

Abstract
The diagnosis of IgA-dominant post-infectious glomerulonephritis (PIGN) may be challenging, as it must be differentiated from that of active IgA nephropathy. Predominant clinicopathologic features of IgA-dominant PIGN substantially overlap with those of active IgA nephropathy. Here, we present a case of a 67-year-old woman with rapidly rising serum creatinine, proteinuria and severe hypertension. The kidney biopsy findings included some features of IgA-dominant PIGN while others were more consistent with classical IgA nephropathy. We describe this patient's immune profile at the time of acute kidney injury and review the literature regarding differentiation of the two entities.
Keywords: IgA, IgA dominant post-infectious glomerulonephritis, IgA nephropathy
Introduction
IgA-dominant post-infectious glomerulonephritis (PIGN), first described in 2003, is a variant of classical PIGN. Its pathological characteristics mimic those of classical PIGN, including endocapillary hypercellularity on light microcopy, mesangial and subepithelial staining for IgG and C3 on immunofluorescence microscopy, and hump-like subepithelial immune deposits on electron microscopy. Other features differ from those of classical PIGN in that presentation is typically in older adults, the dominant or co-dominant immunoglobulin on immunofluorescence is IgA rather than IgG, and the association is with staphylococcal rather than streptococcal infections [1]. Differentiating IgA-dominant PIGN from IgA nephropathy (IgAN) can be challenging. This distinction is further complicated by the fact that IgAN is known to worsen after mucosal infections.
We present an elderly woman who developed acute kidney injury associated with a Clostridium difficile infection. The findings of her kidney biopsy had features of both IgA-dominant PIGN and classical IgAN. We also report the immunologic profile at the time of her acute illness.
Clinical history and initial laboratory data
A 67-year-old woman with no history of kidney disease was diagnosed with adenocarcinoma of the gallbladder; an attempt to fully remove the tumor was unsuccessful and she was referred for further management. Baseline serum creatinine was 88.4 μmol/L (1.0 mg/dL). She subsequently underwent a cystic-duct-stump wedge resection of the gallbladder. Her postoperative course was complicated by right-upper-quadrant pain, nausea, vomiting and fever. The serum creatinine rose to 168 μmol/L (1.9 mg/dL). Urinalysis dipstick showed no protein or blood, and only trace leukocyte esterase. She was started on intravenous piperacillin/tazobactam for a suspected abscess at the surgical site. After volume repletion, the serum creatinine improved to 115 μmol/L (1.3 mg/dL) 15 days after surgery and she was discharged from the hospital. Three days later, the patient became severely hypertensive with worsening edema in her legs and had diarrhea. On readmission to the hospital, her blood pressure was 201/94 mm Hg and oral temperature, 37.5°C (99.5°F). Physical examination was unremarkable except for a healed right-upper-quadrant incision and 2+ pitting pretibial edema. Laboratory studies included serum creatinine 168 μmol/L (1.9 mg/dL), blood urea nitrogen 4.3 mmol/L (12 mg/dL) and serum albumin 27 g/L (2.7 g/dL). Urinalysis revealed 2+ protein and 3+ blood by dipstick, with >25 red blood cells and 11–25 white blood cells per high-power field. The 24-hour protein excretion was 2.4 g. Serum C3, C4 and CH50 levels were normal. Anti-nuclear antibody and serum cryoglobulins were negative. C. difficile cytotoxin on admission was negative; a second test during persistent severe diarrhea was positive. The serum creatinine continued to worsen, and six days after admission a percutaneous kidney biopsy was performed.
Kidney biopsy
The biopsy specimen for light microscopy contained 15 glomeruli, of which three were globally sclerosed and one had a segmental sclerotic lesion. The remaining glomeruli showed moderate to marked expansion of the mesangial matrix, mesangial and segmental endocapillary hypercellularity, and intracapillary neutrophils. There was no focal necrotic lesion. The glomerular basement membranes (GBMs) appeared prominent due to occasional double contours, large subendothelial deposits (Figure 1A) and intracapillary pseudothrombi (Figure 1A). There was a patchy interstitial inflammation without tubulitis. The arterioles showed mild hyaline deposition; the arteries were unremarkable.
Fig. 1.
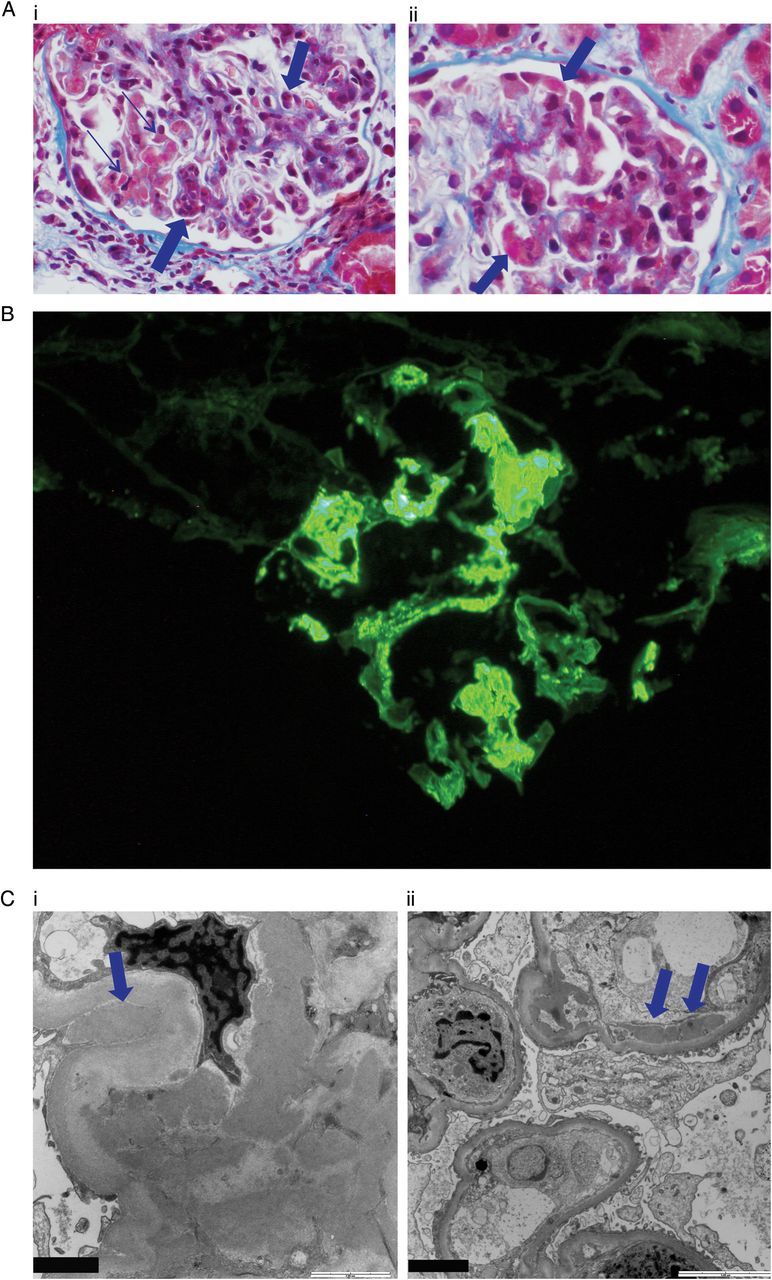
Pathology features in the renal biopsy. (A) (i) Light microscopy: This glomerulus shows mild-to-moderate expansion of the mesangial matrix with mesangial hypercellularity and segmental endocapillary hypercellularity with intracapillary neutrophils (thick arrows). There are several intracapillary pseudothrombi (thin arrows). Trichrome ×600. (A) (ii) This glomerulus shows prominent glomerular basement membranes with large subendothelial eosinophilic deposits and granular mesangial deposits (arrows). Trichrome ×600. (B) Immunofluorescence microscopy. Very strong (4+) staining for IgA is shown in the mesangium and capillary loops. C3 also showed 4+ staining in the capillary loops (not illustrated). (C) (i) Electron microscopy. Numerous large electron-dense immune-complex deposits are present in the mesangium. One large hump-like subepithelial deposit (arrow) in the notch area is shown. The foot processes are completely effaced. (C) (ii) Subendothelial deposits (arrows).
Immunofluorescence microscopy showed three glomeruli, all with diffuse mesangial and segmental capillary-loop staining for IgA (4+, scale 0–4), C3 (4+), IgG (2+), IgM (trace), kappa light chains (3+) and lambda light chains (4+) (Figure 1B). Staining for C1q and albumin was absent. On electron microscopy, GBMs were extremely thickened due to segmental cellular interposition and massive subendothelial immune-complex deposits without substructure. One large subepithelial hump-like deposit was identified (Figure 1C). The mesangial areas were markedly expanded due to increased matrix and numerous large electron-dense deposits. The glomerular foot processes were completely effaced (Figure 1C).
Diagnosis
The diagnosis was diffuse proliferative glomerulonephritis with membranoproliferative features and dominant IgA deposits. The history of recent infection in conjunction with a light microscopy pattern of diffuse endocapillary hypercellularity with neutrophil infiltration in addition to mesangial and subendothelial deposits raised the possibility of an IgA-dominant PIGN.
Clinical follow-up
Due to the severity of the C. difficile colitis and the diagnosis of possible PIGN, no immunosuppressive therapy was instituted. Her kidney failure worsened and she eventually required hemodialysis. One year later, she remained on hemodialysis.
Discussion
This case demonstrates the difficulties in differentiating IgAN from IgA-dominant PIGN, especially when taking into account persistent or resolving PIGN [2]. Pathologic features that have been associated with a diagnosis of IgA-dominant PIGN, as opposed to IgAN, include a clinical history of recent Staphylococcus aureus infection, hypocomplementemia, predominant intracapillary neutrophilic infiltrate and large subepithelial humps [3]. Our patient was elderly and had no previous diagnosis of IgAN but did have a history of microscopic hematuria. She lacked clinical features typically associated with IgA-dominant PIGN such as hypocomplementemia and a preceding staphylococcal infection, and did not have diabetes mellitus. On biopsy, she had subepithelial humps, IgA dominance with equally strong C3 staining on immunofluorescence microscopy, as well as neutrophilic infiltrate, and endocapillary hypercellularity, findings that favor a diagnosis of IgA-dominant PIGN. However, many of these features can be seen with active IgAN as well. Subepithelial humps, specifically, favor the diagnosis of IgA-dominant PIGN, but can also be present in active IgAN [4]. The number of subepithelial or intramembranous deposits per glomerulus may be a differentiating factor, as 13/13 cases with IgA-dominant PIGN in one series had more than five subepithelial or intramembranous deposits per glomerulus, with 10/13 having >10 per glomerulus [2]. Our patient had only one large subepithelial deposit in one of three glomeruli available for electron microscopy, suggesting a diagnosis of IgAN. Hypocomplementemia cannot be used as the distinguishing feature either. Nasr et al. found that only 72% of elderly patients with biopsy-proven PIGN (IgA-dominant and classical) had low serum levels of C3 or C4 [1]. Finally, stronger mesangial staining for lambda light chains than kappa light chains may point to IgAN [2]. Overall, clinical distinction between the two entities can be difficult.
The microbe classically associated with IgA-dominant PIGN is coagulase-positive staphylococcus [1, 3, 5]. Balb/c mice when immunized with S. aureus have been shown to develop mesangial deposits of IgA, IgG and C3 [6]. The S. aureus cell-envelope antigen has been identified as a possible causative antigen and, in one study, was present in 75% of cases with IgA-dominant PIGN and 68% of patients with IgAN [7]. However, these findings do not explain the association of IgA-dominant PIGN with coagulase-negative staphylococcus and Gram-negative organisms such as Escherichia coli and Enterobacter cloacae [1, 3]. Mucosal infections are known to coincide with active IgAN. C. difficile as the inciting organism for glomerular IgA deposition has also been reported twice [8, 9]. Thus, neither the lack nor the presence of S. aureus in the clinical history fully eliminates or supports the diagnosis of IgA-dominant PIGN versus IgAN.
We performed immune profiling at the time of acute kidney injury. Central to the development of IgAN is the aberrant glycosylation of a portion of the circulating IgA1, characterized by galactose deficiency of the hinge-region O-linked glycans. This structural feature is more important than levels of total IgA1, as shown by patients with IgA1 myeloma who only rarely develop glomerular deposits of IgA [10]. The O-glycosylation nature of IgA1 has not been described previously in IgA-dominant PIGN. In our patient, ELISA demonstrated markedly elevated serum levels of total IgA. Western blot after SDS–PAGE separation of serum IgA under non-reducing conditions showed a normal polymer-to-monomer ratio. Serum level of galactose-deficient IgA1 was markedly elevated, to that normally found in patients with IgAN.
In IgAN, the circulating galactose-deficient IgA1 serves as the autoantigen target of circulating anti-glycan-specific IgG and/or IgA1, leading to formation of IgG-IgA1 immune complexes that deposit in the mesangium [11, 12]. To gauge the nephritogenicity of our patient's circulating IgA1, serum (native or IgA- or IgG-depleted) was fractionated by size-exclusion chromatography, fractions added to cultures of human primary mesangial cells, and cellular proliferation assessed by thymidine incorporation. Our patient had circulating IgG- and IgA1-containing immune complexes. Large complexes (>800 kDa) consisting of IgG3 and IgA1 stimulated proliferation of the mesangial cells whereas smaller complexes (700–800 kDa) did not (Figure 2), as has been described in patients with IgAN [13]. Depletion of IgA or IgG from serum removed stimulatory activity from the large-molecular-mass fractions, suggesting that IgA-IgG complexes were responsible for the stimulatory activity. Depletion of IgA, but not IgG, removed the inhibitory activity from fractions 36–42, indicating that the small-molecular-mass inhibitory complexes contained IgA (Figure 2). IgG3 was the only IgG subclass identified in the stimulatory complexes (Table 1). These results indicate that the pathogenesis of the kidney injury was similar to that of active IgAN.
Fig. 2.

Circulating IgA1-containing complexes were isolated from serum (native or IgA- or IgG-depleted) by size-exclusion chromatography on a calibrated Superose 6 column, fractions added to cultures of primary human mesangial cells, and cellular proliferation assessed by thymidine incorporation. High-molecular-mass fractions of serum induced proliferation of the mesangial cells. Fractions 26 and 30 stimulated cellular proliferation in an IgA- and IgG-dependent manner, whereas fractions 36–42 were inhibitory in an IgA-dependent manner. Fractions 30, 36 and 42 (boxed) were analyzed for total IgG and IgG subclasses (see Table 1).
Table 1.
Analysis of IgG subclasses in stimulatory (30) and inhibitory (36, 42) fractions of serum
| Fractions | 30 | 36 | 42 |
|---|---|---|---|
| Total IgG | + | + | +++ |
| IgG1 | − | + | +++ |
| IgG2 | − | − | + |
| IgG3 | + | + | +++ |
| IgG4 | − | − | + |
Results of western blot analyses are expressed as negative (−), positive (+) or strongly positive (+++).
In conclusion, the clinical and pathologic distinction of IgA-dominant PIGN and active IgAN can be very difficult. We have shown that in this patient the mechanisms of disease for the glomerular deposition of IgA associated with colitis induced by C. difficile were similar to those of patients with IgAN. Based on these molecular diagnoses and pathologic review, a diagnosis of active IgAN is favored; however, significant clinicopathological overlap clearly exists between these two entities. Further studies are needed to investigate the IgA1 profiles in patients with more classical IgA-dominant PIGN to confirm similarities or differences with the pathogenesis with IgAN. If such differences exist, they could be exploited to provide a molecular means to differentiate these two entities.
Concise methods
Immune complexes were isolated by fractionation of serum samples by size-exclusion chromatography and their biological activities were tested with cultured primary human mesangial cells [13]. ELISA and immunoblotting, including detection with subclass-specific antibodies after non-reducing SDS–PAGE, were performed, as described [14].
Conflict of interest statement
None declared.
Acknowledgements
This work was supported in part by grants from the National Institutes of Health DK078244, DK082753 and GM098539, and by a gift from the IGA Nephropathy Foundation of America.
References
- 1.Nasr SH, Fidler ME, Valeri AM, et al. Postinfectious glomerulonephritis in the elderly. J Am Soc Nephrol. 2011;22:187–195. doi: 10.1681/ASN.2010060611. [DOI] [PubMed] [Google Scholar]
- 2.Haas M, Racusen LC, Bagnasco SM. IgA-dominant postinfectious glomerulonephritis: a report of 13 cases with common ultrastructural features. Hum Pathol. 2008;39:1309–1316. doi: 10.1016/j.humpath.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 3.Nasr SH, D'Agati VD. IgA-dominant postinfectious glomerulonephritis: a new twist on an old disease. Nephron Clin Pract. 2011;119:c18–c26. doi: 10.1159/000324180. [DOI] [PubMed] [Google Scholar]
- 4.Yoshikawa N, Ito H, Nakahara C, et al. Glomerular electron-dense deposits in childhood IgA nephropathy. Virchows Arch A Pathol Anat Histopathol. 1985;406:33–43. doi: 10.1007/BF00710555. [DOI] [PubMed] [Google Scholar]
- 5.Nasr SH, Markowitz GS, Whelan JD, et al. IgA-dominant acute poststaphylococcal glomerulonephritis complicating diabetic nephropathy. Hum Pathol. 2003;34:1235–1241. doi: 10.1016/s0046-8177(03)00424-6. [DOI] [PubMed] [Google Scholar]
- 6.Sharmin S, Shimizu Y, Hagiwara M, et al. Staphylococcus aureus antigens induce IgA-type glomerulonephritis in Balb/c mice. J Nephrol. 2004;17:504–511. [PubMed] [Google Scholar]
- 7.Koyama A, Sharmin S, Sakurai H, et al. Staphylococcus aureus cell envelope antigen is a new candidate for the induction of IgA nephropathy. Kidney Int. 2004;66:121–132. doi: 10.1111/j.1523-1755.2004.00714.x. [DOI] [PubMed] [Google Scholar]
- 8.Arrich J, Sodeck GH, Sengolge G, et al. Clostridium difficile causing acute renal failure: case presentation and review. World J Gastroenterol. 2005;11:1245–1247. doi: 10.3748/wjg.v11.i8.1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaughan WJ, Hassan MH, McCue PA, et al. Association of IgA nephropathy with Clostridium difficile colitis. Am J Kidney Dis. 1999;34:E1–E4. doi: 10.1053/AJKD034000e16. [DOI] [PubMed] [Google Scholar]
- 10.Zickerman AM, Allen AC, Talwar V, et al. IgA myeloma presenting as Henoch-Schönlein purpura with nephritis. Am J Kidney Dis. 2000;36:1–5. doi: 10.1053/ajkd.2000.16221. [DOI] [PubMed] [Google Scholar]
- 11.Tomana M, Novak J, Julian BA, et al. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J Clin Invest. 1999;104:73–81. doi: 10.1172/JCI5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suzuki H, Fan R, Zhang Z, et al. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J Clin Invest. 2009;119:1668–1677. doi: 10.1172/JCI38468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Novak J, Tomana M, Matousovic K, et al. IgA1-containing immune complexes in IgA nephropathy differentially affect proliferation of mesangial cells. Kidney Int. 2005;67:504–513. doi: 10.1111/j.1523-1755.2005.67107.x. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki H, Moldoveanu Z, Hall S, et al. IgA1-secreting cell lines from patients with IgA nephropathy produce aberrantly glycosylated IgA1. J Clin Invest. 2008;118:629–639. doi: 10.1172/JCI33189. [DOI] [PMC free article] [PubMed] [Google Scholar]