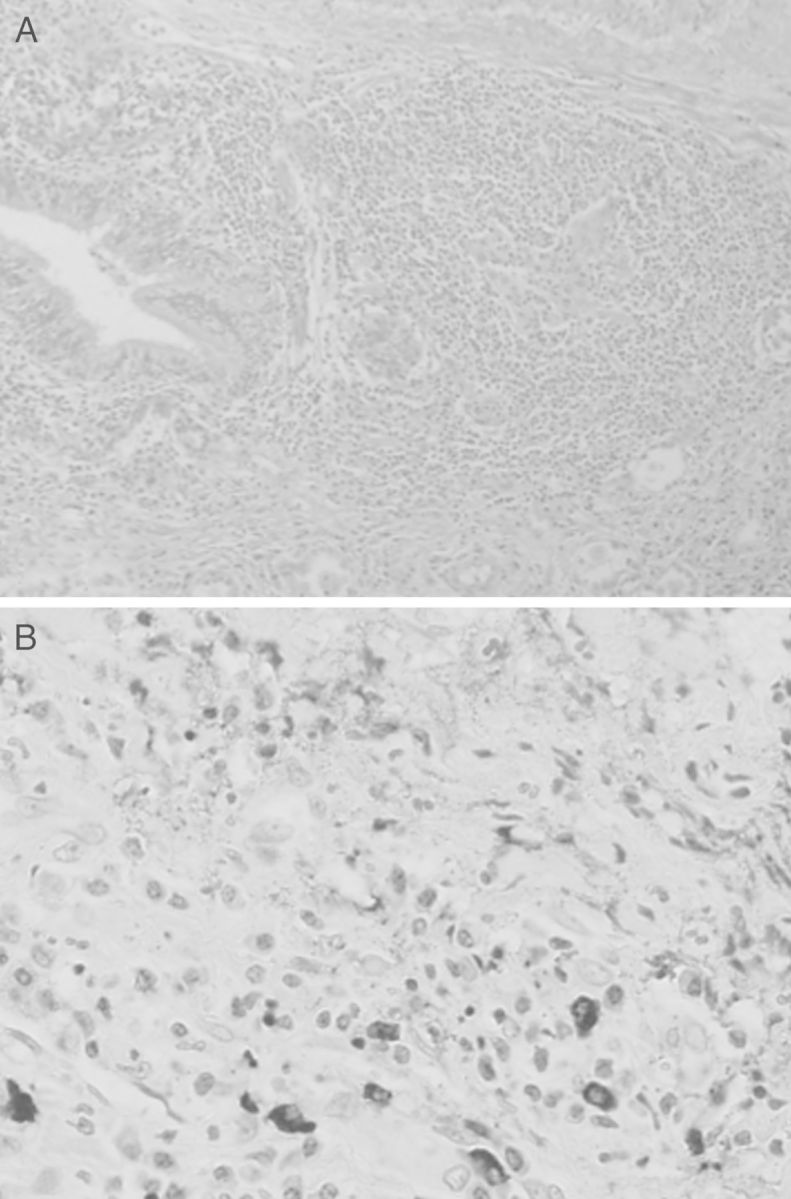
Light microscopy of the resected lung tumor shows heavy plasma cells and lymphocyte proliferation, and fibrotic changes of the interstitium. No malignant change is found (A). Immunohistochemistry microscopy shows IgG4-positive plasma cells in the specimen (B).
From these findings, she was diagnosed with PGNMID (membranous features) complicated with IgG4-related lung disease, according to the criteria for IgG4-related disease [9]. Steroid therapy was initiated by 3 days of intravenous methyl-prednisolone of 250 mg, followed by 30 mg/day of prednisolone. Following the treatment, proteinuria improved gradually, while serum creatinine level showed little change. At the 1-year follow-up, the patient was found to be well with 10 mg/day of prednisolone. The urinary protein was 0.9 g/g creatinine, serum total protein 5.8 g/dL, albumin 3.8 g/dL and creatinine 3.23 mg/dL. No abnormality in the chest X-ray was found during the follow-up period.
Discussion
PGNMID is a disease entity distinguished by monoclonal immunoglobulin deposition restricted to a single heavy and light chain. To date, reports for IgG heavy chain was restricted to IgG1, IgG2 and IgG3. In the largest series reported by Nasr et al. [2], the most common IgG subclass found in the glomeruli was IgG3. Although the reason why IgG3 cases are dominant is yet to be determined, in previous reports it is suggested that it may be due to the characteristics of IgG3; it is the most positively charged subclass, has the highest molecular weight making it more size-restricted by the glomerular barrier, and is the greatest compliment-fixing subclass, that could lead to glomerulonephritis [1, 2]. However, our case presented monoclonal IgG4 deposition, which has never been reported. IgG4 is the least abundant IgG subclass and does not activate the classical complement pathway [8], which partially contradicts the previous explanation regarding IgG3 deposits in PGNMID.
There are several reports of membranous nephropathy (MN) associated with IgG4-related disease, with dominant IgG4 deposits in the glomeruli, but without light chain monoclonality [10, 11]. Interestingly, all of these cases lacked glomerular staining by anti-PLA2R. Since anti-PLA2R staining of the glomeruli is a sensitive examination to determine primary MN [12], the reports concluded that their MN cases were not ‘primary’, but secondary to IgG4-related disease [10, 11]. In our case, anti-PLA2R staining of the glomeruli was also negative, and this confirms that the pathological features of the renal biopsy are not the results of ‘primary membranous nephropathy’. Although the relationship between PGNMID and IgG4-related disease is elusive, there may be common mechanisms resembling the previous reports [10, 11], to form both membranous type PGNMID of monoclonal IgG4 deposits and IgG4-related disease, simultaneously.
In summary, we experienced a case of PGNMID of membranous features with monoclonal IgG4-κ deposition, along with IgG4-related disease of the lung. For this patient, steroid therapy was effective for both kidney and lung lesions. This is the first case of PGNMID with IgG4 deposits since the first literature report [1], and accumulation of cases is essential for further determination of pathogenesis, clinical features and outcome.
Conflict of interest statement
None declared.
References
- 1.Nasr SH, Markowitz GS, Stokes MB, et al. Proliferative glomerulonephritis with monoclonal IgG deposits: a distinct entity mimicking immune-complex glomerulonephritis. Kidney Int. 2004;65:85–96. doi: 10.1111/j.1523-1755.2004.00365.x. [DOI] [PubMed] [Google Scholar]
- 2.Nasr SH, Satoskar A, Markowitz GS, et al. Proliferative glomerulonephritis with monoclonal IgG deposits. J Am Soc Nephrol. 2009;20:2055–2064. doi: 10.1681/ASN.2009010110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Komatsuda A, Masai R, Ohtani H, et al. Monoclonal immunoglobulin deposition disease associated with membranous features. Nephrol Dial Transplant. 2008;23:3888–3894. doi: 10.1093/ndt/gfn363. [DOI] [PubMed] [Google Scholar]
- 4.Masai R, Wakui H, Komatsuda A, et al. Characteristics of proliferative glomerulonephritis with monoclonal IgG deposits associated with membranoproliferative features. Clin Nephrol. 2009;72:46–54. doi: 10.5414/cnp72046. [DOI] [PubMed] [Google Scholar]
- 5.Komatsuda A, Wakui H, Ohtani H, et al. Steroid-responsive nephrotic syndrome in a patient with proliferative glomerulonephritis with monoclonal IgG deposits with pure mesangial proliferative features. NDT Plus. 2010;3:257–359. doi: 10.1093/ndtplus/sfq076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nasr SH, Sethi S, Cornell LD, et al. Proliferative glomerulonephritis with monoclonal IgG deposits recurs in the allograft. Clin J Am Soc Nrphrol. 2011;6:122–132. doi: 10.2215/CJN.05750710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fujiwara T, Komatsuda A, Ohtani H, et al. Proliferative glomerulonephritis with monoclonal IgG deposits in a patient with autoimmune hemolytic anemia. Clin Nephrol. 2013;79:494–498. doi: 10.5414/cn107267. [DOI] [PubMed] [Google Scholar]
- 8.Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366:539–551. doi: 10.1056/NEJMra1104650. [DOI] [PubMed] [Google Scholar]
- 9.Umehara H, Okazaki K, Masaki Y, et al. Comprehensive diagnostic criteria for IgG4-related disease (IgG4-RD), 2011. Mod Rheumatol. 2012;22:21–30. doi: 10.1007/s10165-011-0571-z. [DOI] [PubMed] [Google Scholar]
- 10.Alexander MP, Larsen CP, Gibson IW, et al. Membranous glomerulonephritis is a manifestation of IgG4-related disease. Kidney Int. 2013;83:455–462. doi: 10.1038/ki.2012.382. [DOI] [PubMed] [Google Scholar]
- 11.Wada Y, Saeki T, Yoshita K, et al. Development of IgG4-related disease in a patient diagnosed with idiopathic membranous nephropathy. Clin Kidney J. 2013;6:486–490. doi: 10.1093/ckj/sft062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Debiec H, Ronco P. PLA2R autoantibodies and PLA2R glomerular deposits in membranous nephropathy. N Engl J Med. 2011;364:689–690. doi: 10.1056/NEJMc1011678. [DOI] [PubMed] [Google Scholar]