

ABSTRACT
In this study, we present results demonstrating that mechanotransduction by vascular endothelial cadherin (VE-cadherin, also known as CDH5) complexes in endothelial cells triggers local cytoskeletal remodeling, and also activates global signals that alter peripheral intercellular junctions and disrupt cell–cell contacts far from the site of force application. Prior studies have documented the impact of actomyosin contractile forces on adherens junction remodeling, but the role of VE-cadherin in force sensation and its ability to influence endothelial cell and tissue mechanics globally have not been demonstrated. Using mechanical manipulation of VE-cadherin bonds and confocal imaging, we demonstrate VE-cadherin-based mechanotransduction. We then demonstrate that it requires homophilic VE-cadherin ligation, an intact actomyosin cytoskeleton, Rho-associated protein kinase 1 (ROCK1) and phosphoinositide 3-kinase. VE-cadherin-mediated mechanotransduction triggered local actin and vinculin recruitment, as well as global signals that altered focal adhesions and disrupted peripheral intercellular junctions. Confocal imaging revealed that VE-cadherin-specific changes appear to propagate across cell junctions to disrupt distant inter-endothelial junctions. These results demonstrate the central role of VE-cadherin adhesions and the actomyosin cytoskeleton within an integrated, mechanosensitive network that both induces local cytoskeletal remodeling at the site of force application and regulates the global integrity of endothelial tissues.
KEY WORDS: VE-cadherin, Mechanotransduction, Vinculin, Magnetic twisting cytometry
INTRODUCTION
The vascular endothelium forms a semi-permeable barrier that functions in a highly dynamic mechanical environment. Endothelial cells experience a variety of dynamic and steady state mechanical forces, which can alter the integrity of adherens junctions – key components in the regulation of endothelial barrier permeability (Dejana et al., 2008; Komarova and Malik, 2010; Taddei et al., 2008). Endothelial hormones (Hong et al., 2013; Ranscht and Dours-Zimmermann, 1991; van Nieuw Amerongen et al., 2000) or rigid extracellular matrices (de Rooij et al., 2005), such as in aged arteries (Huynh et al., 2011) or at sites of atherosclerosis, trigger changes in endogenous, actomyosin contractile forces. Alternatively, exogenous forces due to fluid shear stress (Hahn and Schwartz, 2009), inspiration in the lung (Groeneveld, 2002; Birukova et al., 2010), hypertension (Chien et al., 1998; Lee et al., 1998) or changes in smooth muscle contraction can also impinge on adherens junctions to alter endothelial permeability, which is a diagnostic marker of vascular disease. In acute lung injury, aberrant mechanical stimuli can induce pathological vascular leakage (Dudek and Garcia, 2001). VE-cadherin (also known as CDH5) complexes – the primary cohesive proteins at inter-endothelial junctions – are targets of inflammatory mediators and key regulators of endothelial permeability (Dejana et al., 2008; Mehta and Malik, 2006). Adherens junctions connect cytoskeletons of adjacent cells, and are likely sites of force transduction in vascular endothelium.
In epithelia, type 1 classical E-cadherin complexes at intercellular junctions are force sensitive (le Duc et al., 2010; Yonemura et al., 2010), and exogenous forces at cadherin adhesions trigger junctional cytoskeletal remodeling (Barry et al., 2014; le Duc et al., 2010; Leckband et al., 2011; Yonemura et al., 2010). Previous nanomechanical measurements have directly demonstrated mechanotransduction by E-cadherin complexes, which involves force-triggered vinculin accumulation at F-actin-anchored E-cadherin adhesions (Barry et al., 2014; le Duc et al., 2010).
In endothelia, endogenous tugging forces on endothelial cell junctions trigger an increase in the junction size (Liu et al., 2010). Endothelial hormones similarly trigger cell contractility and inter-endothelial junction remodeling (Huveneers et al., 2012). Fluid shear stress induces endothelial cell alignment in a process involving altered inter-endothelial cell tension. However, the inter-endothelial flow sensor identified was PECAM-1, rather than VE-cadherin (Hahn and Schwartz, 2009; Tzima et al., 2005). Such findings might raise questions about the capacity of VE-cadherin complexes to regulate endothelial barriers and tissue mechanics in response to force. Although studies have documented the impact of endogenous force on adherens junction remodeling (Huveneers et al., 2012; Liu et al., 2010), the latter behavior could be due to force transduction by proteins other than VE-cadherin.
Results presented here demonstrate directly that VE-cadherin complexes are endothelial force transducers, and identify early force-activated molecular cascades in pulmonary endothelial cells. We also demonstrate that mechanotransduction by both VE-cadherin and PECAM-1 is ligand and epitope specific. The latter finding suggests that force-transduction by these adhesion receptors is allosterically regulated. Findings also reveal that localized forces on VE-cadherin receptors activate signals that alter global cell mechanics, and that those signals generate force fluctuations at peripheral junctions, which in turn trigger the remodeling of endothelial junctions away from the immediately affected cell. These results reveal that VE-cadherin complexes are central force transducers in a mechanically integrated, force-sensitive network that enables rapid, global changes in endothelial barrier integrity in response to force. These data provide new insights into mechanisms regulating endothelial integrity.
RESULTS
VE-cadherin complexes remodel in response to mechanical force
Direct mechanical stimulation of VE-cadherin bonds triggered force-dependent cytoskeletal remodeling, as it does for their type I classical cadherin counterparts. VE-cadherin-coated beads bound to VE-cadherin receptors at the cell surface were force-loaded using magnetic twisting cytometry (MTC, Fig. 1A) (Wang et al., 1993). In MTC measurements, a continuously oscillating field orthogonal to the bead magnetization axis induces a twisting torque on the beads and VE-cadherin bonds (Fig. 1A), and the amplitude of the resulting bead displacement reflects the viscous and elastic properties of the bead–cell junction. In these studies, changes in bead displacement during bead loading were due mainly to changes in the elastic moduli of the junctions.
Fig. 1.

Endothelial cells actively reinforce VE-cadherin junctions in response to locally applied external force. (A) Schematic of the MTC experiment. Ferromagnetic beads (4.5–4.9 µm diameter) were coated with VE-cadherin-Fc and allowed to adhere on the apical surface of endothelial cell monolayers expressing VE-cadherin. Beads were magnetized with a magnetic moment (M) parallel to the substrate, and a 0.3 Hz oscillating magnetic field (H) was applied. The orthogonal applied field generates a twisting torque (T) on the beads, inducing a bead displacement that is inversely proportional to the stiffness of the local bead–cell junction. (B) A continuously oscillating field of 20 Gauss, corresponding to 2.4 Pa of shear stress, was applied for 2 min to VE-cadherin-Fc-coated beads bound to endothelial cells (n>1100 bead–cell pairs for non-treated cells, and n>500 bead–cell pairs for cytochalsin D-treated cells, from more than six independent experiments). (C) Stiffening response of endothelial cell monolayers that were pretreated with inhibitors as indicated in the Materials and Methods (n>400 bead–cell pairs per condition except for blebbistatin, n = 172, and LY294002, n = 279, from more than four independent experiments). *P<0.01, **P<0.001. Results are mean±s.e.m. of percentage change.
To determine whether endothelial cells reinforce VE-cadherin junctions in proportion to the applied shear – a signature of force sensation – the magnetic field strength (H) was increased stepwise (10 s intervals from 0.3–75 Gauss, equivalent to a shear stress of 0.036–9 Pa), with no pause between successive increases in the field, and hence in the applied load. The increase in the elastic modulus of VE-cadherin junctions with increasing applied shear stress (supplementary material Fig. S1A) demonstrates the force sensitivity of VE-cadherin complexes. This increase in stiffness with applied shear was reversible: in experimental controls for potential hysteresis, the modulus of VE-cadherin junctions decreased with decreasing applied shear stress (supplementary material Fig. S1B). VE-cadherin based mechanotransduction required organized actin. Cytochalasin D, an F-actin disruptor, diminished the force-dependent stiffening by 58% at 1.2 Pa applied shear, and up to 75% at 9 Pa applied shear (P<0.001, n>300 bead–cell pairs) (supplementary material Fig. S1A,B).
VE-cadherin mechanotransduction depends on actin, ROCK1 and PI3K
In order to compare VE-cadherin-mediated force transduction with that mediated by type I classical cadherins (Barry et al., 2014; le Duc et al., 2010; Tabdili et al., 2012b), and to identify molecular cascades triggered by VE-cadherin bond shear, a continuously oscillating field (20 Gauss, equivalent to 2.4 Pa shear stress) was applied for 2 min, as described previously (Barry et al., 2014; le Duc et al., 2010). The bead displacement amplitudes decreased with forcing time, indicative of force-activated VE-cadherin-mediated junction stiffening (Fig. 1B). In these acute measurements, the stiffness increased 22% within 2 min, relative to unperturbed adhesions (Fig. 1B,C). In agreement with prior reports, actin disruption with cytochalasin D decreased the stiffening response to only 4% in the 2 min interval (Fig. 1B,C). This was significantly lower than stiffening by non-treated cells (P<0.001).
Inter-endothelial complexes comprise VE-cadherin, PECAM-1 and VEGFR2. PECAM-1-mediated adaptive stiffening requires Rho-associated protein kinase (ROCK1) and phosphoinositide 3-kinase (PI3K) (Collins et al., 2012; Conway et al., 2013). To determine whether VE-cadherin-based force transduction involves a similar signaling pathway, we tested the impact of PI3K and ROCK1 inhibitors. The inhibitor LY294002 targets the PI3K 110α subunit, which has been implicated in tumor necrosis factor α (TNFα)-dependent perturbations of endothelial junctions (Cain et al., 2010). Cell treatment with LY294002 decreased the stiffening response to −14%, relative to untreated cells, and the ROCK1 inhibitor Y-27632 reduced the stiffening response to −7% (Fig. 1C). In cells treated with blebbistatin, the overall stiffening response was 13% (Fig. 1C; P = 0.33 relative to untreated cells). Blebbistatin similarly does not completely ablate E-cadherin-mediated stiffening (le Duc et al., 2010). Microtubule disruption is known to increase contractility (Bershadsky et al., 1996). In cells treated with the microtubule disruptor nocodazole, cell stiffness increased to 35% (Fig. 1C, P = 0.26 relative to untreated cells).
VE-cadherin-based mechanotransduction depends on ligand identity
In a prior report, tugging on VE-cadherin receptors with beads coated with blocking, anti-VE-cadherin antibody failed to activate integrins (Tzima et al., 2005). That finding might suggest that VE-cadherin complexes are not endothelial force transducers. However, force transduction by type I classical cadherins requires specific ligation (le Duc et al., 2010; Tabdili et al., 2012b). Here, we assessed whether VE-cadherin-based mechanotransduction was epitope-specific, by using beads coated with either blocking or non-blocking anti-VE-cadherin antibodies, with poly-L-lysine (PLL) or with recombinant VE-cadherin ectodomains (VE-cadherin-Fc). It is not possible to compare the density of active VE-cadherin-Fc with active antibody on the beads. However, the expected similar coverage of blocking and non blocking IgG antibodies enabled us to directly compare cell responses to the different biochemical probes. A positive control used beads coated with human fibronectin, because integrin-based adhesions are well-established force transducers (Wang and Ingber, 1995).
Fig. 2 shows endothelial cell stiffening responses measured with different bead coatings. Beads coated with VE-cadherin-Fc, fibronectin or a non-blocking anti-VE-cadherin antibody triggered comparable stiffening responses in 2 min (Fig. 2A), with increases of 23%, 25% or 14%, respectively (Fig. 2C). Controls with VE-cadherin blocking antibody in solution (Fig. 2B) confirmed that the VE-cadherin-Fc beads adhere to cells through homophilic VE-cadherin bonds. However, MTC measurements with beads coated with a blocking anti-VE-cadherin antibody did not induce cell stiffening (Fig. 2A,C; P<0.05 compared to the non-blocking antibody), consistent with prior reports (le Duc et al., 2010; Tabdili et al., 2012b; Tzima et al., 2005). Control beads coated with PLL also did not trigger cell stiffening (Fig. 2A,C).
Fig. 2.
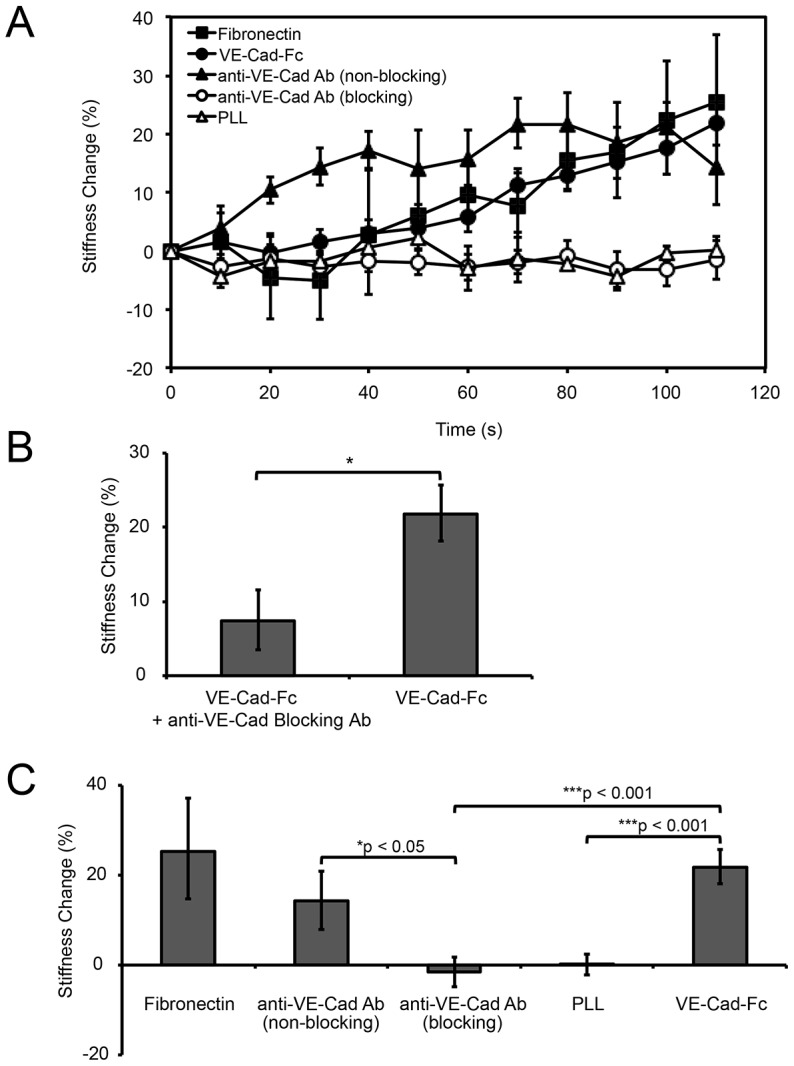
VE-cadherin mechanotransduction is ligand and epitope specific. (A) Time dependence of force-actuated junction stiffening (percentage change relative to the initial stiffness at t = 0 s). Beads bound to endothelial cells were coated with fibronectin, VE-cadherin-Fc, anti-VE-cadherin non-blocking or blocking antibodies, or PLL (n>300 bead–cell pairs per condition from four or more independent experiments). (B) Bar graph of the stiffness change (%) induced by twisting VE-cadherin-Fc coated beads on endothelial cells pre-incubated with blocking anti-VE-cadherin antibody (left) and the non-treated control (right). Displayed is the percentage change in junction stiffness after 2 min of applied shear (n>300 bead–cell pairs per condition). *P<0.001. (C) Bar graph of the percentage change in junction stiffness, relative to unperturbed cells, after 2 min of applied shear, corresponding to data in part A. *P<0.05, ***P<0.001. Results are mean±s.e.m. percentage change.
These results demonstrate that VE-cadherin complexes transduce applied force, similar to type I classical cadherins. Importantly, force transduction is both ligand and epitope specific. The latter finding suggests that force transduction through VE-cadherin receptors is allosterically regulated.
PECAM-1-activated junction remodeling is epitope specific
PECAM-1, but not VE-cadherin, has been identified as the fluid shear sensor at inter-endothelial junctions (Conway et al., 2013; Tzima et al., 2005). To establish whether PECAM-1 also exhibits force-dependent junction remodeling in MTC measurements, we used beads coated with one of two different anti-PECAM-1 antibodies: a commercially available anti-PECAM-1 antibody that binds extracellular domain 6 and the PECAM-1.3 antibody (gift from Peter J. Newman, Blood Center of Wisconsin, Milwaukee, WI) that binds extracellular domain 1 (Kim et al., 2015). Prior studies demonstrating PECAM-1 mechanotransduction used beads coated with PECAM-1.3 antibody (Collins et al., 2012; Tzima et al., 2005).
In MTC studies, the commercial anti-PECAM-1 antibody failed to induce a significant stiffening response (3% after 2 min of loading; P = 0.008, compared to VE-cadherin-Fc beads) (Fig. 3A,B). By contrast, twisting anti-PECAM-1.3-coated beads induced a 17% stiffening increase within 2 min of loading, comparable to VE-cadherin-Fc-induced stiffening (22%, P = 0.29) (Fig. 3A,B). As with VE-cadherin (Fig. 2), force transduction was epitope specific and was not activated merely by pulling on PECAM-1.
Fig. 3.

PECAM-1 mechanotransduction is epitope specific. (A) Time dependence of force-actuated cell stiffening (percentage change relative to the initial stiffness at t = 0 s). Beads were coated with anti-PECAM-1 or anti-PECAM-1.3 antibodies. Data obtained with beads coated with VE-cadherin-Fc or anti-VE-cadherin antibody (from Fig. 2A) are shown here for comparison (n>300 bead–cell pairs per condition except for anti-PECAM-1.3 antibody coated beads, where n = 188. (B) Bar graph of the percentage change in junction stiffness after 2 min of applied shear, corresponding to measurements in A. Data for beads coated with VE-cadherin-Fc or anti-VE-cadherin antibody are the same as in Fig. 2B. *P<0.05, **P<0.01, ***P<0.001. Results are mean±s.e.m. of percentage change.
VE-cadherin-specific receptor loading induces local, force-dependent cytoskeletal remodeling
In order to identify the molecular cascades triggered by VE-cadherin loading, immunofluorescence images of cells fixed before or after VE-cadherin bond shear were used to quantify changes in F-actin, vinculin and VE-cadherin localized at bead–cell junctions. Studies were performed under identical conditions as those used to obtain data in Figs 2 and 3, and the background-subtracted, population-averaged fluorescence intensities at regions of interest at bead–cell junctions (n>30; see Fig. 4A) were compared before and after force loading. Because sample conditions were otherwise identical, intensity differences after loading were attributed to force-actuated biochemical changes.
Fig. 4.

Force-actuated accumulation of VE-cadherin, vinculin and F-actin at bead endothelial cell adhesions, as a function of bead coating. (A) DIC and immunofluorescence images at bead–endothelial-cell adhesions before (top) and after (bottom) bead twisting. Beads were coated with VE-cadherin-Fc. Yellow circles (top left) indicate the ROI used to quantify the mean fluorescence intensities (MFIs) of accumulated protein around beads at the apical surface. (B) Beads were also coated with blocking anti-VE-cadherin antibody or (C) PLL. Images represent >30 bead–cell pairs per condition from more than two independent experiments. Scale bars: 5 µm. (D–F) Bar graphs of the background-subtracted MFIs in ROIs surrounding beads (see panel A), normalized to the initial VE-cadherin MFI, prior to applying bond shear. Data show the normalized MFI values before and after applying shear stress (2.4 Pa). Beads were coated with VE-cadherin-Fc (D), with blocking anti-VE-Cadherin antibody (E), and with PLL (F). n>30 bead–cell pairs per condition. Results are the mean±s.e.m.
We quantified the mean background-subtracted protein accumulation in response to bond shear, based on changes in the mean fluorescence intensity (MFI) in a region of interest (ROI) surrounding bead–cell junctions (n>30 for each condition) (Fig. 4A, yellow rings in DIC image). In unperturbed cells, F-actin, vinculin and VE-cadherin were present at VE-cadherin bead–cell junctions (Fig. 4A, top). Within 2 min of VE-cadherin loading, the fluorescence intensities and thicknesses of the F-actin and vinculin rings increased significantly (n>30, P<0.001; Fig. 4A, bottom). VE-cadherin also increased at bead–cell junctions, analogous to the thickening of the cadherin-associated β-catenin zone at cell–cell junctions, in response to endogenous tugging forces (Liu et al., 2010). The protein accumulation required homophilic VE-cadherin ligation: within experimental error, controls with blocking anti-VE-cadherin-coated beads did not induce force-dependent accumulation of VE-cadherin, F-actin or vinculin, and the levels appeared to decrease (Fig. 4B). Twisting control PLL coated beads also did not induce cytoskeletal remodeling at bead–cell junctions (Fig. 4C). Fig. 4D–F displays the mean fluorescence intensities before and after applying shear, normalized to the initial VE-cadherin MFI prior to loading. The force-dependent recruitment of VE-cadherin, F-actin and vinculin to stressed VE-cadherin junctions occurred on the same time scale as adaptive stiffening (Fig. 1B).
VE-cadherin and PECAM-1 form a flow-sensitive complex with VEGFR2. To test for adventitious mechanical coupling between PECAM-1 and VE-cadherin receptors, we investigated whether PECAM-1 loading could trigger the α-catenin-dependent recruitment of vinculin to VE-cadherin–β-catenin complexes. Beads coated with anti-PECAM-1 antibody did not induce vinculin recruitment or local cytoskeletal remodeling (Fig. 5A,C). The latter results argue against mechanical coupling between VE-cadherin and PECAM-1, although these receptors could be coupled through biochemical signals. Loading anti-PECAM-1.3-antibody-coated beads appeared to slightly increase F-actin accumulation, but neither of the anti-PECAM antibodies used activated vinculin recruitment (Fig. 5A–D; P = 0.025). Loading PECAM-1 receptors also triggered much less F-actin recruitment than VE-cadherin perturbations (compare Figs 4 and 5).
Fig. 5.

PECAM-1 receptor loading does not trigger VE-cadherin, vinculin or F-actin accumulation. DIC and immunofluorescence images of VE-cadherin, vinculin and F-actin at bead–endothelial-cell adhesions before (top) and after (bottom) bond shear. Beads were coated with (A) anti-PECAM-1 antibody or with (B) anti-PECAM-1.3 antibody. Images represent >30 bead–cell pairs per condition from more than two independent experiments. Scale bars: 5 µm. (C,D) Bar graphs of the background-subtracted mean fluorescence intensities corresponding to VE-cadherin, vinculin and F-actin, before and after bond shear (2.4 Pa). Beads were coated with (C) anti-PECAM-1 antibody and (D) with anti-PECAM-1.3 antibody. *P<0.05. Results are the mean±s.e.m.
The effects of different inhibitors on VE-cadherin-mediated stiffening (Fig. 1C) corresponded with their impact on shear-dependent junction remodeling (supplementary material Fig. S2). Treatment with the actin disruptor, cytochalasin D, or with the myosin II ATPase inhibitor, blebbistatin, abolished force-dependent protein accumulation (supplementary material Fig. S2A,B). Thus, force-activated cell stiffening and recruitment of VE-cadherin, vinculin and F-actin to junctions required organized actin and myosin II, similar to during E-cadherin mechanotransduction (Barry et al., 2014). In unperturbed cells treated with nocodazole, F-actin and vinculin accumulated around beads, and bond shear further increased their levels at bead–cell junctions (supplementary material Fig. S2C). By contrast, inhibiting ROCK1 or PI3K abolished F-actin and vinculin accumulation following receptor loading, compared to controls (supplementary material Fig. S2D,E).
Local VE-cadherin mechanotransduction triggers global signals that propagate rapidly through endothelial monolayers
Prior studies of cadherin-mediated force-transduction focused on reactive local cytoskeletal remodeling at perturbed cadherin adhesions (Barry et al., 2014; Huveneers et al., 2012; le Duc et al., 2010; Tabdili et al., 2012a; Thomas et al., 2013; Twiss et al., 2012; Yonemura et al., 2010). At cadherin adhesions, α-catenin is the postulated force transducer, and its main effector, vinculin, alters local cytoskeletal remodeling (Barry et al., 2014; Leerberg et al., 2014; Miyake et al., 2006; Yonemura et al., 2010). By contrast, PECAM-1-mediated force transduction in endothelial cells activates signals that stimulate global cell stiffening (Collins et al., 2012).
We next tested whether VE-cadherin-mediated force transduction triggers global signals, by visualizing changes at peripheral junctions triggered by apical VE-cadherin loading. In unperturbed cell monolayers laden with VE-cadherin-coated beads (∼1 bead/cell), endothelial cells displayed the typical cobblestone morphology with intact, linear adherens junctions and cortical F-actin bundles at the cell periphery (Fig. 6A, top). However, loading VE-cadherin receptors triggered the disruption of peripheral junctions, as shown by punctate VE-cadherin staining, the formation of radial actin fibers and concomitant inter-endothelial gap formation (Fig. 6A, bottom). These changes are phenotypically similar to those induced by pro-inflammatory stimuli such as thrombin (Huveneers et al., 2012; Komarova and Malik, 2010; Verin et al., 2001). Additionally, immunofluorescence images of paxillin in the basal plane revealed corresponding focal adhesion remodeling (supplementary material Fig. S3A). In endothelial monolayers, VE-cadherin force transduction correlated with changes in the focal adhesion number, but not focal adhesion size (supplementary material Fig. S3A, bottom). These force-induced subcellular changes coincided with a significant increase in inter-endothelial gaps (Fig. 6E).
Fig. 6.

VE-cadherin mechanotransduction triggers intercellular junction remodeling. (A) DIC and confocal immunofluorescence images of endothelial cell monolayers bound with VE-cadherin-Fc beads, before (top) and after (bottom) applied shear stress. Scale bars: 20 µm. The immunofluorescence images show the VE-cadherin (green), actin (red) and vinculin (purple) distributions. (B–D) Confocal immunofluorescence images of cells laden with beads, before (top) and after (bottom) applying bond shear. The images display VE-cadherin (green) and F-actin (red) organization at the basal plane. The beads were coated with (B) blocking anti-VE-cadherin antibody, (C) PLL or (D) anti-PECAM-1.3 antibody (images represent >12 images per condition from three independent experiments). Scale bars: 5 µm. (E) Bar graph of the quantified gap areas between endothelial cells, after applying shear stress for 2 min (n>8 images per condition from two independent experiments). Beads were coated with VE-cadherin-Fc or with blocking anti-VE-cadherin antibody. The data show the mean±s.e.m. for the inter-endothelial gap area. *P<0.001.
Specific VE-cadherin ligation actuated the greatest changes in peripheral junctions. Beads coated with blocking anti-VE-cadherin also triggered some junctional disruption (Fig. 6B), but specific VE-cadherin ligand triggered much greater disruption, based on quantitative analyses of total gap areas (Fig. 6E). PLL-coated beads failed to induce similar peripheral junction remodeling (Fig. 6C). Similarly, PECAM-1 loading with anti-PECAM-1.3-coated beads triggered global cell stiffening and peripheral gap formation (Fig. 3A, Fig. 6D; supplementary material Fig. S4A), but use of beads coated with the blocking anti-PECAM-1 antibody did not activate cell stiffening (Fig. 3) or intercellular junction remodeling (supplementary material Fig. S4B).
Gap formation at peripheral cell junctions could activate force fluctuations between neighboring cells that in turn propagate signals away from the immediately perturbed cells. To test this, we imaged endothelial monolayers treated with a low density of VE-cadherin-Fc-coated beads confined to a discrete section of the monolayer (DIC image, Fig. 7A). Specifically, we asked whether local VE-cadherin mechanotransduction could affect cells that did not share a boundary with the perturbed cell or its immediate neighbors. Immunofluorescence images at the bead front were then compared before and after bead twisting, and this sparse bead distribution enabled visualization of several cells that were not in direct contact with bead-laden cells or their neighbors (Fig. 7A). This approach differed from measurements depicted in Fig. 6A where densities were ∼1 bead/cell. Results were compared with measurements with blocking anti-VE-cadherin-coated beads (Fig. 7B).
Fig. 7.
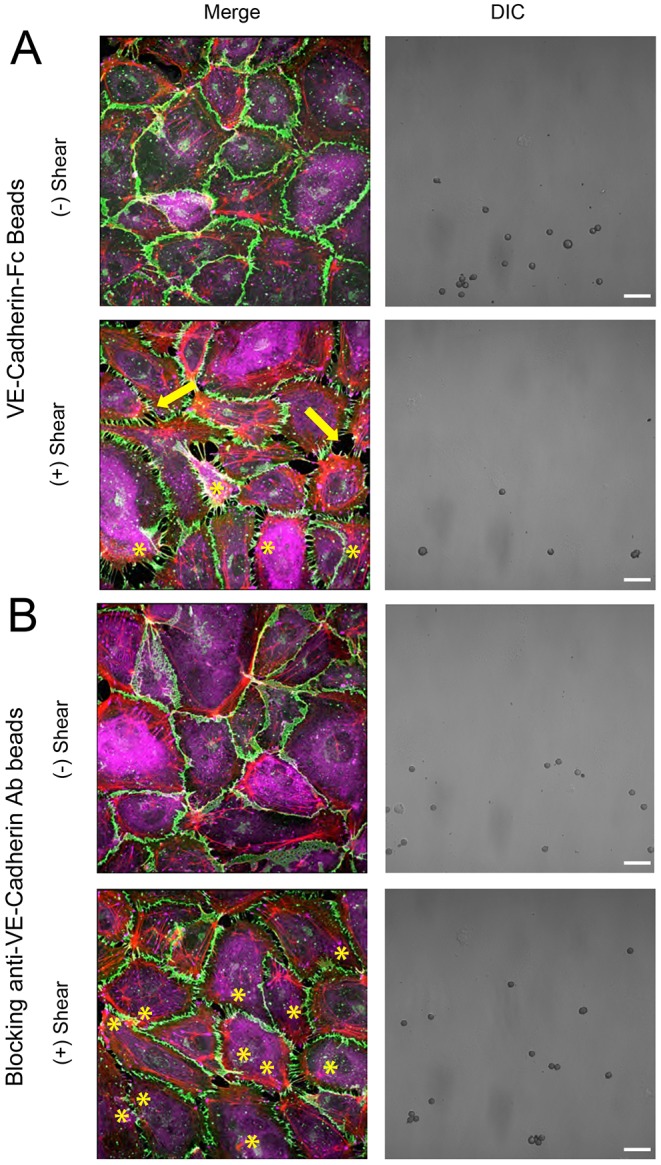
Local VE-cadherin receptor loading induces remodeling of distant junctions between unstimulated cells. Displayed are DIC images and merged immunofluorescence images of VE-cadherin (green), vinculin (purple) and F-actin (red). (A) DIC and merged immunofluorescence images of endothelial cell monolayers seeded with a low density of VE-cadherin-Fc-coated beads confined to a discrete section of the monolayer, evident in the DIC images. Yellow asterisks mark the bead locations (merged image, bottom), and arrows indicate disrupted junctions away from the bead-laden cells. Confocal immunofluorescence images show VE-cadherin (junctions, green), vinculin (purple) and F-actin (red) before (top) and after (bottom) bead twisting. (B) DIC and immunofluorescence images of endothelial cell monolayers laden with a low density of beads coated with the blocking anti-VE-cadherin antibody. The merged confocal immunofluorescence images are of VE-cadherin (green), vinculin (purple) and F-actin (red), before (top) and after (bottom) bead twisting. The yellow asterisks in the merged image (bottom) indicate bead-laden cells. Images are representative of more than three independent experiments. Scale bars: 20 µm.
Somewhat surprisingly, the localized, mechanical stimuli induced changes not only at junctions with bead-laden cells, but also between distal cells that did not share a boundary with the perturbed cells (Fig. 7A, bottom merged image). These distal junctions exhibited features similar to those between cells stimulated directly with VE-cadherin-coated beads (see Fig. 6A): namely, radial fiber formation, junction disruption and the appearance of punctate vinculin that co-localized with VE-cadherin. Blocking anti-VE-cadherin antibody beads caused some perturbations, but use of VE-cadherin beads resulted in much more substantial monolayer disruption, and the formation of gaps that were greater in number and larger (Fig. 7A).
DISCUSSION
Here, we report three major findings. First, these results show directly that VE-cadherin complexes are force transducers in endothelial cells. Second, VE-cadherin and PECAM-1 mechanotransduction are both ligand and epitope specific, suggesting that force transduction by these proteins is conformation specific and might be allosterically regulated. Third, the results show that, in addition to unfurling α-catenin (Rangarajan and Izard, 2012; Yonemura et al., 2010), VE-cadherin-based mechanotransduction also activates global signals, which increase cell stiffness and alter peripheral junctions and focal adhesions. The disruption of peripheral junctions also appears to activate signals that propagate mechanical information through the monolayer to destabilize inter-endothelial junctions away from the site of applied force.
Ligand-specific, VE-cadherin receptor loading triggered local vinculin recruitment and cytoskeletal remodeling, similar to that observed with type I classical cadherins (Barry et al., 2014; le Duc et al., 2010). These local changes are analogous to intercellular junction remodeling in response to endogenous myosin-II-dependent tugging forces triggered by Rho activators, growth factors, or inflammatory mediators (de Rooij et al., 2005; Huveneers et al., 2012; Liu et al., 2010; Lum and Malik, 1994; Miyake et al., 2006). However, contractile forces increase tension throughout intercellular junctions (Liu et al., 2010; Maruthamuthu et al., 2011), and other proteins like PECAM-1 could transduce forces (Chen and Tzima, 2009; Collins et al., 2012). The recruitment of actin and vinculin to force-loaded VE-cadherin-coated beads, but not to beads coated with PECAM-1.3 antibody, is analogous to punctate vinculin staining and junction thickening, following myosin II activation (Huveneers et al., 2012; Liu et al., 2010). The similar changes following both acute cadherin loading and endogenous tugging forces suggests that they reflect the same VE-cadherin-mediated mechanisms.
The demonstration that VE-cadherin complexes are force transducers might be anticipated because α-catenin, which is a force transducer at type I classical cadherin adhesions (Barry et al., 2014; Thomas et al., 2013; Yonemura et al., 2010), binds β-catenin at VE-cadherin adhesions. Although VE-cadherin binds either β-catenin or γ-catenin (Bazzoni and Dejana, 2004), α-catenin is present at both resting and stressed endothelial-cell–bead junctions. The load-induced vinculin recruitment to those junctions is consistent with the postulated force-dependent exposure of the cryptic vinculin site in α-catenin (Barry et al., 2014; Thomas et al., 2013; Yonemura et al., 2010).
The ligand and epitope specificity of VE-cadherin mechanotransduction accounts for the absence of VE-cadherin force transduction in previous studies (Tzima et al., 2005). Here, use of either VE-cadherin-Fc or non blocking antibody-coated beads induced both local cytoskeletal remodeling at bead–cell junctions and global cell stiffening. Blocking antibodies could impair VE-cadherin functions by competitive inhibition or by allosteric regulation. However, because beads coated with blocking antibody bind to VE-cadherin, their inability to induce cell stiffening suggests that VE-cadherin force transduction requires an allosteric ligation- or binding-induced conformational change. PECAM-1 mechanotransduction triggered with antibody-coated beads is similarly epitope specific. Interestingly, anti-PECAM-1 antibodies against different domains also reportedly have different effects on PECAM-1 affinity and adhesion (Mei et al., 2014), consistent with the allosteric regulation of PECAM-1 binding. The behavior of both proteins is analogous to that of integrins, which require specific ligation or conformation-selective antibodies for activation (Hynes, 2002).
Importantly, these results demonstrate that VE-cadherin mechanotransduction also triggers global PI3K and ROCK1 signaling that alters global cell mechanics and affects other adhesion receptors in the cell. VE-cadherin force transduction therefore appears to involve more than α-catenin unfurling (Rangarajan and Izard, 2012; Yonemura et al., 2010). There are intriguing parallels with PECAM-1 force transduction, which activates PI3K and global cell contraction through integrins (Collins et al., 2012; Conway et al., 2013). Thus, VE-cadherin and PECAM-1 might share a common mechanotransduction pathway, but there are differences. PECAM-1.3 beads triggered much less actin accumulation than VE-cadherin-coated beads, and they did not recruit vinculin, which associates with α-catenin at stressed cadherin adhesions (Barry et al., 2014; Thomas et al., 2013; Yonemura et al., 2010). These closely associated receptors are not mechanically coupled, although they might interact biochemically. VE-cadherin-mediated cell stiffening and peripheral junction disruption could similarly involve VEGFR2-dependent PI3K activation and downstream, integrin-dependent activation of ROCK1 (Conway et al., 2013).
The disruption of intercellular junctions stimulated by VE-cadherin perturbations is phenotypically similar to perturbations induced by inflammatory mediators such as thrombin, which activate RhoA. VE-cadherin-mediated activation of ROCK1 and cell contractility, similar to PECAM-1, could thus account for peripheral junction disruption, as well as focal adhesion remodeling. Although beads coated with blocking anti-VE-cadherin antibody also caused some junction disruption, beads coated with VE-cadherin-Fc ligand triggered much more extensive inter-endothelial gap formation (Fig. 6E).
The studies with sparse beads further revealed that these global signals and consequent force fluctuations at junctions propagate signals to affect distant cells in the monolayer. These observed long-ranged effects of localized VE-cadherin stimulation differ from reports of glassy tissue dynamics (Krishnan et al., 2012; Tambe et al., 2011), which focused on material properties of the cytoskeletal network when entire cell monolayers were stretched or migrating. Localized forces, applied to specific VE-cadherin receptors triggered the changes reported here. The mechanism underlying the longer-ranged perturbations beyond immediate neighbors of bead-laden cells remains to be established. Determining whether the latter propagation involves Ca2+ signaling (Batra et al., 2012; Junkin et al., 2013), force propagation through VE-cadherin or PECAM-1 complexes, or another mechanism such as stress focusing (Hu et al., 2003) is a subject of ongoing work, but is beyond the scope of this study.
To summarize, local VE-cadherin-specific force transduction not only triggers cytoskeletal remodeling at stressed adhesions, but also activates global signals that alter cell mechanics, remodel focal adhesions and disrupt peripheral cadherin-mediated junctions. Surprisingly, mechanical perturbations of VE-cadherin complexes appear to trigger signaling cascades that propagate across peripheral junctions to disrupt inter-endothelial contacts distant from the applied force. These results demonstrate that VE-cadherin complexes are central mechanical and signaling loci in an integrated, force sensitive tissue network. These results further revealed mechano-chemical coupling that rapidly propagates cadherin-actuated mechanical cues through the endothelium, and suggest a force-dependent mechanism of rapid endothelial barrier regulation.
MATERIALS AND METHODS
VE-cadherin-Fc production
The Fc region of the pcDNA3.1-VE-cadherin extracellular domain-Fc-His vector (P. Vincent, Albany Medical College, Albany, NY) was generated from the HEEC1-5-Fc-pEE14 construct (provided by Barry Gumbiner, University of Virginia, Charlottesville, VA). This full-length human VE-cadherin extracellular domain (VE-cadherin-Fc) contains the Fc region of human IgG with a C-terminal hexahistidine tag, and is stably expressed as a soluble Fc-dimer in Chinese hamster ovary (CHO) cells. CHO-K1 cells were transfected with the plasmid containing the tagged VE-cadherin insert, using Fugene 6 (Roche, Indianapolis, IN). Soluble VE-cadherin-Fc protein was harvested from a stably transfected CHO cell clone that was selected and cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% (v/v) fetal bovine serum (FBS) and 800 µg/ml G418 (VWR International). The soluble VE-cadherin-Fc–His6 protein (VE-cadherin-Fc) was affinity purified with a Ni-NTA column, followed by a protein-A Affigel column (Bio-Rad, Hercules, CA). Protein purity was assessed by SDS-PAGE.
Cell culture and reagents
Human pulmonary artery endothelial cells were purchased from Lonza (Allendale, NJ) and cultured using the manufacturer's recommended medium (Endothelial Basal Medium-2; EBM-2) supplemented with defined growth factors and supplements (Endothelial Growth Medium-2 SingleQuot Kit, Lonza). Cells were cultured at 37°C in a 5% CO2 humidified atmosphere. Endothelial cells at passages 6–9 were used in experiments.
Prior to magnetic twisting cytometry experiments, a suspension of 30,000 endothelial cells was plated on the 13-mm diameter well of 35-mm glass-bottomed dishes (Cell E&G) that had been preincubated with 20 µg/ml human fibronectin (Sigma-Aldrich, St. Louis, MO) in PBS at 4°C. Endothelial cells were grown to confluence for 2–3 days before performing experiments.
Preparation of ferromagnetic beads for magnetic twisting cytometry
Carboxyl ferromagnetic beads [1.0% (w/v), 4.0–4.9 µm diameter] from Spherotech (Lake Forest, IL) were covalently modified with VE-cadherin-Fc and poly-L-lysine (PLL) (Sigma-Aldrich, St. Louis, MO). Beads were modified with human fibronectin (Sigma-Aldrich, St. Louis, MO) or with antibodies. The blocking anti VE-cadherin antibody (clone 75, BD Transduction Laboratories) binds an epitope within the first two domains (amino acids 1-194) of the VE-cadherin ectodomain. The non blocking, monoclonal rat IgG2a anti-VE-cadherin antibody (Clone VE84.1), which binds to extracellular domain 3, was a gift from Dietmar Vestweber (Max Planck Institute, Muenster, Germany). We also used anti-PECAM-1 antibody (sc-31045, Santa Cruz Biotechnology) or anti PECAM-1.3 antibody (from Peter J. Newman, Blood Center of Wisconsin, Milwaukee, WI).
Beads were first chemically activated by mixing 50 µl of bead stock solution with 20 mg/ml (final concentration) of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (Sigma-Aldrich, St. Louis, MO) and 20 mg/ml N-hydroxysuccinimide (Thermo Scientific, Rockford, IL) in 1 ml 2-(N-morpholino)ethanesulfonic acid buffer (50 mM MES, 100 mM NaCl, pH 5.0) with an orbital shaker at 25°C for 15 min. Beads were then centrifuged at 11,000 g for 10 min at 25°C. The supernatant was aspirated, and the activated beads were then mixed with 20 µg of the protein of interest (VE-cadherin-Fc, PLL, fibronectin, or antibody), in coupling buffer (20 mM HEPES, 100 mM NaCl, 5 mM CaCl2, pH 8.0) for 2 h at 4°C, with continuous mixing on an orbital shaker. The reaction was stopped by mixing with quenching buffer (3.3 mM Tris-HCl, 100 mM NaCl, and 5 mM CaCl2 at pH 8.0) on an orbital shaker for 30 min at 4°C. The modified beads were centrifuged at 11,000 g for 10 min at 25°C. The supernatant was aspirated, and the beads were washed with HEPES buffer. The beads were resuspended in MCDB 131 medium (Gibco) supplemented with 1% (v/v) penicillin-streptomycin and 0.1% (v/v) FBS for MTC experiments. Modified beads were used for experiments immediately after protein binding, in order to minimize aggregation.
Magnetic twisting cytometry
The MTC experiment has been previously described in detail (le Duc et al., 2010; Wang et al., 1993). The instrument exerts shear stress on cell surface receptors, by twisting magnetized beads bound to the cell surface. A twisting field induces a torque on the beads that causes bead displacements, which reflect the viscoelastic properties of the bead–cell junction.
Before adding the modified beads to cells, small bead aggregates were disbursed by sonication for 3 s. Endothelial cell monolayers plated on glass-bottomed dishes were rinsed with MCDB 131 medium, and then an aliquot of the beads was allowed to settle on the cells. The beads were allowed to adhere to the endothelial cell monolayer during incubation at 37°C for 20 min. Dishes of cells were then placed within magnetic coils on a 37°C heated microscope stage. The bead magnetic moment of 0.12 Pa/Gauss (magnetic field×magnetic moment = applied stress) was calibrated as described previously (Wang et al., 1993). The beads were magnetized parallel to the cell monolayer, by applying a short (<100 µs) magnetic field pulse of 1 Tesla. A magnetic field oscillating at a frequency of 0.3 Hz was applied for 2 min perpendicular to the cell monolayer, in order to generate a twisting torque on the beads.
Two types of measurements were performed. In the first, the applied field was increased stepwise (in 10 s intervals from 0.3–75 Gauss, equivalent to a shear stress of 0.036–9 Pa, with no pause between successive increases in the field). In the second, the oscillating field (20 Gauss, equivalent to 2.4 Pa at a frequency of 0.3 Hz) was applied continuously for 120 s. The induced torque causes bead displacements, which were imaged with an inverted microscope (Leica) equipped with a 20× 0.6 NA objective lens and a charge-coupled device camera (ORCA2; Hamamatsu Photonics). The measured complex modulus of the bead–cell junction is the torque divided by the bead displacement, or G = T/D (Wang et al., 1993), and is a function of the viscous and elastic moduli of the bead-cell junction.
Cell treatments for MTC experiments
In order to inhibit actin polymerization, myosin II ATPase, or microtubule polymerization, cells were treated with, respectively, 4 µM cytochalasin D for 20 min, 100 µM blebbistatin for 20 min, or 20 µM nocodazole for 30 min before magnetic twisting experiments (all from Sigma-Aldrich, St. Louis, MO). Rho activity was inhibited by treating cells with a selective inhibitor of the Rho-associated protein kinase ROCK1 (Y-27632) at 10 µM for 1 h (Uehata et al., 1997) (Tocris Bioscience, Ellisville, MO). Phosphatidylinositol-3-kinase (PI3K) was inhibited by treating cells with 30 µM LY294002 for 20 min (Cell Signaling Technology). In order to block VE-cadherin receptors expressed on the cell surface, cells were treated with 12.5 µg/ml anti VE-cadherin antibody (clone 75, BD Transduction Laboratories) for 2 h.
Immunofluorescence
Subcellular remodeling in response to cadherin bond shear was visualized by immunofluorescence. Endothelial cells prior to shear or immediately after applying bond shear were washed in PBS and fixed for 15 min in 4% (w/v) paraformaldehyde at pH 7.4. Endothelial cells were then permeabilized for 5 min with 0.1% (v/v) Triton X-100 in PBS and blocked for 30 min in 2% (w/v) BSA in PBS at pH 7.4 (blocking buffer). Rhodamine–phalloidin (Invitrogen), primary and secondary antibodies were diluted 1∶100 in blocking buffer and incubated with cells for 1 h. Primary antibodies included goat polyclonal anti-VE-cadherin (sc 6458, Santa Cruz Biotechnology) and mouse monoclonal anti-vinculin (V9131, Sigma-Aldrich, St. Louis, MO). Secondary antibodies included anti-goat-IgG conjugated to FITC (F7367, Sigma-Aldrich, St. Louis, MO) and anti-mouse Alexa Fluor 647 (Invitrogen). Cells were mounted with ProLong Gold Antifade Reagent (Invitrogen).
Microscopy was performed with a Zeiss LSM 700 laser-scanning confocal microscope equipped with a Plan Apochromat 63× 1.4 NA oil immersion objective lens (Zeiss). Fluorescence was excited with 405 nm, 488 nm, 555 nm and 633 nm lasers. Eight-bit 512×512 images were acquired with a digital camera, a two-channel spectral detection system, and sequential line-by-line scanning using ZEN 2008 software (Zeiss). Sections along the optical (z) axis were acquired. Inter-endothelial gap area was quantified using ImageJ software (version 1.44, National Institutes of Health).
Confocal image analyses
In order to compare force-induced changes in protein accumulation at bead–cell junctions, mean fluorescence intensities of VE-cadherin, vinculin and F-actin were quantified from cells fixed before and after bond shear by MTC. Mean fluorescence intensities (MFIs) were quantified from optical z-axis sections displaying beads in focus, using ImageJ software. Fluorescence intensities were quantified in regions of interest defined by drawing a ring extending 1.0–1.5 µm from the bead edge, and a mask designating a filled outline of the bead (the region of interest, ROI) was defined based on DIC images (Fig. 4A). The mean fluorescence intensities of VE-cadherin, vinculin and F-actin surrounding for >30 beads (∼one bead per cell) for each condition had the respective background fluorescence channel subtracted, as defined by the mean intensity of a user-defined area near each bead, excluding pixels within the defined ROI surrounding each bead. Averages of MFIs surrounding each bead–cell pair, and the s.e.m. were calculated in Microsoft Excel and plotted in histograms as the percentage change after applying shear stress. The absence of VE-cadherin around beads identified beads that did not engage cell surface cadherins. The latter beads were excluded from the analyses.
Line-scan analyses were performed to display spatial variations in the proteins around beads. For each bead–cell pair (n>30), a straight line was drawn across the bead center and extended 3 µm beyond the bead edges. The background fluorescence level was defined as the mean intensity of a user defined cytoplasmic region near each bead. The background-subtracted fluorescence intensity profile was averaged for each data set and plotted using Microsoft Excel 2011 software.
Statistical analysis
The reported modulus (junction stiffness) from magnetic twisting experiments is expressed as the geometric mean±s.d. of a population of different bead–cell pairs that follows a log normal distribution. The number, n, of different bead–cell pairs is stated in the Results or figure legends. Averages reflect data from more than three independent experiments, as specified in the Results. All other results are expressed as the mean±s.e.m. of multiple independent experiments (n is specified in Results). Two-tailed Student's t-tests were performed with Microsoft Excel 2011 software, in order to compare the means of data from two different experimental groups. In this work, P<0.05 is considered statistically significant, at the 95% confidence level.
Supplementary Material
Acknowledgments
We thank Saiko Rosenberger for technical assistance, Prof. Peter Newman (Medical College of Wisconsin Blood Center) for the anti-PECAM-1.3 antibody, and Prof. Dieter Westveber for the non blocking anti-VE-cadherin antibody (Clone VE84.1). The authors thank Drs Dolly Mehta, Steven Dudek and Joe G.N. Garcia for technical assistance and helpful discussions.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
A.K.B. designed, conducted and analyzed experiments; N.W. assisted with MTC studies; D.E.L. designed experiments and analyzed data; A.K.B. and D.E.L. wrote the paper.
Funding
This work was supported by the National Institutes of Health [grant number 5 RO1 GM097443 to D.E.L. and N.W.); the National Science Foundation [grant number CMMI 10-29871 to D.E.L.]; a Beckman Institute Graduate Fellowship (to A.K.B.); and a American Heart Association Predoctoral Fellowship [grant number 10PRE3840004 to A.K.B.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.159954/-/DC1
References
- Barry A. K., Tabdili H., Muhamed I., Wu J., Shashikanth N., Gomez G. A., Yap A. S., Gottardi C. J., de Rooij J., Wang N. et al. (2014). α-catenin cytomechanics – role in cadherin-dependent adhesion and mechanotransduction. J. Cell Sci. 127, 1779–1791 10.1242/jcs.139014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra N., Burra S., Siller-Jackson A. J., Gu S., Xia X., Weber G. F., DeSimone D., Bonewald L. F., Lafer E. M., Sprague E. et al. (2012). Mechanical stress-activated integrin α5β1 induces opening of connexin 43 hemichannels. Proc. Natl. Acad. Sci. USA 109, 3359–3364 10.1073/pnas.1115967109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzoni G., Dejana E. (2004). Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol. Rev. 84, 869–901 10.1152/physrev.00035.2003 [DOI] [PubMed] [Google Scholar]
- Bershadsky A., Chausovsky A., Becker E., Lyubimova A., Geiger B. (1996). Involvement of microtubules in the control of adhesion-dependent signal transduction. Curr. Biol. 6, 1279–1289 10.1016/S0960-9822(02)70714-8 [DOI] [PubMed] [Google Scholar]
- Birukova A. A., Fu P., Xing J., Yakubov B., Cokic I., Birukov K. G. (2010). Mechanotransduction by GEF-H1 as a novel mechanism of ventilator-induced vascular endothelial permeability. Am. J. Physiol. 298, L837–L848 10.1152/ajplung.00263.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain R. J., Vanhaesebroeck B., Ridley A. J. (2010). The PI3K p110alpha isoform regulates endothelial adherens junctions via Pyk2 and Rac1. J. Cell Biol. 188, 863–876 10.1083/jcb.200907135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Tzima E. (2009). PECAM-1 is necessary for flow-induced vascular remodeling. Arterioscler. Thromb. Vasc. Biol. 29, 1067–1073 10.1161/ATVBAHA.109.186692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien S., Li S., Shyy Y. J. (1998). Effects of mechanical forces on signal transduction and gene expression in endothelial cells. Hypertension 31, 162–169 10.1161/01.HYP.31.1.162 [DOI] [PubMed] [Google Scholar]
- Collins C., Guilluy C., Welch C., O'Brien E. T., Hahn K., Superfine R., Burridge K., Tzima E. (2012). Localized tensional forces on PECAM-1 elicit a global mechanotransduction response via the integrin-RhoA pathway. Curr. Biol. 22, 2087–2094 10.1016/j.cub.2012.08.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway D. E., Breckenridge M. T., Hinde E., Gratton E., Chen C. S., Schwartz M. A. (2013). Fluid shear stress on endothelial cells modulates mechanical tension across VE-cadherin and PECAM-1. Curr. Biol. 23, 1024–1030 10.1016/j.cub.2013.04.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij J., Kerstens A., Danuser G., Schwartz M. A., Waterman-Storer C. M. (2005). Integrin-dependent actomyosin contraction regulates epithelial cell scattering. J. Cell Biol. 171, 153–164 10.1083/jcb.200506152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejana E., Orsenigo F., Lampugnani M. G. (2008). The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 121, 2115–2122 10.1242/jcs.017897 [DOI] [PubMed] [Google Scholar]
- Dudek S. M., Garcia J. G. (2001). Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. 91, 1487–1500. [DOI] [PubMed] [Google Scholar]
- Groeneveld A. B. (2002). Vascular pharmacology of acute lung injury and acute respiratory distress syndrome. Vascul. Pharmacol. 39, 247–256 10.1016/S1537-1891(03)00013-2 [DOI] [PubMed] [Google Scholar]
- Hahn C., Schwartz M. A. (2009). Mechanotransduction in vascular physiology and atherogenesis. Nat. Rev. Mol. Cell Biol. 10, 53–62 10.1038/nrm2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S., Troyanovsky R. B., Troyanovsky S. M. (2013). Binding to F-actin guides cadherin cluster assembly, stability, and movement. J. Cell Biol. 201, 131–143 10.1083/jcb.201211054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S., Chen J., Fabry B., Numaguchi Y., Gouldstone A., Ingber D. E., Fredberg J. J., Butler J. P., Wang N. (2003). Intracellular stress tomography reveals stress focusing and structural anisotropy in cytoskeleton of living cells. Am. J. Physiol. 285, C1082–C1090 10.1152/ajpcell.00159.2003 [DOI] [PubMed] [Google Scholar]
- Huveneers S., Oldenburg J., Spanjaard E., van der Krogt G., Grigoriev I., Akhmanova A., Rehmann H., de Rooij J. (2012). Vinculin associates with endothelial VE-cadherin junctions to control force-dependent remodeling. J. Cell Biol. 196, 641–652 10.1083/jcb.201108120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh J., Nishimura N., Rana K., Peloquin J. M., Califano J. P., Montague C. R., King M. R., Schaffer C. B., Reinhart-King C. A. (2011). Age-related intimal stiffening enhances endothelial permeability and leukocyte transmigration. Sci. Transl. Med. 3, 112ra122 10.1126/scitranslmed.3002761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes R. O. (2002). Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687 10.1016/S0092-8674(02)00971-6 [DOI] [PubMed] [Google Scholar]
- Junkin M., Lu Y., Long J., Deymier P. A., Hoying J. B., Wong P. K. (2013). Mechanically induced intercellular calcium communication in confined endothelial structures. Biomaterials 34, 2049–2056 10.1016/j.biomaterials.2012.11.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T. J., Zheng S., Sun J., Muhamed I., Wu J., Lei L., Kong X., Leckband D. E., Wang Y. (2015). Dynamic visualization of α-catenin reveals rapid, reversible conformational switching between tension states. Curr. Biol. 25, 218–224 10.1016/j.cub.2014.11.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova Y., Malik A. B. (2010). Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu. Rev. Physiol. 72, 463–493 10.1146/annurev-physiol-021909-135833 [DOI] [PubMed] [Google Scholar]
- Krishnan R., Canovic E. P., Iordan A. L., Rajendran K., Manomohan G., Pirentis A. P., Smith M. L., Butler J. P., Fredberg J. J., Stamenovic D. (2012). Fluidization, resolidification, and reorientation of the endothelial cell in response to slow tidal stretches. Am. J. Physiol. 303, C368–C375 10.1152/ajpcell.00074.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- le Duc Q., Shi Q., Blonk I., Sonnenberg A., Wang N., Leckband D., de Rooij J. (2010). Vinculin potentiates E-cadherin mechanosensing and is recruited to actin-anchored sites within adherens junctions in a myosin II-dependent manner. J. Cell Biol. 189, 1107–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leckband D. E., le Duc Q., Wang N., de Rooij J. (2011). Mechanotransduction at cadherin-mediated adhesions. Curr. Opin. Cell Biol. 23, 523–530 10.1016/j.ceb.2011.08.003 [DOI] [PubMed] [Google Scholar]
- Lee K. M., Tsai K. Y., Wang N., Ingber D. E. (1998). Extracellular matrix and pulmonary hypertension: control of vascular smooth muscle cell contractility. Am. J. Physiol. 274, H76–H82. [DOI] [PubMed] [Google Scholar]
- Leerberg J. M., Gomez G. A., Verma S., Moussa E. J., Wu S. K., Priya R., Hoffman B. D., Grashoff C., Schwartz M. A., Yap A. S. (2014). Tension-sensitive actin assembly supports contractility at the epithelial zonula adherens. Curr. Biol. 24, 1689–1699 10.1016/j.cub.2014.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., Tan J. L., Cohen D. M., Yang M. T., Sniadecki N. J., Ruiz S. A., Nelson C. M., Chen C. S. (2010). Mechanical tugging force regulates the size of cell-cell junctions. Proc. Natl. Acad. Sci. USA 107, 9944–9949 10.1073/pnas.0914547107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum H., Malik A. B. (1994). Regulation of vascular endothelial barrier function. Am. J. Physiol. 267, L223–L241. [DOI] [PubMed] [Google Scholar]
- Maruthamuthu V., Sabass B., Schwarz U. S., Gardel M. L. (2011). Cell-ECM traction force modulates endogenous tension at cell-cell contacts. Proc. Natl. Acad. Sci. USA 108, 4708–4713 10.1073/pnas.1011123108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta D., Malik A. B. (2006). Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 86, 279–367 10.1152/physrev.00012.2005 [DOI] [PubMed] [Google Scholar]
- Mei H., Campbell J. M., Paddock C. M., Lertkiatmongkol P., Mosesson M. W., Albrecht R., Newman P. J. (2014). Regulation of endothelial cell barrier function by antibody-driven affinity modulation of platelet endothelial cell adhesion molecule-1 (PECAM-1). J. Biol. Chem. 289, 20836–20844 10.1074/jbc.M114.557454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake Y., Inoue N., Nishimura K., Kinoshita N., Hosoya H., Yonemura S. (2006). Actomyosin tension is required for correct recruitment of adherens junction components and zonula occludens formation. Exp. Cell Res. 312, 1637–1650 10.1016/j.yexcr.2006.01.031 [DOI] [PubMed] [Google Scholar]
- Rangarajan E. S., Izard T. (2012). The cytoskeletal protein α-catenin unfurls upon binding to vinculin. J. Biol. Chem. 287, 18492–18499 10.1074/jbc.M112.351023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranscht B., Dours-Zimmermann M. T. (1991). T-cadherin, a novel cadherin cell adhesion molecule in the nervous system lacks the conserved cytoplasmic region. Neuron 7, 391–402 10.1016/0896-6273(91)90291-7 [DOI] [PubMed] [Google Scholar]
- Tabdili H., Barry A. K., Langer M. D., Chien Y-H., Shi Q., Lee K. J., Lu S., Leckband D. E. (2012a). Cadherin point mutations alter cell sorting and modulate GTPase signaling. J. Cell Sci. 125, 3299–3309 10.1242/jcs.087395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabdili H., Langer M., Shi Q., Poh Y-C., Wang N., Leckband D. (2012b). Cadherin-dependent mechanotransduction depends on ligand identity but not affinity. J. Cell Sci. 125, 4362–4371 10.1242/jcs.105775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei A., Giampietro C., Conti A., Orsenigo F., Breviario F., Pirazzoli V., Potente M., Daly C., Dimmeler S., Dejana E. (2008). Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat. Cell Biol. 10, 923–934 10.1038/ncb1752 [DOI] [PubMed] [Google Scholar]
- Tambe D. T., Hardin C. C., Angelini T. E., Rajendran K., Park C. Y., Serra-Picamal X., Zhou E. H., Zaman M. H., Butler J. P., Weitz D. A. et al. (2011). Collective cell guidance by cooperative intercellular forces. Nat. Mater. 10, 469–475 10.1038/nmat3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas W. A., Boscher C., Chu Y. S., Cuvelier D., Martinez-Rico C., Seddiki R., Heysch J., Ladoux B., Thiery J. P., Mege R. M. et al. (2013). α-Catenin and vinculin cooperate to promote high E-cadherin-based adhesion strength. J. Biol. Chem. 288, 4957–4969 10.1074/jbc.M112.403774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twiss F., le Duc Q., van der Horst S., Tabdili H., van Der Krogt G., Wang N., Rehmann H., Huveneers S., Leckband D., de Rooij J. (2012). Vinculin-dependent cadherin mechanosensing regulates efficient epithelial barrier formation. Biol. Open 1, 1128–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzima E., Irani-Tehrani M., Kiosses W. B., Dejana E., Schultz D. A., Engelhardt B., Cao G., DeLisser H., Schwartz M. A. (2005). A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437, 426–431 10.1038/nature03952 [DOI] [PubMed] [Google Scholar]
- Uehata M., Ishizaki T., Satoh H., Ono T., Kawahara T., Morishita T., Tamakawa H., Yamagami K., Inui J., Maekawa M. et al. (1997). Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 389, 990–994 10.1038/40187 [DOI] [PubMed] [Google Scholar]
- van Nieuw Amerongen G. P., van Delft S., Vermeer M. A., Collard J. G., van Hinsbergh V. W. (2000). Activation of RhoA by thrombin in endothelial hyperpermeability: role of Rho kinase and protein tyrosine kinases. Circ. Res. 87, 335–340 10.1161/01.RES.87.4.335 [DOI] [PubMed] [Google Scholar]
- Verin A. D., Birukova A., Wang P., Liu F., Becker P., Birukov K., Garcia J. G. (2001). Microtubule disassembly increases endothelial cell barrier dysfunction: role of MLC phosphorylation. Am. J. Physiol. 281, L565–L574. [DOI] [PubMed] [Google Scholar]
- Wang N., Ingber D. E. (1995). Probing transmembrane mechanical coupling and cytomechanics using magnetic twisting cytometry. Biochem. Cell Biol. 73, 327–335 10.1139/o95-041 [DOI] [PubMed] [Google Scholar]
- Wang N., Butler J. P., Ingber D. E. (1993). Mechanotransduction across the cell surface and through the cytoskeleton. Science 260, 1124–1127 10.1126/science.7684161 [DOI] [PubMed] [Google Scholar]
- Yonemura S., Wada Y., Watanabe T., Nagafuchi A., Shibata M. (2010). alpha-Catenin as a tension transducer that induces adherens junction development. Nat. Cell Biol. 12, 533–542 10.1038/ncb2055 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.