

Abstract
Mechanisms underlying airway hyperresponsiveness are not yet fully elucidated. One of the manifestations of airway inflammation is leakage of diverse plasma proteins into the airway lumen. They include fibrinogen and thrombin. Thrombin cleaves fibrinogen to form fibrin, a major component of thrombi. Fibrin inactivates surfactant. Surfactant on the airway surface maintains airway patency by lowering surface tension. In this study, immunohistochemically detected fibrin was seen along the luminal surface of distal airways in a patient who died of status asthmaticus and in mice with induced allergic airway inflammation. In addition, we observed altered airway fibrinolytic system protein balance consistent with promotion of fibrin deposition in mice with allergic airway inflammation. The airways of mice were exposed to aerosolized fibrinogen, thrombin, or to fibrinogen followed by thrombin. Only fibrinogen followed by thrombin resulted in airway hyperresponsiveness compared with controls. An aerosolized fibrinolytic agent, tissue-type plasminogen activator, significantly diminished airway hyperresponsiveness in mice with allergic airway inflammation. These results are consistent with the hypothesis that leakage of fibrinogen and thrombin and their accumulation on the airway surface can contribute to the pathogenesis of airway hyperresponsiveness.
Introduction
Multiple cytokines and signaling pathways of inflammation have been implicated in the pathogenesis of asthma (1–3). The etiology of airway hyperresponsiveness, a cardinal feature of asthma, has not been fully elucidated. Much research in this area focuses on processes that can lead to exaggerated airway narrowing such as alterations in smooth muscle structure or function (4–6) and effects of airway remodeling (7–11). Studies of human subjects with asthma indicate that airway hyperresponsiveness is attributable not only to exaggerated airway narrowing but also to exaggerated airway closure (12–16).
Distal airways remain open in part because of the surface tension–lowering properties of surfactant, a bipolar phospholipid found on the distal airway luminal surface (17). Airway inflammation, also a cardinal feature of asthma, causes leakage of numerous, diverse plasma proteins onto the airway surface (18). Studies in vitro have demonstrated that plasma proteins can inactivate surfactant (19). Reduced surfactant function is seen in patients with asthma (20, 21). Conversely, inhalation of surfactant diminishes airway hyperresponsiveness (22).
The most powerful protein inactivator of surfactant, to the best of our knowledge, is fibrin (23). Fibrin is formed when thrombin, generated by the coagulation cascade factors and cofactors, cleaves fibrinopeptides A and B from fibrinogen. Fibrin is a major structural component of thrombi. Fibrin is formed typically at sites of vascular damage. Extravascular thrombin (24), fibrinogen (25), and fibrin (26) have been found in the sputum of patients with asthma. We hypothesized that airway hyperresponsiveness seen in asthma is largely the result of decreased stability of airways and subsequent airway closure secondary to the formation of fibrin on the distal airway surface. In our observational work that led to this hypothesis we found robust amounts of immunohistochemically detectable, extravascular fibrin on distal airway surfaces in a patient who died of status asthmaticus. To directly test our hypothesis we then studied mice with induced allergic airway inflammation.
Methods
Immunohistochemistry.
Sections from autopsy specimens from a patient who died with status asthmaticus were characterized initially. Subsequently, we characterized sections from lungs of mice with induced allergic airway inflammation (see below). The sections of mouse lungs were obtained after fixation of the lungs at an inflation pressure of 30 cm H2O using 10% buffered formalin (Sigma-Aldrich, St. Louis, Missouri, USA) paraffin embedding. All lung sections were 10 ∝m thick. The lung sections were deparaffinized and then immunostained using the alkaline phosphatase system (Vector Laboratories, Burlingame, California, USA) and mouse IgG Ab against the β chain of fibrin that recognizes both murine and human fibrin (Accurate Chemical & Scientific Corp., Westbury, New York, USA). The secondary Ab that was used (goat anti-mouse Alexa Fluor 647; Molecular Probes Inc., Eugene, Oregon, USA) results in a fluorescent red reaction product. SYTOX green (Molecular Probes Inc.) was used as a counterstain. Fluorescent images were obtained with the use of a confocal laser scanning microscope (BioRad MRC1024ES; Bio-Rad Laboratories Inc., Hercules, California, USA). Serial sections were stained with isotype-matched Ab’s and H&E. Serial sections were stained also in the absence of primary Ab and showed no red reaction product.
Plasminogen activator and plasminogen activator inhibitor activity.
Activities of plasminogen activator (PA) and plasminogen activator inhibitor (PAI) of lungs homogenized in PBS, pH 7.4, were determined by using a chromogenic substrate assay (CoAtest; Chromogenix, Milan, Italy). PA activity was expressed as international units. PAI activity was normalized with respect to inhibition of a standard amount of tissue plasminogen activator (tPA) of known and verified specific activity. The assays were linear from 0 to 40 AU (arbitrary units per milliliter).
Mice studies.
All protocols and procedures had been approved by the University of Vermont’s Institutional Animal Care and Use Committee. BALB/c (Jackson Laboratories, Bar Harbor, Maine, USA) mice were anesthetized with 90 mg/kg of pentobarbital given intraperitoneally. A tracheostomy was performed, and a modified 18-gauge IV adaptor was inserted and firmly secured. Mice were then placed on a computer-controlled, piston ventilator (flexiVent; SCIREQ Inc., Montreal, Quebec, Canada). They were ventilated at a tidal volume of 0.2 ml with a positive end expiratory pressure of 3 cm H2O applied by a water trap.
Nebulization of fibrinogen, thrombin, and tPA.
Fibrinogen, fraction I from bovine plasma (Sigma-Aldrich), bovine thrombin (Sigma-Aldrich), and recombinant human tPA (Genentech Inc., South San Francisco, California, USA) were dissolved in PBS to yield concentrations of 10 mg/ml, 16 U/ml, and 500 U/ml, respectively. Each solution was delivered as an aerosol to the mouse airway by channeling the inspiratory flow from the ventilator through an aerosol chamber attached to an ultrasonic nebulizer. The driving volume of the ventilator was changed from the typical 0.2 ml to 0.8 ml, and the respiratory rate was decreased to 30 breaths/min. Aerosol was delivered for 40 seconds in each case.
Determination of the concentration of D-dimer.
Bronchoalveolar lavage (BAL) fluid was assayed for the concentration of cross-linked fibrin breakdown products (D-dimer) by ELISA using a commercially available kit (Diagnostica-Stago Inc., Parsippany, New Jersey, USA) that detects murine D-dimer (27). The assay was linear throughout the range of concentration of D-dimer from 5 to100 ng/ml.
Determination of the concentration of fibrin.
The concentration of fibrin in lavage fluid was determined using a modified established method (28). To prevent the formation of fibrin during the incubation step described below, a buffer composed of 0.05 M Tris, 0.15 M NaCL, and 500 IU/ml heparin at a pH 7.6 was used as the BAL solution. BAL obtained immediately following nebulization of fibrinogen followed by thrombin in naive mice was compared with BAL obtained from mice with allergic airway inflammation. The concentration of D-dimer was determined in each sample (preincubation sample). An aliquot from the same sample was then incubated at 37°C for 4 hours after the addition of a volume of 0.5 AU/ml plasmin (Sigma-Aldrich) to the same buffer was added to achieve plasmin concentration of 0.32 AU/ml. After this digestion with plasmin, the concentration of D-dimer was determined (postincubation sample). The concentration of D-dimer was then corrected for the dilution. The relative amount of intact fibrin present in the BAL was then determined by subtracting the preincubation sample concentration of D-dimer from the postincubation sample concentration of D-dimer.
Allergic airway inflammation protocol.
Allergic airway inflammation was induced by sensitization on days 1 and 14 by injection of 100 ∝l of a 200-∝g/ml mixture of chicken OVA (Sigma-Aldrich) in an adjuvant (Imject Alum; Pierce Chemical Co., Rockford, Illinois, USA) into the peritoneum. On the day 21, 22, and 23 of each study the mice were exposed to aerosolized 1% OVA mixed with sterile PBS for 30 minutes. Aerosols were delivered by a jet nebulizer while the mice were in a compartmentalized Plexiglas chamber. Airway inflammation was confirmed by increased cell counts in BAL fluid that was obtained by instilling 1 ml of cold PBS into the trachea and removing it gently.
Respiratory mechanics.
Respiratory mechanics were determined using the forced oscillation technique as previously described (29). The computer-controlled piston ventilator was programmed to deliver a 2-second pseudorandom perturbation that consisted of waveforms of mutually prime frequencies. Multiple linear regression was used to fit impedance spectra derived from measured pressure and volume in each individual mouse to the constant phase model of the lung (30): Z(f) = Rn + JωI + (Gti + jHti)/ωa. Model fits that resulted in a coefficient of determination that was less than 0.85 were excluded. From this analysis the following physiological properties of the respiratory system were derived: Newtonian resistance (Rn), tissue resistance (Gti), and elastance (Hti).
Determination of airway hyperresponsiveness.
Dose-response curves with aerosolized methacholine were determined as follows. Methacholine (Sigma-Aldrich) was mixed with sterile PBS and delivered by a nebulizer in successively increasing concentrations (3.125, 12.5, and 50 mg/ml). After each dose, the time course of response was followed by applying a 2-second perturbation as described above every 10 seconds for a total of 3 minutes. The peak response for each variable was determined. The percentage of change from baseline measured at the beginning of the protocol was calculated. Linear interpolation was then used to determine the provocative concentration of methacholine at which a 200% (PC200) increase in the variables Rn and Gti, or a 50% increase in the variable Hti (PC50) occurred.
In the mice that were exposed to tPA a single dose of methacholine was used in order to allow time for the enzymatic effect of tPA to occur. In these mice the percentage of change from baseline of the end-point variable was determined for each individual mouse.
Quantification of subepithelial collagen.
Lung sections, 10-∝m thick, were stained with Sirius red. This agent selectively stains collagen (31), yielding birefringence under polarized light. Slide identities were then masked. Each of three independent observers simultaneously scored slides from naive BALB/c mice, mice that had allergic airway inflammation and had been given only saline while on the ventilator, or mice with allergic airway inflammation that had been given tPA while on the ventilator. The scoring scale used was from 1 to 3. All three scores were averaged to create a composite score for each individual slide.
Pressure volume characteristics.
Pressure volume (PV) curves were determined as previously described (32). Increments of volume of 0.1 ml were used with a pressure limit of 25 cm H2O. Plateau values of airway pressure at each stepwise increment were obtained and plotted against volume. Hysteresis was determined as the area subtended by the inspiratory and expiratory limbs of the PV curve.
Administration of anticoagulant.
A group of mice was treated with pegylated hirudin (PEG-hirudin) during the induction of allergic airway inflammation. PEG-hirudin (1 mg/kg) was administered subcutaneously once daily (33) beginning 6 hours before the start of the aerosolized OVA challenges and continued throughout the protocol. Bleeding times were determined the day before the determination of airway hyperresponsiveness. Aside from slight bruising at the sites of injection, no adverse complications attributable to the administration of PEG-hirudin were apparent.
Statistics.
Data were expressed as means plus or minus SE. Comparisons between groups were made using of two-tailed Student’s t tests. P values less than 0.05 indicate significance.
Results
As part of our initial observational work, immunostaining of lung sections from a patient who had died from status asthmaticus revealed red reaction product indicative of fibrin along the airway surface of the alveoli and distal airways (Figure 1C). In mice with allergic airway inflammation intense red reaction product was seen also along the airway surface of the alveoli and distal airways (Figure 1D). Lung sections from mice without allergic airway inflammation did not exhibit appreciable immunoreactivity (data not shown).
Figure 1.

Fibrin immunostaining. H&E staining of a human lung from a patient who died due to status asthmaticus (A) (magnification ×10) and a lung from a mouse with allergic airway inflammation (B) (magnification ×20) are shown as a structural reference for the immunostaining. Immunostaining with a mouse IgG that selectively binds to the β chain of both mouse and human fibrin is shown (C and D). Red reaction product indicating the presence of fibrin (white arrowheads) is seen within the parenchyma and airways in both the human and mouse lung sections. Immunostaining in which a nonspecific mouse IgG was used is shown as an isotype control (E and F).
PAI activity in the homogenates of lung tissue from mice with allergic airway inflammation was significantly increased compared with that in untreated mice (P < 0.01) (Figure 2). The increase was accompanied by a marked decrease in PA activity (P < 0.01).
Figure 2.

PAI activity in lung tissue homogenates of mice with airway inflammation. PAI activity with respect to protein content was significantly increased in mice with allergic airway inflammation (n = 9) compared with controls (n = 9): 2.34 ± 0.3 AU/μg compared with 0.12 ± 0.1 AU/μg, P < 0.01. A concurrent decrease in PA activity was observed in mice with allergic airway inflammation compared with controls: 9.28 ± 0.4 AU/μg compared with 4.6 ± 0.4 AU/μg, P < 0.01.
BAL from naive mice exposed to fibrinogen followed by thrombin had higher preplasmin digestion concentrations of D-dimer compared with mice with allergic airway inflammation. Naive mice exposed to fibrinogen followed by thrombin had postplasmin digestion concentrations of D-dimer that were similar to those found in mice with allergic airway inflammation (Figure 3). The difference between the predigestion and postdigestion BAL was greatest in the OVA mice.
Figure 3.

Quantification of fibrin. Relative amounts of intact fibrin were determined by subjecting the lavage samples to fibrin digestion with plasmin. The graph depicts predigestion (Before) and postdigestion (After) concentrations of D-dimer in lavage fluid. Mice that been exposed to nebulized fibrinogen followed by thrombin (Fibrin) (n = 8) are compared with mice that had allergic airway inflammation (OVA) (n = 10). Predigestion D-dimer concentrations are the highest in the fibrin mice (9.7 ± 2.1; 5.61 ± 1.0). Postdigestion concentration of D-dimer in BAL from OVA mice (12.6 ± 0.8) was similar to postdigestion D-dimer concentration in fibrin mice (13.1 ± 2.0). OVA mice had a larger difference between postdigestion and predigestion BAL D-dimer concentration (7.0 ± 1.4) compared with BAL from fibrin mice (3.39 ± 2.7). This indicates that greater amounts of intact fibrin is present on the airway surface of OVA mice.
Naive mice exposed to aerosolized fibrinogen plus thrombin exhibited airway hyperresponsiveness in terms of Rn, Gti, and Hti (Figure 4) that was significantly different (P < 0.01) compared with values in controls exposed to fibrinogen, thrombin, or saline alone (Figures 4 and 5). Mice exposed to fibrinogen alone exhibited airway hyperresponsiveness in terms of Hti that was less than that seen with fibrinogen plus thrombin but greater than that seen with saline or thrombin (Figure 5). This response was not significantly different from the response in the controls.
Figure 4.

Effect of fibrinogen followed by thrombin on airway hyperresponsiveness. Mice that received fibrinogen followed by thrombin (n = 7) had significantly increased airway hyperresponsiveness in terms of all parameters compared with mice that received saline alone (n = 7): Rn (saline alone, 30.5 ± 5.5, versus fibrinogen followed by thrombin, 9.8 ± 2.9; P < 0.01), Gti (saline alone, 18.5 ± 3.8, versus fibrinogen followed by thrombin, 8.5 ± 1.3; P < 0.01), and Hti (saline alone, 32.7 ± 12, versus fibrinogen followed by thrombin, 5.5 ± 1.7; P < 0.01). The degree of airway hyperresponsiveness seen in fibrinogen followed by thrombin mice was similar to that seen in mice with allergic airway inflammation (OVA) (n = 10): Rn (fibrinogen followed by thrombin, 9.8 ± 2.9, versus OVA, 5.95 ± 0.82), Gti (fibrinogen followed by thrombin, 8.5 ± 1.3, versus OVA, 6.57 ± 0.64), Hti (fibrinogen followed by thrombin, 5.5 ± 1.7, versus OVA, 1.83 ± 0.37).
Figure 5.

Effect of fibrinogen or thrombin alone on airway hyperresponsiveness. Mice given nebulized fibrinogen followed by thrombin (n = 7) had significantly increased airway hyperresponsiveness to methacholine in all parameters compared with mice that received fibrinogen alone (n = 6) or thrombin alone (n = 6): Rn (fibrinogen followed by thrombin, 9.8 ± 2.9; fibrinogen alone, 23.8 ± 4.8, P < 0.03; thrombin alone, 23.2 ± 5.0, P < 0.04), Gti (fibrinogen followed by thrombin, 8.5 ± 1.3; fibrinogen alone, 12.5 ± 1.0, P < 0.04; thrombin alone, 12.6 ± 1.3, P < 0.05), Hti (fibrinogen followed by thrombin, 5.5 ± 1.7; fibrinogen alone, 10.4 ± 1.0, P < 0.04; thrombin alone, 26 ± 8.2, P < 0.03).
Mice with allergic airway inflammation that were exposed to the aerosolized fibrinolytic agent (tPA) exhibited a significant decrease in the response to methacholine with respect to all three physiologic variables (Rn: P < 0.03; Gti: P < 0.01; Hti: P < 0.05) compared with results in mice with allergic airway inflammation exposed to saline alone (Figure 6). Mice given tPA had a near doubling of concentrations of D-dimer (Figure 7) in the BAL fluid, indicating that fibrinolysis had occurred. Subepithelial collagen deposition was significantly increased in mice with allergic airway inflammation (P < 0.01), but the increase was not modified by the acute nebulization of tPA (Figure 8). Furthermore, mice with allergic airway inflammation exposed to aerosolized tPA exhibited a significant decrease in PV curve hysteresis (Figure 9) compared with that evident in curves from those exposed to aerosolized saline alone. The change in PV curve hysteresis following aerosolized tPA was the result of a leftward shift of the upper portion of the inspiratory limb of the PV curve (Figure 10).
Figure 6.

The effect of nebulized fibrinolytic agent, tPA, on the response to methacholine in mice with allergic airway inflammation. The response to a single dose of 12.5 mg/ml of nebulized methacholine administered 15 minutes after administration of saline (black bars) or tPA (white bars) is shown on the ordinate as the percentage of increase from baseline. An increased value indicates an increased response to methacholine. Mice with allergic airway inflammation (n = 7) that had been exposed to tPA had a significantly reduced response to methacholine compared with those exposed to saline alone (n = 7): Rn, 99.2% ± 16% compared with 267% ± 62% (P < 0.03); Gti, 102% ± 23% compared with 225% ± 30% (P < 0.01); and Hti, 71% ± 30% compared with 284% ± 92% (P < 0.05). In terms of Rn and Gti, the response to methacholine in mice with airway inflammation that had been exposed to nebulized tPA (n = 7) was similar to the response in mice without airway inflammation exposed to saline (n = 10): Rn, 99.2% ± 16% compared with 119% ± 14%; Gti, 102% ± 23% compared with 137% ± 21%. Nebulized tPA did not completely reduce the response in terms of Hti to the level seen in controls: 71.2% ± 30% compared with 25.6% ± 2.7%.
Figure 7.

Concentrations of fibrin degradation products (D-dimer) in the BAL fluid of mice that had been exposed to aerosolized tPA. The concentration of D-dimer increased significantly in the BAL from mice with airway inflammation exposed to tPA (n = 9) compared with those exposed to saline (n = 8, 17.8 ± 2.7 ng/ml compared with 9.05 ± 0.6 ng/ml).
Figure 8.
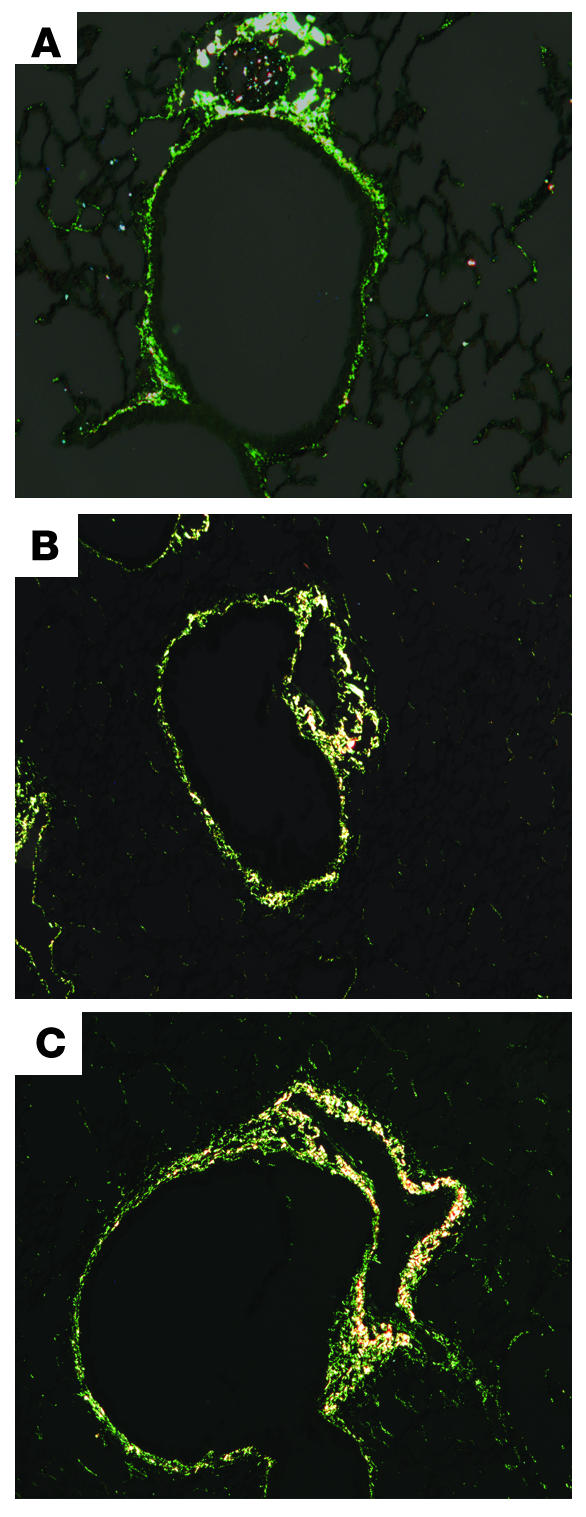
Subepithelial fibrosis in mice with allergic airway inflammation. Staining with Sirius red of lung sections from naive BALB/c mice (A) and those with allergic airway inflammation (B) is shown. Sirius red is a dye that stains collagen specifically. Scoring by masked observers demonstrated a significant increase in subepithelial collagen deposition in mice with allergic airway inflammation (score: 1.14 ± 0.1 naive, n = 7; 1.96 ± 0.26 inflamed, n = 8; P < 0.01). Inflamed mice that received nebulized tPA (C) showed no reduction in subepithelial collagen deposition compared with inflamed mice that received saline only (score: 1.96 ± 0.2, n = 8, inflamed + tPA; 1.96 ± 0.26, n = 8, inflamed + saline).
Figure 9.

The effect of the fibrinolytic agent, tPA, on PV curve hysteresis in mice with inflamed airways. The percentage of increase in the area subtended by the inspiratory and expiratory limb of a PV curve obtained 40 minutes after nebulization of saline or tPA was determined for each individual mouse, and results from all mice in each group were then averaged. Mice exposed to tPA (n = 6) had significantly smaller PV curve areas compared with those in mice exposed to saline alone (n = 9): 66.3% ± 15% compared with 130% ± 25%.
Figure 10.

PV curves from mice with allergic airway inflammation that had been exposed to saline or tPA. PV curves that were generated with a stepwise volume insufflation of an in vivo mouse preparation are shown here with pressure on the abscissa and volume on the ordinate. Mice exposed to tPA exhibited curves characterized by lower pressures at each volume increment. The inspiratory curve is shifted to the left in the mice exposed to tPA, demonstrating an increase in lung compliance consistent with enhanced function of surfactant.
Administration of PEG-hirudin during the induction of allergic airway inflammation (n = 8) significantly reduced airway hyperresponsiveness compared with mice with allergic airway inflammation that did not receive PEG-hirudin (n = 8) in terms of Rn PC200 (inflamed, 5.95 ± 0.82; inflamed plus PEG-hirudin, 12.2 ± 2.7; P < 0.05) and Hti PC50 (inflamed, 1.83 ± 0.37; inflamed plus PEG hirudin, 3.77 ± 0.5; P < 0.01). There was no significant reduction in terms of Gti. Bleeding times were elevated in the mice that received PEG-hirudin. There was no difference in BAL cell counts between mice that received PEG-hirudin and mice that did not (data not shown).
Discussion
Results from our study implicate coagulation system and fibrinolytic system proteins in the pathogenesis of airway hyperresponsiveness in asthma. In our initial observational work lung tissue was evaluated from a patient who had succumbed with status asthmaticus. Subsequently, lung tissue from mice with induced allergic airway inflammation was characterized. In both, robust amounts of fibrin were evident along the surfaces of distal airways (Figure 1). In the lungs of mice with allergic airway inflammation PAI activity was found to be increased (Figure 2), thereby potentially promoting the accumulation of fibrin by suppressing fibrinolysis. Delivery of nebulized coagulation system proteins, fibrinogen followed by thrombin, to naive mice was found to result in airway hyperreponsiveness (Figures 4 and 5). Delivery of nebulized fibrinolytic agent to the airway surface of mice with allergic airway inflammation was found to reduce airway hyperresponsiveness and reduce PV curve hysteresis (Figures 6 and 9). Anticoagulating mice with allergic airway inflammation with a thrombin inhibitor resulted in reduced airway hyperresponsiveness.
The airway surface of normal lung exhibits active fibrinolysis (34). We found that the airway surface of mice with allergic airway inflammation exhibited conditions that potentiate the accumulation of fibrin. Plasmin, a fibrinolytic protein, is formed when plasminogen activators cleave the inert precursor zymogen, plasminogen. Plasminogen activator activity is constrained by plasminogen activator inhibitors. The physiologically prominent PAI in blood and tissues is type 1 (PAI-1). Activity of plasmin is constrained by another naturally occurring inhibitor, α-2 antiplasmin. Mast cells from individuals with asthma produce PAI-1 (35), and a genetic linkage between PAI-1 polymorphisms and asthma has been suggested (36). The concentration of PAI-1 protein (37) is increased in the lavage fluid of mice with chronic allergic airway inflammation. In the present study we found increases in PAI activity in mice with acute allergic airway inflammation (Figure 2). In addition, we observed an apparent decrease in fibrinolytic activity on the airway surfaces in these mice reflected by the presence of fibrin on the surfaces of the distal airways and alveoli (Figure 1). The constrained fibrinolysis is likely to be the result of increased PAI-1 activity. TGF-β is a powerful agonist of PAI-1 synthesis (38). Its concentration is increased in association with allergic airway inflammation (39). It seems reasonable that TGF-β and other cytokines elaborated in association with inflammation convert the airway surfaces from a state that favors breakdown of fibrin to one that favors accumulation of fibrin.
The observed presence of fibrin on surfaces of distal airways and alveoli in patients with asthma or mice with allergic airway inflammation, to the best of our knowledge, is a novel observation. D-dimer, a breakdown product of fibrin, has been found in the sputum of patients with asthma (26). This finding, however, does not point to any specific locus of accumulation of fibrin because induced sputum includes material from the entire airway tree. As noted above in the setting of inflammation proteins within the circulation leak into the airway lumen (18). Previous work in brown Norway rats indicates that protein leakage in the setting of allergic airway inflammation is found only in the distal airways (40). The authors who identified this feature suggest that it indicates that distal airways are particularly susceptible to leakage induced by inflammation. Our results suggest that the same was the case in the patient with asthma that we studied and in mice with allergic airway inflammation.
We hypothesized that the increased concentration of fibrin on the surface of distal airways would result in increased surface tension secondary to inactivation of surfactant, consequently leading to enhanced airway instability and subsequent airway closure following the administration of methacholine. We have recently shown that a response in the variable Hti following methacholine represents increased airway closure (41). Accordingly, we expected the exaggerated Hti response following the fibrinogen/thrombin treatment that was indeed observed (Figure 4). Airway closure is not a feature confined to this mouse model of asthma. It has been demonstrated in human subjects with asthma in multiple studies (12–16).
Inactivation of surfactant does not explain the observed exaggerated response in Rn. Because surfactant is found in minimal concentrations in central airways, its role in the narrowing or closure of central airways is controversial. Alternatively, thrombin or fibrinogen could individually have effects on airway smooth muscle. Thrombin is known to increase airway smooth muscle contraction ex vivo (42), and fibrin degradation products increase pulmonary vascular smooth muscle contraction (43). We did not, however, see an exaggerated response when airways were exposed to thrombin. If fibrin degradation products affect airway smooth muscle, delivery of tPA to mice with induced allergic airway inflammation would have resulted in an increased response to methacholine, not the decrease that we observed (Figure 6). Thrombin and fibrin breakdown products may not penetrate to the extent that they can interact in a substantial way with airway smooth muscle. The observed exaggerated Rn response is perhaps best explained by aerosol deposition dynamics. When airways constrict or close, more of a given aerosol deposits in the lung, and the distribution of aerosol deposition is changed such that more aerosol deposits in the central airways (44). Constriction or closure of the distal airways after exposure to methacholine aerosol, as we observed, will therefore have the effect of enhancing methacholine aerosol deposition in central airways, resulting in an exaggerated central airways, or Rn, response.
Fibrinogen can inactivate surfactant, but much less so as compared with fibrin (45). Accordingly, we were not surprised by the modestly increased Hti response following aerosolized fibrinogen alone (Figure 5). In fact, the response in Hti following nebulization of fibrinogen alone was not significantly different from that seen with saline or thrombin alone.
Our data indicate that concentrations of fibrin in BAL fluid from mice exposed to fibrinogen plus thrombin were similar or less than those observed in mice with induced allergic airway inflammation (Figure 3). In essence, we simulated the effects of leakage of fibrinogen and thrombin onto to the airway surface. This experimental design enabled us to assess the potential importance of the presence of these two proteins and their end product, fibrin. Other coagulation system and plasma proteins present on the airway surface in the setting of inflammation were, of course, not simulated. Nevertheless, the accumulation of fibrin that we observed in the lung from the asthmatic patient and from mice with allergic airway inflammation (Figure 1) indicates that generation of extravascular fibrin occurs under such conditions.
Mice with allergic airway inflammation exhibit a pattern of airway hyperresponsiveness that is similar to the pattern seen following nebulization of fibrinogen plus thrombin (Figure 4). To determine whether accumulated fibrin on the airway surface contributed to airway hyperresponsiveness in mice with allergic airway inflammation, we reduced the extent of accumulation of fibrin on the airway surface using nebulized tPA (Figure 6). Airway hyperresponsiveness decreased significantly with respect to the three physiological variables characterized, Rn, Gti, and Hti (Figure 6). The reduction does not appear to be a reflection of an acute effect of tPA on the amount of subepithelial fibrosis (Figure 8). It is, however, conceivable that long-term administration of aerosolized tPA could diminish subepithelial fibrosis by activating or generating matrix metalloproteinases, including collagenase, from zymogens (46).
PV curve hysteresis decreased in mice with allergic airway inflammation after administration of nebulized tPA (Figure 9). PV curve hysteresis results from the opening of closed lung units and elaboration of surfactant when the lung is stretched (47). Airway closure is manifested by a rightward shift in the early portion of the inspiratory PV curve (48) such that a lower inflection point is observed. We did not see a lower inflection point in the PV curves from mice with allergic airway inflammation (Figure 10). We did, however, observe a rightward shift of the upper portion of the inspiratory PV curve that decreased when aerosolized tPA was administered (Figure 10). This is consistent with a decrease in distal airway surface tension (49) that is likely to reflect enhanced function of surfactant. Plasmin does reverse inactivation of surfactant caused by fibrin in vitro (23). We interpret the decreased Hti response to methacholine following the administration of nebulized tPA in mice with allergic airway inflammation as being a reflection of decreased airway closure, in turn attributable to less fibrin being available to inactivate surfactant.
We have demonstrated that anticoagulation results in a decrease in airway hyperresponsiveness. Heparin is known to have anti-inflammatory properties (50). Accordingly, we chose to use a direct thrombin inhibitor, hirudin. We used a pegylated form of hirudin to facilitate once-a-day dosing. We demonstrated a significant reduction in airway hyperresponsiveness reflected by Hti and Rn in mice with allergic airway inflammation that were treated with PEG-hirudin. We did not suppress airway hyperresponsiveness to an extent comparable to that seen in mice without allergic airway inflammation. This could be a result of incomplete anticoagulation. More likely it represents, at least in part, an effect of extravascular fibrinogen. Inhibiting thrombin would have no effect on fibrinogen, and, as we demonstrated, aerosolized fibrinogen did have some effect on airway hyperresponsiveness, especially in terms of Hti (Figure 5). Our results demonstrate that alteration of the activity of the coagulation system can have an impact upon airway hyperresponsiveness. This suggests that plasma exudation plays at least a partial role in the development of airway hyperresponsiveness.
Our results are consistent with those from previous work that have implicated cytokines in the pathogenesis of asthma. Expression of the cytokine IL-13 mRNA is increased in the airways of patients with asthma (51–53), and IL-13 is known to increase the elaboration of VEGF (54). Increased concentrations of VEGF have been found in the sputum and airway mucosa of individuals with asthma (55, 56). VEGF is known to increase vascular permeability (57) and is therefore likely to at least contribute to the plasma exudation and the subsequent accumulation of coagulation proteins on the airway surface.
Attempts to block cytokine-signaling pathways therapeutically have met with limited success, probably in part because of redundancy of such pathways. A therapeutic strategy that targets a common end point of multiple pathways is likely to be more successful. Inflammation is a common end point of multiple cytokine pathways. Accordingly, systemic steroid therapy is one of the most effective treatments for asthma. Unfortunately, systemic steroid therapy is limited by undesirable side effects. We have focused on a common consequence of inflammation — exudation of plasma proteins. More specifically, we have focused on a consequence of plasma exudation — the formation of extravascular fibrin. A paradigm of therapeutically targeting the exudation of plasma proteins has been suggested previously (58), but has not been accepted widely. Our data raise the intriguing possibility that extravascular pulmonary fibrin or the conditions that promote its formation could be a promising therapeutic target.
Acknowledgments
The authors thank Omer Iqbal of Loyola University for providing the PEG-hirudin that was used. This work was funded by National Center for Research Resources, Centers of Biomedical Research Excellence grant P20RR15557.
Footnotes
See the related Commentary beginning on page 20.
Nonstandard abbreviations used: arbitrary unit(s) (AU); bronchoalveolar lavage (BAL); elastance (Hti); impedance (Z); Newtonian resistance (Rn); pegylated hirudin (PEG-hirudin); plasminogen activator (PA); plasminogen activator inhibitor (PAI); plasminogen activator inhibitor type-1 (PAI-1); pressure volume (PV); provocative concentration of methacholine resulting in a 200% increase from baseline (PC200); provocative concentration of methacholine resulting in a 50% increase from baseline (PC50); tissue resistance (Gti); tissue-type plasminogen activator (tPA).Conflict of interest: The authors have declared that no conflict of interest exists.
References
- 1.McLane MP, et al. Interleukin-9 promotes allergen-induced eosinophilic inflammation and airway hyperresponsiveness in transgenic mice. Am. J. Respir. Cell Mol. Biol. 1998;19:713–720. doi: 10.1165/ajrcmb.19.5.3457. [DOI] [PubMed] [Google Scholar]
- 2.Foster PS, Hogan SP, Ramsay AJ, Matthaei KI, Young IG. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J. Exp. Med. 1996;183:195–201. doi: 10.1084/jem.183.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wills-Karp M, et al. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–2261. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 4.Roberts JA, Raeburn D, Rodger IW, Thomson NC. Comparison of in vivo airway responsiveness and in vitro smooth muscle sensitivity to methacholine in man. Thorax. 1984;3:837–843. doi: 10.1136/thx.39.11.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O’Byrne PM, Dolovich M, Dirks R, Roberts RS, Newhouse MT. Lung epithelial permeability: relation to nonspecific airway responsiveness. J. Appl. Physiol. 1984;57:77–84. doi: 10.1152/jappl.1984.57.1.77. [DOI] [PubMed] [Google Scholar]
- 6.Vincenc KS, et al. Comparison of in vivo and in vitro responses to histamine in human airways. Am. Rev. Respir. Dis. 1983;128:875–879. doi: 10.1164/arrd.1983.128.5.875. [DOI] [PubMed] [Google Scholar]
- 7.Macklem PT. A theoretical analysis of the effect of airway smooth muscle load on airway narrowing. Am. J. Respir. Crit. Care Med. 1996;153:83–89. doi: 10.1164/ajrccm.153.1.8542167. [DOI] [PubMed] [Google Scholar]
- 8.Moreno RH, Hogg JC, Pare PD. Mechanics of airway narrowing. Am. Rev. Respir. Dis. 1986;133:1171–1180. doi: 10.1164/arrd.1986.133.6.1171. [DOI] [PubMed] [Google Scholar]
- 9.James AL, Pare PD, Hogg JC. The mechanics of airway narrowing in asthma. Am. Rev. Respir. Dis. 1989;139:242–246. doi: 10.1164/ajrccm/139.1.242. [DOI] [PubMed] [Google Scholar]
- 10.Irvin CG, Pak J, Martin RJ. Airway-parenchyma uncoupling in nocturnal asthma. Am. J. Respir. Crit. Care Med. 2000;161:50–56. doi: 10.1164/ajrccm.161.1.9804053. [DOI] [PubMed] [Google Scholar]
- 11.Wiggs BR, Bosken C, Pare PD, James A, Hogg JC. A model of airway narrowing in asthma and in chronic obstructive pulmonary disease. Am. Rev. Respir. Dis. 1992;145:1251–1258. doi: 10.1164/ajrccm/145.6.1251. [DOI] [PubMed] [Google Scholar]
- 12.Kraft M, Pak J, Martin RJ, Kaminsky D, Irvin CG. Distal lung dysfunction at night in nocturnal asthma. Am. J. Respir. Crit. Care Med. 2001;163:1551–1556. doi: 10.1164/ajrccm.163.7.2008013. [DOI] [PubMed] [Google Scholar]
- 13.Kaminsky DA, Bates JH, Irvin CG. Effects of cool, dry air stimulation on peripheral lung mechanics in asthma. Am. J. Respir. Crit. Care Med. 2000;162:179–186. doi: 10.1164/ajrccm.162.1.9806079. [DOI] [PubMed] [Google Scholar]
- 14.Samee S, et al. Imaging the lungs in asthmatic patients by using hyperpolarized helium-3 magnetic resonance: assessment of response to methacholine and exercise challenge. J. Allergy. Clin. Immunol. 2003;111:1205–1211. doi: 10.1067/mai.2003.1544. [DOI] [PubMed] [Google Scholar]
- 15.Woolcock AJ, Read J. Lung volumes in exacerbations of asthma. Am. J. Med. 1966;41:259–273. doi: 10.1016/0002-9343(66)90021-0. [DOI] [PubMed] [Google Scholar]
- 16.Wagner EM, Bleecker ER, Permutt S, Liu MC. Direct assessment of small airways reactivity in human subjects. Am. J. Respir. Crit. Care Med. 1998;157:447–452. doi: 10.1164/ajrccm.157.2.9611043. [DOI] [PubMed] [Google Scholar]
- 17.Hohlfeld JM. The role of surfactant in asthma. Respir. Res. 2002;3:4. doi: 10.1186/rr176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yager D, Kamm RD, Drazen JM. Airway wall liquid. Sources and role as an amplifier of bronchoconstriction. Chest. 1995;107(Suppl.):105S–110S. doi: 10.1378/chest.107.3_supplement.105s. [DOI] [PubMed] [Google Scholar]
- 19.Enhorning G, Holm BA. Disruption of pulmonary surfactant’s ability to maintain openness of a narrow tube. J. Appl. Physiol. 1993;74:2922–2927. doi: 10.1152/jappl.1993.74.6.2922. [DOI] [PubMed] [Google Scholar]
- 20.Kurashima K, Fujimura M, Matsuda T, Kobayashi T. Surface activity of sputum from acute asthmatic patients. Am. J. Respir. Crit. Care Med. 1997;155:1254–1259. doi: 10.1164/ajrccm.155.4.9105063. [DOI] [PubMed] [Google Scholar]
- 21.Jarjour NN, Enhorning G. Antigen-induced airway inflammation in atopic subjects generates dysfunction of pulmonary surfactant. Am. J. Respir. Crit. Care Med. 1999;160:336–341. doi: 10.1164/ajrccm.160.1.9806155. [DOI] [PubMed] [Google Scholar]
- 22.Babu KS, et al. Inhaled synthetic surfactant abolishes the early allergen-induced response in asthma. Eur. Respir. J. 2003;21:1046–1049. doi: 10.1183/09031936.03.00069202. [DOI] [PubMed] [Google Scholar]
- 23.Seeger W, Stohr G, Wolf HR, Neuhof H. Alteration of surfactant function due to protein leakage: special interaction with fibrin monomer. J. Appl. Physiol. 1985;58:326–338. doi: 10.1152/jappl.1985.58.2.326. [DOI] [PubMed] [Google Scholar]
- 24.Gabazza EC, et al. Thrombin in the airways of asthmatic patients. Lung. 1999;177:253–262. doi: 10.1007/pl00007645. [DOI] [PubMed] [Google Scholar]
- 25.Pizzichini E, et al. Indices of airway inflammation in induced sputum: reproducibility and validity of cell and fluid-phase measurements. Am. J. Respir. Crit. Care Med. 1996;154:308–317. doi: 10.1164/ajrccm.154.2.8756799. [DOI] [PubMed] [Google Scholar]
- 26.Banach-Wawrzenczyk E, Dziedziczko A, Rosc D. [Fibrinolytic system in bronchial asthma after prednisone treatment] Pol. Merkuriusz Lek. 2000;7:9–11. [PubMed] [Google Scholar]
- 27.Schorer AE. Discordant effects on eicosanoids and fibrin degradation products in two murine models of antiphospholipid antibody. Thromb. Res. 1997;85:295–304. doi: 10.1016/s0049-3848(97)00015-7. [DOI] [PubMed] [Google Scholar]
- 28.Hattori N, et al. Bleomycin-induced pulmonary fibrosis in fibrinogen-null mice. J. Clin. Invest. 2000;106:1341–1350. doi: 10.1172/JCI10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tomioka S, Bates JH, Irvin CG. Airway and tissue mechanics in a murine model of asthma: alveolar capsule vs. forced oscillations. J. Appl. Physiol. 2002;93:263–270. doi: 10.1152/japplphysiol.01129.2001. [DOI] [PubMed] [Google Scholar]
- 30.Hantos Z, Adamicza A, Govaerts E, Daroczy B. Mechanical impedances of lungs and chest wall in the cat. J. Appl. Physiol. 1992;73:427–433. doi: 10.1152/jappl.1992.73.2.427. [DOI] [PubMed] [Google Scholar]
- 31.Junqueira LC, Bignolas G, Brentani RR. Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem. J. 1979;11:447–455. doi: 10.1007/BF01002772. [DOI] [PubMed] [Google Scholar]
- 32.Wagers S, Lundblad L, Moriya HT, Bates JH, Irvin CG. Nonlinearity of respiratory mechanics during bronchoconstriction in mice with airway inflammation. J. Appl. Physiol. 2002;92:1802–1807. doi: 10.1152/japplphysiol.00883.2001. [DOI] [PubMed] [Google Scholar]
- 33.Marty I, et al. Amelioration of collagen-induced arthritis by thrombin inhibition. J. Clin. Invest. 2001;107:631–640. doi: 10.1172/JCI11064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bowen RM, Hoidal JR, Estensen RD. Urokinase-type plasminogen activator in alveolar macrophages and bronchoalveolar lavage fluid from normal and smoke-exposed hamsters and humans. J. Lab. Clin. Med. 1985;106:667–673. [PubMed] [Google Scholar]
- 35.Cho SH, Tam SW, Demissie-Sanders S, Filler SA, Oh CK. Production of plasminogen activator inhibitor-1 by human mast cells and its possible role in asthma. J. Immunol. 2000;165:3154–3161. doi: 10.4049/jimmunol.165.6.3154. [DOI] [PubMed] [Google Scholar]
- 36.Cho SH, et al. Possible role of the 4G/5G polymorphism of the plasminogen activator inhibitor 1 gene in the development of asthma. J. Allergy Clin. Immunol. 2001;108:212–214. doi: 10.1067/mai.2001.117260. [DOI] [PubMed] [Google Scholar]
- 37.Oh CK, Ariue B, Alban RF, Shaw B, Cho SH. PAI-1 promotes extracellular matrix deposition in the airways of a murine asthma model. Biochem. Biophys. Res. Commun. 2002;294:1155–1160. doi: 10.1016/S0006-291X(02)00577-6. [DOI] [PubMed] [Google Scholar]
- 38.Gerwin BI, Keski-Oja J, Seddon M, Lechner JF, Harris CC. TGF-beta 1 modulation of urokinase and PAI-1 expression in human bronchial epithelial cells. Am. J. Physiol. 1990;259:L262–L269. doi: 10.1152/ajplung.1990.259.4.L262. [DOI] [PubMed] [Google Scholar]
- 39.Tanaka H, et al. The effect of allergen-induced airway inflammation on airway remodeling in a murine model of allergic asthma. Inflamm. Res. 2001;50:616–624. doi: 10.1007/PL00000243. [DOI] [PubMed] [Google Scholar]
- 40.Schmiedl A, et al. Increase of inactive intra-alveolar surfactant subtypes in lungs of asthmatic brown Norway rats. Virchows Arch. 2003;442:56–65. doi: 10.1007/s00428-002-0720-z. [DOI] [PubMed] [Google Scholar]
- 41.Wagers S, Lundblad LK, Ekman M, Irvin CG, Bates JH. The allergic mouse model of asthma: normal smooth muscle in an abnormal lung? J. Appl. Physiol. 2004;96:2019–2027. doi: 10.1152/japplphysiol.00924.2003. [DOI] [PubMed] [Google Scholar]
- 42.Panettieri RA, Jr, Hall IP, Maki CS, Murray RK. alpha-Thrombin increases cytosolic calcium and induces human airway smooth muscle cell proliferation. Am. J. Respir. Cell Mol. Biol. 1995;13:205–216. doi: 10.1165/ajrcmb.13.2.7626288. [DOI] [PubMed] [Google Scholar]
- 43.Kern DF, Saldeen K, Saldeen T, Malik AB. Pulmonary vascular effects of a fibrin(ogen)-derived vasoactive peptide. Thromb. Res. 1986;42:783–788. doi: 10.1016/0049-3848(86)90114-3. [DOI] [PubMed] [Google Scholar]
- 44.Kim CS, Eldridge MA, Garcia L, Wanner A. Aerosol deposition in the lung with asymmetric airways obstruction: in vivo observation. J. Appl. Physiol. 1989;67:2579–2585. doi: 10.1152/jappl.1989.67.6.2579. [DOI] [PubMed] [Google Scholar]
- 45.Seeger W, Elssner A, Gunther A, Kramer HJ, Kalinowski HO. Lung surfactant phospholipids associate with polymerizing fibrin: loss of surface activity. Am. J. Respir. Cell Mol. Biol. 1993;9:213–220. doi: 10.1165/ajrcmb/9.2.213. [DOI] [PubMed] [Google Scholar]
- 46.Nagase H, Suzuki K, Enghild JJ, Salvesen G. Stepwise activation mechanisms of the precursors of matrix metalloproteinases 1 (tissue collagenase) and 3 (stromelysin) Biomed. Biochim. Acta. 1991;50:749–754. [PubMed] [Google Scholar]
- 47.Kaminsky DA, et al. Hyperpnea-induced changes in parenchymal lung mechanics in normal subjects and in asthmatics. Am. J. Respir. Crit. Care Med. 1997;155:1260–1266. doi: 10.1164/ajrccm.155.4.9105064. [DOI] [PubMed] [Google Scholar]
- 48.Salmon RB, Primiano FP, Jr, Saidel GM, Niewoehner DE. Human lung pressure-volume relationships: alveolar collapse and airway closure. J. Appl. Physiol. 1981;51:353–362. doi: 10.1152/jappl.1981.51.2.353. [DOI] [PubMed] [Google Scholar]
- 49.Stamenovic D, Smith JC. Surface forces in lungs. III. Alveolar surface tension and elastic properties of lung parenchyma. J. Appl. Physiol. 1986;60:1358–1362. doi: 10.1152/jappl.1986.60.4.1358. [DOI] [PubMed] [Google Scholar]
- 50.Ahmed T, Gonzalez BJ, Danta I. Prevention of exercise-induced bronchoconstriction by inhaled low-molecular-weight heparin. Am. J. Respir. Crit. Care Med. 1999;160:576–581. doi: 10.1164/ajrccm.160.2.9812076. [DOI] [PubMed] [Google Scholar]
- 51.Huang SK, et al. IL-13 expression at the sites of allergen challenge in patients with asthma. J. Immunol. 1995;155:2688–2694. [PubMed] [Google Scholar]
- 52.Humbert M, et al. Elevated expression of messenger ribonucleic acid encoding IL-13 in the bronchial mucosa of atopic and nonatopic subjects with asthma. J. Allergy Clin. Immunol. 1997;99:657–665. doi: 10.1016/s0091-6749(97)70028-9. [DOI] [PubMed] [Google Scholar]
- 53.Kotsimbos TC, Ernst P, Hamid QA. Interleukin-13 and interleukin-4 are coexpressed in atopic asthma. Proc. Assoc. Am. Physicians. 1996;108:368–373. [PubMed] [Google Scholar]
- 54.Corne J, et al. IL-13 stimulates vascular endothelial cell growth factor and protects against hyperoxic acute lung injury. J. Clin. Invest. 2000;106:783–791. doi: 10.1172/JCI9674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Asai K, et al. Increased levels of vascular endothelial growth factor in induced sputum in asthmatic patients. Clin. Exp. Allergy. 2003;33:595–599. doi: 10.1046/j.1365-2222.2003.01576.x. [DOI] [PubMed] [Google Scholar]
- 56.Hoshino M, Takahashi M, Aoike N. Expression of vascular endothelial growth factor, basic fibroblast growth factor, and angiogenin immunoreactivity in asthmatic airways and its relationship to angiogenesis. J. Allergy Clin. Immunol. 2001;107:295–301. doi: 10.1067/mai.2001.111928. [DOI] [PubMed] [Google Scholar]
- 57.Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9–22. [PubMed] [Google Scholar]
- 58.Barnes PJ, et al. Effects of treatment on airway microvascular leakage. Eur. Respir. J. Suppl. 1990;12:663s–670s. [PubMed] [Google Scholar]