

Abstract
This study was designed to understand the contribution of the inflammasome and IL-1β activation in otitis media (OM). We examined the middle ear (ME) response to non-typeable Haemophilus influenzae (NTHi) in wild type (WT) mice using gene microarrays and a murine model of acute OM. Expression of members of the NOD domain-like receptor family of inflammasome genes was significantly up-regulated early in NTHi infection of the ME, potentially activating specific downstream regulatory cascades that contribute to the proliferative inflammatory response observed during OM. Expression of the pro-forms of the inflammasome targets IL-1β and IL-18 were also up-regulated. To evaluate the role of inflammasome-mediated cytokine maturation, NTHi-induced OM was examined in Asc−/−-deficient mice and compared with that seen in WT mice. Mice lacking the Asc gene showed near absence of IL-1β maturation in the ME and a reduction in leukocyte recruitment and infiltration to the cavity, and their macrophages exhibited reduced phagocytosis of NTHi. These inflammatory defects were linked to an increase in the degree and duration of mucosal epithelial hyperplasia in the ME of Asc−/− mice, as well as a delay in bacterial clearance from their MEs. These data demonstrate an important role for the inflammasome and cytokine processing in the course and resolution of OM.
Keywords: IL-1β, cytokine, middle ear, inflammation, macrophage, NTHi
Introduction
Initiation of the innate immune response against invading microbes is largely dependent on host PRRs that trigger release and activation of cytokines to facilitate a series of immune responses leading to the resolution of infection.1–4 The inflammasome is an innate immune complex that has been shown to play a major role in the host immune response to many infections.5–9 This large, multi-protein complex regulates the maturation of several pro-inflammatory cytokines.10,11 Upon activation by ligands of an infectious agent, innate immune receptors of the NLR family, primarily the nacht-domain, leucine-rich repeat and PYD-containing proteins (NLRPs), recruit the scaffolding protein apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and procaspase-1 to assemble the inflammasome. Activation of the inflammasome results in cleavage of procaspase-1, which, in turn, cleaves and releases the active forms of a subset of cytokines, including IL-1β and IL-18. Once released, these mature pro-inflammatory cytokines can play an important role in orchestrating the inflammatory response and defense against infection and injury.8,12–14 Some of the major inflammasome pathways in otitis media (OM) are illustrated schematically in Figure 1.
Figure 1.
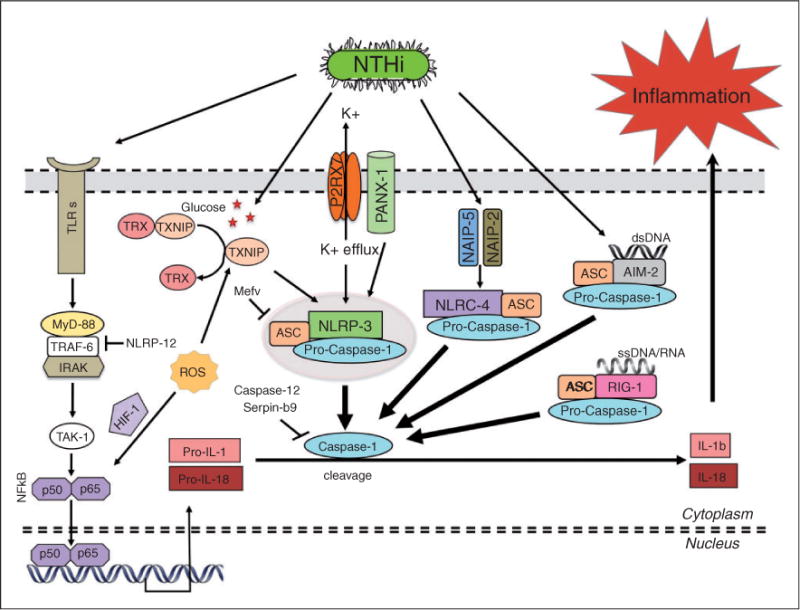
A schematic representation of inflammasome activation and signaling in NTHi-mediated OM. TLR signaling in response to pathogen molecules triggers the initial synthesis of the pro-forms of the inflammatory cytokines IL-1β and IL-18. Pathogen moieties and other danger signals can also trigger the activation of inflammasomes based on various innate immune receptors (NLRP-3, NLRC-4/IPAF, RIG-1 and AIM-2). However, the adaptor molecule ASC mediates assembly of most inflammasomes. The inflammasome then recruits and activates caspase-1, which, in turn, cleaves the pro-forms of IL-1β and IL-18, releasing them in their mature/active forms. As IL-1β is prominent in OM,50,61 inflammasomes seem likely to play a role in inflammatory signaling in the ME during NTHi-induced ME infection in our mouse model. The best-characterized inflammasome is based on NLRP-3, which can be activated by various bacterial molecules and danger signals. However, there are other mechanisms of NLRP-3 activation, including the P2RX7 ATP receptor, ROS formation during oxidative stress, and glucose-mediated release of TXNIP from TRX. The NLRC-4/IPAF inflammasome is activated via its upstream receptors NAIP-2 and NAIP-5, while the AIM-2 and RIG-1 inflammasomes act as sensors for cytosolic bacterial, viral and host dsDNA and RNA, respectively.
IL-1β has been implicated as being crucial to the immune responses of several pathogens, and is strongly regulated at the transcriptional level.15–17 However, its maturation and secretion, which are under the control of the inflammasome, play an equally important post-transcriptional role in regulating its function.18 Cleaved IL-1β has been shown to not only contribute to innate immunity,19,20 but also to aid in the modulation of immune responses and pathogen clearance.21,22 Genetic mutations of NLRP-3, leading to its constitutive activation, have identified the resultant IL-1β activation and secretion as central to the development of the cryopyrin-associated periodic syndromes.23 Activation of IL-1β has also been implicated in gout, Alzheimer’s disease and type-2 diabetes, among others.7,17,24
To date, the involvement of the inflammasome in OM, an extremely common pediatric disease, has not been well studied. Non-typeable Haemophilus influenzae (NTHi), a Gram-negative bacterium, has become the most common pathogen in OM following the introduction of vaccines against Streptococcus pneumonia.25,26 Previous studies in our laboratory, and others, have implicated TLRs in mediating the innate immune response, as TLR deficiencies have been shown to be responsible for abnormalities in NTHi-induced OM pathogenesis and recovery.27–30 TNF-α (TNF), TLR2, TLR4 and TLR9 activation all contribute to OM eradication using a murine model.29,31,32 A major effector of TLR activation is the production of IL-1β and IL-18.12,33,34 However, as noted above, these cytokines—once produced—are inactive until cleaved by the inflammasome. This implicates a potential role for the NLRs in OM. Therefore, to elucidate the role of the inflammasome and IL-1β processing in NTHi-induced OM, we used a mouse model to evaluate the role of NLRs in OM. As numerous NLRs can participate in the inflammasome, we chose to focus our study on ASC, the obligatory component of several inflammasomes, and its major target IL-1β. Our results revealed that lack of ASC and inflammasome activation resulted in defective IL-1β processing, as well as delays in neutrophil recruitment and bacterial clearance of the infected middle ear (ME) cavity. These findings support a role for the inflammasome in OM pathogenesis and recovery.
Materials and methods
Animals
All animal studies were performed in strict accordance to the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, and carried out in strict accordance with an approved Institutional Animal Care and Use Committee protocol. All surgeries were performed under anesthesia, and all efforts were made to minimize suffering. The Asc−/− knockout mice on a C57BL/6 background (more than 10 × generation crossed) were generously supplied by John Bertin, Ethan Grant, and Anthony Coyle and Millenium Pharmaceuticals. Age-matched wild type (WT) C57BL/6 mice were purchased from Jackson Laboratory (Bar Harbor, ME, USA). The NTHi inoculate and the surgeries were preformed as described previously.29,35 In brief, to induce a ME infection, the bullae were bilaterally exposed via a ventral approach and injected with ~ 5 μl saline containing ~ 104 CFUs of H. influenzae strain 3655 (non-typeable NTHi/biotype II), a clinical isolate recovered from the ME of an OM patient.36 It was identified as biotype II based on indole production, urease activity and ornithine decarboxylase reaction. Biotypes I, II and III are the most common upper respiratory forms of NTHi, with II and III being generally non-invasive.37 Following the inoculation, the tympanic membranes were confirmed visually to be intact. The incision was then stapled, and the mice were given Lactated Ringer’s solution and buprenorphine postoperatively.
Histology
The mice used were sacrificed under general anesthesia by intracardiac perfusion. PBS was injected first, followed by 4% paraformaldehyde (PFA). Time points collected were 0h, 6h, and 1, 2, 3, 5, 7 and 10d after NTHi inoculation and infection. The 0 h time point was collected from un-inoculated ears. Dissection of the ear was followed by decalcification in 8% EDTA and 4% PFA for 14 d. The MEs were then embedded into paraffin, cut in 10-μm sections, and stained with hematoxylin and eosin. Digital images of standardized regions from the largest area of the ME cavity were then assessed using SPOT (Sterling Heights, MI, USA) advance image analysis software. Mucosal thickness was measured by averaging the thickness of the epithelium and subepithelium. The lumen area was measured, as well as the area of leukocytic infiltration. The percent area of the ME lumen occupied by inflammatory cells was calculated from several sections and averaged. The numbers of neutrophils versus macrophages were counted at five randomly chosen sites at 400× magnification for MEs that contained infiltrates. This was performed independently by two blinded observers.38
Bacterial clearance
Bacterial presence in the ME was evaluated by obtaining a sample from the ME lumen using a sterile 1-μl loop. This culture was streaked sequentially onto four quadrants of a chocolate agar plate. The plates were then incubated overnight (16–18 h) at 37°C. The isolated bacteria were verified to be NTHi by Gram-staining and ability to grow on chocolate agar versus blood agar. Plates were scored as positive or negative based on the observation of any NTHi growth on chocolate agar plates. In addition, a colonization score (CS) was used to assess semi-quantitatively the degree of colonization of each positive plate: 0 indicated no CFUs on the plate; 1 indicated CFUs in one quadrant; 2 indicated CFUs in two quadrants; 3 indicated CFUs in three quadrants; and 4 indicated CFUs in all four quadrants on the plate.35 The high number of bacteria typically recovered in the initial days after NTHi inoculation is indicative of replication and persistence, namely infection of the ME.
DNA microarrays
The profile of changes in gene expression in the mucosal epithelium during the course of OM inflammatory response in mice was evaluated using DNA microarrays and described elsewhere.29,32,39 In summary, C57BL/6:CB F1 hybrid mice (60–90 d old) were purchased from Jackson Laboratories. Twenty mice per time point were inoculated bilaterally with H. influenzae strain 3655. Un-inoculated (time 0) and shaminoculated (saline) animals served as controls. The ME mucosa were harvested and combined at different intervals: 0 (no treatment), 3 h, 6 h, and 1, 2, 3, 5 and 7 d after NTHi infection. Total RNA was extracted using TRIzol (Invitrogen, Carlsbad, CA, USA). RNA quality was assessed using the RNA 6000 Labchip Kit on an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) to ensure the integrity of 18S and 28S ribosomal RNA. Reverse transcription of the mRNA was done using a T7-oligod T primer and T7 RNA polymerase to generate biotinylated cRNA probes that were hybridized onto two Affymetrix MU430 2.0 (Santa Clara, CA, USA) microarrays per time point sample. This procedure was then duplicated for each time point to obtain a second, independent replication. The raw data of gene expression levels were median normalized, and statistical differences in gene transcript expression levels were evaluated using a variance-modeled posterior inference approach (VAMPIRE).40 Individual transcript fold-level changes were visualized using Genespring (Agilent Technologies). The changes in gene expression were scored as follows: changes over 10-fold gene expression were considered high or strong, changes between 5- and 10-fold were considered moderate, while 2- to 5-fold changes were considered modest. Similarly, down-regulation of genes below 0.5-fold was considered strong, while changes between 0.7- and 0.5-fold were defined as moderate. The gene expression data per time after NTHi inoculation are shown in Supplementary Table S1.
Tissue lysis, protein extraction and immunoblotting
ME mucosal (MEM) tissue from at least five mice was surgically removed, and whole cell extraction was carried out by suspending the tissues in 150 μl of T-PER lysis buffer (Pierce, Rockford, IL, USA) supplemented with protease inhibitors (Roche, Indianapolis, IN, USA) and sonicating briefly on ice. The concentration of proteins in the cell lysates was assessed using BCA assay (Pierce). Proteins (20 μg) were then separated by gel electrophoresis on 4–12% gradient NUPAGE gels (Invitrogen). The proteins were transferred electrophoretically onto polyvinylidene difluoride membranes using iBlot (Invitrogen). After blocking with 5% BSA in PBS-T, the membranes were incubated with goat anti-IL-1β-cleaved m118 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or polyclonal goat anti-pro-IL-1β (R&D Systems, Minneapolis, MN, USA) Abs (1:1000 dilution) at 4°C overnight. Subsequently, the membranes were probed with HRP-conjugated anti-goat (1:10,000 dilution; BioRad, Hercules, CA, USA) at room temperature (20–23°C) for 1 h. Signals were detected using an ECL-Plus kit (Pierce), according to the manufacturer instructions.
Macrophage phagocytosis and NTHi killing assay
For in vitro macrophage/NTHi phagocytosis and killing activities, macrophages were assessed using an established in vitro assay.35,41 Primary peritoneal macrophages were obtained from six Asc−/− mice and control mice (WT/C57BL/6) by i.p. injection of 3 ml thioglycolate medium. Cells were harvested 3 d later by peritoneal lavage with cold RPMI 1640 containing 10% FBS, 50 U/ml penicillin, and 50 μg/ml streptomycin and β-mercaptoethanol, washed with media, enumerated and seeded into 48-well plates at 5 × 105 cells per well in triplicate for each test condition and time point. NTHi were grown to mid-exponential phase, then harvested and re-suspended in PBS, and then added at a titer of 5 × 107 per well—a titer that does not saturate the cells. The tissue culture plates were centrifuged at 200 g for 5 min to enhance contact between the bacteria and macrophage cells, and then incubated for 1 or 3 h at 37°C. Following that, extracellular bacteria were removed by washing with fresh DMEM, and then by DMEM containing 10% FCS, macrophage-colony stimulating factor. Gentamicin (50 μg/ml) was then added to kill the remaining extracellular bacteria. After 1 or 3 h, the cells were rinsed and lysed using 0.5 ml pyrogen-free water followed by aspiration of the lysate five times through a 23-gauge syringe. Lysates were plated on chocolate agar plates in serial dilutions of 1:1 up to 1:105, and incubated overnight at 37°C. Six wells were used per time point and mouse strain condition. The recovery of bacteria after macrophage treatment with gentamicin for 1 h was used to represent phagocytosis. The ratio of bacteria recovered after gentamicin treatment after 3 h compared with bacteria recovered at the 1 h treatment time point was considered to represent intracellular killing, similar to other published assays.35,41
Statistical analysis
All data were analyzed using StatView (version 5.0; JMP-SAS Institute, Cary, NC, USA), a two-tailed t-test was done to compare WT mice with Asc−/− mice data. This was done for each time point comparing mucosal thickness, ME inflammatory cell infiltration (neutrophils and macrophages), and for the assessment of macrophage phagocytic and bacteriocidal capacity. Differences between the two groups were considered to be significant at P < 0.05. The two ears from each mouse were analyzed separately as they were found to be independent from each other on the basis of the following points:
Individual mice were from an inbred strain; hence, there were no individual genetic differences between subjects. Therefore, from a genetic point of view, all ears could be considered to be from the same subject.
The coefficients of variance for the left and right ears were equivalent to the coefficients of variance for all mice in all measures, suggesting that there were no systematic differences between the two sides. The degree of variation between the two ears of the mice was similar to that measured between individual mice.
No differences between MEs were observed on the basis of paired t-tests for each variable.
There was no significant main effect for side (i.e. left versus right) and no significant day/side interaction in two-way ANOVA for each d.
There was no major effect for side in a three-way ANOVA across all d, and there were no significant interaction terms. Descriptive statistics, such as means, were used to prepare the data obtained from the bacterial load and recovery seen in Table 1. As noted above, VAMPIRE was used for analysis of gene array data as presented in Figures 2 and 7, and in Supplementary Table S1.
Table 1.
Impaired bacterial clearance of MEs of the Asc−/− knockout mice. No CFUs were detected by d 5 after NTHi infection in C57BL/6 mice. Bacterial clearance was impaired in Asc−/− mice through 10 d after infection. Bacterial colonization of chocolate-agar plates was evaluated using a semi-quantitative technique to generate a CS: 0 indicated no CFUs; 1 indicated one quadrant with CFUs; 2 indicated two quadrants with CFUs; 3 indicated three quadrants with CFUs; and 4 indicated four quadrants with CFUs. Six MEs were scored per time point.
| Time after NTHi inoculation | C57BL/6 WT culture positive plates | C57BL/6 WT mean bacterial CS | Asc−/− mean positives plates | Asc−/− mean bacterial CS |
|---|---|---|---|---|
| Day 1 | 4/6 | 4.00 | 3/6 | 1.67 |
| Day 2 | 6/6 | 3.00 | 3/6 | 2.33 |
| Day 3 | 3/6 | 1.00 | 6/6 | 4.00 |
| Day 5 | 0/6 | 0.00 | 4/6 | 2.50 |
| Day 10 | 0/6 | 0.00 | 2/6 | 1.00 |
Figure 2.
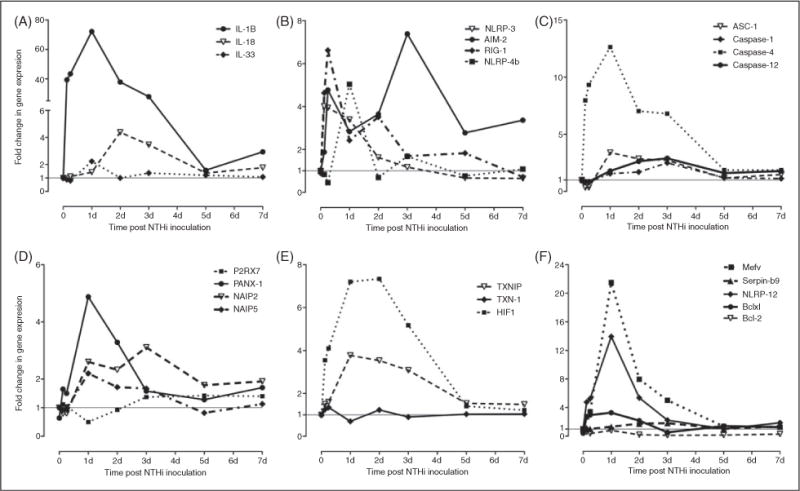
Assessment of changes in the gene expression of the different inflammasomes and their related components in MEM after NTHi infection. Mining gene chip microarray data showed significant and early time-dependent increases in transcripts of the inflammasome targets IL-1β and IL-18 after NTHi infection (A). This is accompanied by up-regulation of various inflammasome receptor mRNAs (B), including NLRP-3, RIG-1, AIM-2 and NLRP-4. Up-regulation of the mRNA for the adaptor molecule ASC and various caspases during NTHi induced OM (C) provides the remaining substrates for the inflammasome. The transcripts of various molecules known to activate the different inflammasomes are also increased in (D) and (E), as are those of inflammasome regulators (F). Data are represented numerically in Supplementary Table S1.
Figure 7.
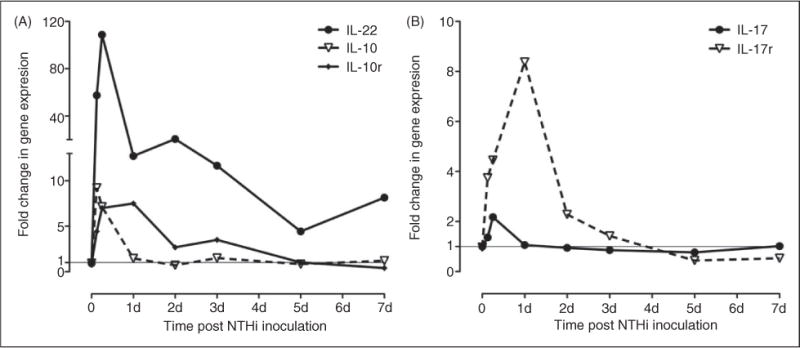
ME expression of genes related to Th17 cell differentiation. There is evidence that caspase-1 activation can play a role in tissue proliferation, mediated by Th17 cytokines. Others have noted these genes are down-regulated in caspase-1 knockout mice in stomach mucosa.46 Gene array data indicated that mRNA up-regulation of Th17 cytokines and receptors possibly play an early role in the innate immune response of the ME. This provided evidence that these genes may play a role in stimulating mucosal hyperplasia in the ME, and would be hypothesized to be reduced in mice unable to activate caspase-1, as in the case of the Asc−/− knockout mice used in this study.
Results
Regulation of inflammasome-related genes during OM
Undertaking a gene-array survey approach, we isolated mRNA from the MEM of uninfected WT mice and compared that to the mRNA profile from mice in which OM had been induced by NTHi inoculation (i.e. injection) into the ME. NTHi injection into the ME cavities resulted in ME infection (OM) where bacterial replication and persistence occurred, triggering the innate immune system to respond. The relative mRNA abundance of genes known to be involved in inflammasome function (see Figure 1) was compared at 3, 6 and 12 h, and 1 d, 2 d, 5 d or 7 d post-inoculation with that in control, untreated (0 h) MEs to identify genes regulated differentially during OM. Expression of a number of these inflammasome-related genes was significantly altered at the mRNA level by NTHi infection (Figure 2), while few were regulated differentially in saline-injected sham mice. Specifically, no significant changes in gene expression (mRNA level) were seen for IL-1β, NLRP-3, AIM-2 and RIG-1 in sham animals over the 7 d period. There was a modest 2-fold upregulation of IL-18 at 6 h. Similarly, sham injection increased ASC gene expression 2-fold at 72 h, and caspase-1 by 2-fold on d 2. However, NLRP-4b gene expression was down-regulated in sham animals to 0.45 at 3 h. These observed changes were compared with the NTHi-infected animals to evaluate the significance of the differences seen across the experimental groups. The fold changes in mRNA gene expression relative to controls per time after inoculation are shown in Supplementary Table S1.
Expression of genes encoding the effector cytokines that could be targeted by the inflammasome pathway were found to be increased in response to NTHi infection (Figure 2A). IL-1β shows a rapid and dramatic increase after NTHi inoculation between time points 3 h and 3 d, and peaks to over 70-fold induction at 24 h. Gene expression of IL-33 peaked more modestly at 24 h, while IL-18, known to be involved in macrophage recruitment, peaked later, at 72 h. Figure 2B and C highlight the many genes encoding potential inflammasome components, as illustrated in Figure 1. NLRP-3, a well studied inflammasome component and innate immune sensor,13 was part of the initial transcriptional response being modestly up-regulated 3 h after infection, with persistent up-regulation noted at 24 h. RIG-1 was moderately up-regulated early (6 h) after NTHi infection and remained over-expressed until d 5. Meanwhile, modest gene up-regulation was also seen for AIM-2, another pathogen DNA-sensing module. IPAF (NLRC-4) and NLRP-1 were not present in the Affymetrix genechip. Additional inflammasome components, including NLRP-4b, the adapter molecule ASC, and caspases-1 and -12 were mainly modestly up-regulated, peaking at 24 h after NTHi infection, with the exception of caspase-4, which was strongly up-regulated 12-fold by 24 h.
Figure 2D illustrates the regulation of upstream inflammasome activators.13 While an upstream ion channel, purinergic receptor ligand-gated ion channel 7 (P2RX7) showed no significant changes in gene expression with time, pannexin, another cell membrane ion channel shown to be involved in activating the inflammasome42 is up-regulated 5-fold by 24 h after NTHi challenge. The NLRP-3 inflammasome can also be activated by reactive oxidative species (ROS) under cellular stress and injury. Under homeostasis, thioredoxin-interacting protein (TXNIP) is bound to thioredoxin (TRX). However, the TXNIP-TRX complex dissociates as ROS concentration increases and TXNIP activates NLRP-3, triggering the inflammasome.42 In the gene array, TXNIP and TRX (also known as TXN-1) gene expression were modestly up-regulated within 6 h of NTHi infection. In addition, gene expression of the transcription factor hypoxiainducible factor-1 (HIF-1), which is related to chronic OM43 and is also regulated by ROS activation and involved in inflammasome signaling via cross-talk with NFkB, was moderately up-regulated early in the course of OM.
Finally, Figure 2F illustrates the gene expression of different inflammasome inhibitors and regulators. NLRP-12, which is a non-inflammasome NLR and plays a regulatory role in TLR-dependent NFkB signaling,44 was strongly up-regulated at 24 h. Similarly, the MefV gene, which modulates ASC activity and interaction by encoding the pyrin protein,42 was up-regulated by 24 h after NTHi infection. Serpin-b9, a direct inhibitor of caspase-1 proteolytic activity, was modestly up-regulated between d 3 and 5. Finally, it has been suggested that Bcl-2 and Bcl-xl proteins inhibit NLRP oligomerization.42 Bcl-xl was modestly up-regulated by 24 h. However, Bcl-2 was strongly down-regulated throughout the time course of NTHi infection.
Taken together, gene-array profiling of the different inflammasome components and their related pathways during the course of OM indicates that multiple genes of the inflammasome pathways are strongly regulated; thus, inflammasomes are likely to play a key role in inflammatory signaling in the ME during NTHi-induced OM.
Lack of the Asc gene alters the processing of IL-1β during OM
Having observed that NTHi infection regulates the different NLRs and inflammasome components at the gene level, we sought to further elucidate the mechanism leading to inflammasome activation by NTHi and the functional role of the inflammasome in OM. Therefore, we investigated the ME response to NTHi infection in mice lacking the obligatory inflammasome component ASC compared with WT C57BL/6 controls. To validate this model, we first confirmed the presence of IL-1β protein in the ME, as well as inflammasome functional deficiency in Asc−/− mice (Figure 3). Western blot analysis of WT ME tissue confirmed NTHi-dependent induction of pro-IL-1β expression at the protein level, as well as its activation and processing into its mature form, 12 h after induction of an NTHi infection. IL-1β is synthesized as a 34-kDa precursor molecule (pro-IL-1β) that is processed by activated caspase-1 into the shorter, mature form. Pro-IL-1β was induced at similar levels in WT and Asc−/− mice. However, lack of ASC protein resulted, as expected, in severely reduced pro-IL-1β processing into mature IL-1β in Asc−/− mice (Figure 3, middle panel).
Figure 3.
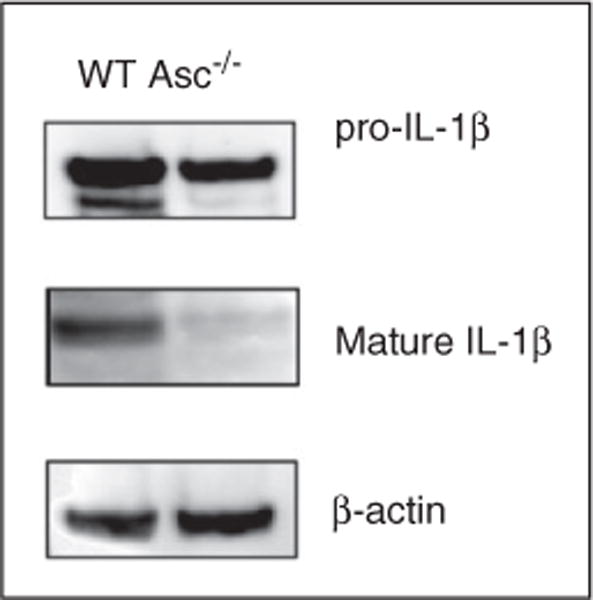
Impaired IL-1β processing in Asc−/− mice. Immunoblotting of mouse MEM tissue 12 h after NTHi infection confirmed the NTHi-dependent induction of pro-IL-1β expression at the protein level, as well as its processing into its shorter, mature, active form. Pro-IL-1β was induced at similar levels in WT and Asc−/− mice. However, lack of ASC protein resulted in severely reduced pro-IL-1β processing into mature IL-1β in Asc−/− mice.
ME bacterial clearance is delayed in Asc−/− mice during OM
To determine if ASC plays a role in bacterial clearance from the ME, the titers of bacteria recovered from the MEs of Asc−/− and WT mice were scored and compared semi-quantitatively (Table 1). In WT mice, the maximum number of animals with infected MEs was observed at d 2 after NTHi inoculation where the number of culture-positive animals increased from 4/6 on d 1 to 6/6 on d 2. By d 5, no viable NTHi was recovered from WT MEs, indicating that none of the animals remained infected (0/6). In contrast, the Asc−/− mice experienced prolonged bacterial persistence in the ME, with some mice being infected even at d 10 after NTHi inoculation (2/6). In Asc−/− mice, bacterial CS peaked later than in the WTs, at d 3. These results indicate that bacterial clearance is significantly compromised in Asc-deficient mice when compared with WT animals.
Asc−/− mice show prolonged ME inflammatory response to NTHi
OM in Asc−/− mice differed significantly from that observed in WT animals. Figure 4 shows the ME hyperplasia and histology of WT and Asc−/− -deficient mice during the course of OM, while Figure 5 presents a quantitative analysis of leukocyte infiltration into the ME lumen, respectively. In WT mice, the inflammatory response to NTHi is characterized by mucosal thickening and robust leukocytic infiltration of the ME cavity. As we have observed previously,41,45 MEM hyperplasia is noted within 24 h of NTHi exposure and reaches a maximum at 2–3 d, after which the hyperplastic response transitions to recovery and healing, with a return to normal thickness observed by d 10. This classic pattern of MEM proliferation was altered in the Asc knockout mice. As indicated in Figure 4A, the mucosal thickness of Asc−/− mice was somewhat greater than that of their WT counterparts prior to inoculation with NTHi. However, this difference was not statistically significant. Moreover, upon being exposed to NTHi, Asc−/− mice displayed significantly greater mucosal proliferation between days 1 to 3 than that seen in WT animals (Figure 4B). Mucosal thickness then decreased by d 5. The histological appearance of the mucosa are shown in Figure 4B for the selected d. In contrast, leukocyte infiltration of the ME lumen of the Asc−/− -deficient mice was delayed when compared with WT mice (Figure 5A).
Figure 4.
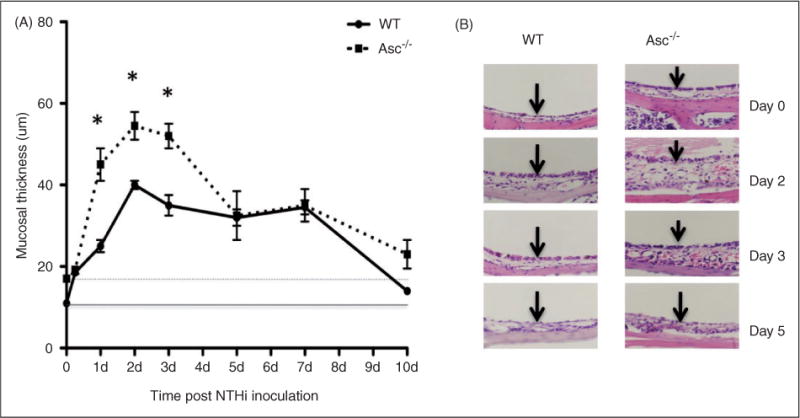
Lack of the Asc gene results in enhanced mucosal hyperplasia. (A) Quantitative comparison of mucosal hyperplasia and (B) histology (hematoxylin and eosin) staining of WTand Asc-deficient mice during the course of OM. In Asc−/− mice mucosal thickness was significantly greater in the first few days after NTHi infection, until full recovery was reached by d 10. *Significantly different than WT mice (P < 0.05), n = 6 ears.
Figure 5.
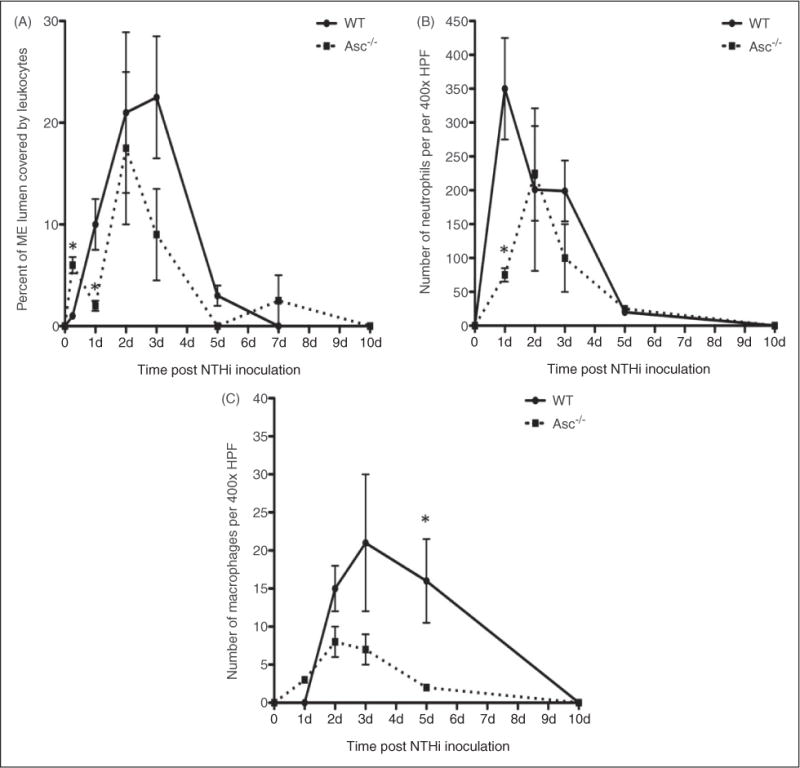
Lack of the Asc gene alters leukocyte recruitment to the ME. (A) Area of the ME lumen occupied by inflammatory cells was used to quantify leukocyte infiltration of the ME cavity (n = 6 ears). The accumulation of ME leukocytes was delayed in Asc−/− mice. The ME infiltrate was examined at 400 × to identify the neutrophils and macrophages. The recruitment of neutrophils (B) was also delayed, and the number of macrophages (C) was significantly decreased in Asc−/− mice. HPF: high-power field. *Significantly different from WT mice (P < 0.05), n = 6 ears.
Figure 5 (B, C) illustrates the leukocyte types present in the ME lumen. In both, WT and Asc−/− mice, neutrophils were the dominant leukocyte type observed in the ME lumen during the initial response to infection. However, in the Asc−/− ME significantly fewer neutrophils were present at 24 h than in WT mice, indicating delayed infiltration. Generally, macrophages enter the ME at a later stage than neutrophils, with their numbers peaking at d 2 and 3 post-infection in the WT, although comprising a significantly smaller number of cells compared with neutrophils. While initially similar in numbers to WT, fewer macrophages infiltrated the MEs of Asc−/− mice at d 1–5; at this latter time point nearly all macrophages had been cleared from the ME in Asc−/− mice, while WT mice continued to exhibit a robust macrophage presence. However, both leukocyte types were completely cleared by d 10 in both WT and Asc−/− animals.
ASC and IL-1β play a role in bacterial phagocytosis during OM
Macrophages function primarily in the recognition and elimination of pathogens. Therefore, we evaluated the NTHi phagocytic and intracellular killing capacity of peritoneal macrophages derived from Asc−/− mice in vitro (Figure 6) to explore whether inflammasomerelated deficiencies in macrophage function might also play a role in the observed OM phenotype. Macrophages were incubated for 1 h with NTHi, followed by gentamicin treatment to kill all extracellular bacteria. The number of pathogens recovered after macrophage lysis was used as a measure of phagocytosis. To evaluate intracellular killing competence, macrophages were treated identically, but were incubated for 3 h in gentamicin. The number of viable NTHi present intracellularly was then compared with that seen after 1 h of gentamicin treatment, and used as a measure of NTHi killing within the cells. Compared with macrophages obtained from WT mice, macrophages from Asc−/− animals showed decreased phagocytic function (Figure 6), demonstrated by a decrease in intracellular NTHi recovery observed after 1 h incubation with bacteria followed by 1 h gentamicin treatment to kill extracellular organisms (WT mean of 5.1 × 105 CFU compared with Asc−/− mean of 3.3 × 105 CFU, P<0.05). After an additional 2 h within WT macrophages, the number of viable NTHi bacteria decreased dramatically, consistent with ~ 70% killing of the intracellular pathogen burden. While the number of bacteria within Asc-deficient macrophages was initially lower, as noted above, intracellular killing was also ~ 70%, suggesting an unimpaired ability to eliminate bacteria once internalized.
Figure 6.
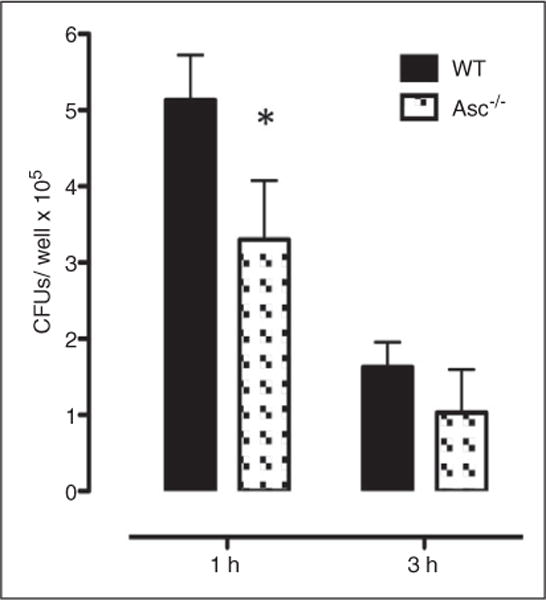
Assessment of macrophages phagocytosis and bacterial clearance. Phagocytosis and killing of NTHi bacteria was assessed in peritoneal macrophages from WT and Asc mice by quantifying the colony counts remaining after loading with NTHi and then 1 h (phagocytosis) or 3 h (killing) incubation with gentamicin to destroy all extracellular bacteria. Bars represent bacteria recovered after lysis of the macrophages. Asc−/− macrophages exhibited less phagocytic uptake of NTHi (1 h), but once inside the cell a similar proportion of bacteria were killed (3 h). Experiments were performed in triplicate and expressed as mean ± SEM. *Significantly different than WT mice (P < 0.05).
A potential role for IL-17 signaling in regulating MEM hyperplasia
In this study, we observed significantly greater mucosal hyperplasia in the Asc−/− mice infected with NTHi on d 1–3. This suggested, paradoxically, that inflammasome activity not only contributes to inflammation in the ME, but also plays a significant role in moderating epithelial hyperplasia. At d 1 and 2 after NTHi infection, we did not see higher levels of bacterial persistence in Asc−/− mice (Table 1). Thus, the increased tissue response did not appear to be related to increased bacterial titers at those times. Further, the preponderance of evidence suggests that the ILs, processed by the inflammasome, themselves are unlikely to inhibit tissue responses. However, there is recent evidence that activated caspase-1 can play a regulatory role in tissue proliferative responses to infection, mediated by Th17 T-helper cells. For example, reduced Th17 responses have been shown to increase reactions to Helicobactor pylori in stomach epithelium.46,47 To determine whether Th17 responses might play a role in OM, we evaluated the mRNA expression of genes encoding IL-17, IL-17 receptor (IL-17r) and IL-22, which are produced by Th17 cells during OM. We also looked at IL-10 mRNA levels over the 7 d period. There was significant regulation of each of these genes; specifically, IL-22 mRNA increased more than 100-fold early in OM (Figure 7) in WT mice. As ASC is important for the autocatalytic activation of caspase-1,8,48 in Asc knockout mice the processing of procaspase-1 is inhibited by the lack of the Asc gene. We have observed that the subsequent processing of Pro-IL-1β by activated caspase-1 was blocked (Figure 3). Therefore, lack of caspase-1 activation in Asc−/− mice could be hypothesized to account for the increased hyperplasia phenotype.
Discussion
OM induced by NTHi inoculation into the mouse ME is characteristically associated with thickening of the mucosa and with ME lumen cellular infiltration peaking by d 2–3, followed, generally, by resolution of the infection by d 5.35 In this study, we observed early up-regulation of inflammasome-related genes, which was consistent with a role for their gene products in OM pathogenesis and recovery. When we compared ME responses to NTHi between Asc−/− and WT mice, we noted prolonged inflammation and delayed bacterial clearance from the ME cavity of the former, in addition to reduced leukocyte recruitment into the ME and impaired macrophage phagocytosis.
Thus, our results suggest that the processing of IL-1β, and possibly IL-18, is important for normal pathogenesis and timely resolution of OM. This is consistent with previous reports showing that IL-1β, along with other cytokines, is present in human ME effusions. Moreover, IL-1β expression has been linked to increases in the number of ciliated cells50 and elevation of mucin gene expression, which would be expected to aid in bacterial clearance. Moreover, exogenous IL-1β expression has been observed to induce MEM inflammation and hyperplasia, in addition to inducing gene expression of several mucin genes.52,53 Thus, it is not surprising that inflammasome defects would alter OM. However, our results identify those components of the ME response to bacteria that are dependent upon the processing of inflammasome targets. Those components regulate both the recruitment of leukocytes to the ME lumen and the phagocytic function of macrophages.
Asc−/− mice did not process IL-1β, and presumably IL-18, both of which play critical roles in chemotaxis. IL-1β has been shown to both increase phagocytosis and decrease apoptosis of leukocytes by up-regulating surface receptors required for bacterial engulfment and reducing reactive oxygen generation.54 IL-18 is itself chemotactic.55 Thus, it is not surprising that reduced processing of these two important cytokines would lead to a reduction in the recruitment of leukocytes to inflamed ME tissue, and reduced phagocytic capability. Others have reported similar observations of impaired IL-1β secretion in Asc−/− -deficient mice using other models of inflammatory disorders.7,16,17,21 A link between IL-1β polymorphisms and acute OM in humans is already established.56 Nonetheless, whether IL-1β-targeted therapy provides potential new therapies for OM remains to be further explored.17
While NLRP-3 and caspase-1-dependent activation are important in inflammasome activation as noted, it has been shown previously that IL-1β cleavage can also occur through caspase-1-independent mechanisms.57,58 Whether these secondary pathways played a parallel role in our studies remains unexplored. The observation of only very small amounts of mature IL-1β in the infected MEs of Asc−/− mice suggests that while such ASC-independent mechanisms may occur at this site, they are much less efficient than ASC-mediated activation. Moreover, the fact that Asc−/− knockout mice exhibited prolonged inflammation and significantly decreased neutrophil infiltration indicates that inflammasome activation of cytokine targets plays a major, if not dominant, role.
Inflammasome-independent pathways involving other cytokines and inflammatory mediators are also activated by NTHi. We, and others, have highlighted the importance of TLR receptors and MyD88 signaling in OM.27,30,32,35,59,60 These pathways provide mechanisms for induction of downstream molecules, such as IL-6 and TNF-α, amongst others, that do not require processing by the inflammasome. However, our observations in bacterial clearance delays and reduction in neutrophil recruitment phenotype, indicate that these mediators alone are insufficient to permit normal timely resolution of OM in the absence of inflammasome activation followed by subsequent processing of target cytokines.
Supplementary Material
Acknowledgments
We acknowledge John Bertin, Ethan Grant, Anthony Coyle and Millenium Pharmaceuticals for providing the Asc−/− knockout mice. We also thank Matthew McGeough and Carla Pena for technical assistance.
Funding
This research was supported by grants DC006279, DC012595 and DC000129 from the NIH/NIDCD, and by the Research Service of the VA.
References
- 1.Beutler BA. TLRs and innate immunity. Blood. 2009;113:1399–1407. doi: 10.1182/blood-2008-07-019307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baxt LA, Garza-Mayers AC, Goldberg MB. Bacterial sub-version of host innate immune pathways. Science. 2013;340:697–701. doi: 10.1126/science.1235771. [DOI] [PubMed] [Google Scholar]
- 3.Gill N, Wlodarska M, Finlay BB. The future of mucosal immunology: Studying an integrated system-wide organ. Nat Immunol. 2010;11:558–560. doi: 10.1038/ni0710-558. [DOI] [PubMed] [Google Scholar]
- 4.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 5.Muruve DA, Petrilli V, Zaiss AK, et al. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature. 2008;452:103–107. doi: 10.1038/nature06664. [DOI] [PubMed] [Google Scholar]
- 6.Franchi L, Nunez G. Immunology. Orchestrating inflammasomes. Science. 2012;337:1299–1300. doi: 10.1126/science.1229010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zaki MH, Lamkanfi M, Kanneganti TD. The nlrp3 inflammasome: Contributions to intestinal homeostasis. Trends Immunol. 2011;32:171–179. doi: 10.1016/j.it.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broz P, Monack DM. Molecular mechanisms of inflammasome activation during microbial infections. Immunol Rev. 2011;243:174–190. doi: 10.1111/j.1600-065X.2011.01041.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinon F, Burns K, Tschopp J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proil-beta. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 10.Martinon F, Mayor A, Tschopp J. The inflammasomes: Guardians of the body. Annu Rev Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 11.Dagenais M, Skeldon A, Saleh M. The inflammasome: In memory of Dr. Jurg Tschopp. Cell Death Differ. 2012;19:5–12. doi: 10.1038/cdd.2011.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mariathasan S, Monack DM. Inflammasome adaptors and sensors: Intracellular regulators of infection and inflammation. Nat Rev Immunol. 2007;7:31–40. doi: 10.1038/nri1997. [DOI] [PubMed] [Google Scholar]
- 13.Rathinam VA, Vanaja SK, Fitzgerald KA. Regulation of inflammasome signaling. Nat Immunol. 2012;13:333–332. doi: 10.1038/ni.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241–247. doi: 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dinarello CA. A clinical perspective of il-1beta as the gatekeeper of inflammation. Eur J Immunol. 2011;41:1203–1217. doi: 10.1002/eji.201141550. [DOI] [PubMed] [Google Scholar]
- 16.Nagarajan UM, Sikes JD, Yeruva L, Prantner D. Significant role of il-1 signaling, but limited role of inflammasome activation, in oviduct pathology during chlamydia muridarum genital infection. J Immunol. 2012;188:2866–2875. doi: 10.4049/jimmunol.1103461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoffman HM, Wanderer AA. Inflammasome and il-1beta-mediated disorders. Curr Allergy Asthma Rep. 2010;10:229–235. doi: 10.1007/s11882-010-0109-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 19.Catanzaro A, Ryan A, Batcher S, Wasserman SI. The response to human RIL-1, RIL-2, and RTNF in the middle ear of guinea pigs. Laryngoscope. 1991;101:271–275. doi: 10.1288/00005537-199103000-00008. [DOI] [PubMed] [Google Scholar]
- 20.Gabay C, Lamacchia C, Palmer G. Il-1 pathways in inflammation and human diseases. Nat Rev Rheumatol. 2010;6:232–241. doi: 10.1038/nrrheum.2010.4. [DOI] [PubMed] [Google Scholar]
- 21.Cai S, Batra S, Wakamatsu N, et al. NLRC4 inflammasome-mediated production of il-1beta modulates mucosal immunity in the lung against Gram-negative bacterial infection. J Immunol. 2012;188:5623–5635. doi: 10.4049/jimmunol.1200195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lamkanfi M, Dixit VM. Modulation of inflammasome pathways by bacterial and viral pathogens. J Immunol. 2011;187:597–602. doi: 10.4049/jimmunol.1100229. [DOI] [PubMed] [Google Scholar]
- 23.Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–550. doi: 10.1146/annurev.immunol.021908.132612. [DOI] [PubMed] [Google Scholar]
- 24.Li J, Moran T, Swanson E, et al. Regulation of IL-8 and IL-1beta expression in crohn’s disease associated NOD2/CARD15 mutations. Hum Mol Genet. 2004;13:1715–1725. doi: 10.1093/hmg/ddh182. [DOI] [PubMed] [Google Scholar]
- 25.Leibovitz E, Jacobs MR, Dagan R. Haemophilus influenzae: A significant pathogen in acute otitis media. Pediatr Infect Dis J. 2004;23:1142–1152. [PubMed] [Google Scholar]
- 26.Fletcher MA, Fritzell B. Pneumococcal conjugate vaccines and otitis media: An appraisal of the clinical trials. Int J Otolaryngol. 2012;2012:312935. doi: 10.1155/2012/312935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leichtle A, Hernandez M, Pak K, et al. TLR4-mediated induction of tlr2 signaling is critical in the pathogenesis and resolution of otitis media. Innate Immun. 2009;15:205–215. doi: 10.1177/1753425909103170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Melhus A, Ryan AF. Expression of cytokine genes during pneumococcal and nontypeable Haemophilus influenzae acute otitis media in the rat. Infect Immun. 2000;68:4024–4031. doi: 10.1128/iai.68.7.4024-4031.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ebmeyer J, Leichtle A, Hernandez M, et al. TNFa deletion alters apoptosis as well as caspase 3 and 4 expression during otitis media. BMC Immunol. 2011;12:12. doi: 10.1186/1471-2172-12-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leichtle A, Hernandez M, Lee J, et al. The role of DNA sensing and innate immune receptor tlr9 in otitis media. Innate Immun. 2012;18:3–13. doi: 10.1177/1753425910393539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leichtle A, Lai Y, Wollenberg B, et al. Innate signaling in otitis media: Pathogenesis and recovery. Curr Allergy Asthma Rep. 2011;11:78–84. doi: 10.1007/s11882-010-0158-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leichtle A, Hernandez M, Pak K, et al. The toll-like receptor adaptor trif contributes to otitis media pathogenesis and recovery. BMC Immunol. 2009;10:45. doi: 10.1186/1471-2172-10-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dunne A, O’Neill LA. The interleukin-1 receptor/toll-like receptor super-family: Signal transduction during inflammation and host defense. Sci STKE. 2003;2003:re3. doi: 10.1126/stke.2003.171.re3. [DOI] [PubMed] [Google Scholar]
- 34.O’Neill LA. The interleukin-1 receptor/toll-like receptor superfamily: 10 years of progress. Immunol Rev. 2008;226:10–18. doi: 10.1111/j.1600-065X.2008.00701.x. [DOI] [PubMed] [Google Scholar]
- 35.Hernandez M, Leichtle A, Pak K, et al. Myeloid differentiation primary response gene 88 is required for the resolution of otitis media. J Infect Dis. 2008;198:1862–1869. doi: 10.1086/593213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Melhus A, Ryan AF. A mouse model for acute otitis media. APMIS. 2003;111:989–994. doi: 10.1034/j.1600-0463.2003.1111012.x. [DOI] [PubMed] [Google Scholar]
- 37.Alrawi AM, Chern KC, Cevallos V, et al. Biotypes and serotypes of Haemophilus influenzae ocular isolates. Br J Ophthalmol. 2002;86:276–277. doi: 10.1136/bjo.86.3.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ebmeyer J, Furukawa M, Pak K, et al. Role of mast cells in otitis media. J Allergy Clin Immunol. 2005;116:1129–1135. doi: 10.1016/j.jaci.2005.07.026. [DOI] [PubMed] [Google Scholar]
- 39.Husseman J, Palacios SD, Rivkin AZ, et al. The role of vascular endothelial growth factors and fibroblast growth factors in angiogenesis during otitis media. Audiol Neurootol. 2012;17:148–154. doi: 10.1159/000333805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hsiao A, Ideker T, Olefsky JM, Subramaniam S. Vampire microarray suite: A web-based platform for the interpretation of gene expression data. Nucleic Acids Res. 2005;33:W627–632. doi: 10.1093/nar/gki443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Leichtle A, Hernandez M, Ebmeyer J, et al. CC chemokine ligand 3 overcomes the bacteriocidal and phagocytic defect of macrophages and hastens recovery from experimental otitis media in TNF−/− mice. J Immunol. 2010;184:3087–3097. doi: 10.4049/jimmunol.0901167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davis BK, Wen H, Ting JPY. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–735. doi: 10.1146/annurev-immunol-031210-101405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheeseman MT, Tyrer HE, Williams D, et al. HIF-VEGF pathways are critical for chronic otitis media in junbo and jeff mouse mutants. PLoS Genet. 2011;7:e1002336. doi: 10.1371/journal.pgen.1002336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Allen IC, Wilson JE, Schneider M, et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-kappaB signaling. Immunity. 2012;36:742–754. doi: 10.1016/j.immuni.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ryan AF, Ebmeyer J, Furukawa M, et al. Mouse models of induced otitis media. Brain Res. 2006;1091:3–8. doi: 10.1016/j.brainres.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 46.Hitzler I, Sayi A, Kohler E. Caspase-1 has both proinflammatory and regulatory properties in helicobacter infections, which are differentially mediated by its substrates IL-1beta and IL-18. J Immunol. 2012;188:3594–3602. doi: 10.4049/jimmunol.1103212. [DOI] [PubMed] [Google Scholar]
- 47.Netea MG, Simon A, van de Veerdonk F, et al. IL-1beta processing in host defense: Beyond the inflammasomes. PLoS Pathog. 2010;6:e1000661. doi: 10.1371/journal.ppat.1000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mariathasan S. Asc, IPAF and cryopyrin/NALP3: Bona fide intracellular adapters of the caspase-1 inflammasome. Microbes Infect. 2007;9:664–671. doi: 10.1016/j.micinf.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 49.Skotnicka B, Hassmann E. Proinflammatory and immunoregulatory cytokines in the middle ear effusions. Int J Pediatr Otorhinolaryngol. 2008;72:13–17. doi: 10.1016/j.ijporl.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 50.Samuel EA, Burrows A, Kerschner JE. Cytokine regulation of mucin secretion in a human middle ear epithelial model. Cytokine. 2008;41:38–43. doi: 10.1016/j.cyto.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.MacArthur CJ, Pillers DA, Pang J, et al. Altered expression of middle and inner ear cytokines in mouse otitis media. Laryngoscope. 2011;121:365–371. doi: 10.1002/lary.21349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Watanabe T, Hirano T, Suzuki M, et al. Role of interleukin-1beta in a murine model of otitis media with effusion. Ann Otol Rhinol Laryngol. 2001;110:574–580. doi: 10.1177/000348940111000613. [DOI] [PubMed] [Google Scholar]
- 53.Kerschner JE, Khampang P, Erbe CB, et al. Mucin gene 19 (muc19) expression and response to inflammatory cytokines in middle ear epithelium. Glycoconj J. 2009;26:1275–1284. doi: 10.1007/s10719-009-9245-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Grutkoski PS, D’Amico R, Ayala A, Simms HH. Il-1beta stimulation induces paracrine regulation of pmn function and apoptosis. Shock. 1999;12:373–381. doi: 10.1097/00024382-199911000-00006. [DOI] [PubMed] [Google Scholar]
- 55.Ruth JH, Park CC, Amin MA, et al. Interleukin-18 as an in vivo mediator of monocyte recruitment in rodent models of rheumatoid arthritis. Arthritis Res Ther. 2010;12:R118. doi: 10.1186/ar3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCormick DP, Grady JJ, Diego A, et al. Acute otitis media severity: Association with cytokine gene polymorphisms and other risk factors. Int J Pediatr Otorhinolaryngol. 2011;75:708–12. doi: 10.1016/j.ijporl.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mayer-Barber KD, Barber DL, Shenderov K, et al. Caspase-1 independent IL-1beta production is critical for host resistance to Mycobacterium tuberculosis and does not require tlr signaling in vivo. J Immunol. 2010;184:3326–3330. doi: 10.4049/jimmunol.0904189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kono H, Orlowski GM, Patel Z, Rock KL. The IL-1-dependent sterile inflammatory response has a substantial caspase-1-independent component that requires cathepsin C. J Immunol. 2012;189:3734–3740. doi: 10.4049/jimmunol.1200136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee HY, Takeshita T, Shimada J, et al. Induction of beta defensin 2 by nthi requires tlr2 mediated myd88 and irak-traf6-p38mapk signaling pathway in human middle ear epithelial cells. BMC Infect Dis. 2008;8:87. doi: 10.1186/1471-2334-8-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hirano T, Kodama S, Fujita K, et al. Role of toll-like receptor 4 in innate immune responses in a mouse model of acute otitis media. FEMS Immunol Med Microbiol. 2007;49:75–83. doi: 10.1111/j.1574-695X.2006.00186.x. [DOI] [PubMed] [Google Scholar]
- 61.Kerschner JE, Horsey E, Ahmed A, et al. Gene expression differences in infected and noninfected middle ear complementary DNA libraries. Arch Otolaryngol Head Neck Surg. 2009;135:33–39. doi: 10.1001/archoto.2008.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.