

Abstract
Lipoprotein (a) [Lp(a)] is an independent risk factor for atherosclerosis-related events that is under strong genetic control (heritability = 0.68–0.98). However, causal mutations and functional validation of biological pathways modulating Lp(a) metabolism are lacking. We performed a genome-wide association scan to identify genetic variants associated with Lp(a)-cholesterol levels in the Old Order Amish. We confirmed a previously known locus on chromosome 6q25-26 and found Lp(a) levels also to be significantly associated with a SNP near the APOA5–APOA4–APOC3–APOA1 gene cluster on chromosome 11q23 linked in the Amish to the APOC3 R19X null mutation. On 6q locus, we detected associations of Lp(a)-cholesterol with 118 common variants (P = 5 × 10−8 to 3.91 × 10−19) spanning a ∼5.3 Mb region that included the LPA gene. To further elucidate variation within LPA, we sequenced LPA and identified two variants most strongly associated with Lp(a)-cholesterol, rs3798220 (P = 1.07 × 10−14) and rs10455872 (P = 1.85 × 10−12). We also measured copy numbers of kringle IV-2 (KIV-2) in LPA using qPCR. KIV-2 numbers were significantly associated with Lp(a)-cholesterol (P = 2.28 × 10−9). Conditional analyses revealed that rs3798220 and rs10455872 were associated with Lp(a)-cholesterol levels independent of each other and KIV-2 copy number. Furthermore, we determined for the first time that levels of LPA mRNA were higher in the carriers than non-carriers of rs10455872 (P = 0.0001) and were not different between carriers and non-carriers of rs3798220. Protein levels of apo(a) were higher in the carriers than non-carriers of both rs10455872 and rs3798220. In summary, we identified multiple independent genetic determinants for Lp(a)-cholesterol. These findings provide new insights into Lp(a) regulation.
Introduction
Cardiovascular disease (CVD) is a leading cause of human morbidity and mortality throughout the world. Dyslipidemia is an important independent risk factor for CVD (1). Recently, Lp(a) has been identified as a causal role in atherosclerotic cardiovascular disease (2–4). Lp(a), which consists of a cholesterol-laden LDL particle and a plasminogen-like glycoprotein [apo(a), encoded by the LPA gene], may contribute to the processes of both atherosclerosis (5–8) and thrombosis (9–11). Lp(a) concentrations are under strict genetic control that are minimally influenced by age, weight and diet (4). The heritability of Lp(a) is estimated to range from 0.68 to 0.98 (12–14). The pattern of inheritance indicates that a major gene, as well as polygenic factors likely, contribute to plasma Lp(a) concentrations (15–17). Candidate gene studies indicate that the copy number of kringle IV-2 repeats and single nucleotide polymorphisms (SNPs) in the LPA gene may be determinants of Lp(a) levels (2,14,18,19). Recently, Lim et al. (20) identified two splice variants (loss of function) in the LPA gene that were associated with lower plasma Lp(a) levels and protection from cardiovascular disease. Genome-wide linkage analyses strongly suggests that the LPA locus on chromosome 6q25-27 is a major determinant of Lp(a) levels (LOD score from 9 to 108) and also suggests other new genetic determinants on chromosomes 1, 2, 11, 12, 13, 14, 15 and 19 (21,22). Ober et al. (23) performed a genome-wide association study (GWAS) of plasma Lp(a) levels in 386 Hutterite subjects and identified SNPs in at least six genes on chromosome 6q in addition to LPA that are significantly associated with Lp(a) levels independent of each other and of the number of KIV-2 repeats. However, causal mutations and functional validation of biological pathways modulating Lp(a) metabolism are lacking. A greater understanding of Lp(a) regulation and metabolism may lead to novel targets for pharmacologic lowering of Lp(a) levels for treatment and/or prevention of CVD.
To better define the genetic architecture of Lp(a), we performed a GWAS for Lp(a)-cholesterol in the Old Order Amish (OOA), a population relatively homogenous in genetic ancestry and lifestyle characteristics. We then sequenced the 40 exons, all intron–exon boundaries and 2 kb of the promoter region of the LPA gene in 24 Amish subjects. To further cover the LPA gene region, we also genotyped five tag SNPs from sequencing results and investigated the previously described KIV-2 repeat polymorphism in LPA. Finally, we investigated whether genetic variants significantly associated with variation in Lp(a)-cholesterol levels were also associated with Lp(a) mRNA and protein levels in human liver.
Results
Clinical characteristics of OOA subjects
OOA subjects who were phenotyped for Lp(a)-cholesterol and related traits were recruited in this study. The study population of 1376 OOA represented a group of relatively healthy adults. The mean age was 49.8 years with women slightly older than men (P = 0.025) (Table 1). The mean level of Lp(a)-cholesterol in this sample was 7.13 mg/dl, similar to the white population in the US. The heritability for Lp(a)-cholesterol in the OOA was 0.68 (P < 0.0001). Mean levels of BMI, triglycerides (TG), high density lipoprotein- (HDL) and Lp(a)-cholesterol were significantly higher in women than in men (age adjusted P < 0.01).
Table 1.
Clinical characteristics of the OOA subjects
| Female | Male | |
|---|---|---|
| Number | 676 | 700 |
| Age (years) | 50.9 ± 16.6 | 48.8 ± 17.4 |
| BMI (kg/m2) | 27.8 ± 5.5 | 26.1 ± 3.6 |
| TG (mg/dl) | 83.6 ± 58.5 | 73.8 ± 45.5 |
| HDL (mg/dl) | 59.6 ± 15.4 | 51.8 ± 13.0 |
| LDL (mg/dl) | 143.1 ± 47.6 | 138.5 ± 40.6 |
| Lp(a) (mg/dl) | 7.7 ± 4.0 | 6.6 ± 3.5 |
Genome-wide association analysis for Lp(a)-cholesterol
We completed a GWAS of Lp(a)-cholesterol in the OOA using the Affymetrix 500 K SNP array after imputing all autosomal SNPs on HapMap by MACH (Supplementary Material, Table S1). We identified two loci significantly associated with Lp(a)-cholesterol on chromosome 6q25-q26 and 11q23 (P < 5 × 10−8) (Fig. 1, Supplementary Material, Fig. S1). The SNPs most strongly associated with Lp(a)-cholesterol are shown in Supplementary Material, Table S2. An imputed SNP rs2305695 on chromosome 16 also reached GWAS significance [P = 2.11 × 10−9, minor allele frequency (MAF) = 0.005]; however, when genotyped, this SNP was found not to be polymorphic in Amish samples, suggesting an imputation error that may be due to low MAF. Previous observations have shown that imputation error rate increases as the MAF decreases (24,25).
Figure 1.
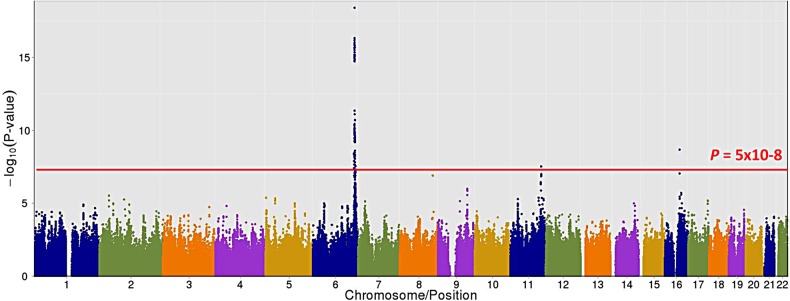
Association results of Lp(a)-cholesterol across all autosomal chromosomes. SNPs are plotted on the x-axis according to their position on each chromosome against association with Lp(a)-cholesterol on the t-axis (shown as –log10 P-value).
We identified an imputed SNP near the APOA5–APOA4–APOC3–APOA1 gene cluster on chromosome 11 to be significantly associated with increased Lp(a)-cholesterol levels (P = 8 × 10−8) (Fig. 2). This SNP, rs12787909, is located in intron 3 of DSCAML1 and has a MAF of 0.045 in the Amish and a MAF of 0.07 in HapMap-CEU population, but is not polymorphic in HapMap-HCB, JPT or YRI samples. SNP rs12787909 was also associated with decreased TG (P = 2 × 10−7), increased HDL-C (P = 4 × 10−9) and decreased LDL-C (P = 0.0001) in the OOA. Since the OOA, due to a founder effect, exhibit a high frequency of the rare null R19X mutation in nearby APOC3 (rs76353203, r2 in Amish with rs12787909 = 0.75) that is also associated with increased Lp(a), decreased TG, increased HDL-C and decreased LDL-C, a conditional analysis was performed. The association of APOC3 rs76353203 not only explained the signal at rs12787909, but was also more significant (Table 2) thus, indicating rs12787909 is a within-Amish association (i.e. linkage) rather than a population association.
Figure 2.

Association of SNPs on chromosome 11 with Lp(a)-cholesterol levels. SNPs are plotted by position on chromosome 11q23 against association with Lp(a)-cholesterol (–log10 P-value). The SNP name shown on the plot was the most significant SNP. Estimated recombination rates (from HapMap) are plotted in cyan to reflect the local LD structure. The SNPs surrounding the most significant SNP are color-coded to reflect their LD with this SNP (taken from pairwise r2 values from the HapMap-CEU database, www.hapmap.org). Genes, position of exons, and direction of transcription from UCSC genome browser (http://genome.ucsc.edu) are noted. Regional plots were generated using LocusZoom (http://csg.sph.umich.edu/locuszoom).
Table 2.
Associations of the DSCAML1 SNPs andAPOC3 null variant with Lp(a) and Lipid Traits
| Trait | N | APOC3 R19X |
DSCAML1 irs12787909 (imputation quality = 0.38) | DSCAML1 irs12787909 adjusted for APOC3 R19X | DSCAML1 irs12787909 adjusted for lnTG | ||
|---|---|---|---|---|---|---|---|
| Beta | P-value | Beta | P-value | P-value | P-value | ||
| LnLp(a) | 1098a | 0.419 | 3 × 10−8 | 0.459 | 8 × 10−8 | 0.28 | 0.014 |
| Ln[Lp(a)+1] | 1113 | 0.396 | 4 × 10−8 | 0.430 | 2 × 10−7 | 0.32 | 0.037 |
| TG | 1113 | −43.340 | 3 × 10−10 | −40.681 | 2 × 10−7 | 0.62 | |
| lnTG | 1113 | −0.637 | 4 × 10−18 | −0.645 | 1 × 10−14 | 0.66 | |
| HDL-C | 1113 | 13.545 | 6 × 10−10 | 14.607 | 4 × 10−9 | 0.29 | |
| LDL-C | 1113 | −22.391 | 0.0004 | −27.571 | 0.0001 | 0.13 | |
aFifteen subjects had Lp(a) = 0.
We identified 118 common variants significantly associated with Lp(a)-cholesterol levels (P = 5 × 10−8 to 3.91 × 10−19) that spanned ∼5.3 Mb on chromosome 6q25-26 in a region encompassing 26 genes, including LPA. Common variants within six genes (SNX9, SERAC3, TAGAP, FNDC1, PLG and AGPAD4) were significantly associated with Lp(a)-cholesterol levels (Fig. 3A).
Figure 3.

Association of SNPs on chromosome 6 with Lp(a)-cholesterol levels. SNPs are plotted by position on chromosome 6q25-26 against association with Lp(a)-cholesterol (–log10 P-value). (A) Highlights the most significant SNP in association analysis adjusted age, age2 and sex; (B) The most significant SNP after conditional analysis where included age, age2 sex and KIV-2 numbers as a covariate; (C) The most significant SNP after conditional analysis where included age, age2 sex, KIV-2 numbers, rs3798220 and rs10455872 as a covariate. (Arrow marked LPA location.)
Rare variants in LPA are associated with Lp(a)-cholesterol levels
Variants in LPA have previously been associated with Lp(a) levels (14). The common variants located in LPA gene in our GWAS were only modestly associated with Lp(a)-cholesterol levels with the lowest P-value = 9.70 × 10−5 (for imputed SNP rs7770685). To further evaluate DNA variation within LPA, we sequenced the 40 exons, all intron–exon boundaries and 2 kb of the LPA promoter region in 24 Amish subjects who were in the extreme high or low tail of the Lp(a)-cholesterol distribution, or who had different genotypes than the most significant association SNP with Lp(a)-cholesterol in GWAS. We identified 23 variants, including 6 missense variants falling within 5 exons: exon 26 (rs7765803 L1358V, rs7765781 L1372V), exon 32 (rs1801693 M1679T), exon 37(rs3798220 I1891M), exon 39 (rs41267809 L1961P) and exon 40 (rs3124784 R2016C) (Supplementary Material, Fig. S2 and Table S3). Selected based on the LD structure, we genotyped five SNPs in the LPA gene region that had not been well covered by our GWAS. Two of these variants (rs3798220 I1891M in exon 37 and rs10455872 A>G in intron 25) were significantly associated with Lp(a)-cholesterol levels (P values: 1.07 × 10−14 and 1.85 × 10−12, respectively) (Table 3). Neither of these SNPs was significantly associated with LDL-C, HDL-C or triglycerides (Table 3).
Table 3.
The association of top variants with lipid traits
| Variants | CHRa | Gene | Location | MAF | LnLP(a) |
TG |
HDL-C |
LDL-C |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Beta | P-value | Beta | P-value | Beta | P-value | Beta | P-value | |||||
| rs76353203 | 11 | APOC3 | Exon2 | 0.045 | 0.419 | 3.00E−08 | −43.340 | 3.00E−10 | 13.545 | 6.00E−10 | −22.391 | 4.00E−03 |
| rs3798220 | 6 | LPA | Exon 37 | 0.009 | 0.843 | 1.07E−14 | −10.194 | 2.94E−01 | −2.233 | 4.60E−01 | 9.887 | 2.64E−01 |
| rs10455872 | 6 | LPA | intron25 | 0.022 | 0.556 | 1.85E−12 | −7.630 | 2.69E−01 | 2.501 | 2.47E−01 | −4.600 | 4.71E−01 |
| KIV-2 | 6 | LPA | Exons | NAa | −0.168 | 2.28E−09 | −1.378 | 5.75E−01 | −0.575 | 4.54E−01 | −2.553 | 2.45E−01 |
aCHR, chromosome; NA, no data available.
Determination of kringle IV-2 number in the LPA gene
KIV-2 copy number variation in LPA is strongly associated with plasma Lp(a) levels and risk of CVD (26,27). Therefore, we measured KIV-2 copy number using quantitative real-time polymerase chain reaction (qPCR) (28). Real-time PCR signals of the two probe sets used for qPCR targeting exons four and five, respectively, were strongly correlated (r2 = 0.76, P = 2.98 × 10−259, Supplementary Material, Fig. S3). Numbers of KIV-2 were inversely correlated with plasma Lp(a)-cholesterol levels (r2 = 0.06, P = 9.44 × 10−17, Supplementary Material, Fig. S4). The KIV-2 copy numbers were significantly associated with Lp(a)-cholesterol levels (P = 2.28 × 10−9), although not with LDL-C, HDL-C or triglycerides (Table 2). The rare alleles of both the rs10455872 and rs3798220 SNPs were each correlated with lower KIV-2 copy numbers (Supplementary Material, Fig. S5).
Conditional analysis
We performed linkage disequilibrium (LD) analysis for chromosome 6q25-26 SNPs associated with Lp(a)-cholesterol using Haploview. The significantly associated SNPs fell into different blocks suggesting that multiple variants/genes in this region may regulate Lp(a)-cholesterol levels (Supplementary Material, Fig. S6). To further assess the number of independent associations on chromosome 6q25-26, we performed conditional association analysis using the most significantly associated SNPs as covariates. We first estimated independence between rs3798220 and rs10455872 in the LPA gene. We performed association analysis of one SNP with Lp(a)-cholesterol including the other SNP as a covariate. In this analysis, the effect sizes (i.e. betas) and statistical evidence for association (i.e. P-values) for each SNP were only minimally changed, suggesting that these two SNPs influenced variation in Lp(a)-cholesterol levels independently from each other (Table 4). Next, we assessed independence of KIV-2 numbers and these two SNPs (rs3798220, rs10455872). We tested the association of one variant with Lp(a)-cholesterol levels while adjusting for the other two variants simultaneously. These results indicated that both SNPs remained significantly associated with Lp(a)-cholesterol levels independently of KIV-2 numbers. KIV-2 numbers also remained significantly associated with Lp(a)-cholesterol levels independently of both SNPs, although the magnitude of the association was reduced due to partial correlation of KIV-2 numbers with the two SNPs (Table 4). We also reciprocally examined the association of SNPs on chromosome 6q25-q26 by adjusting for KIV-2 numbers. These SNPs remained highly associated with Lp(a)-cholesterol levels with only a modest reduction in strength of association (the best P value from 3.91 × 10−19 to 1.49 × 10−17) (Fig. 3B). Finally, we performed conditional analysis of SNPs on chromosome 6q25-26 adjusted for all three LPA variants (KIV-2 CNV, rs3798220 and rs10455872). The magnitude of the association of SNPs on 6q25-q26 with Lp(a)-cholesterol levels was largely reduced (the best P = 3.01 × 10−6) (Fig. 3C). These three variants in LPA gene explained 12.0% of the variation in Lp(a)-cholesterol levels in the Old Order Amish.
Table 4.
Conditional analyses adjusted by age, age2, sex and variants
| Variants | Chromosome | Gene | Location | Beta | SE | P-value |
|---|---|---|---|---|---|---|
| rs3798220a | 6 | LPA | Exon37 | 8.59E−01 | 1.07E−01 | 2.02E−15 |
| rs10455872a | 6 | LPA | intron25 | 5.77E−01 | 7.65E−02 | 9.67E−14 |
| rs3798220b | 6 | LPA | Exon37 | 8.23E−01 | 1.07E−01 | 3.62E−14 |
| rs10455872b | 6 | LPA | intron25 | 5.10E−01 | 7.85E−02 | 1.19E−10 |
| KIV-2b | 6 | LPA | Exons | −1.35E−01 | 2.78E−02 | 1.31E−06 |
aThe effect of rs3798220 is adjusted for the effect of rs10455872; the effect of rs10455872 is adjusted for the effect of 3798220.
bThe effect of rs3798220 is adjusted for KIV-2 and rs10455872; the effect of rs10455872 is adjusted for rs3798220 and KIV-2; the effect of KIV-2 is adjusted for the effect of rs3798220 and rs10455872.
Regulation of LPA gene expression
To gain more insights into the associations between LPA SNPs rs3798220 (I1891M) or rs10455872 (A>G in intron 25) and Lp(a)-cholesterol levels, we tested whether these were also associated with LPA gene expression. Since apo(a) is synthesized predominantly in the liver, we obtained liver samples from 78 white subjects undergoing bariatric weight loss procedures for extreme obesity or related comorbid medical conditions (29,30). We selected samples according to patient's genotype at rs3798220 (N = 20 subjects each with the CT and TT genotypes) and rs10455872 (N = 3, 20 and 20 subjects with the GG, AG and AA genotypes, respectively), matching patients across genotypes by age and gender. We measured LPA gene expression by quantitative PCR (qPCR) and found that LPA mRNA levels were significantly higher in carriers (GG and AG genotypes) than non-carriers (AA genotype) of rs10455872 (P = 0.0001, Fig. 4A)). In contrast, there was no difference in LPA mRNA levels when rs3798220 carriers and non-carriers were compared (P > 0.05, Fig. 4B). We measure protein levels of Apo(a) in these liver samples (rs3798220. N = 18 subjects each for the CT and TT genotypes; rs10455872, N = 12 subjects each for the AG and AA genotypes). Apo(a) protein levels are higher in carriers than non-carriers of either rs10455872 and rs3798220 (P = 0.004, Fig. 5A and P = 0.048, Fig. 5B respectively). These findings suggest that rs10455872 within LPA may play a key role in influencing LPA transcription levels or mRNA stability, while rs3798220 may influence Lp(a) levels through other mechanisms including effects on translation or post-translation.
Figure 4.

mRNA levels for LPA were assessed by qPCR in liver samples with different genotypes. (A) LPA expression was increased in the A>G variant of rs10455872; (B) The T>C variant of rs3798220 did not affect LPA mRNA expression. *P = 0.70054; **P = 0.000182; ***P = 0.000111.
Figure 5.

Western blot analysis of Lp(a) expression in liver samples with different genotypes. Representative western blots were shown for Lp(a) expression in rs10455872 (A; N = 12: 12 for AG: AA genotypes) and in rs3798220 (B; N = 18: 18 for CT: TT genotypes). Densitometric analysis of Lp(a) versus vinculin was performed for each genotype. A mixture of recombinant human Lp(a) isoforms (A10, A14, A22 and A34; provided by Dr Eduardo Angles-Cano) and HiMark™ Pre-stained Protein Standard (Life Technologies, Grand Island, NY) were applied in western blot. Data are presented as means ± SEM. A.U., arbitrary units. *P = 0.048; **P = 0.004.
Discussion
Recent evidence has convincingly shown a causal role for Lp(a) in atherosclerotic cardiovascular disease (2–4). Lp(a) levels in human plasma are under primarily genetic rather than environmental control. Here we present a systematic search for genetic variants with Lp(a)-cholesterol levels and find two significant association signals located on chromosome 6q25-q26 and chromosome 11q23. We further identify two SNPs (rs3798220, rs10455872) and KIV-2 in the LPA gene that are strongly associated with Lp(a)-cholesterol levels. Conditional analyses indicate that these variants act largely independently of one another to influence Lp(a)-cholesterol levels, although the mechanisms by which they do so are different, e.g. intronic variant rs10455872 is associated with reduced LPA mRNA levels while exonic SNP rs3798220 acts through other mechanisms.
We conducted a GWAS in the OOA, a culturally and genetically homogenous population originating from a small founder population. We identified a SNP on chromosome 11q23 that was significantly associated with increased Lp(a)-cholesterol and also with decreased TG, increased HDL-C and decreased LDL-C levels in the OOA. This association is mostly likely attributable to the APOC3 R19X variant, which exists at high frequency in the Amish due to a founder effect and is linked in the Amish to (but not in LD in the outbred population with) rs12787909. The APOC3 R19X is a null allele that reduces apoC-III production in heterozygotes by 50% and is associated with reduced triglycerides and increased HDL-C, LDL-C and cardioprotection as measured by less coronary calcification (31) and a lower rate of myocardial infarction (32,33). While it may seem counterintuitive at first glance that an otherwise cardioprotective variant would be associated with increased Lp(a) levels, a strong inverse correlation between Lp(a) levels and TG levels has been observed by us (r = −0.38, P = 6 × 10−27) and others (r = −0.22, P = 0.001) (34). Since the APOC3 variant is associated with TG and Lp(a) levels but the LPA variants are associated with Lp(a) but not TG levels, this finding provides strong evidence that the Lp(a)/TG correlation is driven by a causal effect of TG on Lp(a). That Lp(a) can be both a risk factor for CVD at very high levels and correlated with cardioprotective factors speaks to the limited understanding of the underlying biology of Lp(a) that our research is designed to address.
We confirmed a strong association of Lp(a)-cholesterol levels with the LPA locus on chromosome 6q25-26. Previous genome-wide linkage analyses have identified linkage of this region with Lp(a) levels with robust LOD scores ranging from 9 to 108 (21,22). The significant variants span ∼5.3 Mb region on chromosome 6q25-26 region that are within or flanking 26 genes including LPA. The LPA gene is an obvious and important candidate gene for influencing Lp(a) levels (26). Although the initial GWAS identified variants within LPA that were modestly associated with Lp(a)-cholesterol levels, sequence analysis identified two rare SNPs (rs3798220, rs10455872) in LPA that were strongly associated with Lp(a)-cholesterol levels. These SNPs (rs3798220, rs10455872) have been associated with Lp(a) levels and with cardiovascular diseases in previous studies used the HumanCVD BeadChip or direct genotyping (3,35,36). These rare SNPs were not tagged well by SNPs on the GWAS panel nor were they tagged by imputed SNPs. Systematically capturing the underlying causal variants within GWAS signals is an important approach for moving beyond GWAS.
The copy number variation of kringle IV-2 in LPA is highly polymorphic and a predictor of Lp(a) levels (26). We measured KIV-2 copy numbers using qPCR and found that the KIV-2 numbers were significantly associated with Lp(a)-cholesterol levels. As reported by others (37), smaller copy numbers of KIV-2 were associated with higher plasma Lp(a)-cholesterol levels. The number of KIV-2 repeats is variable ranging from 2 to >50 in each allele. Most individuals have two alleles that contain different numbers of KIV-2 repeats (38). The qPCR assay we employed measures the average number from both alleles. Our KIV-2 number represents the sum of KIV-2 number from both alleles that may be potentially limiting. This is because studies have demonstrated variable expressions of different KIV-2 numbers (39). Despite this limitation, we detected strong association between the sum of KIV-2 repeats from both alleles and Lp(a)-cholesterol levels consistent with earlier observations (3).
Our studies confirmed that multiple genetic variants on chromosome 6q25-26 are associated with Lp(a)-cholesterol levels. We found SNP rs3798220 was not in linkage disequilibrium with SNP rs10455872 (r2 = 0). Conditional analysis also revealed that these two SNPs influenced variation in Lp(a)-cholesterol levels independently from each other. We measured KIV-2 copy numbers with qPCR. A weakness of this approach is that we cannot assess LD between genotyped SNPs with KIV-2 copy numbers. We found that the carriers of both rs10455872 and rs3798220 had a lower KIV-2 copy numbers than non-carriers. A previous study has shown similar results in that rare alleles of both rs10455872 and rs3798220 are each correlated with a smaller apolipoprotein(a) isoform (as assessed by immunoblotting) and a lower copy number (as assessed by quantitative PCR assay) (3). However, conditional analysis revealed that the most significant SNPs on chromosome 6q25-26 remained strongly associated with Lp(a)-cholesterol levels after adjusting for KIV-2 numbers, suggesting these SNPs were significantly associated with Lp(a)-cholesterol levels independently of KIV-2 copy numbers. Previous studies indicated that KIV-2 copy numbers are not in strong LD with any biallelic markers in LPA (23,40). Therefore, two variants (rs3798220, rs10455872) were independently associated with Lp(a)-cholesterol levels. Our conditional analysis revealed that KIV-2 copy numbers were significantly associated with levels independently of both SNPs, although the magnitude of the association was reduced. We also found some SNPs were significantly associated with Lp(a)-cholesterol levels adjusted for KIV-2 numbers, rs3798220 and rs10455872. Additional rare variants in the LPA non-coding region or other gene region remain to be identified.
We further investigated the effect of rs3798220 and rs10455872 on LPA gene expression. To our knowledge, the higher transcription level of LPA in carriers of rs10455872 has not been previously reported. We also found that a few carriers of rs10455872 have lower LPA gene expression and a few non-carriers have higher LPA gene expression (Supplementary Material, Fig. S7). The SNP rs10455872 may thus be a surrogate for, or ‘tags,’ the underlying causal variant within a linkage block to influence gene expression. Deep sequencing will be necessary to discover functional variants in this region. There are no differences in LPA transcription levels between carriers and non-carriers of rs3798220. However, the protein levels of Apo(a) in liver samples are higher in carrier than non-carrier of either rs10455872 or rs3798220. The substitution from isoleucine to methionine in rs3798220 may affect apo(a) and Lp(a) catabolism to increase Lp(a) levels (41). We used SIFT (42) and PolyPhen-2_HDIV (43) to predict whether this substitution may alter the function of apo(a). The substitution of isoleucine with methionine is predicted to be a functionally deleterious substitution (SIFT: 0.01 and PolyPen-2_HDIV: 0.998).
In summary, we performed a systematic analysis of genetic variants with Lp(a)-cholesterol levels and identified multiple genetic determinants. We confirmed at least three sequence variants on chromosome 6q25-26 that were associated with Lp(a)-cholesterol levels and appear to act largely independent from one another. We also found the APOC3 R19X null mutation on chromosome 11q23 to be associated with increased Lp(a)-cholesterol levels. Our results suggest rs10455872 within LPA may influence mRNA levels of LPA (transcription or stability), while rs3798220 may influence Lp(a) levels through effects on translation or protein stability. Future work will be required to precisely elucidate the complex genetic architecture and regulation of Lp(a) and the contribution of genetic variation to risk factors for cardiovascular diseases.
Materials and Methods
Subjects
The OOA subjects of Lancaster Pennsylvania are relatively homogenous in terms of both genetic ancestry and lifestyle characteristics (44). Subjects (n = 1376) included in this study were relatively healthy adults aged 20 years or older, who participated in the Heredity and Phenotype Intervention (HAPI) Heart Study (45) and the Amish Family Longevity Study (AFLS) (46). Details of the study design, recruitment, and phenotyping have been described previously (45,46). For Lp(a)-cholesterol specifically, quantitative measurements were made with Vertical Auto Profile-II (VAP-II) method (Atherotech, Birmingham, AL, USA) (47). The VAP method measures cholesterol concentration of Lp(a) particles instead of apo(a) or Lp(a) particle concentration that will be a advantage of VAP. Measuring cholesterol concentration enables VAP testing of Lp(a) without influence of apo(a) size, which is a major problem influencing the accuracy of almost all immunoassay-based methods (47,48). The VAP method provides the continuous enzymatic analysis of cholesterol using a continuous flow VAP analyzer in lipoprotein classes separated by single vertical spin density-gradient ultracentrifugations that may be the potential for misclassification between different categories of LDL-particles. Informed consent was obtained from all participants and the study protocol was approved by the Institutional Review Board at the University of Maryland School of Medicine.
Genotyping and sequence
GWAS samples were genotyped using the Affymetrix GeneChip® Human Mapping 500 K array set including a total of 500 568 SNPs (Santa Clara, California). The GeneChip Genotyping Analysis Software (GTYPE 4.0) was used for automated genotype calling as part of the GeneChip Operating Software (GCOS) platform. The GTYPE-generated chip files were re-analyzed using the BRLMM genotype calling algorithm which provided improved call accuracy compared with the DM algorithm (http://www.affymetrix.com/support/technical). A confidence threshold, a measure of the call quality, of 0.33 was used for this analysis. As an initial quality control (QC) measure, BRLMM-generated chip files with call rates <90% for both enzymes were excluded from further analysis. Following quality control, Mendel and HWE checks, 382 935 informative SNPs were included in our analyses. We then used the 382 935 genotyped SNPs to impute all autosomal SNPs on HapMap (∼2.5 million SNPs) by MACH (version 1.0.15), using the publicly available phased haplotypes from HapMap (release 22, build 36, CEU population) as a reference panel (49). Follow-up genotyping was performed using TaqMan Allelic Discrimination Assay (Applied Biosystems).
Direct sequencing (50) was used to screen the 40 exons, exon–intron boundaries, and 2 kb of the promoter region of LPA for genetic variation in 24 Amish participants. We selected subjects according to their genotype at rs3805766 (top SNP from GWAS) or their Lp(a)-cholesterol levels. Both strands were sequenced on an ABI 3730xl DNA sequencer (Applied Biosystems) and analyzed using Sequencher software (GeneCodes).
KIV-2 copy number genotyping
LPA KIV-2 copy number was determined by quantitative real-time polymerase chain reaction (qPCR) as previously described (37). Briefly, a multiplexed qPCR was carried out using TaqMan: probes for LPA KIV-2 and an endogenous single-copy control gene in the Applied Biosystems 7900HT Fast Real-Time PCR system. Experiments were performed using probes targeted to exons 4 and 5 of LPA, each of which is found once within every KIV-2 repeat at the genomic level (37). TaqMan® TERT control reagent was used as single-copy reference gene (part number 44 01 633). Reaction volumes contained: 5 μl of Applied Biosystems Taqman Genotyping Master Mix (part number 43 71 357), 0.5 μl of 20× TaqMan®: primer/probe mix for LPA, 0.5 μl of 20× TaqMan®: primer/probe mix for TERT, 2 μl of water and 2 μl of genomic DNA at a concentration of 3–5 ng/μl. The average of ΔCT4 and ΔCT5 was then used for further analysis as the relative kringle repeat number.
LPA gene expression analyses
Liver samples
Seventy-eight wedge biopsy liver samples based on the carrier and non-carrier status for rs3798220 (N = 20: 20 for CT: TT genotypes) and rs10455872 (N = 3: 20: 20 for GG: AG: AA genotypes) matched by age, sex and BMI were obtained from white patients undergoing open or laparoscopic Roux-en-Y gastric bypass operations or laparoscopic adjustable gastric banding procedures for extreme obesity or its comorbid medical problems at Geisinger Medical Center, Danville, Pennsylvania (29,30).
RT-qPCR
Liver total RNA was extracted by TRIzol® reagent (Invitrogen) according to the manufacturer's instruction. The resulting RNAs were subjected to DNAse digestion using RNase-free DNase Set (Qiagen) and purification using RNeasy® MinElute® Cleanup Kit (Qiagen). The RNA quantity and quality were determined using a nanodrop spectrophotometer. cDNA was produced using Transcriptor First Strand cDNA Synthesis Kit (Roche). Real-time PCR was performed using TaqMan gene expression assay primers and probes [assay ID: Hs00916691-m1 for human LPA, Hs99999903_m1 for human Beta Actin (ACTB) as the endogenous control] and LightCycler 480 Probes Master (Roche) on LightCycler 480 Real-Time PCR system (Roche) using standard conditions and cycle times/temperatures. Relative expression of mRNAs was determined after normalization with reference gene levels using LightCycler 480 software 1.5. We normalized expression of mRNAs to a sample that had been tested in both SNPs.
Immunoblotting
Liver samples were homogenized in radioimmunoprecipitation buffer (Teknova, Hollister, CA) containing 150 mM sodium chloride, 1% Triton X-100, 1% deoxycholic acid-sodium salt, 0.1% sodium dodecyl sulfate, 50 mM Tris–HCL (pH 7.5), 2 mM EDTA, 50 mM NaF, 100 µM Na3VO4, and a pill of complete inhibitor (Roche Diagnostics, Indianapolis, Indiana). Liver tissue lysates were centrifuged at 16 000g for 30 min at 4°C and the supernatant was saved for subsequent immunoblotting. Protein concentrations were measured with the Pierce BCA kit (Thermo Scientific, Rockford, IL). Of note, 66 µg of liver protein was subjected to electrophoresis on NuPage 3–8% Tris–Acetate Gel (Life Technologies, Grand Island, NY), samples were transferred to nitrocellulose membrane, and then immunoblotted with Lp(a) sheep polyclonal antibody (provided by Dr Eduardo Angles-Cano, Institut national de la santé et de la recherche médicale, France) at a dilution of 1:20 000 for 2 h at room temperature (51). Then the blots were incubated with a donkey anti-sheep IgG secondary antibody conjugated with peroxidase (Santa Cruz Biotechnology, Dallas, Texas) at a dilution of 1:20 000 for 1 h at room temperature. The blots were subsequently incubated with monoclonal anti-vinculin mouse antibody (Sigma-Aldrich, St. Louis, MO) used as an internal control. The immunoblot signals were visualized with the Pierce ECL Western Blotting Substrate (Thermo Scientific, Rockford, IL). Image quantitative analysis was performed using ImageJ Software (52).
Statistical analysis
The natural log of plasma Lp(a)-cholesterol was used for association analyses. Association analyses were performed using a variance component model that includes a polygenic component to account for phenotypic correlation due to relatedness. The polygenic component was modeled using the relationship matrix derived from the complete 14-generation Amish pedigree structure (53,54). A one-degree of freedom likelihood ratio test was used to assess significance of the measured genotype under an additive model. We included age, age2 and sex as covariates in the association models. Pairwise linkage-disequilibrium (LD) statistics (D′ and r2) were calculated with Haploview version 4.2 (http://www.broadinstitute.org/haploview/).
LPA gene expression data were normalized to the quantity of beta-actin mRNA present in each sample. This allowed us to account for any variability in the initial template concentration as well as the conversion efficiency of the reverse transcription reaction. Student's t test was used to compare expression levels between carriers and non-carriers.
Supplementary Material
Funding
This work was supported in part by American Heart Association grant (10SDG2690004 to M.F.); American Diabetes Association grant (7-12-CT-26 to M.F.); National Institutes of Health (U01 HL072515), (U01 HL84756), (R01 AG18728), (R01 DK088231) the Mid-Atlantic Nutrition Obesity Research Center (grant P30 DK72488); the VA BLR&D Career Development award (Y.-C.C.).
Supplementary Material
Acknowledgements
We thank the Amish Research Clinic Staff for their energetic efforts in study subject recruitment and characterization as well as all the volunteers for their participation in these studies. This study would not have been possible without the outstanding cooperation of the Amish community. We thank Alexis Gorden for helping to get clinic information from Geisinger Clinic bariatric surgery patients.
Conflict of Interest statement. None declared.
References
- 1.(2001) Executive summary of the third report of the National Cholesterol Education Program (NCEP) expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III). JAMA, 285, 2486–2497. [DOI] [PubMed] [Google Scholar]
- 2.Kamstrup P.R., Tybjaerg-Hansen A., Steffensen R., Nordestgaard B.G. (2009) Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA, 301, 2331–2339. [DOI] [PubMed] [Google Scholar]
- 3.Clarke R., Peden J.F., Hopewell J.C., Kyriakou T., Goel A., Heath S.C., Parish S., Barlera S., Franzosi M.G., Rust S., et al. (2009) Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N. Engl. J. Med., 361, 2518–2528. [DOI] [PubMed] [Google Scholar]
- 4.Tregouet D.A., Konig I.R., Erdmann J., Munteanu A., Braund P.S., Hall A.S., Grosshennig A., Linsel-Nitschke P., Perret C., DeSuremain M., et al. (2009) Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat. Genet., 41, 283–285. [DOI] [PubMed] [Google Scholar]
- 5.Haberland M.E., Fless G.M., Scanu A.M., Fogelman A.M. (1992) Malondialdehyde modification of lipoprotein(a) produces avid uptake by human monocyte-macrophages. J. Biol. Chem., 267, 4143–4151. [PubMed] [Google Scholar]
- 6.Longenecker J.C., Klag M.J., Marcovina S.M., Powe N.R., Fink N.E., Giaculli F., Coresh J. (2002) Small apolipoprotein(a) size predicts mortality in end-stage renal disease: The CHOICE study. Circulation, 106, 2812–2818. [DOI] [PubMed] [Google Scholar]
- 7.Grainger D.J., Kirschenlohr H.L., Metcalfe J.C., Weissberg P.L., Wade D.P., Lawn R.M. (1993) Proliferation of human smooth muscle cells promoted by lipoprotein(a). Science, 260, 1655–1658. [DOI] [PubMed] [Google Scholar]
- 8.Enas E.A., Chacko V., Senthilkumar A., Puthumana N., Mohan V. (2006) Elevated lipoprotein(a)—a genetic risk factor for premature vascular disease in people with and without standard risk factors: a review. Dis. Mon., 52, 5–50. [DOI] [PubMed] [Google Scholar]
- 9.Caplice N.M., Panetta C., Peterson T.E., Kleppe L.S., Mueske C.S., Kostner G.M., Broze G.J., Jr., Simari R.D. (2001) Lipoprotein (a) binds and inactivates tissue factor pathway inhibitor: a novel link between lipoproteins and thrombosis. Blood, 98, 2980–2987. [DOI] [PubMed] [Google Scholar]
- 10.Marcovina S.M., Koschinsky M.L. (2003) Evaluation of lipoprotein(a) as a prothrombotic factor: progress from bench to bedside. Curr. Opin. Lipidol., 14, 361–366. [DOI] [PubMed] [Google Scholar]
- 11.McLean J.W., Tomlinson J.E., Kuang W.J., Eaton D.L., Chen E.Y., Fless G.M., Scanu A.M., Lawn R.M. (1987) cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature, 330, 132–137. [DOI] [PubMed] [Google Scholar]
- 12.Ober C., Abney M., McPeek M.S. (2001) The genetic dissection of complex traits in a founder population. Am. J. Hum. Genet., 69, 1068–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rainwater D.L., Kammerer C.M., VandeBerg J.L., Hixson J.E. (1997) Characterization of the genetic elements controlling lipoprotein(a) concentrations in Mexican Americans. Evidence for at least three controlling elements linked to LPA, the locus encoding apolipoprotein(a). Atherosclerosis, 128, 223–233. [DOI] [PubMed] [Google Scholar]
- 14.Kraft H.G., Kochl S., Menzel H.J., Sandholzer C., Utermann G. (1992) The apolipoprotein (a) gene: a transcribed hypervariable locus controlling plasma lipoprotein (a) concentration. Hum. Genet., 90, 220–230. [DOI] [PubMed] [Google Scholar]
- 15.Hasstedt S.J., Wilson D.E., Edwards C.Q., Cannon W.N., Carmelli D., Williams R.R. (1983) The genetics of quantitative plasma Lp(a): analysis of a large pedigree. Am. J. Med. Genet., 16, 179–188. [DOI] [PubMed] [Google Scholar]
- 16.Morton N.E., Berg K., Dahlen G., Ferrell R.E., Rhoads G.G. (1985) Genetics of the Lp lipoprotein in Japanese-Americans. Genet. Epidemiol., 2, 113–121. [DOI] [PubMed] [Google Scholar]
- 17.Hasstedt S.J., Williams R.R. (1986) Three alleles for quantitative Lp(a). Genet. Epidemiol., 3, 53–55. [DOI] [PubMed] [Google Scholar]
- 18.Rosby O., Berg K. (2000) LPA gene: interaction between the apolipoprotein(a) size (‘kringle IV’ repeat) polymorphism and a pentanucleotide repeat polymorphism influences Lp(a) lipoprotein level. J. Intern. Med., 247, 139–152. [DOI] [PubMed] [Google Scholar]
- 19.Ogorelkova M., Kraft H.G., Ehnholm C., Utermann G. (2001) Single nucleotide polymorphisms in exons of the apo(a) kringles IV types 6 to 10 domain affect Lp(a) plasma concentrations and have different patterns in Africans and Caucasians. Hum. Mol. Genet., 10, 815–824. [DOI] [PubMed] [Google Scholar]
- 20.Lim E.T., Wurtz P., Havulinna A.S., Palta P., Tukiainen T., Rehnstrom K., Esko T., Magi R., Inouye M., Lappalainen T., et al. (2014) Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genet., 10, e1004494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lopez S., Buil A., Ordonez J., Souto J.C., Almasy L., Lathrop M., Blangero J., Blanco-Vaca F., Fontcuberta J., Soria J.M. (2008) Genome-wide linkage analysis for identifying quantitative trait loci involved in the regulation of lipoprotein a (Lpa) levels. Eur. J. Hum. Genet., 16, 1372–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barlera S., Specchia C., Farrall M., Chiodini B.D., Franzosi M.G., Rust S., Green F., Nicolis E.B., Peden J., Assmann G., et al. (2007) Multiple QTL influence the serum Lp(a) concentration: a genome-wide linkage screen in the PROCARDIS study. Eur. J. Hum. Genet., 15, 221–227. [DOI] [PubMed] [Google Scholar]
- 23.Ober C., Nord A.S., Thompson E.E., Pan L., Tan Z., Cusanovich D., Sun Y., Nicolae R., Edelstein C., Schneider D.H., et al. (2009) Genome-wide association study of plasma lipoprotein(a) levels identifies multiple genes on chromosome 6q. J. Lipid. Res., 50, 798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M., et al. (2007) A second generation human haplotype map of over 3.1 million SNPs. Nature, 449, 851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchini J., Howie B. (2010) Genotype imputation for genome-wide association studies. Nat. Rev. Genet., 11, 499–511. [DOI] [PubMed] [Google Scholar]
- 26.Kronenberg F., Utermann G. (2013) Lipoprotein(a): resurrected by genetics. J. Intern. Med., 273, 6–30. [DOI] [PubMed] [Google Scholar]
- 27.Erqou S., Thompson A., Di Angelantonio E., Saleheen D., Kaptoge S., Marcovina S., Danesh J. (2010) Apolipoprotein(a) isoforms and the risk of vascular disease: systematic review of 40 studies involving 58,000 participants. J. Am. Coll. Cardiol., 55, 2160–2167. [DOI] [PubMed] [Google Scholar]
- 28.Lanktree M.B., Anand S.S., Yusuf S., Hegele R.A. (2009) Replication of genetic associations with plasma lipoprotein traits in a multiethnic sample. J. Lipid Res., 50, 1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Still C.D., Wood G.C., Chu X., Erdman R., Manney C.H., Benotti P.N., Petrick A.T., Strodel W.E., Mirshahi U.L., Mirshahi T., et al. (2011) High allelic burden of four obesity SNPs is associated with poorer weight loss outcomes following gastric bypass surgery. Obesity (Silver Spring), 19, 1676–1683. [DOI] [PubMed] [Google Scholar]
- 30.Kumashiro N., Erion D.M., Zhang D., Kahn M., Beddow S.A., Chu X., Still C.D., Gerhard G.S., Han X., Dziura J., et al. (2011) Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA, 108, 16381–16385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pollin T.I., Damcott C.M., Shen H., Ott S.H., Shelton J., Horenstein R.B., Post W., McLenithan J.C., Bielak L.F., Peyser P.A., et al. (2008) A null mutation in human APOC3 confers a favorable plasma lipids profile and apparent cardioprotection. Science, 322, 1702–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute, Crosby J., Peloso G.M., Auer P.L., Crosslin D.R., Stitziel N.O., Lange L.A., Lu Y., Tang Z.Z., Zhang H., Hindy G., et al. (2014) Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N. Engl. J. Med., 371, 22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jørgensen A.B., Frikke-Schidt R., Nordestgaard B.G., Tybjærg-Hansen A. (2014) Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N. Engl. J. Med., 371, 32–41. [DOI] [PubMed] [Google Scholar]
- 34.Rainwater D.L. (1996) Lp(a) concentrations are related to plasma lipid concentrations. Atherosclerosis, 127, 13–18. [DOI] [PubMed] [Google Scholar]
- 35.Koch W., Mueller J.C., Schrempf M., Wolferstetter H., Kirchhofer J., Schomig A., Kastrati A. (2013) Two rare variants explain association with acute myocardial infarction in an extended genomic region including the apolipoprotein(A) gene. Ann. Hum. Genet., 77, 47–55. [DOI] [PubMed] [Google Scholar]
- 36.Zabaneh D., Kumari M., Sandhu M., Wareham N., Wainwright N., Papamarkou T., Hopewell J., Clarke R., Li K., Palmen J., et al. (2011) Meta analysis of candidate gene variants outside the LPA locus with Lp(a) plasma levels in 14,500 participants of six White European cohorts. Atherosclerosis, 217, 447–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lanktree M.B., Rajakumar C., Brunt J.H., Koschinsky M.L., Connelly P.W., Hegele R.A. (2009) Determination of lipoprotein(a) kringle repeat number from genomic DNA: copy number variation genotyping using qPCR. J. Lipid Res., 50, 768–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marcovina S.M., Hobbs H.H., Albers J.J. (1996) Relation between number of apolipoprotein(a) kringle 4 repeats and mobility of isoforms in agarose gel: basis for a standardized isoform nomenclature. Clin. Chem., 42, 436–439. [PubMed] [Google Scholar]
- 39.Kraft H.G., Lingenhel A., Kochl S., Hoppichler F., Kronenberg F., Abe A., Muhlberger V., Schonitzer D., Utermann G. (1996) Apolipoprotein(a) kringle IV repeat number predicts risk for coronary heart disease. Arterioscler. Thromb. Vasc. Biol., 16, 713–719. [DOI] [PubMed] [Google Scholar]
- 40.Crawford D.C., Peng Z., Cheng J.F., Boffelli D., Ahearn M., Nguyen D., Shaffer T., Yi Q., Livingston R.J., Rieder M.J., et al. (2008) LPA and PLG sequence variation and kringle IV-2 copy number in two populations. Hum. Hered., 66, 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luke M.M., Kane J.P., Liu D.M., Rowland C.M., Shiffman D., Cassano J., Catanese J.J., Pullinger C.R., Leong D.U., Arellano A.R., et al. (2007) A polymorphism in the protease-like domain of apolipoprotein(a) is associated with severe coronary artery disease. Arterioscler. Thromb. Vasc. Biol., 27, 2030–2036. [DOI] [PubMed] [Google Scholar]
- 42.Ng P.C., Henikoff S. (2003) SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res., 31, 3812–3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cross H.E. (1976) Population studies and the Old Order Amish. Nature, 262, 17–20. [DOI] [PubMed] [Google Scholar]
- 45.Mitchell B.D., McArdle P.F., Shen H., Rampersaud E., Pollin T.I., Bielak L.F., Jaquish C., Douglas J.A., Roy-Gagnon M.H., Sack P., et al. (2008) The genetic response to short-term interventions affecting cardiovascular function: rationale and design of the Heredity and Phenotype Intervention (HAPI) Heart Study. Am. Heart J., 155, 823–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sorkin J., Post W., Pollin T.I., O'Connell J.R., Mitchell B.D., Shuldiner A.R. (2005) Exploring the genetics of longevity in the Old Order Amish. Mech. Ageing Dev., 126, 347–350. [DOI] [PubMed] [Google Scholar]
- 47.Kulkarni K.R., Garber D.W., Marcovina S.M., Segrest J.P. (1994) Quantification of cholesterol in all lipoprotein classes by the VAP-II method. J. Lipid Res., 35, 159–168. [PubMed] [Google Scholar]
- 48.Kulkarni K.R. (2006) Cholesterol profile measurement by vertical auto profile method. Clin. Lab. Med., 26, 787–802. [DOI] [PubMed] [Google Scholar]
- 49.Heard-Costa N.L., Zillikens M.C., Monda K.L., Johansson A., Harris T.B., Fu M., Haritunians T., Feitosa M.F., Aspelund T., Eiriksdottir G., et al. (2009) NRXN3 is a novel locus for waist circumference: a genome-wide association study from the CHARGE Consortium. PLoS Genet., 5, e1000539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fu M., Sabra M.M., Damcott C., Pollin T.I., Ma L., Ott S., Shelton J.C., Shi X., Reinhart L., O'Connell J., et al. (2007) Evidence that Rho guanine nucleotide exchange factor 11 (ARHGEF11) on 1q21 is a type 2 diabetes susceptibility gene in the Old Order Amish. Diabetes, 56, 1363–1368. [DOI] [PubMed] [Google Scholar]
- 51.Angles-Cano E., Loyau S., Cardoso-Saldana G., Couderc R., Gillery P. (1999) A novel kringle-4 number-based recombinant apo[a] standard for human apo[a] phenotyping. J. Lipid Res., 40, 354–359. [PubMed] [Google Scholar]
- 52.Schneider C.A., Rasband W.S., Eliceiri K.W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods, 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Agarwala R., Biesecker L.G., Hopkins K.A., Francomano C.A., Schaffer A.A. (1998) Software for constructing and verifying pedigrees within large genealogies and an application to the Old Order Amish of Lancaster County. Genome Res., 8, 211–221. [DOI] [PubMed] [Google Scholar]
- 54.Agarwala R., Biesecker L.G., Tomlin J.F., Schaffer A.A. (1999) Towards a complete North American Anabaptist genealogy: A systematic approach to merging partially overlapping genealogy resources. Am. J. Med. Genet., 86, 156–161. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.