

Abstract
Sialyltransferases are responsible for the synthesis of a diverse range of sialoglycoconjugates predicted to be pivotal to deuterostomes’ evolution. In this work, we reconstructed the evolutionary history of the metazoan α2,3-sialyltransferases family (ST3Gal), a subset of sialyltransferases encompassing six subfamilies (ST3Gal I–ST3Gal VI) functionally characterized in mammals. Exploration of genomic and expressed sequence tag databases and search of conserved sialylmotifs led to the identification of a large data set of st3gal-related gene sequences. Molecular phylogeny and large scale sequence similarity network analysis identified four new vertebrate subfamilies called ST3Gal III-r, ST3Gal VII, ST3Gal VIII, and ST3Gal IX. To address the issue of the origin and evolutionary relationships of the st3gal-related genes, we performed comparative syntenic mapping of st3gal gene loci combined to ancestral genome reconstruction. The ten vertebrate ST3Gal subfamilies originated from genome duplication events at the base of vertebrates and are organized in three distinct and ancient groups of genes predating the early deuterostomes. Inferring st3gal gene family history identified also several lineage-specific gene losses, the significance of which was explored in a functional context. Toward this aim, spatiotemporal distribution of st3gal genes was analyzed in zebrafish and bovine tissues. In addition, molecular evolutionary analyses using specificity determining position and coevolved amino acid predictions led to the identification of amino acid residues with potential implication in functional divergence of vertebrate ST3Gal. We propose a detailed scenario of the evolutionary relationships of st3gal genes coupled to a conceptual framework of the evolution of ST3Gal functions.
Keywords: β-galactoside α2,3-sialyltransferases; molecular evolution; phylogenetics; genomics; molecular modeling; glycobiology; zebrafish; specificity determining position; evolution rates; coevolved amino acid
Introduction
Sialyltransferases are biosynthetic enzymes of the sialic acid metabolic pathway that mediate the transfer of sialic acid residues to terminal nonreducing positions of a variety of oligosaccharide chains found on glycoproteins and glycolipids. Their activities lead to the formation of the so-called sialome (Cohen and Varki 2010) specific of each tissues of all the biological systems of the vertebrate species (Varki 2006, 2011). Owing to their anionic charge and their peripheral position in glycans at the cell surface, sialic acids are crucial players modulating cell functions and regulating cell communications. For instance, sialylated glycans represent specific receptors for various vertebrate-binding proteins such as selectins mediating leukocytes and platelets trafficking, and siglecs involved in immune cell regulation. Likewise, a number of pathogenic agents such as viruses (influenza virus A, myxoviruses) and bacteria (Helicobacter pylori, Pseudomonas aeruginosa) also evolved this ability to read a specific sialylated sugar code (Gabius 2000; Gabius et al. 2002) distinguishing α2,3- or α2,6-linked sialic acids to dock in the vertebrate host tissues (Lehmann et al. 2006). Therefore, variability of the cognate α2,3/6-sialyltransferases is likely to have played major roles in the coevolution of these crucial self and nonself interactions, although this remains a poorly debated issue. Twenty sialyltransferases have been described and characterized mostly in the human, mouse, and chicken tissues (reviewed in Harduin-Lepers 2010, 2013). Classically, vertebrate sialyltransferases are classified into four families depending on the glycosidic linkage formed (i.e., α2,3-, α2,6-, or α2,8-) and their primary monosaccharide acceptor (i.e., galactose [Gal], N-acetylgalactosamine [GalNAc] or another sialic acid residue), and are named ST3Gal, ST6Gal, ST6GalNAc, and ST8Sia, accordingly. At the protein level, vertebrate sialyltransferases show comparable architecture with a short cytoplasmic tail, a unique transmembrane domain, a stem region of variable length, and a C-terminal catalytic domain of about 260 amino acids oriented within the trans-Golgi network of the cell (Harduin-Lepers 2013). Despite low overall protein sequence identities (15–57% for human sialyltransferase paralogs), sialyltransferase proteins share four conserved peptide motifs located in the catalytic domain known as sialylmotifs Large, Small, III, and Very Small (L, S, III, and VS, respectively) essential in substrates binding (Datta and Paulson 1995; Geremia et al. 1997; Datta et al. 1998) and in the catalytic activity (Jeanneau et al. 2004; Kim et al. 2010; Audry et al. 2011; Rakic et al. 2013; Takashima et al. 2013). In addition, five family peptide motifs (noted motif a through e) of 4–20 amino acids further differentiate each family in metazoa (Patel and Balaji 2006; Harduin-Lepers 2010, 2013). Altogether, these conserved peptide motifs represent useful hallmarks for animal sialyltransferases identification. At the gene level, each sialyltransferase family shows an evolutionary conserved gene structure made up of multiple exons. In addition, they are widely dispersed in mammalian genomes (Harduin-Lepers, Krzewinski-Recchi, et al. 2001; Harduin-Lepers, Vallejo-Ruiz, et al. 2001; Harduin-Lepers et al. 2005; Takashima 2008; Harduin-Lepers 2010). A seminal work reported on the ancient occurrence of sialyltransferase-related genes in “basal” metazoan species, much sooner than previously anticipated and the sudden emergence of several new sialyltransferase sequences more or less retained in the various vertebrate lineages (Harduin-Lepers et al. 2005; Varki 2006, 2011). Two of the four sialyltransferase multigene families (e.g., ST8Sia and ST6Gal) were subjected to phylogenetic reconstructions in the context of the two rounds of whole-genome duplication (2R-WGD) events that occurred in early vertebrates (Ohno 1970) shedding new light into the key genetic events underpinning the establishment of α2,6- and α2,8-sialylation machineries (Harduin-Lepers et al. 2008; Petit et al. 2010, 2013). Almost nothing is known pertaining to the origin and evolutionary history of two remaining sialyltransferase families (e.g., ST6GalNAc and ST3Gal).
In this study, we examined in deep details the ST3Gal family in an effort to understand the forces that shaped the human genome content of st3gal-related genes. As reviewed recently, six β-galactoside α2,3-sialyltransferases denoted ST3Gal I through ST3Gal VI belonging to the ST3Gal family have been cloned from mouse and human genomes (Harduin-Lepers 2010, 2013). Genes encoding these subfamilies were arbitrarily denoted by numbers (e.g., ST3GAL1–ST3GAL6 in human and st3gal1–st3gal6 in mice) according to a systematic nomenclature (Tsuji et al. 1996) used in table 1. However, information on invertebrate st3gal-related sequences is scarce. A unique cDNA homolog called ST3Gal I/II (Harduin-Lepers et al. 2005) was cloned from the tunicate Ciona intestinalis (Lehmann et al. 2008) and from the amphioxus Branchiostoma belcheri (Guérardel et al. 2012) suggesting that the ST3Gal family is present in a few copy number in tunicates and in cephalochordates.
Table 1.
Vertebrate α2,3-Sialyltransferases-Related Sequences.
| 2,3-Sialyltransferase | Vertebrate Species | Accession Number | Length (AA) | % of Identities to Human Ortholog |
|---|---|---|---|---|
| ST3Gal I | Homo sapiens | L29555 | 340 | 100 |
| Mus musculus | X73523 | 337 | 81.5 | |
| Gallus gallus | X80503 | 342 | 66.7 | |
| Silurana tropicalis | FN550106 | 334 | 56.7 | |
| Danio rerio | AJ864512 | 321 | 38.4 | |
| AJ864513 | 330 | 41.4 | ||
| AM287261 | 317 | 38.6 | ||
| AM287262 | 317 | 38.9 | ||
| ST3Gal II | Homo sapiens | X96667 | 350 | 100 |
| Mus musculus | X76989 | 350 | 93.4 | |
| Gallus gallus | AJ585761 | 349 | 84.4 | |
| XM_417321 | 313 | 43.0 | ||
| Silurana tropicalis | XM_002931660 | 351 | 46.5 | |
| AJ585763 | 332 | 43.2 | ||
| Danio rerio | AJ783741 | 374 | 67.1 | |
| AJ783740 | 341 | 43.1 | ||
| ST3Gal III | Homo sapiens | L23768 | 375 | 100 |
| Mus musculus | X84234 | 374 | 96.5 | |
| Gallus gallus | AJ865086 | 374 | 90.7 | |
| Silurana tropicalis | AJ626823 | 358 | 83.2 | |
| Danio rerio | AJ626821 | 356 | 65.6 | |
| AJ626820 | 372 | 62.9 | ||
| ST3Gal IV | Homo sapiens | L23767 | 333 | 100 |
| Mus musculus | X95809 | 333 | 91 | |
| Gallus gallus | AJ866777 | 328 | 76.0 | |
| XM_004945803 | 393 | 27.7 | ||
| Silurana tropicalis | AJ622908 | 330 | 59.5 | |
| Danio rerio | AJ744809 | 329 | 49.9 | |
| ST3Gal V- GM3synthase | Homo sapiens | AB018356 | 362 | 100 |
| Mus musculus | Y15003 | 359 | 85.6 | |
| Gallus gallus | AY515255 | 360 | 69.4 | |
| Silurana tropicalis | FN550108 | 372 | 53.9 | |
| Danio rerio | AJ619960 | 364 | 45.8 | |
| AJ783742 | 383 | 28.7 | ||
| ST3Gal VI | Homo sapiens | AF119391 | 331 | 100 |
| Mus musculus | AF119390 | 331 | 74.6 | |
| Gallus gallus | AJ585767 | 329 | 63.4 | |
| Silurana tropicalis | AJ626744 | 331 | 55.3 |
Note.—Orthologous sequences to six known human α2,3-sialyltransferase subfamilies (ST3Gal I–VI) are indicated with their accession number in GenBank, length of the deduced amino acid (AA) sequence, and percentage of identities to the human ortholog. In addition, gray background highlights other α2,3-sialyltransferase-related sequences with lower percentage of identities corresponding to new ST3Gal paralogs whose origin, evolutionary relationships, name and predicted function are to be established in this study.
The first aim of this study was to provide information on the mechanisms involved in st3gal gene expansion in metazoan. We identified several new st3gal-related sequences inferred from genome and transcriptome sequencing projects available in public databases that disappeared in mammals. We further analyzed the evolutionary history of this family in the context of the 2R- and teleost-specific 3R-WGD (Jaillon et al. 2004) genetic events using molecular phylogeny and synteny analysis including the use of comparative genomic programs (Catchen et al. 2009; Louis et al. 2012). We also took advantage of the ancestral genome reconstruction concept as an independent way to definitively infer orthologous/paralogous relationships of the vertebrate st3gal-related sequences (Kasahara et al. 2007; Nakatani et al. 2007; Putnam et al. 2008), according to a strategy illustrated recently by Yegorov and Good (2012). Furthermore, we present a conceptual framework to understand the specific forces influencing st3gal-related gene copy evolution. In particular, we brought attention to the events that might have led to st3gal gene losses in some vertebrate lineages. It has been argued that genes with a higher propensity to be lost along evolution showed 1) higher substitution rates under relaxed selection (Lynch and Conery 2000) and 2) low levels of expression in a limited number of tissues (Krylov et al. 2003; Wolf et al. 2006). Toward this aim, we determined the evolution rates of ST3Gal proteins, and compared the expression pattern of st3gal genes in vertebrate lineages, through functional genomics analysis of st3gal genes in the teleostean Danio rerio and the mammal Bos taurus.
The second goal of the study was to characterize the functional diversification underpinning the evolution of the various ST3Gal subfamilies. The six mammalian ST3Gal enzymes share similar molecular functions catalyzing the transfer of sialic acid residues to the terminal Gal residue of either the type 1, type 2, or type 3 disaccharides (Galβ1,3GlcNAc; Galβ1,4GlcNAc; or Galβ1,3GalNAc, respectively) resulting in the formation of α2-3 glycosidic linkages (Harduin-Lepers 2010). Briefly, ST3Gal I and ST3Gal II synthesize preferentially α2,3-sialylated structures on type 3 disaccharide as found on the mucin-type O-glycans of glycoproteins and on glycosphingolipids of the ganglioside series. ST3Gal III, ST3Gal IV, and ST3Gal VI enzyme activities result in α2,3-sialylated structures on type 1 or type 2 disaccharides leading to the formation of sialyl Lewis epitopes found on cell surface-expressed glycoproteins and glycolipids with binding activity to selectins. Finally, ST3Gal V uses mainly Lac-Cer (Galβ1,4Glc-Cer) and to a lesser extent Gal-Cer glycosphingolipid substrates leading to the formation of the α2,3-monosialylated gangliosides named GM3 and GM4, respectively. However, several in vitro studies conducted with recombinant ST3Gal enzymes have pointed to their slightly different and overlapping enzymatic specificity based on the underlying carbohydrate chain and glycan class (Kitagawa and Paulson 1994; Kojima et al. 1994; Kono et al. 1997; Rohfritsch et al. 2006). This observation further suggested their common ancestry and functional divergence, although the underlying molecular bases remained unknown. A partial three-dimensional (3D)-structure of the pig ST3Gal I obtained recently (Rao et al. 2009) provided the first detailed structural and mechanistic insights into a mammalian sialyltransferase. Conserved amino acid residues interacting with both donor and acceptor substrates were identified resulting in a better understanding of the substrate specificity of ST3Gal. Interestingly, the presence of a disordered loop located next to the catalytic center was identified further suggesting significant conformational changes during catalysis in which a flexible loop acts as a lid covering the bound donor substrate. In this study, we predicted specificity determining positions (SDPs) and coevolved amino acid residues that might be involved in functional divergence of the various ST3Gals. Those functionally important sites were mapped on the unique available ST3Gal 3D structure to propose the residues changes that might be responsible for the different ST3Gal molecular functions.
This integrated study focusing on molecular evolution and expression of st3gal-related genes enabled us to identify four additional st3gal subfamilies in vertebrates. The ten vertebrate ST3Gal subfamilies originated from genome duplication events that occurred before or during the emergence of vertebrates. We further discuss the involvement of block duplication events that took place before the 2R-WGD and we propose an evolutionary scenario in which members of the ST3Gal family are organized in three distinct and ancient groups of genes predating the early deuterostomes. We assessed the various hypotheses relative to the gene loss trends, and we highlighted the biological context of these lineage-specific losses. Our study provided convincing evidences that st3gal-related genes evolved by expansion and decline according to various selective forces associated with speciation forces and environmental changes.
Results
Identification of st3gal-Related Genes in Eumetazoa
To identify st3gal-related genes, we carried out Basic Local Alignment Search Tool (BLAST) search in various invertebrate and vertebrate nucleotide databases using the known human ST3Gal sequences and taking advantage of the sialylmotifs L, S, III, and VS described in metazoan sialyltransferases and the family-motifs a, b, c characteristic of β-galactoside α2,3-sialyltransferases (Patel and Balaji 2006; Harduin-Lepers 2010). A broad distribution of st3gal genes was observed in metazoans from the sponge Oscarella carmela, where several expressed sequence tag (EST) sequences are attributable to a unique st3gal1/2 sequence to mammals. In Ambulacraria, we found two copies of st3gal gene in the hemichordate Saccoglossus kowalevskii and in the echinoderm Strongylocentrotus purpuratus and one copy in the sea urchin Hemicentrotus pulcherrimus genome. In chordates, the α2,3-sialyltransferase-related sequence content is variable among the three subphyla cephalochordates, urochordates, and vertebrates. A unique st3gal sequence was previously identified in the tunicates C. intestinalis and C. savignyi genome (Harduin-Lepers et al. 2005) and further enzymatically characterized as an ST3Gal I/II (Lehmann et al. 2008). Several st3gal sequences were identified in Br. floridae and the expression of an amphioxus st3gal1/2 gene ortholog was studied recently in Br. belcheri (Guérardel et al. 2012). However, in spite of an extensive examination of Expressed Sequence Tags (EST) and Whole Genome Shotgun (WGS) sequences in public databases, no homologous st3gal gene could be identified in the cnidarian Nematostella vectentris or protostomes (nematodes, insects, crustacea, annelids, and mollusks).
As summarized in table 1, most examined mammalian genomes contain orthologs of the six previously described α2,3-sialyltransferase members of the ST3Gal family. Interestingly, several paralogous sequences highlighted in gray in table 1, with variable length and lower sequence identities compared with their human counterpart, were identified in nonmammalian vertebrate species. The avian Gallus gallus shows eight α2,3-sialyltransferase-related sequences and the amphibian Silurana tropicalis genome contains seven α2,3-sialyltransferase-related sequences, whereas the axolotl Ambystoma mexicanum genome shows six st3gal-related gene sequences. The coelacanth Latimeria chalumnae shows seven α2,3-sialyltransferase-related sequences. In the Actinopterygii branch, the spotted gar Lepisosteus oculatus shows eight α2,3-sialyltransferase-related sequences, whereas teleost genomes (D. rerio and Gasterosteus aculeatus) show up to 12 β-galactoside α2,3-sialyltransferase-related sequences expressed in the zebrafish liver cell line model ZFL (Vanbeselaere et al. 2012). Finally, six α2,3-sialyltransferase-related sequences were identified in the lamprey Petromyzon marinus. Altogether, this observation suggests the occurrence of multiple β-galactoside α2,3-sialyltransferases paralogs in vertebrates whose origin and evolutionary relationships remain to be established. The accession numbers of the 121 st3gal sequences analyzed in this study are gathered in supplementary tables S1 and S2, Supplementary Material online.
Molecular Phylogeny Analysis: The st3gal Family Encompasses Ten Subfamilies in Vertebrates
As a first step in the molecular phylogeny analysis, we assessed the homology of vertebrate and invertebrate ST3Gal-related protein sequences by multiple sequence alignments (MSA) with MUSCLE (supplementary fig. S1, Supplementary Material online). Following the best model suggested by Bayesian Information Criterion (BIC) for the Maximum Likelihood (ML), the method based on the Whelan and Goldman model (Whelan and Goldman 2001) +G +I was retained (see Materials and Methods). As several positions in the st3gal1/2/8 clade were not robustly supported, we conducted new analyses after removing the divergent D. rerio ST3Gal I sequences (fig. 1). The ML and Minimum Evolution (ME) approaches gave very close topologies with three clearly defined monophyletic clades of α2,3-sialyltransferase-related sequences and an additional group of sequences found only in invertebrate deuterostomes.
Fig. 1.

Maximum-likelihood phylogenetic tree of 121 sialyltransferases of the ST3Gal family. The tree with the highest log likelihood (−17,431.9308) is shown. A discrete Gamma distribution was used to model evolutionary rate differences among sites (five categories [+G, parameter = 1.1543]). The analysis involved 121 amino acid sequences. All positions with less than 95% site coverage were eliminated. There were a total of 228 positions in the final data set. Bootstrap values were calculated from 500 replicates and values greater than 80% are reported at the left of each divergence point. The bootstrap values indicated at the left correspond to ML and the right ones to ME.
The first group of α2,3-sialyltransferase-related sequences named GR1 in figure 1 gathers the well-known subfamilies ST3Gal I and ST3Gal II widely distributed in vertebrates. Interestingly, this GR1 group also includes a new subfamily present in each vertebrate order such as in the cartilaginous fish Callorhinchus milii, in the bony fish D. rerio (AJ783740, in table 1), in the coelacanth L. chalumnae, in the amphibians A. mexicanum and Si. tropicalis (AJ585763, in table 1), in G. gallus (XM_417321, in table 1) and in the nonavian sauropsida A. carolinensis, Python molurus and Chrysemys picta bellii, and notably absent in mammals (although traces of the st3gal8 gene could be detected in the gorilla genome, data not shown). Consequently, we have named this subfamily ST3Gal VIII according to the refined nomenclature of sialyltransferases that reflects their evolutionary relationships proposed in Petit et al. (2013). The genome of the lamprey P. marinus contains a sequence well assigned to ST3Gal II. At the base of these three vertebrate subfamilies, we found well-supported α2,3-sialyltransferase-related sequences originating from early metazoa (O. carmela) and deuterostomia (S. kowalevskii, St. purpuratus, Br. floridae, and C. intestinalis) that are called ST3Gal I/II/VIII (fig. 1). Comparative analysis of the genomic organization of the st3gal genes supports the hypothesis of an early individualization of GR1 group of sequences from GR2 and GR3 groups (data not shown). In the GR1 group, the gene structure is homogeneous and sialylmotif S split between two different exons, whereas it is found onto a unique exon for the genes of the GR2 and GR3 groups.
The second group of α2,3-sialyltransferase-related sequences named GR2 in figure 1 appears to be limited to vertebrates. It is composed of the two subfamilies already described in mammals ST3Gal IV and ST3Gal VI and a new vertebrate subfamily that we called ST3Gal IX. The ST3Gal IV subfamily is present in each vertebrate order, even in the chondrichthyan C. milii and in P. marinus (only a short sequence was retrieved and excluded from further phylogenetic analysis). In contrast, the ST3Gal VI subfamily is absent in teleosts, but is still detected in L. chalumnae and short sequences are found in P. marinus. The presence of ST3Gal IX is sporadic and limited to the turkey Meleagris gallopavo, the chicken G. gallus (XM_004945803, in table 1), the turtles Pelodiscus sinensis and C. pictabellii, the green lizard A. carolinensis, and in the mammal platypus Ornithorhynchus anatinus. This ST3Gal IX sequence has disappeared in frog, fish, and other mammalian genomes.
The third group of α2,3-sialyltransferase-related sequences named GR3 represented in figure 1 contains both invertebrate and vertebrate sequences. Two of the three vertebrate subfamilies, ST3Gal III and ST3Gal V are widely distributed from fish to human. The remaining one renamed ST3Gal VII (Petit et al. 2013) is related to the ST3Gal V subfamily. It is restricted to bony fishes (Le. oculatus and teleosts) and to L. chalumnae further suggesting that this gene has disappeared from tetrapod lineages. The corresponding st3gal7 gene was previously identified in the zebrafish genome (AJ783742, in table 1) (Harduin-Lepers et al. 2005) and more recently enzymatically characterized as a GM4 synthase (Chisada et al. 2009). Finally, several teleost species such as D. rerio possess two ST3Gal III-related sequences named ST3Gal III (AJ626821, in table 1) and ST3Gal III-r (AJ626820, in table 1). Three sequences of sea-urchin St. purpuratus branch out of the tree before the emergence of the three subfamilies and two sequences of Br. floridae are related to the ST3Gal III from ME analysis and to ST3Gal V/VII from ML analysis. We hypothesize that these two amphioxus sequences should be better placed at the base of the three vertebrate subfamilies, as for the three sea-urchin sequences. The genomic organization of the vertebrate genes supports the relationship between st3gal5 and st3gal7 genes as both groups of sequences have lost three exons in regions encoding the stem and beginning of the catalytic domains compared with the other genes of GR1 and GR2 (data not shown).
Interestingly, we delineated an additional group of α2,3-sialyltransferase-related sequences named GRx. These sequences are restricted to the invertebrate deuterostomes in the amphioxus Br. floridae and in the hemichordate S. kowalevskii (fig. 1).
We also observed on the phylogenetic tree that the branch lengths in the various vertebrate ST3Gal subfamilies were extremely variable. We thus assessed the significance of these differences by calculating change rates by site and subfamilies. As expected, the profiles showed contrasting rates in the stem and catalytic domains. As shown in figure 2A, most subfamilies showed high rates of substitution in their stem domain, in contrast to the catalytic domain where the peptide sequence motifs characterizing each sialyltransferase family were highly conserved with variable intervening sections. The two subfamilies ST3Gal II and ST3Gal III constituted an interesting exception as they showed highly conserved stem and catalytic domains with the lowest substitution rates as illustrated by boxplots in figure 2B. Conversely, ST3Gal IX and ST3Gal IV subfamilies showed the most variable stem region, and ST3Gal IV and ST3Gal V subfamilies, the highest rates of evolution in their catalytic domain.
Fig. 2.

Substitution rates in vertebrate ST3Gal polypeptide sequence. (A) The transmembrane and cytoplasmic domains have been discarded from the analysis. For clarity of representation, the profiles of only four ST3Gal subfamilies are shown, that is, ST3Gal II, ST3Gal VIII, ST3Gal IV, and ST3Gal III. Position 1 corresponds to the first amino acid of the stem. (B) Comparisons of mean substitution rates of the metazoan ST3Gal. The left graph corresponds to the stem part and the right one to the catalytic domain. Fifty percent of values are comprised in the limits of boxes and the median is shown with a horizontal line inside the box. The different letters a–f correspond to significant differences (Tukey’s test on log-transformed substitution rates). The three groups of subfamilies GR1, GR2, and GR3 are in brackets.
Sequence similarity network visualization (Atkinson et al. 2009) of relationships across the various extant ST3Gal protein subfamilies in a much larger set of sequences confirmed our phylogenetic analysis (fig. 3). The greatest degree of similarity occurred between sequences belonging to GR1 including the sequences of the newly described subfamily ST3Gal VIII. These sequences remained grouped together even at stringent cutoff values (1e-80 to 1e-100; fig. 3D–F), providing strong support of the proposed classification. GR2 sequences formed distinct clusters for each subfamily at permissive E values (1e-60; fig. 3B), highlighting the comparatively low degree of similarity between its members. Furthermore, ST3Gal IX sequences from the GR2 group showed a closer relationship with ST3Gal IV than ST3Gal VI sequences. On the other hand, sequences belonging to GR3 formed a separated subnetwork at E value 1e-70 (fig. 3C) and ST3Gal VII sequences have greater similarity with ST3Gal V sequences than ST3Gal III. At stringent threshold (1e-100; fig. 3F), sequences of the GR2 and GR3 separated into distinguishable groups, only GR1 sequences remain connected. Interestingly, ST3Gal fish sequences formed separate subnetworks indicating their lower degree of similarity within the different subfamilies, which could not be established in our phylogenetic tree analysis. Permissive thresholds (E value of 1e-55 to 1e-70; fig. 3A–C) pointed to associations between GR2 and GR3 groups (i.e., ST3Gal IV/VI/IX and ST3Gal III/III-r/V/VII protein sequences) and also showed individual groups GR1 and GRx. Altogether, this sequence similarity network analysis allowed making functional inferences of uncharacterized ST3Gal sequences. The outgroup comprising distantly related sequences of the ST6Gal family was used as a negative control. The absence of edges between the outgroup and ST3Gal sequences helped to evaluate whether the most permissive E value threshold used (i.e., 1e-55) implied similarity relationships.
Fig. 3.

Sequence similarity network: The similarity network was constructed as described in Materials and Methods. It included 336 ST3Gal-related sequences identified from vertebrate and invertebrate genomes and 27 ST6Gal I sequences constituting an outgroup. The pairwise relationship between sequences was calculated by a BLASTall search in the custom database with each individual sequence in the set and the E value was taken as a measure of similarity between sequences. Thresholded similarity network represents sequences as nodes (circles) and all pairwise sequence relationships (alignments) better than an E value threshold as edges (lines) between nodes. The same network is shown here at six different thresholds, varying the cutoff value from permissive (A–C) to stringent (D–F). At a permissive E value thresholds 1e-55 (A) sequences belonging to GR2 and GR3 merge together, as the threshold is becoming more stringent and edges associate with more significant relationships, sequences break up into disconnected groups with high similarity within each group. Nodes were colored according to the subfamily to which the sequence belongs, either known (ST3Gal I–ST3Gal VI) or predicted (ST3Gal VII, ST3Gal VIII, and ST3Gal IX). Also ST3Gal-related sequences that belong to intermediate groups have been considerate separately, as invertebrate ST3Gal I/II/VIII, ST3Gal III/V/VII, ST3Gal IV/VI/IX, and ST3Gal III-r sequences, yielding in total to 14 different groups including the control group. The network was visualized using Cytoscape 2.8.3 version (Shannon et al. 2003), default Cytoscape force-directed layout was applied.
Reconstruction of the Genetic Events that Have Led to the Diversification of ST3Gals in Vertebrates
To investigate the dynamic of st3gal genes evolution across vertebrate genomes and to explain the appearance of several new vertebrate st3gal gene subfamilies, we analyzed the evolutionary history of st3gal in the context of 2R- and 3R-WGD genetic events (Ohno 1999; Jaillon et al. 2004) using comparative genomic programs (Catchen et al. 2009; Louis et al. 2012). We also used paleogenomics reconstructions of the vertebrate (Nakatani et al. 2007) and chordate (Putnam et al. 2008) ancestral genomes as an independent approach to study the duplication history of these st3gal genes. As illustrated in figure 4A, 2R-duplicated genes are contained on one of the ten vertebrate ancestral (VA) protochromosomes in the pre-2R genome established on the extant ciona genome and designated A–J in the N-model (Nakatani et al. 2007). Similarly, they are contained on one of the nine chordate linkage groups (CLG) in the pre-2R genome reconstructed from the extant amphioxus genome and named 1–9 in the P-model (Putnam et al. 2008) and on one of the 13 teleost ancestral protochromosomes named a–m in the pre-3R genome reconstructed from the extant fish genomes (Kasahara et al. 2007). After the 2R-WGDs, they are found on four linkage groups with shared synteny (e.g., gnathostome ancestor [GNA] protochromosomes A0, A1, A2, A3 in the N-model and VA protochromosomes 1a, 1b, 1c, 1d in the P-model). Intensive interchromosomal rearrangements (fission, fusion, and translocations), which took place between and after the 2R-WGD genetic events, have led to conserved vertebrate linkage blocks widely distributed throughout the human genome.
Fig. 4.

Ancestral genome reconstruction to assess the ST3Gal subfamily origin. (A) Simplified phylogenetic tree illustrating evolution of chordate genome with ancestral genome reconstruction using the N-model (Kasahara et al. 2007; Nakatani et al. 2007) for the reconstruction of the 550-My-old ancestor of vertebrates just before the 2R WGD events in early vertebrates (VACs, N-model) and the P-model (Putnam et al. 2008) for the reconstruction of the 770-My-old common ancestor of amphioxus and vertebrates + tunicates (CLGs, P-model). (B) Schematic illustration of the data obtained in combination with phylogenetic and syntenic analysis to assess origin and evolutionary relationships of st3gal-related genes in vertebrates. Using the known genomic st3gal gene location in Oryzias latipes (OLA), in G. gallus (GGA), and in H. sapiens (HSA), each st3gal-related gene was mapped to a chromosomal segment (Block ID). The identified chromosomal segments were traced to CLG using P-model and to VAC using N-model. This latter hosting ancestral st3gal-related genes in pre-2R vertebrates confirmed ohnology of st3gal1, st3gal2 and st3gal8 genes from GR1, of st3gal4, st3gal6 and st3gal9 genes from GR2, and of st3gal3, st3gal5 and st3gal7 genes from GR3. Subsequent genome rearrangements for human ST3GAL5 gene on HSA4 and medaka st3gal6 on OLA21 have influenced evolution of these st3gal subfamilies. Crosses indicate gene losses. Tel Anc, pre-3R teleost ancestor; Gn Anc, post-2R GNA; CLG, pre-1R CLG.
Within the α2,3-sialyltransferase-related sequences of GR1, a well-conserved synteny could be established for st3gal1, st3gal2, and st3gal8 gene loci in human (table 2), chicken, medaka, and zebrafish genomes (supplementary fig. S2, Supplementary Material online). As the st3gal8 gene was absent in mammals, we considered the neighboring CPNE1 gene to retrieve the synteny on human chromosome 20 (Hsa20) and mouse chromosome 2 (Mmu2). The paralogy between the segments bearing the three st3gal genes of GR1 was clearly visible in chicken chromosomes Gga2 (st3gal8), Gga11 (st3gal2), and Gga20 (st3gal1) (supplementary fig. S2, Supplementary Material online) and in the zebrafish genome (table 3) and corresponded to the GNA protochromosomes B1, B4, and B0, respectively, to the VA protochromosome B, and to the CLG 3 (fig. 4B).
Table 2.
Number of Human Paralogous Genes Identified from Synteny Database at Uoregon Site (http://syntenydb.uoregon.edu/synteny_db/, last accessed December 2014) Using ST3GAL Loci of GR1 as Seed Genes and Branchiostoma floridae as an Outgroup.
| ST3GAL1 | ST3GAL2 | ST3GAL8 | |
|---|---|---|---|
| Hsa 8 | Hsa 16 | (Hsa 20) | |
| ST3GAL1 | |||
| ST3GAL2 | 11–46 | ||
| ST3GAL8a | 40–85 | 32–34 |
Note.—When an ST3GAL gene was absent in human, but present in other vertebrates, several seed genes were chosen within the synteny common to other vertebrates. Predicted human chromosome bearing lost ST3GAL gene loci is indicated in parenthesis. The first number represents the number of paralogous genes obtained with a window of 100 genes and the second one with a window of 200 genes.
aSeed gene was CPNE1.
Table 3.
Number of Danio rerio Paralogous Genes Identified from Synteny Database at Uoregon Site Using st3gal Loci of GR1 as Seed Genes and Branchiostoma floridae as an Outgroup.
| ST3GAL1 | ST3GAL2 | ST3GAL8 | |
|---|---|---|---|
| Dre19 | Dre18 | Dre11 | |
| ST3GAL1 | |||
| ST3GAL2 | 35 | ||
| ST3GAL8 | 2 | 11 |
Within the α2,3-sialyltransferase-related sequences of GR3, the syntenies including st3gal loci were also highly conserved in the vertebrate lineages (supplementary fig. S3, Supplementary Material online). As st3gal7 was absent in tetrapods, we could retrieve the corresponding block of synteny using hsp1 and hps2e genes widely distributed from fish to human. We found clear correspondences around these genes on human chromosome 10 (Hsa10) as well as mouse chromosome 19 (Mmu19), chicken chromosome 6 (Gga6), and frog chromosome 6 (Str6). The block associated with these genes clearly corresponded to GNA protochromosome C0 and CLG 7. The block harboring the st3gal5 gene seemed to belong to GNA protochromosome C1 and CLG 7. Unexpectedly, its location in the human genome was on human chromosome Hsa4 instead of Hsa2 further suggesting translocation of the st3gal5 gene in the human genome (fig. 4B). Moreover, the block including the st3gal3 gene fitted with GNA protochromosome A2, and did not fit to any CLG further suggesting that it could result from gene duplication and translocation/chromosome rearrangements that took place during the pre-1R to post-2R period (Nakatani et al. 2007; Yegorov and Good 2012).
Similarly, within the α2,3-sialyltransferase-related sequences of GR2, the syntenies including st3gal loci were also highly conserved in the vertebrate lineages (supplementary fig. S4, Supplementary Material online). As the st3gal9 gene was absent from meta- and eutherians, we considered the neighboring genes tlr4, col27A1, and dbc1 widespread from teleosts to birds as seed genes to search the most probable synteny on human chromosome 9 (Hsa9) (supplementary fig. S4, Supplementary Material online). As the st3gal6 and neighbor lnp1 genes were absent in teleosts (medaka, zebrafish, and fugu), we used the same strategy, choosing the COL8A1 and NIT2 genes on human chromosome 3 (Hsa3). We found that the synteny was broken and present on two different chromosomes in teleosts (medaka Ola21 and zebrafish Dre9 for col8a1 block, and Ola 4 and Dre 6 for nit2 block). As a result, the blocks around st3gal4 and st3gal6 loci could be associated with GNA protochromosomes J0 and J1, and VA protochromosomes 9c and 9b, respectively (fig. 4B). As previously observed for the st3gal3 gene, the block including the st3gal9 locus corresponded to GNA protochromosome A0, and did not fit to any CLG, further illustrating the occurrence of inter protochromosome rearrangements during the pre-1R to post-2R period.
Using Synteny database (Catchen et al. 2009), we could obtain similar results calculating the number of paralogs between the different gene combinations of the GR2 and GR3 in the human genome (table 4). As expected for ohnologs, we found paralogies between the segments bearing ST3GAL4 and ST3GAL6 on one hand, corresponding to GNA protochromosomes J0 and J1, and those bearing ST3GAL5 and ST3GAL7 on the other hand (corresponding to GNA protochromosomes C1 and C0). Furthermore, these analyses revealed clear paralogy between chromosomal segments bearing ST3GAL9 from the GR2 and ST3GAL3 from the GR3, corresponding to GNA protochromosomes A0 and A2, and also ST3GAL3 and ST3GAL6 from the GR2 (GNA protochromosomes A2 and J1). Finally, looking at Br. floridae genome in deeper details, we found two segments bearing st3gal sequences. The first one included in the scaffold 210 was syntenic to Hsa11q13 (ST3GAL4), Hsa10q24 (lost ST3GAL7, GR3), Hsa3p21-25 (ST3GAL6), Hsa1p34-q41 (ST3GAL3), that is, members of GR2 and GR3 (supplementary fig. S5, Supplementary Material online). The second one included in the scaffold 67 was syntenic to Hsa3p11 (ST3GAL6) and Hsa2q14 (ST3GAL5), also members of GR2 and GR3. The segment Bfl-V2-127 hosting an st3gal gene of the GR3 group was syntenic to Hsa1p21 (including ST3GAL3, GR3) and Hsa9p13 (lost ST3GAL9, GR2) supporting paralogies between segments bearing genes of GR2 and GR3.
Table 4.
Number of Human Paralogous Genes Identified from Synteny Database at Uoregon Site Using ST3GAL Loci of GR2 and GR3 as Seed Genes and Branchiostoma floridae as an Outgroup.
| ST3GAL3 | ST3GAL5 | ST3GAL7 | ST3GAL4 | ST3GAL6 | ST3GAL9 | |
|---|---|---|---|---|---|---|
| Hsa 1 | Hsa 2 | (Hsa 10) | Hsa 11 | Hsa 3 | (Hsa 9) | |
| ST3GAL3 | ||||||
| ST3GAL5 | 1–8 | |||||
| ST3GAL7a | Not found | 8–67 | ||||
| ST3GAL4 | 1–7 | 1–2 | Not found | |||
| ST3GAL6 | 4–66 | 1–80 | Not found | 10–17 | ||
| ST3GAL9b | 78–78 | Not found | Not found | Not found | Not found |
Note.—When an ST3GAL gene was absent in human, but present in other vertebrates, several seed genes were chosen within the synteny common to other vertebrates. Predicted human chromosome bearing lost ST3GAL gene loci is indicated in parenthesis. The first number represents the number of paralogous genes obtained with a window of 100 genes and the second one with a window of 200 genes.
aSeed gene was CNNM2.
bSeed gene was Col27A1.
Conserved and Coevolving Amino Acids in ST3Gal Sequences: Structure Modeling of ST3Gal Lid Domain
In an attempt to shed light on the molecular mechanisms underpinning vertebrate ST3Gal functional divergence, we further studied conserved and coevolving amino acids in ST3Gal sequences. Indeed, changes in conservation degree in a particular amino acid position may reflect functional innovation after gene duplication, because one copy evolves under relaxed constraints, which allows it to accumulate changes and develop new functions and specificities. Traditionally, two basic types of functional divergence sites have been distinguished according to Gu (2001) (see Materials and Methods). Our bioinformatic program retrieved consensus sequences in the stem region for each vertebrate ST3Gal subfamily, but nearly none for groups of subfamilies (fig. 5A). In contrast, most of the catalytic domain was retrieved as a series of consensus sequences common to groups of subfamilies. From this initial analysis, the GR1 group was clearly apart from the two remaining GR2 and GR3 groups as illustrated by the presence of about 67 type I sites highlighting different evolutionary rates (fig. 5A).
Fig. 5.

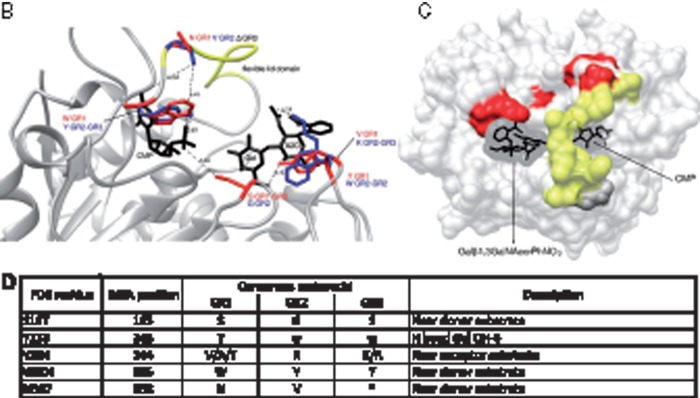
(A) Conservation of amino acid stretches in the vertebrate ST3Gal sequences evidenced by automatic conserved sequence searches. We assessed conservation of the 382 amino acid positions selected from the vertebrate ST3Gal sequences considered for phylogeny analyses (see tables S1 and S2, Supplementary Material online) using an automatic search of consensus sequences described in Materials and Method. For each subfamily, the consensus sequences were aligned with the one generated by MEGA 5.0. The resulting alignment is used to detect those positions called SDP that are conserved within groups of related proteins that perform the same function (specificity groups). SDP type I is variable in one group and conserved in the others, whereas type II positions show different conserved amino acids among the various groups of sequences. Amino acids residues belonging to conserved peptide sequences are shown. Amino acid positions with a yellow background identify conserved amino acid for the α2,3-sialyltransferase-related sequences of the GR1 group, with a green background those of the GR3 (especially the ST3Gal V–VII), with an orange background, those of the GR2, and with a blue background, those of the GR2 and GR3. The sialylmotifs L, S, III, and VS are indicated in black background, and the specific motifs to ST3Gal family –a-, -b- and –c-, in gray background. The positions are numbered from the beginning of the stem, excluding the N-terminus and the transmembrane domain. SDP prediction for GR1, GR2, and GR3 groups. (B) Representation of five SDPs located nearby the active site is represented in the reference structure (PDB: 2WNB). The modeled lid domain is shown in yellow, donor and acceptor substrates are shown in black stick representation, SDPs are shown in red or blue stick, the label indicates the conserved amino acid and the corresponding group (i.e., near CMP a W is conserved in GR1 and it is represented in red, whereas a Y is conserved in GR2 and GR3 at the same position, which is represented in blue).When there is no conserved residue in a group, the position is labeled as Δ in black. (C) SDP in surface representation of the reference structure (PDB: 2WNB). The modeled lid domain is highlighted in yellow, SDP predicted in red, the donor and acceptor substrate are shown in black. Note that the lid domain flexibility can alter the binding site of both substrates, and also the amino acid change in the SDP. (D) Table summarizing SDPs information. First column indicates the corresponding position in the reference structure (PDB: 2WNB); second column the position in the MSA, the third the most conserved residues for each group, the * indicates variable position; in the last column is entered a description that can be related to the function of the position.
Furthermore, amino acid residues that are likely to be responsible for the differential substrate specificity of ST3Gals were predicted using SDPpred. Figure 5B and C illustrates SDP predictions between the three vertebrate α2,3-sialyltransferase sequence groups (GR1–GR3) and describes five SDP positions located in the active site. At the alignment position 195, a Ser (S) was highly conserved in GR1 and GR3, whereas in GR2, a Gly (G) is conserved. Interestingly, this type II site also represented a unique reliable marker of the ancient functional divergence of ST3Gal sequences belonging to GR2 and GR3/GR1 groups. In the reference pST3Gal I structure, this position corresponding to S-197 (fig. 5D) was located at contact distance of the CMP phosphate group and OH group could be involved in H-bond formation helping to phosphate stabilization, whereas G would not have any implication in phosphate stabilization. The Tyr (Y) at position 243 (Y-233 in the structure, fig. 5D) was highly conserved in the GR1, with exception of sequences belonging to O. carmela, which showed a Trp (W) in this position, as all the sequences of the GR2 and GR3 groups defining a type II position. The Y was involved in an H-bond formation with the Gal residue CG3 (fig. 5B), and it has been reported as a determinant of Gal acceptor specificity (Rao et al. 2009; Rakic et al. 2013). At position 244, a type I SDP was predicted (V-234 in the structure, fig. 5D) denoting no altered functional constraints. It was a variable position in GR1 and in the sequences of the GR2 and GR3 groups, a Lys (K) was conserved. In the known pST3Gal I structure, this amino acid position corresponded to V-234 that is at contact distance of the hydroxy oxoammonium of the Gal residue (CG3). Two SDPs corresponding to positions 335 and 338 (residues W-304 and N-307 in the structure; fig. 5D) were predicted in the sialylmotif III and in the flexible lid domain, respectively. The aromatic ring of the amino acid at the 335 position and the donor substrate could stabilize each other by attractive nonbonded interactions known as pi-stacking. Regarding the type I amino acid position 338, an Asn residue (N) was conserved in the GR1 group, but this amino acid is highly variable in the other groups GR2 and GR3. Altogether, these SDP predictions identified several positions indicative of the functional divergence of each group of sequences in early vertebrates.
As a combination of changes in the ST3Gal sequences might also account for specificity novelties, we investigated coevolution of ST3Gal amino acids using mutual information (MI). If two residues share high signal of MI, the two residues most likely are coevolving, meaning that to maintain a given protein function, a mutation of one residue is linked to a specific compensatory mutation of the other residue. The MI network for ST3Gal family members showed that higher MI values (top 10%) are found in amino acid positions that lies within a sialylmotif (L, S, III, or VS) or within a family motif (a, b, or c) and most of the MI was found both within and between motifs (fig. 6A). Moreover, amino acid residues with high MI (top 10% MI values) were found within each known motif (red lines in fig. 6A). The cumulative MI (cMI) characterizes the extent of MI for each position. Information accumulated mainly at particular positions within sialylmotifs and positions 318 and 324, for instance, with high cMI value was more likely to be important within the VS motif. Interestingly, the amino acid residues R-60, F-124, L-331, and F-340 showed high cMI values and were predicted to play important role in the maintenance of the coevolutionary network, even though they are positioned outside the previously described conserved motifs. The top scoring cMI residues were considered for further analysis, the MI subnetwork is shown in figure 6B. Residue G-273 showed the highest cMI value and the highest number of MI interactions (55 lines in the complete network). The top ten scoring cMI residues formed a fully connected subnetwork (fig. 6B), where each residue was connected to all the others (nine lines in every case). Each residue in the subnetwork belonged to a motif, eight to the sialylmotifs L, S and III, and two to the family motifs b and c. Residue N-173 shared the highest MI value with Y-303, both were located in the enzyme active site. N-173 was near donor substrate and participated in phosphate group stabilization, as described previously (Rao et al. 2009). The amino acid residue D-216 belonged to the b family motif and it has been reported to interact with Gal-OH-6 (Rao et al. 2009). This position was the most distant in 3D structure from the rest of the subnet residues and it showed his higher MI values with positions C-145, G-160, G-185 of sialylmotif L, and C-284 of sialylmotif S.
Fig. 6.

Coevolving amino acid positions described in ST3Gal family. (A) MI circos. The information of each circle from outer to inner is the following: Sialylmotifs, highlighted in dark gray, and family motifs highlighted in light gray subfamily motifs, the following internal circle indicates the number residue and amino acid identity of the reference pST3Gal I structure. The colored square boxes of the third circle indicate the conservation degree (highly conserved positions are in red, whereas less conserved ones are in cyan). The fourth and fifth circles show the proximity MI and cMI scores as histograms, facing outwards and inwards, respectively. In the center of the circle, the lines that connect pairs of positions represent a significant MI value (>6.5), highlighted in red are lines with higher MI score (top 5%), black ones are between 70% and 95%, and gray edges account for the remaining 70%. (B) Subnetwork of higher cMI. Nodes represent amino acid residues and lines between nodes, significant MI score. The length of the lines is proportional to MI value, the closest nodes have higher MI. Nodes are colored by cMI from violet to yellow (higher to lower). (C) Predicted coevolved residues position in the reference pST3Gal I 3D structure. Red balls illustrate Cα of the ten amino acid residues with higher cMI score positioned in the pST3Gal I reference structure (PDB 2WNB).
ST3Gal Genes Spatiotemporal Expression in Vertebrates
To understand the evolutionary forces that might have influenced st3gal-related gene number, we assessed their expression profile across vertebrates. Changes in their spatiotemporal profile of expression might be indicative of their functional fate and indeed, several studies have noticed differential expression pattern of α2,3-sialylation and st3gal genes in various mammal species (Kono et al. 1997; Nairn et al. 2008). The expression of each of the six bovine st3gal genes was quantified in five tissues using a Taqman Low Density Array (TLDA). TLDA analyses revealed parallel variations in the different tissues, that is, higher levels in heart and muscle than in liver and thymus except for st3gal1 for which the level recorded in liver is as high as in heart (fig. 7A). We also analyzed zebrafish st3gal genes expression patterns in various adult tissues and embryonic developmental stages by means of reverse transcription polymerase chain reaction (RT-PCR) (fig. 7B) and whole-mount in situ hybridization (ISH) of RNA (supplementary figs. S6–S8, Supplementary Material online). Eleven of the 12 zebrafish st3gal genes were differentially transcribed in the various D. rerio adult tissues tested. The st3gal1D was not expressed at all in any tissues examined, whereas st3gal1C showed almost undetectable gene expression in gills, fins, and gall-bladder. The st3gal2, st3gal3-r, st3gal4, st3gal5, and st3gal7 genes were the most widely expressed with similar spatial distribution in all the tested tissues. St3gal1A was mainly expressed in gonads, large intestine and gills. St3gal4 was expressed to a very low level in brain, heart, gills, kidney, eye and spleen, st3gal8 was highly expressed in gonads and brain, whereas st3gal7 was expressed in small and large intestine and in liver (fig. 7B). Finally, zebrafish st3gal genes were found to be mainly expressed from early somitogenesis to 48 h postfertilization in unique and overlapping territories either with a spatially restricted pattern (st3gal2, st3gal3, st3gal3-r, st3gal7, and st3gal4) observed mainly in vasculature, developing skeletal elements, liver, and pronephric system or with a spatially diffuse pattern (st3gal1A, st3gal1B, st3gal1C, st3gal8, and st3gal5) observed mainly in the head (supplementary figs. S6–S8, Supplementary Material online). Except for the highly conserved st3gal2 and st3gal3 genes, which maintained a high and specific level of expression during zebrafish development, it appeared from these data that the functional fate of st3gal-related gene was not predictable on the basis of gene expression profile alone.
Fig. 7.

Expression pattern of st3gal genes in two model organisms. (A) Expression pattern of bovine st3gal genes in five adult tissues using TLDA approach. Total RNA was extracted from heart, liver, muscle, lung and thymus and retrotranscribed. TLDA was carried out as described in Materials and Methods. The different letters a–d corresponded to significantly different expressions per tissue (ANOVAs on five independent individuals). (B) Expression pattern of the zebrafish st3gal genes in various adult tissues using RT-PCR. Relative expression levels of zebrafish st3gal and β-actin mRNA were evaluated by RT-PCR as described in Materials and Methods, among various zebrafish adult tissues. Oligonucleotide primer sequence specific of each zebrafish st3gal gene is already described in Vanbeselaere et al. (2012). The zebrafish β-actin (378 bp) was amplified as a control of cDNA synthesis and purity. Data gathered each group of st3gal genes, that is, GR1, GR2, and GR3.
To estimate the breadth of st3gal gene expression through vertebrate evolution, we screened various tissue EST libraries from several representative animal species in Unigene site and we completed this survey taking advantage of our own data on B. taurus and D. rerio experiments. We observed that the expression sites and level of st3gal genes were higher in most mammalian tissues than in zebrafish or chicken tissues, as the vectors corresponding to the different tissues in our Principal Component Analysis (PCA) were globally oriented to the right of the projection, in the direction of mammalian genes (supplementary fig. S9, Supplementary Material online). Furthermore, we compared the diversity (Shannon index) of tissues expressing the st3gal-related genes in the set of organisms described previously. The most widely represented genes were st3gal2 from GR1, st3gal6 from GR2, and both st3gal3 and st3gal5 genes from GR3 (supplementary table S3, Supplementary Material online). These data further supported the hypothesis that the most widely expressed genes in a variety of vertebrate tissues were also the most conserved, although this should be confirmed using more vertebrate models.
Discussion
Many biological processes such as host-pathogen recognition or fertilization and development are governed by carbohydrate–protein interactions involving sialic acids expressed on the cell surface (Varki 1992; Schauer and Kamerling 1997; Schauer 2009). Yet, the mechanisms and roles of glycan diversification in metazoa remain extremely challenging issues in Glycoscience (National Research Council 2012). Sialyltransferases of the GT-29 entry of the CAZy classification (Cantarel et al. 2009) are key enzymes in sialoglycoconjugates biosynthesis of utmost interest to gain further insights into the biological relevance of sialic acid containing glycan chains during animal evolution (Varki 2007; Schauer 2009; Petit et al. 2013). This study concentrated on the broad ST3Gal family known to mediate the addition of α2,3-linked sialic acid to Galβ1-4/3GlcNAc and GalNAcβ1-3GalNAc disaccharides in mammals. Taking advantage of numerous genome sequencing projects, several hundreds of new st3gal-related genes were identified in metazoan genomes. Interestingly, sialyltransferases-related sequences were recently identified in plants, archeplastidia, and chromoalveolates (Harduin-Lepers et al. 2005; Giacopuzzi et al. 2012; Moreau et al. 2012) further suggesting that these genes appeared prior the separation of multicellular lineage of Opisthokonta, although no sialyltransferase-related sequences could be identified in fungi. These sequences showed a metazoan taxonomic affiliation based on the occurrence of conserved sialylmotifs L, S, III, and VS and could be related to st3gal gene family based on BLAST analysis, but no st3gal family motif (Patel and Balaji 2006; Harduin-Lepers 2010) was detected in these sequences and thus, they were not included in this study. Although the evolutionary relationships between all the sialyltransferase families are not yet established, the st3gal as well as the st6gal gene (Petit et al. 2010) families could represent the most ancient sialyltransferase families described in animals with major role in metazoan evolution. To obtain the enlarged view on the evolution of the α2,3-sialylation machinery in metazoan illustrated in figure 8, we first reconstructed the molecular phylogeny of ST3Gal-related sequences. Second, we deciphered the mechanisms involved in their expansion using nonsequence-based information, such as exon–intron organization, paralogies and conserved syntenies, and ancestral genome reconstruction. Third, the functional diversification was studied through analyses of conserved amino acid positions of ST3Gals that could be implicated in critical aspect of protein general function using an SDP approach (Teppa et al. 2012), and through analysis spatiotemporal expression of st3gal genes in two model organisms. Fourth, the lineage-specific significance of gene losses was tentatively linked to evolutionary rates and expression profile of duplicated genes. Altogether our data revealed that the ST3Gal family has undergone a complex evolutionary history of gene duplication and gene loss events.
Fig. 8.

Schematic diagram of the st3gal gene evolution in various animal species. This figure depicts the evolution of st3gal genes represented by black rectangles when still detected in genomes or gray rectangles when they are lost. Functional divergence between each group GR1, GR2, GR3, and GRx was acquired very early during deuterostome evolution and new enzymatic activities characteristic of each subfamily were acquired after whole-genome duplication events. Block duplication events and intron loss are indicated by a black circle. O-GP, O-glycosylprotein; N-GP, N-glycosylprotein; GL, glycolipids.
St3gal Genes Expansion
Using whole-genome information and a molecular phylogeny approach, we identified four steps of genomic innovation that yielded st3gal gene expansion from Protometazoa stem to vertebrates illustrated in figure 8. The first genomic event was the duplication of an ancestral st3gal gene giving rise to GR1 and GR2/GR3/GRx groups of α2,3-sialyltransferase-related sequences, before the emergence of the sponge O. carmella, about 700 Ma according to the timescale of life of Kumar and Hedges (2011).
We delineated a second period of expansion that gave rise to the full diversity of α2,3-sialyltransferase groups, namely GR1, GR2, GR3, and GRx groups, after the divergence of sponges and Eumetazoa, as no member of these groups could be identified in available protostome genomes, as previously described for the st8sia gene family (Harduin-Lepers et al. 2008). The st3gal1/2/8 sequence ancestor to GR1 was probably present in the protochromosome 3 of first chordates (CLG 3) and the protochromosome B of ancestral vertebrates (vertebrate ancestral chromosome [VAC] B). We could not establish a clear picture concerning the ancestral chromosomes bearing the st3gal sequences of GR2 and GR3. The involvement of VAC C (CLG 7) and VAC J (CLG 9) is highly probable, but identification of GNA A0 and A2 remains difficult to understand. One possibility is that the segments hosting st3gal3 and st3gal9 gene loci were translocated independently on A2 and A0, respectively, just after the R2 event. Another hypothesis is that these segments have been generated by small block duplications before R1. This last view is supported by 1) the lancelet segment Bfl-V2-210, which shows multiple paralogies associated with both GR2 and GR3 (supplementary fig. S5, Supplementary Material online), and 2) multiple paralogies between segments hosting members of GR2 and GR3 in human (table 4), implying further refinements in the reconstruction of vertebrate protochromosomes. We hypothesize a series of two block duplications, a first one generating the segments bearing the genes of GRx (Bfl-V2-210 and V2-67) and another segment that underwent also duplication before the two rounds of genome doubling (fig. 8).
The third period was obviously linked to the two rounds of genome duplication that occurred in early vertebrates around 500 and 555 Ma, respectively, before the divergence of lampreys from Gnathostomes (Smith et al. 2013). However, in teleosts, the synteny around the GR2 st3gal6 gene was broken into two different chromosomes, which were not issued from the teleost-specific R3 WGD (Meyer and Schartl 1999). In medaka, Ola4 is the “right” chromosome corresponding to GNA J1, in contrast to Ola21, which bears the remaining genes of the synteny. Similarly, the synteny around ST3GAL5 gene in the human genome is broken into two chromosomes, the theoretical one (Hsa2) corresponding to GNA C1, and the actual one bearing the gene (Hsa4) further suggesting the occurrence of a gene translocation, distinct from a gene loss event.
The fourth expansion step of st3gal genes related to the R3 event about 250–300 Ma (Meyer and Schartl 1999) and showed limited consequences in teleosts, as it only gave rise to st3gal3-r, a sister sequence of st3gal3. Interestingly, in ray-finned fish such as zebrafish, tilapia, stickleback or platyfish, (not in fugu, tetraodon or spotted gar), we also detected massive Cis-duplication events of the st3gal1 gene leading to 4 st3gal1 gene copies clustered on the same chromosome (chromosome 19 in zebrafish). Besides this other pulse of diversification specifically detected in certain fish, we noticed a high divergence rate of these ST3Gal I sequences (fig. 3) as previously described for duplicated genes (Robinson-Rechavi and Laudet 2001; Jaillon et al. 2004). This observation further suggested functional evolution of these enzymes in fish lineages that could account for the abundant and unusual mucin-type O-glycosylation described in the developing zebrafish (Baskin et al. 2010) leading to Fucα1-3GalNAcβ1-4(Neu5Ac/Neu5Gcα2-3)Galβ1-3GalNAc structure (Guérardel et al. 2006). Abbreviations used are Neu5Ac, N-acetylneuraminic acid; Neu5Gc, N-glycolylneuraminic acid.)
Functional Fate of Vertebrate st3gal Gene Duplicates
Up to now, adult tissue distribution of st3gal genes has been mainly studied in mouse and human tissues (Gagneux and Varki 1999; Comelli et al. 2006; Takashima 2008) whereas limited sialyltransferase expression studies were carried out in the zebrafish model (Chang et al. 2009; Harduin-Lepers 2010; Petit et al. 2010; Vanbeselaere et al. 2012; Flanagan-Steet and Steet 2013). The zebrafish st3gal gene expression profiles were analyzed in the light of evolution in adult tissues, using RT-PCR and in embryonic zebrafish tissues, using ISH. Remarkably, the evolutionary dynamics of the zebrafish st3gal-duplicated genes appeared to be asymmetrical and spatiotemporally regulated as one duplicate was widely expressed in all tissues, whereas the other was restricted to specific tissues. This observation provided additional evidence that following duplication one resulting duplicate might be evolved faster than the other (Steinke et al. 2006). However, comparing the evolution of these st3gal gene expression profiles in various vertebrate species and looking at associations within these gene expression data using a PCA approach as previously described for st6gal genes (Petit et al. 2010) did not enable us to propose a clear picture of the duplicate fates. The ongoing evolution of st3gal functions in mammals was also illustrated by the variable gene expression levels in the three studied models (human, mouse, and bovine) that might reflect differential transcriptional regulations of these α2,3-sialyltransferases (Nairn et al. 2008; Harduin-Lepers et al. 2012). Indeed, epigenetic control and transcriptional regulation of glycosyltransferases are known to be major mechanisms regulating sialyltransferases and glycosylation on an evolutionary scale (Harduin-Lepers et al. 2012; Horvat et al. 2013; Lauc et al. 2013). It would be informative to investigate evolution of Cis-acting region and regulatory elements controlling expression of these st3gal genes during vertebrates’ evolution. We predict series of transcription factor-binding sites losses in teleosts and gains in mammals, as it has been described previously for FUT7 (Laporte et al. 2012).
In terms of enzymatic activities, the functional fates of ST3Gal duplicates were variable along chordate evolution, even though all the identified enzymes of this family probably catalyze the transfer of sialic acid residues in α2,3-linkage to terminal Gal residues found in glycoproteins or glycolipids (fig. 8). To go a step further toward understanding functional divergence of ST3Gal enzymes, we also carried out analysis at the molecular level looking for ST3Gal features of structural/functional importance that had appeared during vertebrate evolution. In this study, we predicted the existence of conserved amino acid positions in MSA of ST3Gal sequences that could be implicated in critical aspect of protein general function and differential donor and acceptor substrate specificity of these enzymes. We conducted an SDP approach (Teppa et al. 2012) and mapped these SDPs on the unique pig ST3Gal I 3D model (PDB: 2WNB). A first SDP analysis between the three vertebrate groups GR1/GR2/GR3 of ST3Gal sequences identified five SDP located nearby the active site in the reference structure. The first two SDPs corresponding to S-197 and Y-233 in the reference structure were found between sialylmotifs L and S, whereas the three others (V-234, W-304, and N-307) were located within a polypeptide loop that we have modeled, as it was not observed by crystallography. It is believed to be part of binding site and form a closed lid accommodating donor substrate (Rao et al. 2009). We suggest that these three SDPs might play a role in modulating donor and acceptor substrates specificity and ST3Gal enzymatic activity during vertebrate evolution. It is interesting to note that the mammalian ST3Gal III and ST3Gal IV that showed the same stringency toward Gal HO-6 (Rohfritsch et al. 2006) share the same SDP W-243 in the MSA shown in figure 4B, a position that was suggested to be important in establishing hydrogen bound with Gal HO-6 (Rao et al. 2009).
Our coevolution analysis revealed specific sites with high MI within sialylmotifs (L, S, or VS) and family motifs suggesting that at least part of these motifs have evolved in a concerted manner. This analysis pointed out the group of distant residues in the protein sequence that may evolve under a common selection pressure. As every described motif showed residues with high MI, we could interpret the results as a network of coevolving residues that link the highly conserved motifs. Interestingly, the top ten cMI residues formed a fully connected network indicating a high level of redundancy and cohesiveness. In this context, we predicted that each of these residues coevolved with all of the others. All of these residues belong to previously described motifs and two of them are implicated in substrates interactions. Even though it is difficult to draw general conclusions about the functional role of coevolving sites, MI scores obtained highlighted functionally important residues that were different from those detected by conservation within groups. A future challenge will be to establish tools to study the enzymatic activity of these newly identified ST3Gal.
Datation of the functional divergences between ST3Gals (particularly the possibility to transfer sialic acid on mucin-type O-glycans) could be more easily assessed for ST3Gals of GR1 compared with ST3Gals of the GR2–GR3–GRx as these lineages separated in the early metazoa. Biochemical functions pertaining to the ST3Gals of the GR2 and GR3 (especially the ability to form α2,3-monosialylated gangliosides GM3 and GM4) probably emerged in early deuterostomes. However, the amino acid associated with these functional changes probably does not result solely from type I and type II amino acid changes, as demonstrated by our MI approach.
Significance of Gene Losses Following Genetic Duplications
Our study of the evolutionary history of st3gal genes in metazoan indicated that the period from 2R to recent time was controlled by several potential gene loss events. It is well admitted that most of the vertebrate duplicated pairs of genes resulting from the 2R duplication events rapidly underwent extensive genomic rearrangements and gene losses (Wolfe 2001) with moderate functional consequences, as both sister genes are redundant (Lynch and Conery 2000). However, on larger evolutionary scales, which of the duplicated genes will disappear in specific lineages is a less documented issue and consequences of specific-lineages gene loss on the functional fate of the surviving duplicate remain not fully understood (Krylov et al. 2003; Wolf et al. 2006).
In an attempt to correlate gene losses with relaxed gene evolution and reduced gene expression, we investigated substitution rates in each st3gal subfamily and profile of expression of these genes in zebrafish. We noticed significantly higher substitution rates of the newly described st3gal8 gene of the GR1 genes indicating a weaker selective pressure and acquisition of mutations that compromised its function in mammals. Interestingly, numerous tissues expressed this gene in teleosts and xenopus and very few in chicken, indicating a potential role linked to aquatic life. Its expression in zebrafish gills and skin suggested a protective role against bacterial infection, as it has long been known that sialylated O-glycans from skin mucus bind diverse bacteria thus preventing infection of the underlying tissues (Nigam et al. 2012). Similarly, st3gal7 gene from GR3 was specifically found in actinopterygian fish and was lost in tetrapods. The higher substitution rate of st3gal7 compared with st3gal3 showed about the same values as st3gal5. However, zebrafish st3gal7 gene expression was highly restricted to a few tissues indicating specificity linked to aquatic environments, in contrast to st3gal5, which was ubiquitously expressed. The newly described st3gal9 subfamily, restricted to birds and duck-billed platypus, is a sister gene of st3gal4 resulting from the R2 event. Curiously, this gene would have been lost independently in multiple vertebrate lineages as it could not be found in teleosts, amphibians, marsupials, and placental mammals. Substitution rates of st3gal9 genes were more elevated than in the other subfamilies of the GR2 group, at least in the stem region of the sialyltransferase. It is noteworthy that all the vertebrates possessing this st3gal9 gene perform terrestrial egg deposition. Besides, chicken amniotic cells have α2,3- and α2,6-linked sialic acids, allowing experimental cultivation of several strains of influenza viruses (Ito et al. 1997). Amnios sensitivity to viruses is not a problem when eggs are protected from environment by an egg shell. We made the hypothesis that α2,3-sialylation coating the amniotic cells could result from ST3Gal IX enzymatic activity that would have disappeared from fish, amphibians, and most mammals, as it represents a source of contamination in the case of eggs laid in water (lower vertebrates) or for embryos connected to maternal tissues through placenta (most mammals).
Interestingly, we noticed that the st3gal6 genes totally disappeared from fish genomes as it was the case for st8sia4 genes described previously (Harduin-Lepers et al. 2008). Our paleogenomics analysis suggests that the immediate cause of st3gal6 gene loss in teleosts could be due to the fission of a chromosome ancestral to teleosts. One probable consequence of this absence could be associated with their original fertilization process. In human, the lectins of sperm acrosome bind to Sialyl-LewisX structures capping N- and O-glycans found onto the four zona pellucida (ZP) glycoproteins of the coat surrounding the oocyte (Pang et al. 2011; Clark 2013). Once bound, the acrosome releases enzymes that digest the extracellular matrix, enabling the progression of sperm toward oocyte membrane. In teleosts, there is no acrosome and the sperm reaches the oocyte membrane through a micropyle, a sperm guidance system through the ZP (Jamieson 2011), replacing the crucial role of sialyl LewisX to ensure this step of egg fertilization.
In summary, the st3gal genes evolutionary biology offers a suitable example to highlight the contexts that have favored the specific lineage gene loss events. We found that st3gal gene losses were generally linked to high substitution rates, indicative of low selective pressure, and generally to restricted tissue expression in adult. However, in the developing zebrafish, no particular spatiotemporal profile of expression of these st3gal genes could be associated with their fate in higher vertebrates. The replacement of ST3Gal enzymes allowed modifying potential binding sites to viruses and it is possible that some st3gal gene losses in higher vertebrates were linked to the increasing number of organs expressing the st6gal1 gene (Petit et al. 2010). It has long been known, for instance, that there is a switch in the expression of α2,3- to α2,6-linked sialic acids in the respiratory tract of human (Varki 2008; Yu et al. 2011), explaining limited binding of several strains of influenza virus to α2,3-sialylated flu receptor in human and the coevolution of new viral strains to cope with the situation. These general observations do not rule out particular events, such as changes in fertilization process as in teleosts.
Altogether, this integrated study has shown that st3gal genes have arisen through multiple duplication events including probable ancient block duplication that occurred in early deuterostomes and WGD after vertebrate emergence. Furthermore, we showed that st3gal gene losses were major force driving the evolutionary history of this gene family in deuterostomes and that there is not a trend toward a broader range of spatial expression culminating in mammals, but rather a progressive loss of st3gal genes expressed in a limited number of organs in earlier diverging vertebrates.
Materials and Methods
In Silico Sialyltransferase-Related Sequence Retrieval
Only metazoan sequences were considered for this study and st3gal-related sequences were identified using the well-described human ST3Gal sequences as seed sequences, as previously described in Petit et al. (2013). A major source of information came from the GT-29 family of the CAZy database (http://www.cazy.org/GlycosylTransferases.html, last accessed December 2014; Cantarel et al. 2009). In addition, st3gal-related sequences were searched in all genomic and transcriptomic divisions available from general nonredundant databases such as those maintained at the NCBI (Wheeler et al. 2005), DNA Data Bank of Japan (Tateno et al. 2002), ENSEMBL (Flicek et al. 2013) or in specialized databases such as the JGI genome browser for Br. floridae (V2 genome assembly) (Grigoriev et al. 2011) or the genome Sequencing center at the Washington University School of medicine, St Louis, MO for P. marinus (Pruitt 1997) and KEGG GENES (Kanehisa and Goto 2000; Hashimoto et al. 2006, 2009) using BLAST (Altschul et al. 1997) with default parameters (an E value cut off at 0.01 was used in all BLAST searches). The assignation of these sequences to ST3Gal was determined using the specific motifs that are hallmarks of this family (Patel and Balaji 2006; Harduin-Lepers 2010).
Phylogenetic Analysis
The alignment of 121 selected ST3Gal-related sequences was conducted using MUSCLE algorithm included in MEGA5.0 software and refined by hand (Hall 2013). The variable transmembrane domain in the N-terminal part of sequences was removed, as well as the C-terminus because of different lengths of sequences among subfamilies. The final alignment contains 382 sites including most of the stem and the catalytic domains. Phylogeny trees were produced by ML method using MEGA5.0. The models with the lowest BIC scores are considered to describe the substitution pattern the best (Tamura et al. 2011). Nonuniformity of evolutionary rates among sites may be modeled by using a discrete Gamma distribution (+G) with five rate categories and by assuming that a certain fraction of sites are evolutionarily invariable (+I). The ME method was also conducted using the JTT matrix as transition between amino acids. The rate variation among sites was modeled with a Gamma distribution (shape = 1.3, calculated by the software). All positions with less than 95% site coverage were eliminated. That is, fewer than 5% alignment gaps, missing data, and ambiguous bases were allowed at any position. There were a total of 228 positions in the final data set. Evolutionary analyses were conducted in MEGA5.0 (Tamura et al. 2011) and bootstrap percentages were calculated from 500 replicates.
The numbers of site changes in the vertebrate members of each subfamily were calculated with the Protpars program included in the PHYLIP Package (see Felsenstein 1997 and Petit et al. 2006 for details) using 128 sites in the stem part (the first 24 sites were removed to homogenize the length) and 230 in the catalytic domain. As there were variations in the number of sequences between each subfamily, and in the successive sites due to small deletions, the total number of changes for each site was divided by the number of sequences at each site. The change rates were then smoothed by calculating a mean within a window of five successive amino acids running along the whole sequence. This data set was used to draw boxplots, and then normalized with log-transformed values to compare the different subfamilies with Tukey’s test conducted with PAST 2.17c (Hammer et al. 2001).
Synteny Analysis and Paralogon Detection
Synteny between vertebrate st3gal and related genes in invertebrates was assessed by manual chromosomal walking and reciprocal BLAST searches of genes adjacent to st3gal loci in human (Hsa), mouse (Mmu), chicken (Gga), medaka (Ola), zebrafish (Dre), Takifugu rubripes (Tru) (ENSEMBL, release 70, January 2013), and amphioxus (Bfl) (JGI Br. floridae v.2.0) genome databases. The detection of paralogous blocks was also done using the latest ENSEMBL data set (ENS61) at the synteny database site (http://teleost.cs.uoregon.edu/synteny_db/, last accessed December 2014) (Catchen et al. 2009) and visualized at the Genomicus site (version 69.01) (http://www.dyogen.ens.fr/genomicus/, last accessed December 2014) (Louis et al. 2012). When an st3gal gene was absent in a given animal genome, we used one or two genes physically close as seed to identify the syntenic segments.
Ancestral genome reconstruction in conjunction with phylogenetic and syntenic analyses was also used to rapidly assess the dynamic of st3gal genes evolutionary relationships, taking into account the reconstructions of protochromosomes of ancestral vertebrates (Kasahara et al. 2007; Nakatani et al. 2007) and of chordates (Putnam et al. 2008), as previously reported for the case study of RLN/INSL and RXFP genes (Yegorov and Good 2012). Briefly, each block of syntenic genes (ohnologs) common to teleosts, birds, and mammals arose from the 2R genetic events that occurred during early vertebrate evolution and thus could be associated with one of the approximately 40 protochromosomes of gnathostomes (after the 2R events) and hence to one of the approximately ten protochromosomes of vertebrates (before the 2R events). The calibration used for dating the divergence between ST3Gal sequences in metazoa followed the work of Vandepoele et al. (2004) and Kumar and Hedges (2011).
Expression Analysis
For each gene and each organism, Unigene statistics at the NCBI database were used to calculate the Neperian logarithm of the relative number of st3gal ESTs identified by tissue. Well-studied organs with unambiguous homology from zebrafish to human were retained as previously described for st6gal genes (Petit et al. 2010). The following species have been considered: Homo sapiens, Mus musculus, B. taurus, G. gallus, Si. tropicalis, and D. rerio. PCA was carried out according to the previously described method (Petit et al. 2010, 2013) using PAST 2.17c (Hammer et al. 2001). The vectors corresponding to each st3gal gene were oriented toward the highest expression levels. The angle between vectors was related to the correlation between the same genes across the different tissues: High positive correlation with acute angle, but negative one with opposite vectors. The diversity of tissues in which a given gene is expressed for an organism was calculated using H′ Shannon index: H′ = −σ((ni/N).ln(ni/N)) where ni is the number of ESTs found in the each tissue and N is the total number of ESTs.
Animals Maintenance and Whole Mount mRNA In Situ Analysis
Zebrafish (D. rerio) were maintained in our aquatic biology facility, as previously described (Westerfield 1995; Chang et al. 2009). All experimental procedures adhered to the CNRS (Centre National de La Recherche Scientifique) guidelines for animals use in research. Both sense and antisense digoxigenin-labeled RNA probes were synthesized from PCR-amplified template using primers as previously described (Chang et al. 2009; Petit et al. 2010). Whole mount ISH was performed according to standard procedures (Thisse C and Thisse B 1998, 2008; Thisse et al. 2004).
Isolation of RNA, cDNA Synthesis, and PCR Analysis
Total RNA was extracted from various D. rerio tissues using the nucleospin RNA II kit (Macherey-Nagel, Hoerdt, France). A proteinase K digestion step (55 °C, 10 min) and phenol/chloroform extraction were inserted in the protocol after dounce homogenization of the tissues and before column purification of total RNA. Similarly, bovine total RNAs were prepared from various tissues (lung, skeletal muscle, heart, thymus, and liver) of five individuals of B. taurus (Holstein and Charolais-crossbred steers) collected at the slaughter using the RNeasy midi kit (Qiagen Inc, Hilden, Germany) according to the manufacturer’s instructions. Bovine and zebrafish RNAs were quantified using a NanoDrop ND-1000 UV-Vis spectrophotometer (NanoDrop Technologies, Wilmington, DE). RNA integrity was further assessed using the RNA 6000 Nano LabChip Kit on an Agilent Bioanalyzer (Agilent Technologies, Stratagene, La Jolla, CA). For subsequent PCR amplifications, first strand cDNA was synthesized from 5 µg of total zebrafish RNA using an oligo(dT) primer and the AffinityScript Q-PCR cDNA synthesis kit (Agilent Technologies) or from 3 µg of total bovine RNA using random hexamers and the High Capacity cDNA reverse transcription kit (Life Technologies, Saint-Aubin, France), following the supplier’s recommendations.
Oligonucleotide primers were designed (Eurogentec, Herstal, Belgium) in the open-reading frame of the zebrafish sequences (Vanbeselaere et al. 2012). PCR amplifications were carried out with the Taq Core kit DNA polymerase (Qiagen, Courtaboeuf, France). Annealing temperatures ranged from 48 to 55 °C and amplified fragments were subjected to 2% agarose gel electrophoresis, visualized by Ethidium Bromide, gel-extracted and subcloned in the pCR2.1-TOPO vector (TOPO TA Cloning, Invitrogen, Cergy Pontoise, France), and entirely sequenced (GATC Biotech AG, Köln, Germany).
Quantitative PCRs were performed using one TaqMan Gene Expression Assay (Applied Biosystems, Life Technology) made-to-order form for each st3gal gene, with the exception of the st3gal3 gene carried out with two assays. Amplifications of the six mammalian st3gal genes and five independent housekeeping genes used as control genes (18S, ACTAB, B2M, PPIA, Tbp) were done in triplicate according to the manufacturer’s instructions in 96-well microplates and normalized using the 18S RNA, the most stable gene for the five tissues. Twenty nanograms of cDNA was distributed in each well by 2-min centrifugation at 260 × g. Conditions of amplification were 10 min at 95 °C, followed by 40 cycles of amplification (15 s at 95 °C, 1 min at 60 °C). Changes in the fluorescence of the TaqMan probe were monitored on the ABI PRISM 7900HT sequence detector system and quantified using the ΔCt method by the SDS 2.2 software (Applied Biosystems, Life Technology).
Functional Site Prediction Using Conservation between and within Groups
To assess conservation of each groups of α2,3-sialyltransferase subfamilies, we programmed an automatic search of consensus sequences. We considered windows of four consecutive amino acids running along st3gal sequences. Such a four-amino acid peptide was deemed conserved if at least three of four positions could be found in at least 60% of the selected st3gal sequences; otherwise, the position was considered as variable (window length and thresholds on demand). To remove uninformative positions, columns showing more than 50% of gaps were deleted. The final step was to assemble overlapping sequences. The MSA encompassing 382 amino acid positions used for this analysis was the same as the one used for phylogeny analyses. The program written in C++ under Windows environment is available on request. For each subfamily, a consensus sequence was generated using the program described above and then the consensus sequences were aligned with the one generated by MEGA 5.0. The resulting alignment was used to detect two types of functional divergent sites according to Gu (2001): Type I is variable in one group and conserved in the other(s) resulting in altered functional constraints between duplicated genes, whereas type II positions show different conserved amino acids among groups accounting for different functions or specificities. Prediction of SDPs was performed considering the three groups of subfamilies (GR1–GR3) (Kalinina et al. 2004) and these predicted positions located within the active site were mapped into the unique mammalian sialyltransferase 3D structure complexed with CMP and Galβ1,3GalNAcα–PhNO2 (PDB code 2WNB) described up to now (Rao et al. 2009).
Sequence Similarity Network
To refine the relationships between ST3Gal-related proteins, sequence similarity network was constructed using formatdb, from the stand-alone BLAST software and a custom BLAST database was created. The database included 336 ST3Gal-related sequences according to CAZy classification and 27 ST6Gal I sequences constituting an outgroup of negative control. This approach allowed examining the relationships within large, diverse sets of sequences for which the costs of traditional methods of analysis such as phylogenetic trees would have been prohibitive, due to the difficulty in generating accurate multiple alignments. In addition, the networks provided a graphical overview of interrelationships among and between sets of proteins that are not easily discerned from visual inspection of large trees and multiple alignments. The pairwise relationships between sequences were calculated by a BLASTall search in the custom database with each individual sequence in the set and the E value was taken as a measure of similarity between sequences. It should be mentioned that in a particular database of homologous proteins, the E value can be considered as a type of similarity score, rather than a true expectation value. For this reason, the distantly related sequences of ST6Gal I were included and different E value thresholds were considered: Permissive threshold (1e-55, 1e-60, and 1e-70) and stringent threshold (1e-80, 1e-90, and 1e-100). The network was visualized using Cytoscape version 2.8.3 (Shannon et al. 2003), where each sequence was represented as a node and edges were defined between any pair of nodes with an E value less than threshold, also the default Cytoscape force-directed layout was applied. Nodes were colored according to the subfamily to which the sequence belongs, either known (ST3Gal I–ST3Gal VI) or predicted (ST3Gal VII, ST3Gal VIII, and ST3Gal IX).
Prediction of Coevolving Positions
Coevolved positions were predicted using the MISTIC web server with default parameters (http://mistic.leloir.org.ar/index.php, last accessed December 2014) (Simonetti et al. 2013). Coevolving residue-pairs prediction was based on the calculation of MI between pairs of amino acid residues from an MSA. The results included MI between residue pairs, a per-residue cMI score that measured the degree of shared MI of a given residue and the proximity Mutual Information (pMI), indicative of the networks of MI in the 3D proximity of a residue. To calculate this latter score, we provided a pig ST3Gal I structure (PDB: 2WNB). The crystal structure of pST3Gal I showed a 12 amino acid residues flexible loop (positions 305-316) missing from the electron density. This region was modeled using the loop modeling method of MODELLER software (Sali and Blundell 1993). We generated 50 examples of the loop region 305–316, which were subsequently ranked by DOPE statistical potential. The highest ranked models were visually inspected and evaluated by Ramachandran plot. The final model showed all torsional angles of the modeled region in the allowed zone. As it is a mobile loop, the model was intended to represent just a possible position of atoms in space in order to visualize the prediction results in the context of the complete protein for a better understanding of the functional importance of this region.
Supplementary Material
Supplementary tables S1–S3 and figures S1–S9 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors are very grateful to the reviewers’ valuable comments that greatly improved the manuscript; Jean-Michel Petit and Cristina Marino Buslje for helpful discussions and constant interest in the work; Lucie Ducrocq for the excellent technical assistance and Antoine Petit in the writing of C++ program identifying conserved motifs in aligned sequences. Mohcen Benmounah and Virginie Cogez are acknowledged for their help handling sialyltransferase sequences. This work was supported by the Centre National de La Recherche Scientifique (CNRS), Institut National de la Recherche Agronomique (INRA), Institut National de la Santé et de la Recherche Médicale (INSERM), and the PPF-Bionformatique de Lille1. The National Center for Research Resources (5R24RR016344) at The National Institute of Health supported the Ambystoma research resources. A.H.L. work is supported by the ANR-2010-BLAN-120401 grant (project Galfish) from the Agence Nationale de la Recherche and by the Région Nord-Pas de Calais (program Arcir dynamique). B.T. and C.T. were supported by funds from University of Virginia. E.T. received an Erasmus Mundus fellowship for this project.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson HJ, Morris JH, Ferrin TE, Babbitt PC. Using sequence similarity networks for visualization of relationships across diverse protein superfamilies. PLoS One. 2009;4:e4345. doi: 10.1371/journal.pone.0004345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audry M, Jeanneau C, Imberty A, Harduin-Lepers A, Delannoy P, Breton C. Current trends in the structure-activity relationships of sialyltransferases. Glycobiology. 2011;21:716–726. doi: 10.1093/glycob/cwq189. [DOI] [PubMed] [Google Scholar]
- Baskin JM, Dehnert KW, Laughlin ST, Amacher SL, Bertozzi CR. Visualizing enveloping layer glycans during zebrafish early embryogenesis. Proc Natl Acad Sci U S A. 2010;107:10360–10365. doi: 10.1073/pnas.0912081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009;37:D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen JM, Conery JS, Postlethwait JH. Automated identification of conserved synteny after whole-genome duplication. Genome Res. 2009;19:1497–1505. doi: 10.1101/gr.090480.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang LY, Mir AM, Thisse C, Guerardel Y, Delannoy P, Thisse B, Harduin-Lepers A. Molecular cloning and characterization of the expression pattern of the zebrafish alpha2, 8-sialyltransferases (ST8Sia) in the developing nervous system. Glycoconj J. 2009;26:263–275. doi: 10.1007/s10719-008-9165-1. [DOI] [PubMed] [Google Scholar]
- Chisada SI, Yoshimura Y, Sakaguchi K, Uemura S, Go S, Ikeda K, Uchima H, Matsunaga N, Ogura K, Tai T, et al. Zebrafish and mouse alpha2,3-sialyltranserases responsible for synthesizing GM4 ganglioside. J Biol Chem. 2009;284:30534–30546. doi: 10.1074/jbc.M109.016188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark GF. The role of carbohydrate recognition during human sperm-egg binding. Hum Reprod. 2013;28:566–577. doi: 10.1093/humrep/des447. [DOI] [PubMed] [Google Scholar]
- Cohen M, Varki A. The sialome—far more than the sum of its parts. Omics. 2010;14:455–464. doi: 10.1089/omi.2009.0148. [DOI] [PubMed] [Google Scholar]
- Comelli EM, Head SR, Gilmartin T, Whisenant T, Haslam SM, North SJ, Wong NK, Kudo T, Narimatsu H, Esko JD, et al. A focused microarray approach to functional glycomics: transcriptional regulation of the glycome. Glycobiology. 2006;16:117–131. doi: 10.1093/glycob/cwj048. [DOI] [PubMed] [Google Scholar]
- Datta AK, Paulson JC. The sialyltransferase “sialylmotif” participates in binding the donor substrate CMP-NeuAc. J Biol Chem. 1995;270:1497–1500. doi: 10.1074/jbc.270.4.1497. [DOI] [PubMed] [Google Scholar]
- Datta AK, Sinha A, Paulson JC. Mutation of the sialyltransferase S-sialylmotif alters the kinetics of the donor and acceptor substrates. J Biol Chem. 1998;273:9608–9614. doi: 10.1074/jbc.273.16.9608. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. An alternating least squares approach to inferring phylogenies from pairwise distances. Syst Biol. 1997;46:101–111. doi: 10.1093/sysbio/46.1.101. [DOI] [PubMed] [Google Scholar]
- Flanagan-Steet HR, Steet R. “Casting” light on the role of glycosylation during embryonic development: insights from zebrafish. Glycoconj J. 2013;30:33–40. doi: 10.1007/s10719-012-9390-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flicek P, Ahmed I, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, et al. Ensembl 2013. Nucleic Acids Res. 2013;41:D48–D55. doi: 10.1093/nar/gks1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabius HJ. Biological information transfer beyond the genetic code: the sugar code. Naturwissenschaften. 2000;87:108–121. doi: 10.1007/s001140050687. [DOI] [PubMed] [Google Scholar]
- Gabius HJ, Andre S, Kaltner H, Siebert HC. The sugar code: functional lectinomics. Biochim Biophys Acta. 2002;1572:165–177. doi: 10.1016/s0304-4165(02)00306-9. [DOI] [PubMed] [Google Scholar]
- Gagneux P, Varki A. Evolutionary considerations in relating oligosaccharide diversity to biological function. Glycobiology. 1999;9:747–755. doi: 10.1093/glycob/9.8.747. [DOI] [PubMed] [Google Scholar]
- Geremia RA, Harduin-Lepers A, Delannoy P. Identification of two novel conserved amino acid residues in eukaryotic sialyltransferases: implications for their mechanism of action. Glycobiology. 1997;7:v–vii. doi: 10.1093/glycob/7.2.161. [DOI] [PubMed] [Google Scholar]
- Giacopuzzi E, Bresciani R, Schauer R, Monti E, Borsani G. New insights on the sialidase protein family revealed by a phylogenetic analysis in metazoa. PLoS One. 2012;7:e44193. doi: 10.1371/journal.pone.0044193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriev IV, Nordberg H, Shabalov I, Aerts A, Cantor M, Goodstein D, Kuo A, Minovitsky S, Nikitin R, Ohm RA, et al. The genome portal of the Department of Energy Joint Genome Institute. Nucleic Acids Res. 2011;40:D26–D32. doi: 10.1093/nar/gkr947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu X. Maximum-likelihood approach for gene family evolution under functional divergence. Mol Biol Evol. 2001;18:453–464. doi: 10.1093/oxfordjournals.molbev.a003824. [DOI] [PubMed] [Google Scholar]
- Guérardel Y, Chang LY, Fujita A, Coddeville B, Maes E, Sato C, Harduin-Lepers A, Kubokawa K, Kitajima K. Sialome analysis of the cephalochordate Branchiostoma belcheri, a key organism for vertebrate evolution. Glycobiology. 2012;22:479–491. doi: 10.1093/glycob/cwr155. [DOI] [PubMed] [Google Scholar]
- Guérardel Y, Chang LY, Maes E, Huang CJ, Khoo KH. Glycomic survey mapping of zebrafish identifies unique sialylation pattern. Glycobiology. 2006;16:244–257. doi: 10.1093/glycob/cwj062. [DOI] [PubMed] [Google Scholar]
- Hall BG. Building phylogenetic trees from molecular data with MEGA. Mol Biol Evol. 2013;30:1229–1235. doi: 10.1093/molbev/mst012. [DOI] [PubMed] [Google Scholar]
- Hammer Ø, Harper DAT, Ryan PD. PAST: paleontological statistics software package for education and data analyses. Paleontol Electron. 2001;4:9. [Google Scholar]
- Harduin-Lepers A. Comprehensive analysis of sialyltransferases in vertebrate genomes. Glycobiol Insights. 2010;2:29–61. [Google Scholar]
- Harduin-Lepers A. Vertebrate sialyltransferases. In: Tiralongo J, Martinez-Duncker I, editors. Sialobiology: structure, biosynthesis and function. Sialic acid glycoconjugates in health and diseases. Bentham Science; 2013. pp. 139–187. [Google Scholar]
- Harduin-Lepers A, Krzewinski-Recchi MA, Colomb F, Foulquier F, Groux-Degroote S, Delannoy P. Sialyltransferases functions in cancers. Front Biosci (Elite Ed). 2012;4:499–515. doi: 10.2741/e396. [DOI] [PubMed] [Google Scholar]
- Harduin-Lepers A, Krzewinski-Recchi MA, Hebbar M, Samyn-Petit B, Vallejo-Ruiz V, Julien S, Peyrat JP, Delannoy P. Sialyltransferases and breast cancer. Recent Res Dev Cancer. 2001;3:111–126. [Google Scholar]
- Harduin-Lepers A, Mollicone R, Delannoy P, Oriol R. The animal sialyltransferases and sialyltransferase-related genes: a phylogenetic approach. Glycobiology. 2005;15:805–817. doi: 10.1093/glycob/cwi063. [DOI] [PubMed] [Google Scholar]
- Harduin-Lepers A, Petit D, Mollicone R, Delannoy P, Petit JM, Oriol R. Evolutionary history of the alpha2,8-sialyltransferase (ST8Sia) gene family: tandem duplications in early deuterostomes explain most of the diversity found in the vertebrate ST8Sia genes. BMC Evol Biol. 2008;8:258. doi: 10.1186/1471-2148-8-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harduin-Lepers A, Vallejo-Ruiz V, Krzewinski-Recchi MA, Samyn-Petit B, Julien S, Delannoy P. The human sialyltransferase family. Biochimie. 2001;83:727–737. doi: 10.1016/s0300-9084(01)01301-3. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Goto S, Kawano S, Aoki-Kinoshita KF, Ueda N, Hamajima M, Kawasaki T, Kanehisa M. KEGG as a glycome informatics resource. Glycobiology. 2006;16:63R–70R. doi: 10.1093/glycob/cwj010. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Tokimatsu T, Kawano S, Yoshizawa AC, Okuda S, Goto S, Kanehisa M. Comprehensive analysis of glycosyltransferases in eukaryotic genomes for structural and functional characterization of glycans. Carbohydr Res. 2009;344:881–887. doi: 10.1016/j.carres.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Horvat T, Dezeljin M, Redzic I, Barisic D, Herak Bosnar M, Lauc G, Zoldos V. Reversibility of membrane N-glycome of HeLa cells upon treatment with epigenetic inhibitors. PLoS One. 2013;8:e54672. doi: 10.1371/journal.pone.0054672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Suzuki Y, Takada A, Kawamoto A, Otsuki K, Masuda H, Yamada M, Suzuki T, Kida H, Kawaoka Y. Differences in sialic acid-galactose linkages in the chicken egg amnion and allantois influence human influenza virus receptor specificity and variant selection. J Virol. 1997;71:3357–3362. doi: 10.1128/jvi.71.4.3357-3362.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillon O, Aury JM, Brunet F, Petit JL, Stange-Thomann N, Mauceli E, Bouneau L, Fischer C, Ozouf-Costaz C, Bernot A, et al. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature. 2004;431:946–957. doi: 10.1038/nature03025. [DOI] [PubMed] [Google Scholar]
- Jamieson B. Fish evolution and systematics: evidence from spermatozoa: with a survey of lophophorate, echinoderm and protochordate sperm and an account of gamete cryopreservation. Cambridge: Cambridge University Press 1991; 2011. Reissue edition (June 16, 2011) [Google Scholar]
- Jeanneau C, Chazalet V, Auge C, Soumpasis DM, Harduin-Lepers A, Delannoy P, Imberty A, Breton C. Structure-function analysis of the human sialyltransferase ST3Gal I: role of N-glycosylation and a novel conserved sialylmotif. J Biol Chem. 2004;279:13461–13468. doi: 10.1074/jbc.M311764200. [DOI] [PubMed] [Google Scholar]
- Kalinina OV, Novichkov PS, Mironov AA, Gelfand MS, Rakhmaninova AB. SDPpred: a tool for prediction of amino acid residues that determine differences in functional specificity of homologous proteins. Nucleic Acids Res. 2004;32:W424–W428. doi: 10.1093/nar/gkh391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasahara M, Naruse K, Sasaki S, Nakatani Y, Qu W, Ahsan B, Yamada T, Nagayasu Y, Doi K, Kasai Y, et al. The medaka draft genome and insights into vertebrate genome evolution. Nature. 2007;447:714–719. doi: 10.1038/nature05846. [DOI] [PubMed] [Google Scholar]
- Kim KS, Oh SU, Lee JH, Lee YC. Mutational analysis for enzyme activity of mouse Galbeta1,3GalNAc alpha2,3-sialyltransferase (mST3Gal I) Indian J Biochem Biophys. 2010;47:135–140. [PubMed] [Google Scholar]
- Kitagawa H, Paulson JC. Differential expression of five sialyltransferase genes in human tissues. J Biol Chem. 1994;269:17872–17878. [PubMed] [Google Scholar]
- Kojima N, Lee YC, Hamamoto T, Kurosawa N, Tsuji S. Kinetic properties and acceptor substrate preferences of two kinds of Gal beta 1,3GalNAc alpha 2,3-sialyltransferase from mouse brain. Biochemistry. 1994;33:5772–5776. doi: 10.1021/bi00185a014. [DOI] [PubMed] [Google Scholar]
- Kono M, Ohyama Y, Lee YC, Hamamoto T, Kojima N, Tsuji S. Mouse beta-galactoside alpha 2,3-sialyltransferases: comparison of in vitro substrate specificities and tissue specific expression. Glycobiology. 1997;7:469–479. doi: 10.1093/glycob/7.4.469. [DOI] [PubMed] [Google Scholar]
- Krylov DM, Wolf YI, Rogozin IB, Koonin EV. Gene loss, protein sequence divergence, gene dispensability, expression level, and interactivity are correlated in eukaryotic evolution. Genome Res. 2003;13:2229–2235. doi: 10.1101/gr.1589103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Hedges SB. TimeTree2: species divergence times on the iPhone. Bioinformatics. 2011;27:2023–2024. doi: 10.1093/bioinformatics/btr315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte B, Petit D, Rocha D, Boussaha M, Grohs C, Maftah A, Petit JM. Characterization of bovine FUT7 furthers understanding of FUT7 evolution in mammals. BMC Genet. 2012;13:74. doi: 10.1186/1471-2156-13-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauc G, Vojta A, Zoldos V. Epigenetic regulation of glycosylation is the quantum mechanics of biology. Biochim Biophys Acta. 2013;1840:65–70. doi: 10.1016/j.bbagen.2013.08.017. [DOI] [PubMed] [Google Scholar]
- Lehmann F, Kelm S, Dietz F, von Itzstein M, Tiralongo J. The evolution of galactose alpha2,3-sialyltransferase: Ciona intestinalis ST3GAL I/II and Takifugu rubripes ST3GAL II sialylate Galbeta1,3GalNAc structures on glycoproteins but not glycolipids. Glycoconj J. 2008;25:323–334. doi: 10.1007/s10719-007-9078-4. [DOI] [PubMed] [Google Scholar]
- Lehmann F, Tiralongo E, Tiralongo J. Sialic acid-specific lectins: occurrence, specificity and function. Cell Mol Life Sci. 2006;63:1331–1354. doi: 10.1007/s00018-005-5589-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis A, Muffato M, Roest Crollius H. Genomicus: five genome browsers for comparative genomics in eukaryota. Nucleic Acids Res. 2012;41:D700–D705. doi: 10.1093/nar/gks1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- Meyer A, Schartl M. Gene and genome duplications in vertebrates: the one-to-four (-to-eight in fish) rule and the evolution of novel gene functions. Curr Opin Cell Biol. 1999;11:699–704. doi: 10.1016/s0955-0674(99)00039-3. [DOI] [PubMed] [Google Scholar]
- Moreau H, Verhelst B, Couloux A, Derelle E, Rombauts S, Grimsley N, Van Bel M, Poulain J, Katinka M, Hohmann-Marriott MF, et al. Gene functionalities and genome structure in Bathycoccus prasinos reflect cellular specializations at the base of the green lineage. Genome Biol. 2012;13:R74. doi: 10.1186/gb-2012-13-8-r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nairn AV, York WS, Harris K, Hall EM, Pierce JM, Moremen KW. Regulation of glycan structures in animal tissues: transcript profiling of glycan-related genes. J Biol Chem. 2008;283:17298–17313. doi: 10.1074/jbc.M801964200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani Y, Takeda H, Kohara Y, Morishita S. Reconstruction of the vertebrate ancestral genome reveals dynamic genome reorganization in early vertebrates. Genome Res. 2007;17:1254–1265. doi: 10.1101/gr.6316407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Counci. Washington [DC]: The National Academies Press; 2012. Transforming Glycoscience: A Roadmap for the Future. [PubMed] [Google Scholar]
- Nigam AK, Kumari U, Mittal S, Mittal AK. Comparative analysis of innate immune parameters of the skin mucous secretions from certain freshwater teleosts, inhabiting different ecological niches. Fish Physiol Biochem. 2012;38:1245–1256. doi: 10.1007/s10695-012-9613-5. [DOI] [PubMed] [Google Scholar]
- Ohno S. Evolution by gene duplication. Heidelberg: Springer-Verlag; 1970. [Google Scholar]
- Ohno S. Gene duplication and the uniqueness of vertebrate genomes circa 1970-1999. Semin Cell Dev Biol. 1999;10:517–522. doi: 10.1006/scdb.1999.0332. [DOI] [PubMed] [Google Scholar]
- Pang PC, Chiu PC, Lee CL, Chang LY, Panico M, Morris HR, Haslam SM, Khoo KH, Clark GF, Yeung WS, et al. Human sperm binding is mediated by the sialyl-Lewis(x) oligosaccharide on the zona pellucida. Science. 2011;333:1761–1764. doi: 10.1126/science.1207438. [DOI] [PubMed] [Google Scholar]
- Patel RY, Balaji PV. Identification of linkage-specific sequence motifs in sialyltransferases. Glycobiology. 2006;16:108–116. doi: 10.1093/glycob/cwj046. [DOI] [PubMed] [Google Scholar]
- Petit D, Maftah A, Julien R, Petit JM. En bloc duplications, mutation rates, and densities of amino acid changes clarify the evolution of vertebrate α1,3/4-fucosyltransferases. J Mol Evol. 2006;63:353–364. doi: 10.1007/s00239-005-0189-x. [DOI] [PubMed] [Google Scholar]
- Petit D, Mir AM, Petit JM, Thisse C, Delannoy P, Oriol R, Thisse B, Harduin-Lepers A. Molecular phylogeny and functional genomics of beta-galactoside alpha2,6-sialyltransferases that explain ubiquitous expression of st6gal1 gene in amniotes. J Biol Chem. 2010;285:38399–38414. doi: 10.1074/jbc.M110.163931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit D, Teppa RE, Petit JM, Harduin-Lepers A. A practical approach to reconstruct evolutionary history of animal sialyltransferases and gain insights into the sequence-function relationships of Golgi-glycosyltransferases. In: Brochausen I, editor. Glycosyltransferases: methods and protocols. New York: Springer, Humana Press; 2013. pp. 73–97. [DOI] [PubMed] [Google Scholar]
- Pruitt KD. WebWise: the Washington University Genome Sequencing Center’s web site. Genome Res. 1997;7:1118–1121. doi: 10.1101/gr.7.12.1118. [DOI] [PubMed] [Google Scholar]
- Putnam NH, Butts T, Ferrier DE, Furlong RF, Hellsten U, Kawashima T, Robinson-Rechavi M, Shoguchi E, Terry A, Yu JK, et al. The amphioxus genome and the evolution of the chordate karyotype. Nature. 2008;453:1064–1071. doi: 10.1038/nature06967. [DOI] [PubMed] [Google Scholar]
- Rakic B, Rao FV, Freimann K, Wakarchuk W, Strynadka NC, Withers SG. Structure-based mutagenic analysis of mechanism and substrate specificity in mammalian glycosyltransferases: porcine ST3Gal-I. Glycobiology. 2013;23:536–545. doi: 10.1093/glycob/cwt001. [DOI] [PubMed] [Google Scholar]
- Rao FV, Rich JR, Rakic B, Buddai S, Schwartz MF, Johnson K, Bowe C, Wakarchuk WW, Defrees S, Withers SG, et al. Structural insight into mammalian sialyltransferases. Nat Struct Mol Biol. 2009;16:1186–1188. doi: 10.1038/nsmb.1685. [DOI] [PubMed] [Google Scholar]
- Robinson-Rechavi M, Laudet V. Evolutionary rates of duplicate genes in fish and mammals. Mol Biol Evol. 2001;18:681–683. doi: 10.1093/oxfordjournals.molbev.a003849. [DOI] [PubMed] [Google Scholar]
- Rohfritsch PF, Joosten JA, Krzewinski-Recchi MA, Harduin-Lepers A, Laporte B, Juliant S, Cerutti M, Delannoy P, Vliegenthart JF, Kamerling JP. Probing the substrate specificity of four different sialyltransferases using synthetic beta-d-Galp-(1→4)-beta-d-GlcpNAc-(1→2)-alpha-d-Manp-(1→O) (CH(2))7CH3 analogues general activating effect of replacing N-acetylglucosamine by N-propionylglucosamine. Biochim Biophys Acta. 2006;1760:685–692. doi: 10.1016/j.bbagen.2005.12.012. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Schauer R. Sialic acids as regulators of molecular and cellular interactions. Curr Opin Struct Biol. 2009;19:507–514. doi: 10.1016/j.sbi.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schauer R, Kamerling J. Chemistry, biochemistry and biology of sialic acids. In: Montreuil J, Vliegenthart J, Schachter H, editors. Glycoproteins II. Amsterdam: Elsevier; 1997. pp. 243–402. [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonetti FL, Teppa E, Chernomoretz A, Nielsen M, Marino Buslje C. MISTIC: mutual information server to infer coevolution. Nucleic Acids Res. 2013;41:W8–W14. doi: 10.1093/nar/gkt427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JJ, Kuraku S, Holt C, Sauka-Spengler T, Jiang N, Campbell MS, Yandell MD, Manousaki T, Meyer A, Bloom OE, et al. Sequencing of the sea lamprey (Petromyzon marinus) genome provides insights into vertebrate evolution. Nat Genet. 2013;45:415–421. doi: 10.1038/ng.2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinke D, Salzburger W, Braasch I, Meyer A. Many genes in fish have species-specific asymmetric rates of molecular evolution. BMC Genomics. 2006;7:20. doi: 10.1186/1471-2164-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima S. Characterization of mouse sialyltransferase genes: their evolution and diversity. Biosci Biotechnol Biochem. 2008;72:1155–1167. doi: 10.1271/bbb.80025. [DOI] [PubMed] [Google Scholar]
- Takashima S, Matsumoto T, Tsujimoto M, Tsuji S. Effects of amino acid substitutions in the sialylmotifs on molecular expression and enzymatic activities of alpha2,8-sialyltransferases ST8Sia-I and ST8Sia-VI. Glycobiology. 2013;23:603–612. doi: 10.1093/glycob/cwt002. [DOI] [PubMed] [Google Scholar]
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateno Y, Imanishi T, Miyazaki S, Fukami-Kobayashi K, Saitou N, Sugawara H, Gojobori T. DNA Data Bank of Japan (DDBJ) for genome scale research in life science. Nucleic Acids Res. 2002;30:27–30. doi: 10.1093/nar/30.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teppa E, Wilkins AD, Nielsen M, Buslje CM. Disentangling evolutionary signals: conservation, specificity determining positions and coevolution. Implication for catalytic residue prediction. BMC Bioinformatics. 2012;13:235. doi: 10.1186/1471-2105-13-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thisse B, Heyer V, Lux A, Alunni V, Degrave A, Seiliez I, Kirchner J, Parkhill JP, Thisse C. Spatial and temporal expression of the zebrafish genome by large-scale in situ hybridization screening. Methods Cell Biol. 2004;77:505–519. doi: 10.1016/s0091-679x(04)77027-2. [DOI] [PubMed] [Google Scholar]
- Thisse C, Thisse B. Zebrafish Science Monitor. Vol. 5. Eugene (OR): University of Oregon Press; 1998. High resolution whole-mount in situ hybridization. [Google Scholar]
- Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc. 2008;3:59–69. doi: 10.1038/nprot.2007.514. [DOI] [PubMed] [Google Scholar]
- Tsuji S, Datta AK, Paulson JC. Systematic nomenclature for sialyltransferases. Glycobiology. 1996;6:v–vii. doi: 10.1093/glycob/6.7.647. [DOI] [PubMed] [Google Scholar]
- Vanbeselaere J, Chang LY, Harduin-Lepers A, Fabre E, Yamakawa N, Slomianny C, Biot C, Khoo KH, Guerardel Y. Mapping the expressed glycome and glycosyltransferases of zebrafish liver cells as a relevant model system for glycosylation studies. J Proteome Res. 2012;11:2164–2177. doi: 10.1021/pr200948j. [DOI] [PubMed] [Google Scholar]
- Vandepoele K, De Vos W, Taylor JS, Meyer A, Van de Peer Y. Major events in the genome evolution of vertebrates: paranome age and size differ considerably between ray-finned fishes and land vertebrates. Proc Natl Acad Sci U S A. 2004;101:1638–1643. doi: 10.1073/pnas.0307968100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A. Diversity in the sialic acids. Glycobiology. 1992;2:25–40. doi: 10.1093/glycob/2.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A. Nothing in glycobiology makes sense, except in the light of evolution. Cell. 2006;126:841–845. doi: 10.1016/j.cell.2006.08.022. [DOI] [PubMed] [Google Scholar]
- Varki A. Glycan-based interactions involving vertebrate sialic-acid-recognizing proteins. Nature. 2007;446:1023–1029. doi: 10.1038/nature05816. [DOI] [PubMed] [Google Scholar]
- Varki A. Sialic acids in human health and disease. Trends Mol Med. 2008;14:351–360. doi: 10.1016/j.molmed.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A. Evolutionary forces shaping the Golgi glycosylation machinery: why cell surface glycans are universal to living cells. Cold Spring Harb Perspect Biol. 2011;3:a005462. doi: 10.1101/cshperspect.a005462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M. The zebrafish book. A guide for laboratory use of zebrafish (Danio rerio) Eugene (OR): University of Oregon Press; 1995. p. 385. [Google Scholar]
- Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Church DM, DiCuccio M, Edgar R, Federhen S, Helmberg W, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2005;33:D39–D45. doi: 10.1093/nar/gki062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelan S, Goldman N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol. 2001;18:691–699. doi: 10.1093/oxfordjournals.molbev.a003851. [DOI] [PubMed] [Google Scholar]
- Wolf YI, Carmel L, Koonin EV. Unifying measures of gene function and evolution. Proc Biol Sci. 2006;273:1507–1515. doi: 10.1098/rspb.2006.3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe KH. Yesterday’s polyploids and the mystery of diploidization. Nat Rev Genet. 2001;2:333–341. doi: 10.1038/35072009. [DOI] [PubMed] [Google Scholar]
- Yegorov S, Good S. Using paleogenomics to study the evolution of gene families: origin and duplication history of the relaxin family hormones and their receptors. PLoS One. 2012;7:e32923. doi: 10.1371/journal.pone.0032923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JE, Yoon H, Lee HJ, Lee JH, Chang BJ, Song CS, Nahm SS. Expression patterns of influenza virus receptors in the respiratory tracts of four species of poultry. J Vet Sci. 2011;12:7–13. doi: 10.4142/jvs.2011.12.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.