

Abstract
Podophyllotoxin (PPT) exhibited significant activity against P-glycoprotein mediated multidrug resistant (MDR) tumor cell lines; however, due to its poor solubility and high toxicity, PPT cannot be dosed systemically, preventing its clinical use for MDR cancer. We developed a nanoparticle dosage form of PPT by covalently conjugating PPT and polyethylene glycol (PEG) with acetylated carboxymethyl cellulose (CMC-Ac) using one-pot esterification chemistry. The polymer conjugates self-assembled into nanoparticles (NPs) of variable sizes (20–120 nm) depending on the PPT-to-PEG molar ratio (2–20). The conjugate with a low PPT/PEG molar ratio of 2 yielded NPs with a mean diameter of 20 nm and released PPT at ~5%/day in serum, while conjugates with increased PPT/PEG ratios (5 and 20) produced bigger particles (30 nm and 120 nm respectively) that displayed slower drug release (~2.5%/day and ~1%/day respectively). The 20 nm particles exhibited 2- to 5-fold enhanced cell killing potency and 5- to 20-fold increased tumor delivery compared to the larger NPs. The biodistribution of the 20 nm PPT-NPs was highly selective to the tumor with 8-fold higher accumulation than all other examined tissues, while the larger PPT-NPs (30 and 100 nm) exhibited increased liver uptake. Within the tumor, >90% of the 20 nm PPT-NPs penetrated to the hypovascular core, while the larger particles were largely restricted in the hypervascular periphery. The 20 nm PPT-NPs displayed significantly improved efficacy against MDR tumors in mice compared to the larger PPT-NPs, native PPT and the standard taxane chemotherapies, with minimal toxicity.
Keywords: Podophyllotoxin, Multidrug resistance, Nanoparticles, Drug Delivery
Introduction
Tubulin, a major component of the cellular cytoskeleton, plays an important role in the survival and growth of cells. Its functions extend from cellular transport to cell motility and mitosis. The importance of tubulin in mitosis and cell division makes them an attractive target for anticancer therapy. Chemotherapeutic agents that disrupt the normal function of tubulin are amongst the most potent and broadest spectrum anticancer agents in the clinic, and have been used to treat major cancers including lung, breast, prostate, and ovarian cancer [1]. A structurally diverse class of compounds has been found to antagonize tubulin functions with various tubulin binding sites and different mechanisms of action [1].
Anti-tubulin agents can be divided into two major categories, microtubule-destabilizing agents and microtubule-stabilizing agents, based on their effect on microtubule dynamics. Microtubule-destabilizing agents, such as colchicine and the vinca alkaloids, inhibit polymerization and decrease the mass of microtubules. Microtubule-stabilizing agents like taxanes stabilize microtubules, increase microtubule polymer mass, and induce the formation of microtubule bundles in cells. Both classes of anti-microtubule agents function by disrupting the dynamic equilibrium of the microtubules, resulting in arrest of cells in mitosis through blocking cell cycle at the metaphase–anaphase transition and leading to cellular apoptosis [2, 3].
However, clinical success of these anti-microtubule agents has been compromised by the emergence of drug resistance [4]. Often, the resistance renders ineffectiveness to a variety of anticancer agents and is termed multidrug resistance (MDR). The therapeutic options after development of MDR are limited. MDR can be induced by various mechanisms, including decreased drug uptake, increased drug efflux, activation of detoxifying systems, activation of DNA repair mechanisms and evasion of drug-induced apoptosis [5]. Among these, overexpression of P-glycoprotein (Pgp) is the most commonly found mechanism for MDR in clinical samples [6, 7]. Another shortcoming of these anti-tubulin drugs are their significant side effects, including drug induced neutropenia and neurotoxicity [8], as well as hypersensitivity reactions provoked by the surfactants used in the formulation to increase their solubility [9, 10].
The goal of this study was to develop a new treatment for Pgp-mediated MDR tumors that are resistant to the standard taxane chemotherapies. Nanoparticle (NP) drug delivery system has been postulated to enhance activity of standard drugs such as taxanes and anthracyclines against MDR tumors by modulating the cellular uptake pathway [11]. It is hypothesized that NPs can carry drugs into an MDR cell via cellular endocytosis, bypassing the Pgp mediated drug efflux, and thus enhancing the drug activity. However, this class of products has not produced significant clinical success [12]. We aimed to explore an alternative approach: first identifying a potent anti-tubulin compound that is not a Pgp substrate, and then developing a NP formulation to selectively deliver the drug to the tumor. Additionally, NP drug delivery has been compromised by significant uptake by the reticuloendothelial system (RES) and poor tumor penetration, leading to impaired drug delivery and efficacy [13]. In this study, we also focused on optimizing the NP formulation to improve its targeting to tumors and its penetration into tumor core for enhanced safety and therapy.
We first screened a wide range of tubulin inhibitors against different MDR cell lines, and demonstrated that podophyllotoxin (PPT) remained active against those highly resistant lines. However, PPT exhibits poor solubility and a has a very narrow therapeutic window, preventing its systemic use for treating MDR cancer [14]. We hypothesized that PPT could be targeted to MDR tumors by NPs in a detergent and solvent free formulation to exert significant therapeutic activity with reduced side effects. We covalently conjugated PPT and polyethylene glycol (PEG) to an acetylated carboxymethyl cellulose (CMC-Ac) backbone via ester linkages in a one-pot reaction. The resultant polymer conjugates self-assembled into NPs of various sizes (20–120 nm) depending on the PPT-to-PEG ratio. Their drug release kinetics, cytotoxic potency and in vivo biodistribution were analyzed and the optimal formulation was tested for its efficacy and safety against MDR tumor models in mice with comparison to other standard taxane chemotherapies.
Materials and Methods
Reagents and Reference Drugs
Podophyllotoxin (PPT) was obtained from Carbosynth (Berkshire, UK). Docetaxel (DTX), Paclitaxel (PTX) and Cabazitaxel (CBZ) were obtained from LC Laboratories (Woburn, MA). Cholchicin (Cho) and Vinblastin (Vin) were purchased from Sigma Aldrich (Oakville, ON). Carboxymethylcellulose (CMC) sodium salt (CEKOL 30000-P) was purchased from CPKelco (Atlanta, GA). Poly(ethylene glycol) methyl ether (mPEG−OH, MW = 2000), 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide HCl (EDC.HCl), and 4-dimethylaminopyridine (DMAP) were purchased from Sigma Aldrich (Oakville, ON). Hydrophobic fluorescent dye DiI (1,1`-dioctadecyl-3,3,3`,3`-tetramethylindocarbocyanine perchlorate, D-307) was purchased from Invitrogen (Burlington, ON). For all the in vitro studies, free drugs were first dissolved in DMSO and then diluted with DMEM medium.
Synthesis of PPT-CMC-Ac-PEG polymer conjugate
The polymer conjugate was synthesized in a two-step reaction protocol. In the first step, sodium salt of CMC was acetylated as described by Namikoshi et al. [15]. Briefly, sodium CMC was first de-salted using 20% sulfuric acid solution, and the free acid was then acetylated with acetic anhydride to yield acetylated CMC (CMC-Ac). The purified CMC-Ac was subsequently conjugated to PEG and PPT via EDC/DMAP coupling chemistry. CMC-Ac (300 mg, 1.2 mmol acid) was weighed into a 25 mL round bottom flask, and dissolved in a mixture of anhydrous MeCN (9 mL) and DMSO (6 mL). EDC HCl (448 mg, 2.4 mmol) and DMAP (580 mg, 4.8 mmol) were added into that solution followed by addition of variable amount of PPT and m-PEG-OH. The solution was stirred overnight at room temperature with protection from light. The reaction mixture was then precipitated through 135 mL diethyl ether. The precipitate was dried, re-dissolved in MeCN, and the precipitation process was repeated twice. The final precipitate was dried under vacuum, and the fine powder was suspended in water (25 mL) and dialyzed (MW cut-off = 10 kDa) against MilliQ water for 24 h with 3 changes. The product was analyzed by LC-MS for uncoupled PEG and DTX, and washing was repeated if residual reagent was detected. The chemical composition of the polymer conjugate was determined by 1H-NMR using 2-methyl 5-nitro benzoic acid as an internal standard. The NPs were prepared by the nano-precipitation method using nanoAssemblr (Precision Nanosystems, Vancouver, Canada). Thirty mg of the polymer was dissolved in 1 mL MeCN and precipitated into 3 mL of normal saline in the nanoAssemblr at the flow rate of 18 mL/min. The formed particles were dialyzed in a Slide-A-Lyzer 10,000 MWCO cartridge against 0.9% saline overnight to extract solvent. The particles were filtered through a 0.22 µm Millipore PVDF filter, and were concentrated using a Vivaspin unit (10,000 MWCO). Particle size and zeta potential were measured with a Zetasizer (Nano-ZS, Malvern Instruments, Malvern, UK). PPT content of the NPs was determined by 1H-NMR using 2-methyl 5-nitro benzoic acid as an internal standard. DiI loaded NPs were prepared by dissolving 30 mg of the polymer in MeCN (1 mL) containing 0.1 mg/mL DiI and was precipitated into 3 mL of normal saline in the nanoAssemblr at the flow rate of 18 mL/min. DiI content of the NPs was determined by dissolving the NPs in DMSO and assaying for fluorescence (Excitation filter: 535 nm; Emission Filter 590 nm) and comparing to a calibration curve of fluorescence versus DiI concentration, subtracting the background signal of un-loaded particle fluorescence.
Cell culture and animals
Human MDA-MB-231 and PC3 and mouse EMT6 and 4T1 cancer cell lines were obtained from the American Type Culture Collection (ATCC). Resistant EMT6/AR1 cells were a gift from Ian Tannock, Princess Margaret Hospital, Toronto. DTX resistant PC3 and MDA-MB-231 cells were generated from the native phenotype by treating them with gradually increasing concentrations of DTX until the cells become fully resistant to 100 nM and 10 nM of DTX respectively. The cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum. NOD-SCID and BALB/c mice were purchased from Jackson Laboratories (Bar Harbour, ME). All protocols were approved by the Animal Care Committee of the University Health Network.
In vitro release of PPT from the NPs
PPT-NPs were 1:1 (v:v) mixed with fetal bovine serum (FBS) at the final concentration of 100 µg PPT /mL. Samples were incubated at 37 °C, and at selected time points triplicate samples were removed and serum protein was precipitated using 600 µL MeCN containing 1% acetic acid. The sample was centrifuged for 5 min at 10,000 rpm and the supernatant was analyzed for released PPT by a Waters Acquity UPLC/MS system equipped with a PDA and SQ MS detector. The samples were injected into an Agilent XDB-C18 column (1.8 µm, 4.6 × 50 mm) at a flow rate of 0.4 mL/min, with a gradient program of 95/5 to 10/90 water/MeCN over 5 min. A calibration curve for PPT was prepared by spiking known amounts of PPT in a saline/FBS mixture, followed by the same extraction protocol.
Transmission electron microscopy (TEM)
The size and morphology of the NPs were determined by TEM using a Hitachi 7000 microscope (Schaumburg, IL) operating at an acceleration voltage of 75 kV. PPT-NPs were prepared as described previously with slight modification of using double distilled water as the precipitating media instead of normal saline. The NPs were negatively stained with a 1% uranyl acetate (UA) solution immediately prior to analysis. The samples were first deposited on copper grids (Ted Pella Inc., Redding, CA) that had been pre-coated with carbon and negatively charged and then stained with UA. The copper grids were briefly left to stand to allow the solvent to evaporate. The imaging was done at 100,000 magnification.
In vitro analysis of viability
Cell growth inhibition activity of different drugs was analyzed by measuring cell viability with the XTT assay. Briefly, cells were dislodged and re-suspended at a concentration of 1 × 104 cell/mL, and 100 µL of cell suspension per well was added to a 96 well plate. A slight modification (5 × 104 cell/mL) was made for the MDA-MB-231 analysis, as these cells were slow-growing compared to the other lines. The cells were maintained for 24 h in culture (37 °C, 5% CO2, humidified) before treatment. Cells were treated with different concentration of the free drugs (primarily dissolved in DMSO and further diluted in DMEM) or NPs (suspended in normal saline and diluted in DMEM). After 72 h of treatment, viability was assayed by the XTT assay. Briefly, a 1 mg/mL solution of XTT reagent and 1.53 mg/mL solution of phenazine methylsulfate in water were prepared, and 5 µL of phenazine methylsulfate was added to each mL of the XTT solution. Twenty-five µL of the mixture was added to each well, the culture plate was incubated for 2 h at 37 °C, and absorbance of each well at 480 nm was then measured. Wells treated with media represent 100% viable cells, and wells containing no cells represent background signal. The viability data were analyzed in GraphPad Prism, and the IC50 was calculated.
In vivo biodistribution study of DiI labelled NPs
DiI loaded PPT-NPs of different sizes were prepared as described previously. EMT6/AR1 tumor cells (2×105) were s.c. implanted in the right flank of BALB/c mice. When the tumor size reached 6–8 mm in diameter, they were i.v. injected with 1 µg DiI/mL dose of DiI loaded NPs. After different time points, animals were sacrificed and different organs and tissues were collected and imaged by the Xenogen system using 550 nm excitation filter and 570 nm emission filter. The DiI signal was quantified in each tissue using the Xenogen software. Tumors were cryo-sectioned and stained with anti-CD31-FITC antibody. The tumor sections were scanned and analyzed with Tissue Studio (Definiens) software.
Maximum tolerated dose
Maximum tolerated dose (MTD) of drugs in mice was determined in a dose escalation study, and is identified as the maximum dose of a drug that does not induce animal death or >20% body weight loss. The maximum deliverable dose for an i.v. injection of PPT-NPs was 180 mg PPT/kg.
In Vivo Antitumor Efficacy
Anticancer efficacy of PPT-NPs was analyzed against two s.c. tumor models, EMT6/AR1 and PC-3 RES. EMT6/AR1 cells (0.2 × 106 cells) and PC-3 RES cells (5 × 106 cells) were s.c. inoculated into the shaved right lateral flank of female BALB/c and male NOD–SCID mice respectively. When tumors reached 5–7 mm in diameter, the mice were treated i.v. with either saline (control), MTD of native PPT (20 mg/kg, day 0), MTD of DTX (12 mg/kg; day 0, 4 and 8), MTD of CBZ (5 mg/kg; day 0, 4 and 8), maximum deliverable dose of PPT-NPs (180 mg PPT/kg; day 0, 4 and 8). Tumor volume and body weight of the treated animals were monitored. Standard taxane chemotherapies (DTX, CBZ) at their MTDs were included as controls. CBZ is used clinically to treat prostate cancer patients that no longer respond to DTX [16].
Histology and immunohistochemical analysis
Representative animals were sacrificed and tissues were harvested and fixed in 10% formalin for 2 days followed by storage in 70% ethanol. Histology and immunohistochemistry slides were prepared at the Toronto General Hospital Pathology Research Program lab (Toronto, ON). Ki67 (SP6) antibody (ThermoFisher) was used at a 1/1000 dilution in citrate for 1 h. TUNEL staining was performed according to the method of Wijsman [17]. Prepared slides were analyzed on an Aperio Scanner at the Advanced Optical Microscopy Facility (AOMF) at the Princess Margaret Hospital (Toronto, ON). Image analysis was performed with the ImageScope software using Positive Pixel Count algorithm. Image analysis output were positive pixel counts divided by the area analyzed. All data was normalized against the control.
Statistical Analysis
All data are expressed as mean ± SEM. Statistical analysis was conducted with the two-tailed unpaired t test for two-group comparison or one-way ANOVA, followed by the Tukey multiple comparison test by using GraphPad Prism (for three or more groups). A difference with p < 0.05 was considered to be statistically significant.
Results and discussion
Analysis of cytotoxic activity of different anti-tubulin agents against MDR tumor cells
MDR has most often been linked to the overexpression of Pgp, which confers resistance to over 300 compounds [18–20]. Even many of the highly potent “new generation” anticancer compounds (e.g., kinase inhibitors) are also substrates for Pgp [21, 22]. Different approaches, such as identification of Pgp inhibitors, synthesis and evaluation of more active analogues, synthesis of conjugates or prodrugs, as well as combined use with other drugs have been pursued to overcome resistance. Unfortunately, most of them have not produced significant improvements in the clinic [23, 24]. Hence, there is a considerable need for the development of new treatment strategies for MDR cancer.
Although the search for new chemical entities for enhanced treatment of cancer is an ongoing process and has attracted considerable attention, screening of the existing chemotherapeutic candidates for diverse indications can be an alternative [25]. Since anti-tubulin drugs are among the most potent and broadest spectrum chemotherapeutic agents, we first tested the potency of a panel of six anti-tubulin drugs [Docetaxel (DTX), Paclitaxel (PTX), Podophyllotoxin (PPT), Cabazitaxel (CBZ), Colchicine (Col) and Vinblastin (Vin)] against different MDR tumors as well as their native phenotypes (EMT6, PC3 and MDA-MB-231) (Figure 1, Table 1 and Supplementary Figures 1–2). The MDR phenotype of EMT6 cells, (EMT6/AR1) was obtained from Dr. Ian Tannock at Princess Margaret Cancer Centre, Toronto, whose team has demonstrated EMT6-AR1 express a high level of Pgp compared to EMT6 [26]. The resistant phenotype of PC3 and MDA-MB-231 cells were generated in our lab as described in the materials and methods. We tested Pgp expression in all these three cell lines and found >200- to 500-fold overexpression of Pgp in the MDR phenotypes compared to the native cells (Supplementary figure 3). The native cell lines were found to be highly sensitive to all the tested drugs, with IC50s varying between 0.01 nM and 14 nM. CBZ was found to be the most potent: IC50 varied from 0.01 to 1 nM (Table 1). However, PTX, DTX, CBZ, Col and Vin showed a considerable loss in potency against the MDR lines (EMT6/AR1, PC3-RES and MDA-MB-231-RES). PC3-RES cells were found to be the most refractive to these drugs: the resistance index (RI = IC50 in the resistant line divided by IC50 in the parent line) of PTX, DTX, CBZ, Chol and Vin for this cell line varied from 100 to 500 (Table 1, Figure 1). The loss of potency against the MDR phenotype was in the order of PTX (RI: 510) > DTX (450) > Vin (400) > Col (190) > CBZ (100). A similar trend was also noted in the EMT6 and MDA-MB-231 models (Table 1, Supplementary Figures 1–2). However, PPT retained its activity against the MDR cells in all the three models (RI = 1–3). The reason why PPT remained active against the MDR lines is yet to be elucidated, but one of the plausible explanations is that PPT is not a substrate for Pgp [27, 28], which is the major MDR mechanism for these tested cell lines.
Figure 1.

In vitro viability assay of different drugs against PC3 and PC3-RES tumor cells. ● PC3 Δ PC3-RES. Data = mean ± SEM (n=3)
Table 1.
IC50 and resistance index (RI) of different drugs against different cell lines. Data = mean ± SEM (n= 3). RI = IC50 in the resistant cell line divided by IC50 in the parent cell line.
| IC50 (nM) | DTX | PTX | PPT | CBZ | Col | Vin |
|---|---|---|---|---|---|---|
| PC3 | 2 ± 1 | 4 ± 2 | 13 ± 5 | 0.06 ± 0.05 | 14 ± 5 | 0.2 ± 0.1 |
| PC3-RES | 903 ± 50 | 2057 ± 120 | 15 ± 3 | 6.3 ± 2 | 2610 ± 140 | 80 ± 5 |
| Resistance Index | 450 | 510 | 1 | 105 | 190 | 400 |
| EMT6 | 7 ± 2 | 13 ± 5 | 12 ± 2 | 1 ± 2 | 10 ± 5 | 1 ± 1 |
| EMT6 AR1 | 1091 ± 45 | 6667 ± 150 | 13 ± 3 | 43 ± 5 | 2375 ± 230 | 80 ± 4 |
| Resistance Index | 156 | 513 | 1 | 43 | 237 | 80 |
| MDA-MB-231 | 1 ± 2 | 1 ± 3 | 6 ± 5 | 0.01 ± 0.02 | 2 ± 1 | 0.5 ± 0.1 |
| MDA-MB-231-RES | 40 ± 5 | 130 ± 15 | 20 ± 5 | 1 ± 1 | 73 ± 8 | 130 ± 15 |
| Resistance Index | 40 | 130 | 3 | 100 | 37 | 260 |
Despite of its significant potency against tumors, clinical use of PPT for systemic cancer treatment has been unsuccessful, mainly due to its poor solubility and significant toxicity [29, 30]. There are a number of PPT derivatives approved clinically (etoposide, taniposide, ethophos), but these drugs have a different mode of action (topo-isomerase inhibition), are significantly less potent (100–1000 times higher IC50) than PPT, and are substrates for Pgp [31, 32]. Other non-Pgp substrate derivatives of PPT such as YB-1EPN have failed to show increased efficacy in animal models [33]. Instead of pursuing chemical modification of PPT, we focused on developing a tumor targeted delivery system for PPT, aiming to improve the solubility and tissue selectivity of this drug to make it potent and safe therapeutics for MDR tumors.
Preparation of PPT-CMC-Ac-PEG conjugates and evaluation of their physicochemical properties
Not much research has been done to improve delivery of PPT. For example, Greenwald et al. [34] conjugated PPT with PEG to increase the solubility, but failed to demonstrate any improvement in prolonging survival of the tumor-bearing mice. Qin et al. [35] prepared a NP formulation for PPT consisting of layered double hydroxides (Mg-Al-hydrotalcite), which displayed little in vivo efficacy.
Different nanotherapeutic formulations have been developed to provide increased safety and efficacy for cancer chemotherapy [36]. Among them, polymer–drug conjugate have received significant attention due to the advantages in increased drug loading capacity, enhanced stability, and prolonged blood circulation [37]. In the present study, carboxymethyl cellulose (CMC) was selected as the polymer backbone to synthesize a polymer conjugate of PPT due to its documented safety, biocompatibility and multiple conjugation sites (carboxylate groups) for efficient coupling of the drug. However, CMC is highly hydrophilic and PPT is hydrophobic, causing problem for usual coupling chemistries. Modifications in polysaccharides to increase organic solvent solubility include triethylamine (TEA) or tetrabutylammonium (TBA) complexation, but the coupling efficiency of these polymers remained low (15 – 20 wt%) [38, 39].
To improve the solvent solubility of CMC, the hydroxyl groups on the CMC were first acetylated to produce acetylated CMC (CMC-Ac), which was soluble in a number of organic solvents including dimethyl sulfoxide (DMSO), acetonitrile (MeCN) etc. Acetylation of the hydroxyl groups of CMC could also prevent polymer cross-linking during the 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) mediated coupling chemistry. PPT, mPEG-OH and CMC-Ac were reacted in the presence of EDC and 4-Dimethylaminopyridine (DMAP) in an anhydrous solvent mixture of MeCN and DMSO (Figure 2). The product was purified by ether precipitation and water washing. The purity of the conjugate was determined by LC analysis and no more than 0.5% free drug and PEG was detected. The relative mole and weight percentage of each unit (CMC, PEG, and PPT) in the conjugate was estimated by integration of the 1H-NMR spectra using 2-methyl 5-nitro benzoic acid as an internal standard. We prepared conjugates with a range of PPT and PEG loading by varying the feed ratio. The 1H-NMR profiles of these conjugates were found to be identical, varying only in the integration of peaks assigned to PEG and PPT. An inverse linear correlation (R2 = 0.9) was found between the relative mol% of PPT and PEG in the final conjugate i.e., increased PPT loading led to reduction in PEG loading and vice versa (Figure 3B). This indicates competition of PPT and PEG for reacting with the available carboxylate groups in CMC-Ac. Compared to other polysaccharide polymers, increased drug conjugation efficiency was achieved in this CMC-Ac system (~40 mol % of drug and PEG) under the conditions of a water free reaction and a high content of carboxylate groups in the polymer. As discussed in previous studies, increased drug loading in polymer-conjugates often results in controlled drug release (reduced burst release) and improved pharmacokinetics [40].
Figure 2.

Reaction scheme and 1H-NMR spectra of the PPT-CMC-Ac-PEG, CMC-Ac and PPT.
Figure 3.

Physicochemical characteristics of PPT-CMC-Ac-PEG conjugates prepared with variable PPT/PEG ratios. (A) Illustration of the formation of a nanoparticulate structure by the conjugate in an aqueous medium. (B) Variability of PPT and PEG in conjugates synthesized with different PPT/PEG compositions; (C) Relationship of PPT/PEG ratio to the NP size measured by dynamic light scattering; (D – F) TEM images of different PPT-NPs. (D) NPs of PPT/PEG ratio of 20, (E) NPs of PPT/PEG ratio of 5, (F) NPs of PPT/PEG ratio of 2. (G) Relationship of PPT/PEG ratio to the drug release kinetics in serum; (H) Relationship of PPT/PEG ratio to the IC50 (nM) against EMT6/AR1. Data = mean ± SEM (n = 3) for (C, G, H).
A wide range of conjugates with different PPT/PEG compositions (0.01 – 21, mol ratio) were synthesized (Figure 3B, Supplementary Table 1), and all of the conjugates were tested for nanoparticle formation by the nano-precipitation method. Conjugates with the PPT/PEG ratio of less than 1 were water soluble and did not form any observable particles. Conjugates with the PPT/PEG ratio greater than 1 formed distinct nanoparticulate structures in saline (20–120 nm in diameter measured by dynamic light scattering). A significant correlation (R2 = 0.9) was found between the nanoparticle size and the PPT/PEG ratio (Figure 3C). Increased content of PPT in the conjugate led to increased particle size, whereas increased PEG resulted in decreased particle size. The conjugates with a PPT/PEG mol ratio of 2 assembled into ~20 nm particles, whereas the particle size of the conjugate with a PPT/PEG ratio of 20 was found to be ~120 nm (Figure 3C, Supplementary Table 1). This composition dependent size variability may reflect the nature of the self-assembled structure of the particles, in which the hydrophobic PPT aggregates in the aqueous media to form a nanoparticle core (Figure 3A). When the amount of PPT increases, the conjugate assembles into a larger particle to accommodate an increased amount of PPT in the core. Additionally, as the increase in PPT content resulted in a decrease in PEG content, the conjugate became less hydrophilic. To reduce the water exposed surface, the particles may assemble into larger particles with a reduced surface area. On the contrary, the conjugate with a decreased amount of PPT and increased content of PEG would collapse into smaller particles due to the decreased core volume and the increased hydrophilic surface area. The self-assembled particles were fairly mono-disperse with PDIs close to 0.1 and spherical in shape as seen in the TEM images (Figure 3D–F). The PPT/PEG ratio dependent variability in size was also confirmed with the TEM images.
Along with the particle size, the PPT/PEG ratio also affected the drug release rate and cytotoxic potency of the NPs. PPT release profiles in serum from 3 different NPs composed with different PPT/PEG ratios are depicted in Figure 3G. It was found that NPs with an increased PPT/PEG ratio exhibited a reduced drug release rate. All the NPs exhibited linear drug release kinetics: PPT release rate from the NPs with a PPT/PEG mol ratio of 2 was about 5%/day whereas it was ~1%/day from NPs with a PPT/PEG mol ratio of 20. It has also been noted by other investigators that polymer-drug conjugates with an increased amount of drug loading display retarded drug release kinetics [41]. It has been argued that with increased drug loading in a polymer-drug conjugate, the polymer tend to form complex secondary and tertiary structures in an aqueous medium and can self-assemble into core-shell nanostructures with a stable hydrophobic drug core which can reduce the hydrolytic release of the drug. Conjugates with low drug loading are increasingly hydrated, which would expose the polymer–drug linker to the external medium, facilitating hydrolytic cleavage of the bond for increased drug release [42]. In the current study, conjugates with a decreased PPT/PEG ratio had a smaller particle size and a larger surface area compared to the bigger particles. Increase in surface area may also enhance the drug release rate. The NPs with a decreased PPT/PEG ratio also exhibited increased cytotoxic potency which may be due to the enhanced drug release (Figure 3H). IC50 of NPs with a PPT/PEG molar ratio of 2 was ~130 nM PPT equivalent whereas it was ~460 nM for NPs with a PPT/PEG molar ratio of 20.
Although polymer-drug conjugates have been shown to significantly alter the pharmacokinetics of the native drug, in many instances significant RES interaction has been measured with these drug delivery systems, resulting in significant liver and spleen accumulation of the conjugates [43]. One of the most advanced polymer-drug conjugates, Opaxio™ (Poly-glutamic acid-paclitaxel conjugate), which failed to show significant advantage in early phase III clinical trials over native paclitaxel or docetaxel [44, 45] exhibited 5- to 8-fold increased RES uptake compared to the tumor [46]. Doxorubicin–dextran conjugate (DOX-OXD, AD-70) in Phase I clinical trials induced significant hepatotoxicity, ascribed to increased uptake by the RES [47]. Although PEGylation has been employed to improve pharmacokinetics of many NPs [48], it has not been extensively used for polymer-drug conjugates, which may be due to the complexities involved in the chemistry. Again, the water-free EDC coupling chemistry employed in our system allowed efficient conjugation of PEG and PPT onto CMC-Ac, and tuning the PPT/PEG ratio led to production of conjugates and NPs with a range of size, drug release kinetics and cell killing potency. These parameters are important determinants for in vivo pharmacokinetics, biodistribution, tissue penetration and tumor bioavailability [49], and the ability to control these parameters allow us to effectively optimize the conjugates and NPs to maximize their in vivo performance.
Biodistribution analysis of different PPT-NPs
Next we studied the biodistribution of three candidate NPs with different PPT/PEG ratios (20, 5 and 2) and particle size (120 nm, 30 nm, 20 nm respectively) after systemic (i.v.) administration. 1,1`-dioctadecyl-3,3,3`,3`-tetramethylindocarbocyanine perchlorate (DiI) loaded particles were prepared by encapsulating DiI into PPT-NPs during the particle preparation process. Different NPs with the same DiI concentration (300 µg/mL) was i.v. injected into EMT6/AR1 tumor bearing mice, and after selected time points animals were sacrificed and different tissues were collected and imaged by the Xenogen system. A prominent tissue distribution pattern was noticed: the smaller particles with an increased degree of PEGylation showed significantly enhanced tumor accumulation compared to the bigger, less PEGylated particles. The 20 nm particles (PPT/PEG ratio 2) accumulated predominantly in the tumor whereas the 120 nm particles (PPT/PEG ratio 20) congregated in the RES organs like liver and spleen (Figure 4A). Also the kinetics of the tumor accumulation of different particles was different: tumor accumulation of the 20 nm particles reached the maximum value (4.2×108 p/s/cm2/sr) at 24 h post administration and remained constant for 96 h (Figure 4B). On the other hand, tumor accumulation of the 30 nm particles was slower and attained a maximum value at 48 h (3.7×108 p/s/cm2/sr) but significantly reduced to 2.6×108 p/s/cm2/sr at 96 h, signifying elimination. Tumor accumulation of the 120 nm particles was found to be quicker but significantly lower than both 20 nm and 30 nm particles: it reached the maximum value at 6 h (7.9×107 p/s/cm2/sr) but gradually decreased to 4.4×107 p/s/cm2/sr at 96 h. On the contrary, the liver accumulation of the 120 nm particles was significantly higher: at 24 h it reached the maximum value of 4.2×108 p/s/cm2/sr but gradually came down to 2.7×108 p/s/cm2/sr at 96 h, may be due to digestion and elimination (Figure 4C). The liver accumulation of both the 20 and 30 nm particles were significantly lower than that of the 120 nm particles and contrary to the 120 nm particles, a very slow but constant increase in liver accumulation was measured with these two formulations. The liver accumulation was slightly higher with the 30 nm particles compared to the 20 nm: at 96 h post administration it was 1.3×108 p/s/cm2/sr for the 30 nm particles compared to 9.5×107 p/s/cm2/sr with the 20 nm particles. Overall, throughout all of the time points, the 20 nm particles demonstrated a significantly higher tumor/liver uptake ratio compared to the other two formulations, signifying enhanced tumor targeting (Figure 4D). This may be due to the high degree of PEGylation of the 20 nm PPT-NPs, minimizing immune recognition of the particles. Forty-eight hours post treatment all the major organs and tissues were imaged (Supplementary Figures 4). It was found that the 20 nm PPT-NPs were highly selective for tumor with little to minimal uptake in other tissues including the liver. On the other hand, the liver displayed significant uptake of the 30 nm PPT-NPs, and the RES uptake (both liver and spleen) dominated the clearance for the 120 nm PPT-NPs. In the animals treated with the 20 nm NPs, an 8-fold higher signal was detected in the tumor compared to any other organs. With the 30 nm treatment, the tumor accumulation was ~3 fold higher compared to other organs but with the 120 nm NP treatment, tumor accumulation was ~7 fold lower than the liver accumulation. Previously it has been shown that particle size influenced tumor accumulation of a PEG-PLA copolymer and smaller particles displayed increased tumor delivery (80 nm versus 150 nm), however, significant liver and spleen accumulation was still noted [50]. Again, the 20 nm PPT-NPs displayed significantly increased tissue selectivity towards the tumor.
Figure 4.

Biodistribution of differently sized DiI labeled PPT-NPs. DiI loaded PPT-NPs were injected i.v. in the tumor bearing mice. After selected time points, animals were sacrificed and different organs and tissues isolated and imaged in the Xenogen system. (A) Images of tumor (T) liver (L) and spleen (S) uptake at 6, 24, 48 and 96 h; (B) Kinetics of tumor uptake with different NPs; (C) Kinetics of liver uptake with different NPs; (D) Quantitative comparison of tumor/liver ratio with different NPs. Data = mean ± SEM (n=3).
We also cryosectioned the tumors isolated from animals 48 h after treatment to study the intra-tumoral micro-distribution of the particles (Figure 5A). The tumor sections were labelled with FITC-anti-CD31 antibody (blood vessels) and DAPI (nuclei). The sections were scanned with TISSUEscope (TS4000, Huron Technologies). Similar to the Xenogen data, increased tumor accumulation was determined with the 20 nm NPs compared to both the 30 and 120 nm NPs (Figures 5B). Also, a different pattern of intra-tumoral distribution was noticed with differently sized NPs. While the 30 and 120 nm NPs mostly accumulated in the highly vascularised peripheral portion of the tumor, an increased amount of the 20 nm NPs were found to penetrate into the core of the tumor compared to the vascularised periphery. When the images were analyzed using ImageScope software with the positive pixel count algorithm, an overall 20-and 5-fold increased DiI signal was noted in the 20 nm NPs treated tumors compared to that treated with the 120 nm and 30 nm NPs respectively (Figures 5B). When comparing between the rim and core of the tumors, it was found that the 20 nm NPs exhibited 12-fold increased accumulation in the core compared to the periphery. On the contrary, both the 30 nm and 120 nm particles exhibited 5- and 20-fold increased accumulation at the periphery compared to the core (Figures 5C). It has been shown by Yuan at al. [51] that tumor permeability was inversely proportional to nanoparticle radius. Lee et al. [52] also demonstrated that smaller particles penetrate better following extravasation in the tumor. Cabral et al. [53] demonstrated the size dependency of polymer micelles for tumor penetration. They have shown that the 30 nm micelles exhibited better tumor penetration compared to the 70 nm micelles. This enhanced penetration of the smaller particles may improve the efficacy, as efficient extravasation and tumor penetration are prerequisites for targeting cancer cells. Compromised tumor core penetration has been a significant limit for NP drug delivery [54], which could invariably result in disease recurrence. On the contrary, the 20 nm PPT-NPs displayed 12-fold increased uptake in the hypovascular core compared to the highly vascularized periphery, representing a potential advantage over other systems.
Figure 5.
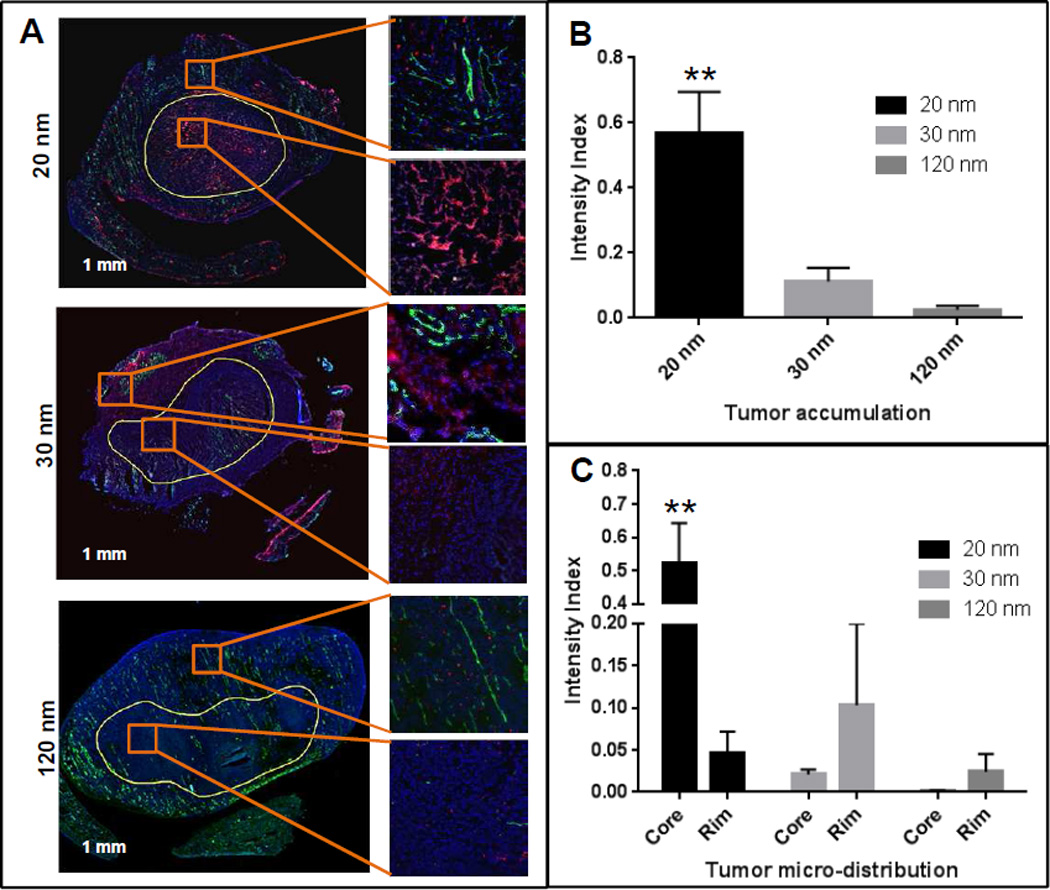
Nanoparticle microdistribution in the tumors treated with the 20, 30 and 120 nm DiI labeled NPs. Tumor sections were stained with FITC-anti CD31 Ab and DAPI and scanned with TISSUEscope. Differential accumulation was found between tumor core and perivascular region. (A) Tumor sections treated with different sized nanoparticles. Red (DiI-PPT-NPs), green (FITC-CD31, vascular endothelium) and blue (DAPI, nuclei). (B) Overall DiI fluorescence intensity (normalized against DAPI) in tumors. ** p < 0.01 vs 120 nm. (C) Microdistribution of DiI signal in the core (hypovascular) and rim (hypervascular) of the tumors. ** p < 0.01 vs Rim. Data = mean ± SEM (n=3).
In this study, Xenogen fluorescence imaging was employed as a robust method to compare the efficiency of tumor delivery and tissue selectivity of the NP candidates. This method has been used intensively in the field for the same purpose [55–57]. It is noted that optical imaging methods cannot provide absolute quantification of in vivo drug delivery due to the limited penetration of light and autofluorescence of tissue, although in this study, we have minimized these factors by excising the tissues, selecting a fluorescent dye with increased wavelengths of spectrum, and subtracting the tissue autofluorescence by the Xenogen imaging program’s correction method. The purpose of this preliminary biodistribution study was to efficiently compare different NP candidates, and therefore, the Xenogen imaging method was used.
Maximum tolerated dose study
Prior to performing an in vivo efficacy study, the maximum tolerated doses (MTDs) of native PPT, native DTX, native CBZ and PPT-NPs were determined in a dose escalation study. The MTDs for DTX and CBZ (day 0, 4, 8) in mice were 12 mg/kg and 5 mg/kg, respectively. The mice tolerated three i.v. doses of PPT-NPs (20 nm, 30 nm and 120 nm formulations) at 180 mg PPT/kg (the maximum deliverable dose) without showing any significant signs of stress or physiological abnormality. Native PPT (formulated with either Tween80/ethanol/saline or DMSO/PBS) was found to be very toxic and could only be dosed once at 20 mg/kg with 2/10 animal death either by i.p. or i.v. Our data with native PPT were consistent with previous reports [34, 58]. These, again, emphasize the importance of developing an improved formulation to deliver PPT systemically.
In vivo antitumor efficacy against MDR tumor models
First, we compared the efficacy of the PPT-NPs in three different sizes with standard taxane therapies against the EMT-6/AR1 model. Female BALB/c mice were s.c. inoculated with EMT-6/AR1 murine breast tumor cells. When tumors reached the size of 50 mm3, the mice were dosed i.v. with either saline (control), DTX (12 mg/kg; day 0, 4, and 8), CBZ (5 mg/kg; day 0, 4 and 8), PPT (20 mg/kg, day 0) or the PPT-NPs (120, 30 and 20 nm; 180 mg PPT/kg; day 0, 4 and 8). CBZ is used clinically for cancer patients after DTX failure [59]. Native DTX, PPT and CBZ were dosed at their maximum tolerated doses (MTDs), but exhibited no significant antitumor activity compared to the saline control (Figure 6 A, B). All the PPT-NPs exhibited significantly improved antitumor efficacy compared to the saline control, native DTX, PPT and CBZ treatment. At day 11, more than 80% tumor inhibition was noted with all the PPT-NPs (20, 30 and 120 nm formulations) whereas tumor growth inhibition was 5%, 40% and 45%, respectively with the DTX, PPT and CBZ treatments. However, tumor rebounded rapidly in the 120 nm PPT-NP treated group at day 14 and all the animals reached the end point by day 19. The 20 nm PPT-NP formulation was found to be highly efficacious in retarding the aggressive growth of this MDR tumor and at day 40, no palpable tumor was found in 7 out of 10 animals. Tumor volumes in the 30 nm particle treated animals were controlled for more than 3 weeks but all eventually rebounded. The median survival of the control, DTX, PPT and CBZ treated animals were 11, 11, 11 and 14 days respectively compared to 19 and 38 days for 120 and 30 nm formulations. In the observation period of 60 days, only 20% of animals treated with the 20 nm PPT-NPs reached the end point; hence the median survival for this group was > 60 days. The larger PPT-NPs (30 and 120 nm) initially retarded the growth of the EMT6/AR1 tumors, but eventually all tumors rebound, whereas the 20 nm PPT-NPs completely regressed tumors in 7/10 mice. The improved efficacy of the 20 nm PPT-NPs could be attributed to the 4-fold increased cytotoxic potency and 20-fold enhanced tumor penetration compared to the 120 nm formulations. As the drug delivery to the tumor rim was comparable among these three NP compositions, the initial antitumor activity was similar (a moderate improvement with the 20 nm PPT-NPs possibly due to the increased cytotoxic potency: 120 nM with 20 nm NPs compared to 200–450 nM with 30 and 120 nm NPs in IC50). The tumors treated with the 120 nm PPT-NPs rebound first by day 11 possibly due to little drug delivery to the tumor core, leaving a substantial faction of the tumor for recurrence. The tumor penetration and the cytotoxic potency of the 30 nm PPT-NPs were significantly improved compared to the 120 nm PPT-NPs and therefore, the tumor recurrence occurred later by day 30. The delivery to the tumor core was 5-fold increased by the 20 nm PPT-NPs with 2-fold improved potency compared to the 30 nm PPT-NPs, which could lead to depletion of the tumor cells in the core, resulting in a decreased rate of recurrence (30%) compared to the larger PPT-NPs (100%).
Figure 6.

In vivo efficacy of the PPT-NPs against multiple tumor models. (A, B): EMT6/AR1 tumor. 2×105 EMT6/AR1 cells were implanted s.c. in the female BALB/c mice. After one week of inoculation, they were treated with saline (control), DTX (12 mg/kg; day 0, 4, and 8), CBZ (5 mg/kg; day 0, 4 and 8), PPT (20 mg/kg, day 0) or PPT-NPs (120, 30 and 20 nm; 180 mg PPT/kg; day 0, 4 and 8). (A) Tumor volume; (B) Percent survival. Data = mean ± SEM (n=10). (C, D): PC3 RES tumor. 5×106 PC3-RES cells were implanted s.c. in the male NOD-SCID mice. After one week of inoculation, they were treated with saline (control), CBZ (5 mg/kg; day 0, 4 and 8), PPT (20 mg/kg, day 0) or PPT-NPs (20 nm, 180 mg PPT/kg; day 0, 4 and 8). (C) Tumor volume; ** p < 0.01 vs CBZ (D) Histology and immunohistochemical analysis (H&E, TUNEL and Ki67) of tumors. Data = mean ± SEM (n=10).
Other controls including Tween80/ethanol/saline and CMC-Ac-PEG polymer showed no antitumor activity (data not shown). Additionally, the mice treated with DTX and CBZ showed consistent and significant weight loss (5–10%) as well as visible signs of pain or distress including piloerection, lethargy, weight loss, and weakness (Supplementary figure 5A). Two out of 10 mice treated with native PPT were dead 3 days post injection possibly due to the drug toxicity.
We further extended the evaluation of antitumor efficacy of the optimal PPT-NPs (20 nm) against another MDR model (PC-3 RES, human prostate cancer). As this MDR model was developed in house, we first validated the models by comparing the efficacy of the standard taxanes in the native and MDR models. It was found that while both DTX and CBZ regressed the native PC3 tumors, the resistant phenotype was completely refractory to DTX treatment and partially resistant to CBZ (Supplementary figure 6). This PC3-RES model represents significant clinical relevance, as CBZ is one of the standard therapies for prostate cancer patients after DTX failure, but only results in moderate improvement. We then focused on comparing the efficacy of the 20 nm PPT-NPs with native PPT and CBZ in the PC3-RES model. Male NOD-SCID mice were inoculated s.c. with PC-3 RES tumor. At 10 days post inoculation the tumors reached 100 mm3 in size, and were divided into 4 groups: saline (control), PPT (20 mg/kg; day 0), CBZ (5 mg/kg; day 0, 4 and 8) and 20 nm PPT-NP group (180 mg PPT/kg; day 0, 4 and 8). At day 17, compared to the initial size, tumor size reduced by 30% in the 20-nm PPT-NP treated animals, compared to 180% and 700% size increase with CBZ and PPT treatment respectively (Figure 6C). Again, no significant weight loss was measured with the mice treated with the 20 nm PPT-NPs, but those receiving free CBZ suffered from significant weight loss (~20%, Supplementary figure 5B). Tumor histology at day 17 revealed that the NPs-treated tumors displayed 8-fold increased apoptosis (TUNEL assay) and 6-fold reduced proliferation (Ki-67 staining) compared to control, whereas CBZ induced only 2-fold increased apoptosis and 1.5- fold reduced proliferation (Figure 6D). There was no significant effect of native PPT against this tumor model at the MTD (Figure 6C, D). The tumor apoptosis mainly occurred in the core after therapy with the 20 nm PPT-NPs as shown in the histology data; however, this phenomenon might not be completely explained by the increased drug delivery to the core relative to the rim. First, tumor cells in the core undergo necrosis and apoptosis regularly due to reduced blood supply [60]. Second, as shown in our previous publication [61], large NPs (120 nm) that did not penetrated the tumor core also induced apoptosis predominantly in the core. Cytotoxic drugs released in the rim could promote antiangiogenesis, further reducing blood supply to the core, leading to enhanced necrosis and apoptosis. Nevertheless, this secondary effect has been shown not efficient to eradicate the cells in the core, which promote aggressive phenotypes of tumors [62]. Drug delivery to the core has been a major challenge in this field, but the 20 nm PPT-NPs have shown significant promise.
Similar to the EMT6/AR1 model, the PPT-NPs treatment did not cause any body weight loss in the animals, rather an increase in body weight was noticed compared to the saline control (Supplementary figure 5B). This may be due to the significantly reduced tumor burden in the PPT-NP treated animals. CBZ treatment showed dose limiting toxicity and significant body weight loss (~20%); this weight loss continued till day 10 and stabilized at day 21. This was compared to a ~5% weight gain with the PPT-NPs treatment.
CONCLUSION
PPT exhibited significant activity against Pgp-mediated MDR cancer cell lines, but its poor solubility and high toxicity prevent its clinical use to treat cancer. We conjugated PPT and PEG onto an acetylated CMC backbone and demonstrated that by varying the PPT/PEG ratio in the conjugates, NPs with different physicochemical properties (size, drug release kinetics, cell killing potency, biodistribution) could be produced. A NP formulation with a mean diameter of 20 nm demonstrated 5- to 20-fold improved tumor-targeted delivery, increased tumor penetration and enhanced efficacy against multiple MDR tumors, with little toxicity. The delivery of the 20 nm PPT-NPs was highly selective to the tumor with minimal uptake in other tissues, and >90% of the 20 nm PPT-NPs penetrated into the hypovascular tumor core. With this new systemic delivery technology, PPT became a highly effective and safe therapy for MDR tumors in animal models.
Supplementary Material
ACKNOWLEDGEMENTS
This work was funded by grants from the Ontario Institute for Cancer Research Intellectual Property Development and Commercialization Fund, the Canadian Institutes of Health Research (PPP-122898) and National Institutes of Health (CA17633901). S.D. Li is a recipient of a Coalition to Cure Prostate Cancer Young Investigator Award from the Prostate Cancer Foundation and a New Investigator Award from Canadian Institutes of Health Research (MSH-130195). Funding for the Ontario Institute for Cancer Research is provided by the Government of Ontario. The authors thank Dr. Jonathan May for reviewing the manuscript, and Dr. Steven Doyle of University of Toronto for the TEM analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pellegrini F, Budman DR. Review: tubulin function, action of antitubulin drugs, and new drug development. Cancer investigation. 2005;23:264–273. doi: 10.1081/cnv-200055970. [DOI] [PubMed] [Google Scholar]
- 2.Das GC, Holiday D, Gallardo R, Haas C. Taxol-induced cell cycle arrest and apoptosis: dose-response relationship in lung cancer cells of different wild-type p53 status and under isogenic condition. Cancer Lett. 2001;165:147–153. doi: 10.1016/s0304-3835(01)00404-9. [DOI] [PubMed] [Google Scholar]
- 3.Rieder CL, Palazzo RE. Colcemid and the mitotic cycle. J Cell Sci. 1992;102(Pt 3):387–392. doi: 10.1242/jcs.102.3.387. [DOI] [PubMed] [Google Scholar]
- 4.Dumontet C, Sikic BI. Mechanisms of action of and resistance to antitubulin agents: microtubule dynamics, drug transport, and cell death. J Clin Oncol. 1999;17:1061–1070. doi: 10.1200/JCO.1999.17.3.1061. [DOI] [PubMed] [Google Scholar]
- 5.Gillet JP, Gottesman MM. Mechanisms of multidrug resistance in cancer. Methods Mol Biol. 2010;596:47–76. doi: 10.1007/978-1-60761-416-6_4. [DOI] [PubMed] [Google Scholar]
- 6.Kamazawa S, Kigawa J, Kanamori Y, Itamochi H, Sato S, Iba T, et al. Multidrug resistance gene-1 is a useful predictor of Paclitaxel-based chemotherapy for patients with ovarian cancer. Gynecologic oncology. 2002;86:171–176. doi: 10.1006/gyno.2002.6738. [DOI] [PubMed] [Google Scholar]
- 7.Baekelandt MM, Holm R, Nesland JM, Trope CG, Kristensen GB. P-glycoprotein expression is a marker for chemotherapy resistance and prognosis in advanced ovarian cancer. Anticancer research. 2000;20:1061–1067. [PubMed] [Google Scholar]
- 8.Cella D, Peterman A, Hudgens S, Webster K, Socinski MA. Measuring the side effects of taxane therapy in oncology: the functional assesment of cancer therapy-taxane (FACT-taxane) Cancer. 2003;98:822–831. doi: 10.1002/cncr.11578. [DOI] [PubMed] [Google Scholar]
- 9.Markman M. Management of toxicities associated with the administration of taxanes. Expert Opin Drug Saf. 2003;2:141–146. doi: 10.1517/14740338.2.2.141. [DOI] [PubMed] [Google Scholar]
- 10.Gelderblom H, Verweij J, Nooter K, Sparreboom A. Cremophor EL: the drawbacks and advantages of vehicle selection for drug formulation. Eur J Cancer. 2001;37:1590–1598. doi: 10.1016/s0959-8049(01)00171-x. [DOI] [PubMed] [Google Scholar]
- 11.Kapse-Mistry S, Govender T, Srivastava R, Yergeri M. Nanodrug delivery in reversing multidrug resistance in cancer cells. Front Pharmacol. 2014;5:159. doi: 10.3389/fphar.2014.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kunjachan S, Blauz A, Mockel D, Theek B, Kiessling F, Etrych T, et al. Overcoming cellular multidrug resistance using classical nanomedicine formulations. European journal of pharmaceutical sciences : official journal of the European Federation for Pharmaceutical Sciences. 2012;45:421–428. doi: 10.1016/j.ejps.2011.08.028. [DOI] [PubMed] [Google Scholar]
- 13.Aggarwal P, Hall JB, McLeland CB, Dobrovolskaia MA, McNeil SE. Nanoparticle interaction with plasma proteins as it relates to particle biodistribution, biocompatibility and therapeutic efficacy. Advanced drug delivery reviews. 2009;61:428–437. doi: 10.1016/j.addr.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beutner KR, von Krogh G. Current status of podophyllotoxin for the treatment of genital warts. Semin Dermatol. 1990;9:148–151. [PubMed] [Google Scholar]
- 15.Namikoshi H. In: US Patent: Carboxyalkyl acetyl celluloses, their salts and a process for the preparation of them. USPTO, editor. USA: Daicel Chemical Industries Ltd; 1985. [Google Scholar]
- 16.Richards L. Prostate cancer: Cabazitaxel boosts post-docetaxel survival. Nat Rev Urol. 2010;7:645. doi: 10.1038/nrurol.2010.200. [DOI] [PubMed] [Google Scholar]
- 17.Wijsman JH, Jonker RR, Keijzer R, van de Velde CJ, Cornelisse CJ, van Dierendonck JH. A new method to detect apoptosis in paraffin sections: in situ end-labeling of fragmented DNA. J Histochem Cytochem. 1993;41:7–12. doi: 10.1177/41.1.7678025. [DOI] [PubMed] [Google Scholar]
- 18.Wang Z, Chen Y, Liang H, Bender A, Glen RC, Yan A. P-glycoprotein substrate models using support vector machines based on a comprehensive data set. J Chem Inf Model. 2011;51:1447–1456. doi: 10.1021/ci2001583. [DOI] [PubMed] [Google Scholar]
- 19.Chen L, Li Y, Yu H, Zhang L, Hou T. Computational models for predicting substrates or inhibitors of P-glycoprotein. Drug Discov Today. 2012;17:343–351. doi: 10.1016/j.drudis.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 20.Bellamy WT. P-glycoproteins and multidrug resistance. Annu Rev Pharmacol Toxicol. 1996;36:161–183. doi: 10.1146/annurev.pa.36.040196.001113. [DOI] [PubMed] [Google Scholar]
- 21.Hegedus T, Orfi L, Seprodi A, Varadi A, Sarkadi B, Keri G. Interaction of tyrosine kinase inhibitors with the human multidrug transporter proteins, MDR1 and MRP1. Biochim Biophys Acta. 2002;1587:318–325. doi: 10.1016/s0925-4439(02)00095-9. [DOI] [PubMed] [Google Scholar]
- 22.Wang XK, Fu LW. Interaction of tyrosine kinase inhibitors with the MDR- related ABC transporter proteins. Curr Drug Metab. 2010;11:618–628. doi: 10.2174/138920010792927316. [DOI] [PubMed] [Google Scholar]
- 23.Thomas H, Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control. 2003;10:159–165. doi: 10.1177/107327480301000207. [DOI] [PubMed] [Google Scholar]
- 24.Crowley E, McDevitt CA, Callaghan R. Generating inhibitors of P-glycoprotein: where to, now? Methods Mol Biol. 2010;596:405–432. doi: 10.1007/978-1-60761-416-6_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turk D, Hall MD, Chu BF, Ludwig JA, Fales HM, Gottesman MM, et al. Identification of compounds selectively killing multidrug-resistant cancer cells. Cancer Res. 2009;69:8293–8301. doi: 10.1158/0008-5472.CAN-09-2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patel KJ, Tannock IF. The influence of P-glycoprotein expression and its inhibitors on the distribution of doxorubicin in breast tumors. BMC cancer. 2009;9:356. doi: 10.1186/1471-2407-9-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Etievant C, Schambel P, Guminski Y, Barret JM, Imbert T, Hill BT. Requirements for P-glycoprotein recognition based on structure-activity relationships in the podophyllotoxin series. Anticancer Drug Des. 1998;13:317–336. [PubMed] [Google Scholar]
- 28.Nguyen TL, Cera MR, Pinto A, Lo Presti L, Hamel E, Conti P, et al. Evading Pgp activity in drug-resistant cancer cells: a structural and functional study of antitubulin furan metotica compounds. Mol Cancer Ther. 2012;11:1103–1111. doi: 10.1158/1535-7163.MCT-11-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.You Y. Podophyllotoxin derivatives: current synthetic approaches for new anticancer agents. Curr Pharm Des. 2005;11:1695–1717. doi: 10.2174/1381612053764724. [DOI] [PubMed] [Google Scholar]
- 30.Li J, Sun H, Jin L, Cao W, Zhang J, Guo CY, et al. Alleviation of podophyllotoxin toxicity using coexisting flavonoids from Dysosma versipellis. PloS one. 2013;8:e72099. doi: 10.1371/journal.pone.0072099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aneja R, Liu M, Yates C, Gao J, Dong X, Zhou B, et al. Multidrug resistance-associated protein-overexpressing teniposide-resistant human lymphomas undergo apoptosis by a tubulin-binding agent. Cancer Res. 2008;68:1495–1503. doi: 10.1158/0008-5472.CAN-07-1874. [DOI] [PubMed] [Google Scholar]
- 32.Li WQ, Wang XL, Qian K, Liu YQ, Wang CY, Yang L, et al. Design, synthesis and potent cytotoxic activity of novel podophyllotoxin derivatives. Bioorg Med Chem. 2013;21:2363–2369. doi: 10.1016/j.bmc.2013.01.069. [DOI] [PubMed] [Google Scholar]
- 33.Chen H, Bi W, Cao B, Yang Z, Chen S, Shang H, et al. A novel podophyllotoxin derivative (YB-1EPN) induces apoptosis and down-regulates express of P-glycoprotein in multidrug resistance cell line KBV200. Eur J Pharmacol. 2010;627:69–74. doi: 10.1016/j.ejphar.2009.10.056. [DOI] [PubMed] [Google Scholar]
- 34.Greenwald RB, Conover CD, Pendri A, Choe YH, Martinez A, Wu D, et al. Drug delivery of anticancer agents: water soluble 4-poly (ethylene glycol) derivatives of the lignan, podophyllotoxin. J Control Release. 1999;61:281–294. doi: 10.1016/s0168-3659(99)00153-4. [DOI] [PubMed] [Google Scholar]
- 35.Qin L, Xue M, Wang W, Zhu R, Wang S, Sun J, et al. The in vitro and in vivo anti-tumor effect of layered double hydroxides nanoparticles as delivery for podophyllotoxin. Int J Pharm. 2010;388:223–230. doi: 10.1016/j.ijpharm.2009.12.044. [DOI] [PubMed] [Google Scholar]
- 36.Moghimi SM, Hunter AC, Murray JC. Nanomedicine: current status and future prospects. FASEB J. 2005;19:311–330. doi: 10.1096/fj.04-2747rev. [DOI] [PubMed] [Google Scholar]
- 37.Maeda H, Seymour LW, Miyamoto Y. Conjugates of anticancer agents and polymers: advantages of macromolecular therapeutics in vivo. Bioconjug Chem. 1992;3:351–362. doi: 10.1021/bc00017a001. [DOI] [PubMed] [Google Scholar]
- 38.Wang X, Li J, Wang Y, Cho KJ, Kim G, Gjyrezi A, et al. HFT-T, a targeting nanoparticle, enhances specific delivery of paclitaxel to folate receptor-positive tumors. ACS Nano. 2009;3:3165–3174. doi: 10.1021/nn900649v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosato A, Banzato A, De Luca G, Renier D, Bettella F, Pagano C, et al. HYTAD1-p20: a new paclitaxel-hyaluronic acid hydrosoluble bioconjugate for treatment of superficial bladder cancer. Urol Oncol. 2006;24:207–215. doi: 10.1016/j.urolonc.2005.08.020. [DOI] [PubMed] [Google Scholar]
- 40.Ernsting MJ, Tang WL, MacCallum N, Li SD. Synthetic modification of carboxymethylcellulose and use thereof to prepare a nanoparticle forming conjugate of docetaxel for enhanced cytotoxicity against cancer cells. Bioconjug Chem. 2011;22:2474–2486. doi: 10.1021/bc200284b. [DOI] [PubMed] [Google Scholar]
- 41.Sugahara S, Kajiki M, Kuriyama H, Kobayashi TR. Carrier effects on antitumor activity and neurotoxicity of AZ10992, a paclitaxel-carboxymethyl dextran conjugate, in a mouse model. Biol Pharm Bull. 2008;31:223–230. doi: 10.1248/bpb.31.223. [DOI] [PubMed] [Google Scholar]
- 42.Roy A, Bhattacharyya M, Ernsting MJ, May JP, Li SD. Recent progress in the development of polysaccharide conjugates of docetaxel and paclitaxel. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2014 doi: 10.1002/wnan.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Markovsky E, Baabur-Cohen H, Eldar-Boock A, Omer L, Tiram G, Ferber S, et al. Administration, distribution, metabolism and elimination of polymer therapeutics. J Control Release. 2012;161:446–460. doi: 10.1016/j.jconrel.2011.12.021. [DOI] [PubMed] [Google Scholar]
- 44.Langer CJ, O'Byrne KJ, Socinski MA, Mikhailov SM, Lesniewski-Kmak K, Smakal M, et al. Phase III trial comparing paclitaxel poliglumex (CT-2103, PPX) in combination with carboplatin versus standard paclitaxel and carboplatin in the treatment of PS 2 patients with chemotherapy-naive advanced non-small cell lung cancer. J Thorac Oncol. 2008;3:623–630. doi: 10.1097/JTO.0b013e3181753b4b. [DOI] [PubMed] [Google Scholar]
- 45.Paz-Ares L, Ross H, O'Brien M, Riviere A, Gatzemeier U, Von Pawel J, et al. Phase III trial comparing paclitaxel poliglumex vs docetaxel in the second-line treatment of non-small-cell lung cancer. Br J Cancer. 2008;98:1608–1613. doi: 10.1038/sj.bjc.6604372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li C, Newman RA, Wu QP, Ke S, Chen W, Hutto T, et al. Biodistribution of paclitaxel and poly(L-glutamic acid)-paclitaxel conjugate in mice with ovarian OCa-1 tumor. Cancer Chemother Pharmacol. 2000;46:416–422. doi: 10.1007/s002800000168. [DOI] [PubMed] [Google Scholar]
- 47.Danhauser-Riedl S, Hausmann E, Schick HD, Bender R, Dietzfelbinger H, Rastetter J, et al. Phase I clinical and pharmacokinetic trial of dextran conjugated doxorubicin (AD-70, DOX-OXD) Invest New Drugs. 1993;11:187–195. doi: 10.1007/BF00874153. [DOI] [PubMed] [Google Scholar]
- 48.Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev. 2001;53:283–318. [PubMed] [Google Scholar]
- 49.Pang X, Du HL, Zhang HQ, Zhai YJ, Zhai GX. Polymer-drug conjugates: present state of play and future perspectives. Drug Discov Today. 2013;18:1316–1322. doi: 10.1016/j.drudis.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 50.Schadlich A, Caysa H, Mueller T, Tenambergen F, Rose C, Gopferich A, et al. Tumor accumulation of NIR fluorescent PEG-PLA nanoparticles: impact of particle size and human xenograft tumor model. ACS Nano. 2011;5:8710–8720. doi: 10.1021/nn2026353. [DOI] [PubMed] [Google Scholar]
- 51.Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, et al. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995;55:3752–3756. [PubMed] [Google Scholar]
- 52.Lee H, Fonge H, Hoang B, Reilly RM, Allen C. The effects of particle size and molecular targeting on the intratumoral and subcellular distribution of polymeric nanoparticles. Mol Pharm. 2010;7:1195–1208. doi: 10.1021/mp100038h. [DOI] [PubMed] [Google Scholar]
- 53.Cabral H, Matsumoto Y, Mizuno K, Chen Q, Murakami M, Kimura M, et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat Nanotechnol. 2011;6:815–823. doi: 10.1038/nnano.2011.166. [DOI] [PubMed] [Google Scholar]
- 54.Li L, Sun J, He Z. Deep penetration of nanoparticulate drug delivery systems into tumors: challenges and solutions. Current medicinal chemistry. 2013;20:2881–2891. doi: 10.2174/09298673113209990004. [DOI] [PubMed] [Google Scholar]
- 55.Cai L, Xu G, Shi C, Guo D, Wang X, Luo J. Telodendrimer nanocarrier for co-delivery of paclitaxel and cisplatin: A synergistic combination nanotherapy for ovarian cancer treatment. Biomaterials. 2015;37:456–468. doi: 10.1016/j.biomaterials.2014.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Luo LM, Huang Y, Zhao BX, Zhao X, Duan Y, Du R, et al. Anti-tumor and anti-angiogenic effect of metronomic cyclic NGR-modified liposomes containing paclitaxel. Biomaterials. 2013;34:1102–1114. doi: 10.1016/j.biomaterials.2012.10.029. [DOI] [PubMed] [Google Scholar]
- 57.Shi W, Mei H, Deng J, Chen C, Wang H, Guo T, et al. A tissue factor targeted nanomedical system for thrombi-specific drug delivery. Biomaterials. 2012;33:7643–7654. doi: 10.1016/j.biomaterials.2012.06.094. [DOI] [PubMed] [Google Scholar]
- 58.Levy RK, Hall IH, Lee KH. Antitumor agents LXII: synthesis and biological evaluation of podophyllotoxin esters and related derivatives. J Pharm Sci. 1983;72:1158–1161. doi: 10.1002/jps.2600721012. [DOI] [PubMed] [Google Scholar]
- 59.Vrignaud P, Semiond D, Lejeune P, Bouchard H, Calvet L, Combeau C, et al. Preclinical antitumor activity of cabazitaxel, a semisynthetic taxane active in taxane-resistant tumors. Clin Cancer Res. 2013;19:2973–2983. doi: 10.1158/1078-0432.CCR-12-3146. [DOI] [PubMed] [Google Scholar]
- 60.McKeage MJ, Baguley BC. Disrupting established tumor blood vessels: an emerging therapeutic strategy for cancer. Cancer. 2010;116:1859–1871. doi: 10.1002/cncr.24975. [DOI] [PubMed] [Google Scholar]
- 61.Ernsting MJ, Tang WL, MacCallum NW, Li SD. Preclinical pharmacokinetic, biodistribution, and anti-cancer efficacy studies of a docetaxel-carboxymethylcellulose nanoparticle in mouse models. Biomaterials. 2012;33:1445–1454. doi: 10.1016/j.biomaterials.2011.10.061. [DOI] [PubMed] [Google Scholar]
- 62.Subarsky P, Hill RP. The hypoxic tumour microenvironment and metastatic progression. Clinical & experimental metastasis. 2003;20:237–250. doi: 10.1023/a:1022939318102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.