

Abstract
We report two siblings with infantile onset seizures, severe developmental delay and spastic paraplegia, in whom whole-genome sequencing revealed compound heterozygous mutations in the AP4S1 gene, encoding the σ subunit of the adaptor protein complex 4 (AP-4). The effect of the predicted loss-of-function variants (p.Gln46Profs*9 and p.Arg97*) was further investigated in a patient's fibroblast cell line. We show that the premature stop mutations in AP4S1 result in a reduction of all AP-4 subunits and loss of AP-4 complex assembly. Recruitment of the AP-4 accessory protein tepsin, to the membrane was also abolished. In retrospect, the clinical phenotype in the family is consistent with previous reports of the AP-4 deficiency syndrome. Our study reports the second family with mutations in AP4S1 and describes the first two patients with loss of AP4S1 and seizures. We further discuss seizure phenotypes in reported patients, highlighting that seizures are part of the clinical manifestation of the AP-4 deficiency syndrome. We also hypothesize that endosomal trafficking is a common theme between heritable spastic paraplegia and some inherited epilepsies.
Introduction
Epilepsy is one of the most common neurological disorders, and in at least 70% of patients genetic factors are thought to play a role (1). Genetic epilepsies can be divided into (i) epilepsy syndromes that exhibit a complex inheritance pattern and (ii) rarer monogenic epilepsy syndromes. Mild monogenic epilepsies are often self-limiting or well treatable. They mostly occur in multigeneration families where a single gene defect segregates with the phenotype through several generations. In contrast, the most severe monogenic epilepsies are often drug resistant, are accompanied by developmental delay and mainly occur in isolated patients carrying de novo mutations (2). Two or more siblings are rarely found to be affected with a phenotype including early-onset seizures, developmental delay and variable additional neurological symptoms. In several siblings, a somatic or germline mosaicism has been confirmed, however, recessive gene defects have also been reported, especially but not exclusively, in consanguineous families (3–5). Here, we identified a non-consanguineous family with infantile onset seizures, severe developmental delay and spastic paraplegia in a genetic study on siblings with epilepsy and intellectual disability (ID). Whole-genome sequencing (WGS) of both siblings and healthy parents revealed compound heterozygous mutations in the AP4S1 gene, encoding the σ subunit of the adaptor protein complex 4 (AP-4).
AP-4 belongs to a highly conserved protein family consisting of five adaptor protein complexes (AP1-5), which are ubiquitously expressed in human tissue (http://www.proteinatlas.org) (6–10). APs are cytosolic proteins that dynamically associate with membranes and function in the regulation of vesicle trafficking throughout the endocytic pathway by recruiting other accessory proteins and selecting cargo. They form independent heterotetrameric complexes with a similar structural organization: two large subunits or adaptins (β 1-5, and γ, α, δ, ε or ζ), which have appendages linked by flexible arms to the central membrane-binding domain, a medium-sized (µ 1-5) and a small-sized (σ 1-5) subunit both located in the core of the complex. Several subunits furthermore occur in different cell-type-specific isoforms generating considerable complexity of AP assembly. Generally, AP complexes are assumed to act as obligate tetramers, where loss of one subunit destabilizes the entire complex. However, the β and μ subunits are in close contact, as are the σ and other large subunit; therefore, they can be considered as two hemicomplexes (10–14).
Mutations in several genes encoding AP subunits have been linked to specific human disorders. To date, 34 families with a genetic defect in one of the subunits of an AP complex have been reported. Nine families are described with dominant mutations in AP2S1 and familial hypocalciuric hypercalcemia type III (MIM 600740); nine other have X-linked syndromic ID type 5 due to AP1S2 mutations (MIM 304340); one family has autosomal recessive (AR) mental retardation, enteropathy, deafness, neuropathy, ichthyosis and keratodermia (MEDNIK) syndrome caused by loss of AP1S1 function (MIM 609313); and one family with Hermansky–Pudlak syndrome 2 has AR mutations in AP3B1 (MIM 608233). Fourteen unrelated families with complex hereditary spastic paraplegia (HSP) were found to carry AR mutations in genes encoding AP-4 subunits (MIM 614066, MIM 613744, MIM 612936 and MIM 614067). The remarkable overlap of clinical characteristics and genetic findings in these families has led to the postulation of the AP-4 deficiency syndrome as a clinical recognizable entity (15). Recently, AR mutations in the latest identified AP-5 complex have also been associated with HSP, more specifically in the AP5Z1 gene for SPG48 (16).
Although the family was recruited for this study because of the combination of seizures and ID, in retrospect the siblings' phenotype was consistent with the AP-4 deficiency syndrome, demonstrating the complex overlapping phenotype in neurodevelopmental disorders. It is the second family described with mutations in AP4S1, notably the first with seizures. Based on the families reported in literature, we give an overview of the different epilepsy phenotypes that can be seen in patients with AP-4 deficiency syndrome. We also further investigated the effect of the variants identified in our family on AP-4 subunit expression and AP-4 complex assembly.
Results
Case report
The two sisters are the only offspring of a Caucasian, non-consanguineous healthy couple (Fig. 1A). They were both born after a normal pregnancy and had an average birth size and weight. The oldest sister, now 14 years old, had a normal early development with head control at 3 months and sitting position at 5 months. Between 5 months and 5 years of age she had five brief generalized febrile seizures (simple FS). She became seizure free after starting valproic acid (VPA) at the age of 5 years and was without treatment between the age of 10 and 13 years. At that time seizures recurred as afebrile focal tonic and tonic-clonic seizures. Seizures remitted for a while after initiation of levetiracetam (LEV) but recurred afterwards; she is now seizure free for 6 months on VPA. A stagnation of development was seen around the period of seizure onset (5 months). She now has profound ID, microcephaly (−3SD), axial hypotonia and spastic diparesis. She started to crawl at the age of 4 years and is now able to walk a few steps independently. Brain MRI showed dilation of the lateral ventricles and hypoplasia of the posterior portion of the corpus callosum (Supplementary Material, Fig. S1A). EEGs showed slow background activity without epileptiform abnormalities.
Figure 1.
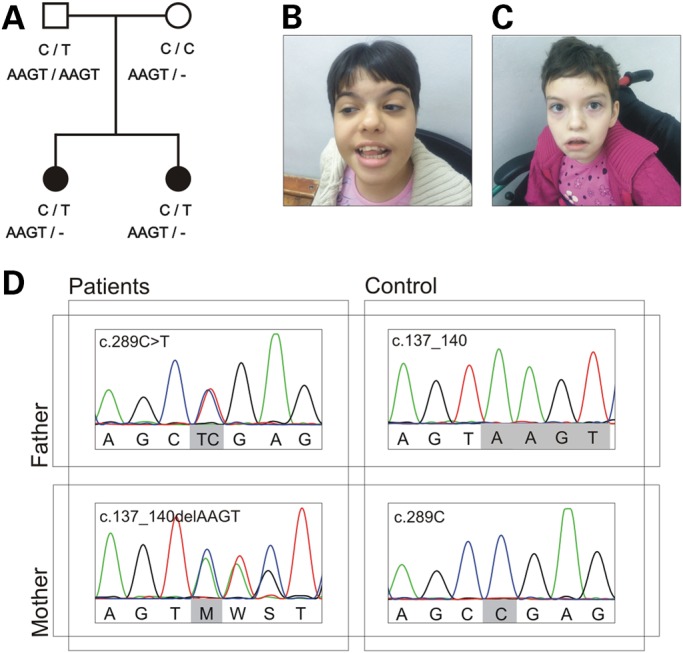
Genetic findings from the family reported here. (A) Pedigree of the family with segregation of the identified AP4S1 mutations. Filled symbols indicate affected individuals. The deletion of four base pairs is noted with a dash (-). (B and C) Pictures of the oldest and youngest sibling to illustrate the facial hypotonia and dysmorphic features consisting of a broad nasal bridge, hypertelorism with telecantus (most prominent in the youngest), arched eyebrows (most prominent in the oldest), bulbous nose, short philtrum, wide mouth, full lips and high palate. (D) Sequence NM_007077.4 was used as reference. Variant c.289C>T is a nonsense mutation (p.Arg97*) inherited from the father. Variant c.137_140delAAGT is a frameshift mutation (p.Gln46Profs*9) inherited from the mother. Parents are homozygous reference for the other variant. The patients carry both recessive variants, each on a different allele and are thus compound heterozygous.
The youngest sister, now 13 years old, had apneic episodes of unknown origin until the age of 3 months. She achieved head control at 3 months but then slowing of development occurred. She had two simple FS at the age of 1 and 2 years. She was treated with VPA since the second FS, but afebrile focal seizures occurred after the age of 9 years. VPA was changed to LEV, and dosage was increased at the age of 12 years due to seizure recurrence. She is now seizure free for >1 year. She started walking at the age of 3 years but has profound ID, axial hypotonia, convergent strabismus and spastic diparesis. EEGs showed slow background activity without epileptiform discharges. MRI of the brain shows a non-progressive hydrocephalus due to congenital stenosis of the aqueduct of Sylvius with severe reduction of subcortical white matter, the absence of visualization of the corpus callosum and a lack of white–gray matter differentiation in basal ganglia (Supplementary Material, Fig. S1B). Ophtalmological exam was normal and, in particular, did not show signs of increased intracranial pressure.
Both sisters have facial hypotonia and dysmorphic features consisting of broad nasal bridge, hypertelorism with telecantus (most prominent in the youngest), arched eyebrows (most prominent in the oldest), bulbous nose, short philtrum, wide mouth, full lips and high palate (Fig. 1B and C). The older sister has clinodactyly of the fifth finger. Neither of the parents showed any sign of dysmorphic features, epilepsy or spasticity.
Identification of AP4S1 mutations
To identify the genetic etiology in the two siblings, WGS was performed for both healthy parents and the affected children. Data analysis resulted in a shortlist of three candidate genes (Supplementary Material, Table S2). Based on the predicted loss-of-function nature and concordant phenotype, the compound heterozygous variants in AP4S1 were considered pathogenic (Fig. 1). Both children inherited a heterozygous frameshift variant (NM_007077.4: c.137_140delAAGT) from the mother and a heterozygous nonsense variant (NM_007077.4: c.289C>T) from the father, resulting in a premature stop codon in both copies of the gene (p.Gln46Profs*9 and p.Arg97*). According to the hg19 refGene annotation, AP4S1 has six isoforms at mRNA level, which encode four different proteins ranging from 135 to 159 amino acids in size. Up to amino acid 98 all four proteins are identical; hence all isoforms will be affected by both variants, without taking notice of possible cell- or tissue-specific expression (Supplementary Material, Fig. S3). The nonsense variant was found in the Exome Variant Server (EVS) with a frequency of 0.015% (rs200440467), whereas the frameshift variant was novel. Screening of the AP4S1 gene in 164 patients with epilepsy and ID but without spastic paraplegia did not reveal any additional homozygous or compound heterozygous mutation carriers.
Molecular characterization of the AP4S1 mutations in patient-derived fibroblasts
Using western blotting and immunoprecipitation we investigated the impact of the predicted loss-of-function mutations on protein formation and complex assembly. To this end, fibroblasts from a control individual and one sibling with AP-4 σ4 loss-of-function mutations (AP4S1*) were cultured. Whole cell lysates were tested for the presence of all AP-4 subunits (Fig. 2A), using clathrin heavy chain (CHC) as a loading control. In the AP4S1* cell line appeared completely absent, and there was a substantial reduction of all other AP-4 subunits, implying that the loss of σ4 leads to the destabilization of the entire AP-4 complex.
Figure 2.

Molecular characterization of the AP4S1 mutations described here. (A) Western blot of whole cell lysates show loss of σ4 and reduced levels of the other subunits in human fibroblasts from a patient with loss-of-function mutations in the AP4S1 gene (AP4S1*) versus a control sample. CHC was used as a loading control and did not show any differences. Bands corresponding to the different subunits are indicated with arrows, whereas cross-reaction bands are noted with a black dot. (B) Native immunoprecipitations (IPs) of control and patient cell lines were performed with antibodies against the β4 and ε subunit. Note the reduction of β4 and ε in their own IP, as well as the loss or reduction of all subunits, suggesting that the assembly of AP-4 is impaired. The interaction with the accessory protein tepsin is also clearly reduced in the AP-4-deficient patient. IgG bands confirm the use of equal amount of antibody in each IP. (C) Supernatant of the first IP (B) was used for a second IP with an antibody against AP-1γ. This shows no reduction of AP-1 complex assembly and is a control for equal amounts of starting material in the primary IP. (D and E) Results of immunofluorescence using an antibody against AP-4ε only (D) and double staining against tepsin and AP-1γ (E) on control- and patient AP4S1* patient-derived cell lines. The control line shows the punctuated perinuclear pattern typical for AP-4 localization. Whereas the patient line shows loss of AP-4 labeling (D) and loss of AP-4-associated tepsin (E). In contrast, the localization of AP-1γ is not affected in AP4S1* patient fibroblasts compared with the control.
We studied this process further using native immunoprecipitations with antibodies against either ε or β4 that allowed us to distinguish between effects on hemicomplex formation and complete complex assembly (Fig. 2B). Comparing patient with control fibroblasts, we not only observed the reduction of β4 or ε in their own immunoprecipitation but also revealed a substantial impairment in the assembly of the hemicomplexes (loss of both σ4 and ε) and also the entire AP-4 complex (because β4 and μ4 are lost too). A control immunoprecipitation using an antibody against γ indicated normal AP-1 complex assembly (Fig. 2C). Similarly, by immunofluorescence, we observed a clear reduction in the membrane association of ε in the AP4S1* patient line, while AP-1 localization was normal (Fig. 2D and E).
The loss of AP-4 assembly can be expected to have an impact on other accessory proteins. To date, tepsin is the only known interactor of AP-4, which binds to the appendage domain of β4 (17). We indeed observed that the membrane association of tepsin is abolished, presumably because it is unable to associate with AP-4 (Fig. 2B and E).
Epilepsy in reported families with AP-4 deficiency syndrome
Based on the clinical overlap of several families with HSP and genetic mutations in any one of the four AP-4 subunits, the AP-4 deficiency syndrome was postulated as an AR-inherited group of neurological disorders. Fourteen families and a total of 31 affected individuals have been described so far (Table 1). Besides HSP, the patients exhibit additional features such as severe ID, facial dysmorphy, microcephaly, shy character or stereotypic laughter and/or growth retardation. Brain MRI often shows white matter loss, dilated ventricles or thinning of the corpus callosum.
Table 1.
Detailed overview of the seizure phenotype of published patients with AP-4 deficiency syndrome
| Ref. | Family | Mutated AP-4 subunit | Original patient identifier | Age at publication | Seizure phenotype |
Brain MRI | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Onset age | Onset type | Later types | AED use and response | EEG | ||||||
| (18) | 1 | B1 | 1 | 14 years | 3.5 years | FS | 1 additional FS | AED started at 6.5 years after second FS, seizure free since then | Slow background | B |
| 2 | 12 years | No seizures | – | – | – | – | B | |||
| (19,20) | 2 | 1 | 9 months | Prolonged FS | Focal | Seizure free on VPA since 4 years of age | A, B, C | |||
| 2 | ? | Prolonged FS | 1 additional FS | B, C | ||||||
| (15) | 3 | 5 | 12.5 years | 9 months | FS, focal | FS, focal, secondary GTCS, SE | Seizure free on VPA since 8 years of age | Rare multifocal sharp waves | A, B, C | |
| 6 | 10.5 years | 6 months | FS | Focal, SE | Temporary response to PB, partial to VPA, seizure free on LEV since 8 years of age | Rare multifocal sharp waves | A, B, C | |||
| (21) | 4 | IDO1 IV-2, 4, 5 |
23, 15, 11 years | No seizures | – | – | – | – | n.d. | |
| 5 | E1 | MR071-III-11 | 6 years | 2 yearsa | ‘Epilepsy’ | Unknown | Unknown | Unknown | n.d. | |
| MR071-III-5 | 11 years | No seizures | – | – | – | – | n.d. | |||
| (22) | 6 | IV-4 | 23 years | 5 years | GTCS | Slow background | A, B, C, D | |||
| IV-5 | 22 years | 15 years | Staring spells | GTCS | Slow background | A (CT scan) | ||||
| (23) | 7 | P1, P2 | 3, 3 years | No seizures | – | – | – | |||
| (24) | 8 | 1161-1 | 15 yearsa | No seizuresa | – | – | – | Normal | n.d. | |
| 1161-2 | 13 yearsa | 1 yeara | FSa | 1 additional FSa | Untreateda | Normal | n.d. | |||
| (25) | 9 | M254 | ||||||||
| 10 | M1 | M004 | ||||||||
| (15) | 11 | 1 | 17 years | 6 months | GTCS | GTCS, atonic | Seizure free on PB and VPA since 8 years | No epileptiform discharges | A, B | |
| 2 | 11 years | 6 months | Febrile GTCS | GTCS, astatic | Seizure free on VPA since 8 years | No epileptiform discharges | A, B | |||
| 12 | 3 | 10.5 years | 11 months | Febrile GTCS | Febrile GTCS | PB, outcome unknown | Normal | A, B, C, D | ||
| 4 | 2.5 years | 10 months | Febrile tonic | 2 additional febrile tonic | Normal | A, B, C | ||||
| (26) | 13 | IV-1, 3, 4, 5, 6 | 24, 23, 22, 1.5 (+), 21 years | No seizures | – | – | – | – | A, B, C, D | |
| (21) | 14 | S1 | MRO61 - V-4, 28, VI-3 | 22, 20, 18 years | No seizures | – | – | – | – | n.d. |
| Current | 15 | 1 | 14.5 years | 5 months | FS | FS, focal | Seizure free for 6 months on VPA | Slow background | A, B | |
| 2 | 13 years | 1 year | FS | FS, focal | Seizure free since 12 years on LEV | Slow background | B, C, non-progressive hydrocephalus due to stenosis of aqueduct of Sylvius | |||
aInformation through personal communication with authors.
dash (-), not applicable; GTCS, generalized tonic-clonic seizures; FS, febrile seizure; SE, status epileptics; VPA, valproic acid; PB, phenobarbital; LEV, levetiracetam; AED, anti-epileptic drugs. For blank field, information was not available; A, dilated ventricles; B, thin corpus callosum; C, white matter loss; D, cerebral atrophy; n.d., not done.
The single previously reported AP4S1 mutation was identified in a large consanguineous Syrian family. All three affected patients had a progressive spasticity with loss of ambulation, severe ID, microcephaly, mild facial dysmorphic features, foot deformities and short stature. They did not present any seizures (21).
Looking at all reported AP-4-deficient families collectively, seizures have been described for 13 of the previously reported patients (42%) and tend to cluster within families. The specific AP-4 subunit defect does not seem to predict seizure prevalence or seizure type. A detailed overview of the seizure phenotype of published patients with AP-4 deficiency syndrome is given in Table 1. For two families (Family 9 and 10), we were unable to gather additional information on the seizure phenotype. In the remaining families and including the one described here, seizure onset ranges from 5 months to 15 years, with a median of 9 months. The majority of the patients (70%) had their first seizure in the first year of life. The most common seizure type at onset is FS (80%). Later in life infrequent afebrile focal or generalized seizures can occur (50%). The epilepsy tends to be mild, and reported patients gain seizure control before puberty with monotherapy or rarely dual therapy (Family 11, Patient 1). Interictal epileptiform discharges on EEG are rare and were only seen in one family in which the siblings had occasional status epilepticus (Family 3).
Discussion
We describe the second independent family with AR-inherited AP4S1 mutations and a phenotype compatible with AP-4 deficiency syndrome [the first family is described in Ref. (21)]. Notably, the family reported here represents the first outbred family with compound heterozygous mutations in a subunit of the AP-4 complex. All 14 previously described families with AP-4 deficiency syndrome carry homozygous mutations in one of the four AP-4 subunits (Fig. 3). Consanguinity combined with the corresponding phenotype is thus indicative for screening of AP-4 subunit encoding genes, but it does not exclude outbred families from an AP-4 deficiency diagnosis. The fact that this family was recruited through a study on epilepsy genetics shows that seizures can be a central feature in these patients. This is also supported by the high prevalence (42%) of seizures in previously reported patients with AP-4 deficiency syndrome. The epilepsy phenotype is milder than usually seen in genetic epilepsies with concomitant ID: AP-4-deficient patients have infantile or childhood onset seizures with frequent fever sensitivity. As commonly seen in mild genetic epilepsy syndromes, differences in genetic background and external factors (e.g. the occurrence of fever) may account for the variable penetrance. We show that AP4S1 mutations do not appear to be a frequent cause of epilepsy and ID without spastic paraplegia, though. We therefore advocate screening of all AP-4 subunit encoding genes in patients with a combination of HSP, ID and (fever-sensitive) seizures.
Figure 3.

Overview of reported AP-4 subunit mutations. In blue the ε subunit (NM_007347.4), in green the β4 subunit (NM_006594.3), in yellow the µ4 subunit (NM_004722.3) and in red the σ4 subunit (NM_007077.4). The scale bar on top indicates the size of the different proteins. Black arrows pinpoint mutation locations. Family IDs and mutations correspond to Table 1. Family 15, underlined, is the family described here; the patients carry both mutations in a compound heterozygous state. All other mutations (Family 1–14) were found homozygous in the patients and heterozygous in the respective parents.
We showed that σ4 is completely absent in AP4S1* patient fibroblasts and that there is a significant reduction of all other AP-4 subunits. These results imply that the loss of σ4 leads to the destabilization of the entire AP-4 complex. The minor pool of ε in the β4 immunoprecipitation, and vice versa, suggests that these two proteins may still be able to interact weakly, but it seems unlikely that such a complex is functional, because we see no membrane association of the residual ε. Loss of the correctly encoded σ4 protein thus results in downregulation of all other AP-4 subunits and significantly impacts the ability of the remaining AP-4 subunits to assemble into a complex. We also confirm that a dysfunctional AP-4 complex loses its ability to recruit accessory proteins such as tepsin. We therefore hypothesize a total loss of function of the AP-4 complex in our patients. Similar effects have been shown for one of the AP4E1 mutations (23) and in patients with loss-of-function mutations in AP4M1 and AP4B1, destabilization of the β4 protein has been reported (11). All these results point to the obligate heterotetrameric nature of the AP-4 complex, because (i) genetic mutations resulting in a loss of one subunit clearly lead to reduction of the other subunits and (ii) loss of one subunit leads to loss of complex assembly; conversely, in the absence of complex assembly, the single subunits are unstable and degrade. They furthermore explain the homogeneity of the clinical phenotype associated with mutations in any of the four AP-4 subunits.
Besides one missense mutation [p.Glu193Lys in AP4M1 (25)], all current disease-associated AP-4 mutations are premature stop variants (Fig. 3). The heterozygous carrier frequency of premature stop variants in AP-4 subunit encoding genes is consistent with the incidence of the phenotype and AR inheritance. The lack of common loss-of-function variants also supports the major role of these proteins in cell homeostasis, but it does not explain how the loss of a functioning AP-4 heterotetramer leads to such a specific neurological phenotype in humans. Several studies have investigated different functional aspects of the AP-4 complex. The AP-4 complex is ubiquitously expressed and includes expression in neurons during embryonic and postnatal development (27). Proper neuronal development and function depends on the regulation and sustainability of synaptic transmission between the presynaptic axon and postsynaptic dendrite. Neurons are therefore highly polarized, and membrane proteins have to be transported to and from precise positions within the neuron. The AP-4 complex participates in membrane sorting between the trans-Golgi Network and the endosomes, independent from the scaffolding protein clathrin (11,28,29). Moreover, it has been shown to regulate trafficking and specific expression of the δ2 glutamate receptor (GluRδ2) and the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptor to the dendritic domain (27,30). Disruption of AP-4-mediated transport of the amyloid precursor protein (APP) furthermore decreases APP localization to the endosomes and subsequently enhances γ-secretase-catalyzed cleavage of APP to the pathogenic amyloid-β peptide (31).
The underlying pathomechanism of HSP is in general described as a length-dependent axonal degeneration of corticospinal tracts, yielding a transport deficiency (32). Loss of AP-4 might cause direct axonal degeneration due to reduction of membrane trafficking or indirectly via the loss of accessory protein recruitment. The microcephaly, white matter loss and thinning of corpus callosum seen in several patients further support a crucial role for AP-4 in early brain development and neuronal integrity. Neuronal loss does, however, not automatically lead to epilepsy (as can be deducted from several neurodegenerative disorders). The underlying pathophysiology for the seizures in our patients must therefore depend on a more specific mechanism. While disturbance of endosomal trafficking is a well-known mechanism in HSP (32), this pathway is little studied in epilepsy. Epilepsy in general is characterized by the recurrence of spontaneous epileptic seizures, and the common underlying pathomechanism is hyperexcitability of neuronal networks. It seems plausible that a disturbance of time- and place-specific receptor expression, due to loss of AP-4-regulated trafficking (30), impairs the ability of neurons to maintain their polarization. In turn, this could lead to dysregulation of synaptic transmission and disruption of the balance between inhibitory and excitatory responses in the brain. A recent study showed that de novo mutations in genes encoding proteins involved in the regulation of synaptic transmission are significantly enriched in patients with an epileptic encephalopathy (33). In AP-4-deficient patients, the specific disturbance of glutamate receptor trafficking could play a role in synapse dysregulation. The glutamatergic system can, however, influence neuronal activity via different receptors and mechanisms (34); thus, the exact mechanism underlying epileptogenesis is not straightforward. Glutamate receptors associated with epilepsy generally carry gain-of-function mutations (35,36). Nevertheless, knock-out mice models also suggest that net brain excitability is a subtle balance depending on many factors, including regional and cell-type-specific input, and interaction with other ion channels (37,38). Still, the seizures in AP-4 deficiency syndrome are generally mild, and remission occurs in all patients, which could be explained by the process of brain maturation resulting in decreased excitability, brain plasticity or the development of specific compensatory mechanisms.
Whether the HSP and epilepsy in AP-4-deficient patients result from the same pathomechanism in different types of neurons will need more in-depth investigation. It is, however, conceivable that endosomal trafficking defects may be a common theme between epilepsy and HSP. Recently, a large whole exome sequencing project failed to identify a significant overlap between HSP-associated disease loci (n = 71) and epilepsy causing gene defects (n = 58) (24). Nevertheless, comorbid seizures, though rare in HSP, have been described for 12 HSP-associated loci (SPG2, 3A, 4, 6, 11, 15, 18, 35, 47, 50, 51 and 52). Four are the AP-4 subunit encoding genes and remarkably, of the eight remaining ‘HSP + epilepsy syndrome genes' half encode for proteins involved in endosome function. Conversely, seizures have been described in 8/14 [AP-4, spatacsin, spastizin, spastin and NIPA1 (28,39–42)] of the HSP-associated protein defects causing endosomal dysfunction (32). Epilepsy is thus a more frequent comorbidity in the subgroup of HSP patients with mutations leading to endosomal dysfunction than previously realized.
With the identification of compound heterozygous loss-of-function mutations in the AP4S1 gene, we have identified the second independent family with AP-4 deficiency syndrome caused by a defective AP-4 σ4 subunit. We furthermore showed a complete loss of expression of the encoded protein and a loss of AP-4 complex assembly. We provided an overview of seizure phenotypes seen in patients with AP-4 deficiency syndrome and highlight the prevalence of infantile and mostly fever-sensitive seizures as part of the clinical spectrum. In conclusion, we hypothesize that endosomal dysfunction is a common theme between HSP and epilepsy pathology, although different neuronal cell types and cellular mechanisms might underlie the specific nature of the symptoms. Additional studies in a neuronal cell system or animal model may allow us to investigate neuronal endosomal (dys)function in more detail, study possible in vivo compensatory mechanisms and cell-type-specific effects of AP-4 subunit. Eventually, this will lead us to a better understanding of the AP-4 pathway, its neuronal function and its role in disease.
Materials and Methods
This study was part of a project within the EuroEPINOMICS RES consortium on recessive epilepsies and approved by the Ethical Committees of the local institutes. Parents or legal guardians of each patient signed an informed consent form for participation.
WGS and analysis
Genomic DNA was isolated from 200 µl total blood using the QIAmp DNA Blood Maxi Kit (Qiagen) according to the manufacturer's instructions. DNA of both parents and the two siblings was sent for WGS to Complete Genomics (Mountain View, California). Their services include library preparation, massively parallel short-read sequence by ligation, sequence mapping, local de novo assembly, as well as structural variant calling and generating alignment and coverage files (43). All variants were annotated and analyzed by two independent WGS variant filtering pipelines. The ‘Antwerp’ pipeline was performed with the GenomeComb tool (44). Prioritization was done by including only high-quality callings (coverage >9, exclusion of repeat regions), excluding variants with a frequency above 1% in control populations (data from 1000Genomes, EVS and 30 in-house WGS projects) and selecting variants present in both patients with a predicted impact on the encoded protein according to the hg19 refGene annotation (missense, nonsense, frameshift and essential splice variants). Segregation analysis of these variants was done under an AR model, retaining both homozygous and compound heterozygous variants. We further looked for de novo variants shared by both patients (due to a germline mosaicism) and X-linked variants in the PCDH19 gene, which is known to cause fever-sensitive epilepsy in females (45). Our filtering strategy resulted in a shortlist of three candidate genes (Supplementary Material, Table S1). Using identical filter parameter settings, the same set of variants was found with the ‘Luxembourg’ pipeline as described previously (46). The compound heterozygous variants in the AP4S1 gene were considered pathogenic and confirmed by Sanger sequencing.
Description of the epilepsy phenotype in AP-4-deficient patients described in literature
The phenotype of all patients with mutations in one of the AP-4 subunits reported in literature was reviewed for clinical features related to epilepsy. When information was missing, the corresponding authors of the publications were contacted to ask additional clinical details.
Genetic follow-up studies
To elucidate whether AP4S1 mutations are present in epilepsy patients with ID, but without spastic paraplegia, we developed a Multiplex Amplification of Specific Targets for Resequencing (MASTR) assay (Multiplicom). The MASTR assay was designed using mPCR (Multiplicom) and covered the complete coding sequences as well as flanking intronic sequences of AP4S1. Multiplex amplifications and subsequently labeling reactions were done on a Veriti AB machine (LifeTechnologies) followed by pooling of the different amplicons. Next-generation sequencing was carried out on a MiSeq platform (Illumina) using the MiSeq Reagent Kit v3 (2 × 300 bp reads). With this assay, we analyzed a total of 164 patients with epilepsy and ID. The generated data were aligned to the reference genome (hg19) using Burrows-Wheeler Aligner (http://bio-bwa.sourceforge.net/). Variants were called using Genome Analysis Toolkit Unified Genotyper (https://www.broadinstitute.org/gatk/) and SAMTools (http://samtools.sourceforge.net/). Annotation and filtering of the data were done using GenomeComb (44).
In patients in whom we only identified one heterozygous variant with the gene panel, we further explored the presence of copy number variants encompassing AP4S1. We used the Multiplex Amplicon Quantification (MAQ) technique (Multiplicom), which comprises a multiplex PCR amplification of fluorescently labeled target and reference amplicons, followed by fragment analysis on the ABI3730 DNA Analyzer. More precisely, we designed seven target amplicons located in the genomic region of AP4S1 and four reference amplicons randomly located on different chromosomes. The comparison of normalized peak areas between the test individual and the average of control individuals results in target amplicon doses indicating the copy number of the target amplicon.
Cell culture
Functional characterization of the variants was done on fibroblasts generated from a 5 mm skin biopsy punch of the oldest affected sister. Primary fibroblasts were cultivated at 37°C in Dulbecco's Modified Eagle's medium supplemented with 10% fetal calf serum, 1% l-glutamine and 1% penicillin/streptomycin.
Immunofluorescence
Fibroblasts were seeded on glass-bottomed dishes, washed with phosphate-buffered saline (PBS: 137 mm NaCl, 2.7 mm KCl, 10 mm Na2HPO4, 1.76 mm KH2PO4) and then fixed with either ice-cold methanol for 5 min at −20°C (ε) or fixed with 3% formaldehyde for 10 min (tepsin). Formaldehyde-fixed cells were permeabilized with 0.1% Triton X-100 (Sigma-Aldrich); all were blocked with PBS–bovine serum albumin (BSA; PBS plus 0.5% BSA) for 10 min. The cells were then incubated with primary antibodies against AP-4ε (BD 612019), tepsin or AP-1γ for 1 h, washed with PBS–BSA, followed by Alex Fluor 488 or Alex Fluor 594 secondary antibodies for 1 h. All incubations were performed at room temperature. All antibodies were used at 1 µg/ml. The cells were imaged with a Zeiss Axiovert 200 inverted microscope with a Zeiss Plan Achromat ×63 oil immersion objective, a Hamamatsu ORCA-ER2 camera and IMPROVISION OPENLAB software.
Western blotting and immunoprecipitation
For western blotting, cells were washed in PBS, scraped and homogenized in lysis buffer (50 mm Tris pH 8 containing 2% sodium dodecyl sulphate) and then passed through a QIAshredder (Qiagen). Protein concentrations were estimated with a BCA assay (Thermo Fisher Scientific); samples were suspended in sample buffer and loaded at equal protein amounts for gel electrophoresis. Proteins were transferred onto nitrocellulose and blocked with PBS containing 0.1% Tween20 and 5% milk. The blots were then incubated with primary antibodies against clathrin, AP-4 ε, β4, µ4, σ4, tepsin, AP-1γ (Mab100.3) and σ1 (all used at 1 µg/ml) and were followed by incubation with anti-rabbit or anti-mouse HRP (Sigma-Aldrich). The bands were detected by enhanced chemiluminescence (ECL Prime; Amersham, GE Healthcare). All antibodies used here were raised in-house and have been described previously (17,28), except for the antibody against σ4, which was made by injecting a GST fusion of the C-terminal 32 amino acid fragment. Although all antibodies are affinity purified and specific, some non-specific bands are present in whole cell lysate western blots.
For immunoprecipitation under native conditions, cell pellets containing equal amounts of starting material were washed in PBS and then lysed in PBS-T (PBS containing 1% Triton X-100 supplemented with protease inhibitors). Lysates were cleared by centrifugation, and immunoprecipitation was then performed with antibodies against ε (of AP-4), β4 (of AP-4) or γ (of AP-1) with recovery using protein A–Sepharose (GE Healthcare).
Note Added in Proof
During the process of proof reading this article an additional family with mutations in one of the AP-4 subunit encoding genes was reported. Two brothers of a consanguineous family were found to carry a homozygous frame shift mutation in AP4M1 and presented the clinical features of AP-4 deficiency syndrome. One of them is described to have childhood onset seizures. The genetic and clinical data reported is in correspondence with the data presented here (47).
Supplementary Material
Funding
This work was supported by the Eurocores program EuroEPINOMICS of the European Science Foundation, the Fund for Scientific Research Flanders (FWO), the Methusalem excellence grant of the Flemish Government, The Wellcome Trust (J.H.), and the University of Antwerp. K.H. and T.D. are PhD fellows of the Institute for Science and Technology (IWT)-Flanders and A.S. is a postdoctoral fellow of the FWO. P.M. is supported as a ISB/LCSB fellow by ‘le plan Technologies de la Santé par le Gouvernment du Grand-Duché de Luxembourg’ through the Luxembourg Centre for Systems Biomedicine (LCSB) at the University of Luxembourg.
Supplementary Material
Acknowledgements
We thank the family for their participation to this study. Dr Rasim Ozgur Rosti, Dr Katalin Štěrbová and Dr Abou Jamra Rami are thanked for providing additional information on the seizure phenotype of their AP-4-deficient patients. We thank Dr Margaret Robinson for the insightful discussions and Richard Nash for establishing the fibroblasts cultures. Dr Leroy Hood and the Family Genomics group at the Institute for Systems Biology deserve thanks for their support and project management of the whole-genome sequencing. Some of the computational results presented in this paper were carried out using the HPC facilities of the University of Luxembourg (http://hpc.uni.lu). We acknowledge the contribution of the VIB Genetic Service Facility (http://www.vibgeneticservicefacility.be) for the genetic follow-up analyses. This research would not have been possible without the collaborative support of the EuroEpinomics RES Consortium and in specific all other members of the AR working group: Zaid Afawi; Nina Barisic; Stéphanie Baulac; Hande Caglayan; Christel Depienne; Carolien G.F. De Kovel; Petia Dimova; Rosa Guerrero-López; Renzo Guerrini; Helle Hjalgrim; Dorota Hoffman-Zacharska; Johanna Jahn; Karl Martin Klein; Bobby P.C. Koeleman; Eric Leguern; Anna-Elina Lehesjoki; Johannes Lemke; Holger Lerche; Carla Marini; Hiltrud Muhle; Felix Rosenow; Jose M. Serratosa; Rikke S. Møller; Ulrich Stephani, Pasquale Striano; Tiina Talvik; Sarah Von Spiczak; Yvonne Weber; Federico Zara.
Conflict of Interest statement. None declared.
References
- 1.Hildebrand M.S., Dahl H.H., Damiano J.A., Smith R.J., Scheffer I.E., Berkovic S.F. (2013) Recent advances in the molecular genetics of epilepsy. J. Med. Genet., 50, 271–279. [DOI] [PubMed] [Google Scholar]
- 2.Allen A.S., Berkovic S.F., Cossette P., Delanty N., Dlugos D., Eichler E.E., Epstein M.P., Glauser T., Goldstein D.B., Han Y., et al. (2013) De novo mutations in epileptic encephalopathies. Nature, 501, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Molinari F., Kaminska A., Fiermonte G., Boddaert N., Raas-Rothschild A., Plouin P., Palmieri L., Brunelle F., Palmieri F., Dulac O., et al. (2009) Mutations in the mitochondrial glutamate carrier SLC25A22 in neonatal epileptic encephalopathy with suppression bursts. Clin. Genet., 76, 188–194. [DOI] [PubMed] [Google Scholar]
- 4.Paciorkowski A.R., Weisenberg J., Kelley J.B., Spencer A., Tuttle E., Ghoneim D., Thio L.L., Christian S.L., Dobyns W.B., Paschal B.M. (2013) Autosomal recessive mutations in nuclear transport factor KPNA7 are associated with infantile spasms and cerebellar malformation. Eur. J. Hum. Genet., 22, 587–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gennaro E., Santorelli F.M., Bertini E., Buti D., Gaggero R., Gobbi G., Lini M., Granata T., Freri E., Parmeggiani A., et al. (2006) Somatic and germline mosaicisms in severe myoclonic epilepsy of infancy. Biochem. Biophys. Res. Commun., 341, 489–493. [DOI] [PubMed] [Google Scholar]
- 6.Gonzalez-Gaitan M., Jackle H. (1997) Role of Drosophila alpha-adaptin in presynaptic vesicle recycling. Cell, 88, 767–776. [DOI] [PubMed] [Google Scholar]
- 7.Zizioli D., Meyer C., Guhde G., Saftig P., von F.K., Schu P. (1999) Early embryonic death of mice deficient in gamma-adaptin. J. Biol. Chem., 274, 5385–5390. [DOI] [PubMed] [Google Scholar]
- 8.Shim J., Lee J. (2000) Molecular genetic analysis of apm-2 and aps-2, genes encoding the medium and small chains of the AP-2 clathrin-associated protein complex in the nematode Caenorhabditis elegans. Mol. Cells, 10, 309–316. [PubMed] [Google Scholar]
- 9.Shim J., Sternberg P.W., Lee J. (2000) Distinct and redundant functions of mu1 medium chains of the AP-1 clathrin-associated protein complex in the nematode Caenorhabditis elegans. Mol. Biol. Cell, 11, 2743–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boehm M., Bonifacino J.S. (2002) Genetic analyses of adaptin function from yeast to mammals. Gene, 286, 175–186. [DOI] [PubMed] [Google Scholar]
- 11.Hirst J., Irving C., Borner G.H. (2013) Adaptor protein complexes AP-4 and AP-5: new players in endosomal trafficking and progressive spastic paraplegia. Traffic, 14, 153–164. [DOI] [PubMed] [Google Scholar]
- 12.Collins B.M., McCoy A.J., Kent H.M., Evans P.R., Owen D.J. (2002) Molecular architecture and functional model of the endocytic AP2 complex. Cell, 109, 523–535. [DOI] [PubMed] [Google Scholar]
- 13.Jackson L.P., Kelly B.T., McCoy A.J., Gaffry T., James L.C., Collins B.M., Honing S., Evans P.R., Owen D.J. (2010) A large-scale conformational change couples membrane recruitment to cargo binding in the AP2 clathrin adaptor complex. Cell, 141, 1220–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson M.S. (2004) Adaptable adaptors for coated vesicles. Trends Cell Biol., 14, 167–174. [DOI] [PubMed] [Google Scholar]
- 15.Tuysuz B., Bilguvar K., Kocer N., Yalcinkaya C., Caglayan O., Gul E., Sahin S., Comu S., Gunel M. (2014) Autosomal recessive spastic tetraplegia caused by AP4M1 and AP4B1 gene mutation: expansion of the facial and neuroimaging features. Am. J. Med. Genet. A, 164, 1677–1685. [DOI] [PubMed] [Google Scholar]
- 16.Slabicki M., Theis M., Krastev D.B., Samsonov S., Mundwiller E., Junqueira M., Paszkowski-Rogacz M., Teyra J., Heninger A.K., Poser I., et al. (2010) A genome-scale DNA repair RNAi screen identifies SPG48 as a novel gene associated with hereditary spastic paraplegia. PLoS Biol., 8, e1000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Borner G.H., Antrobus R., Hirst J., Bhumbra G.S., Kozik P., Jackson L.P., Sahlender D.A., Robinson M.S. (2012) Multivariate proteomic profiling identifies novel accessory proteins of coated vesicles. J. Cell Biol., 197, 141–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abdollahpour H., Alawi M., Kortum F., Beckstette M., Seemanova E., Komarek V., Rosenberger G., Kutsche K. (2014) An AP4B1 frameshift mutation in siblings with intellectual disability and spastic tetraplegia further delineates the AP-4 deficiency syndrome. Eur. J. Hum. Genet., doi 10.1038/ejhg.2014.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bauer P., Leshinsky-Silver E., Blumkin L., Schlipf N., Schroder C., Schicks J., Lev D., Riess O., Lerman-Sagie T., Schols L. (2012) Mutation in the AP4B1 gene cause hereditary spastic paraplegia type 47 (SPG47). Neurogenetics, 13, 73–76. [DOI] [PubMed] [Google Scholar]
- 20.Blumkin L., Lerman-Sagie T., Lev D., Yosovich K., Leshinsky-Silver E. (2011) A new locus (SPG47) maps to 1p13.2–1p12 in an Arabic family with complicated autosomal recessive hereditary spastic paraplegia and thin corpus callosum. J. Neurol. Sci., 305, 67–70. [DOI] [PubMed] [Google Scholar]
- 21.Abou J.R., Philippe O., Raas-Rothschild A., Eck S.H., Graf E., Buchert R., Borck G., Ekici A., Brockschmidt F.F., Nothen M.M., et al. (2011) Adaptor protein complex 4 deficiency causes severe autosomal-recessive intellectual disability, progressive spastic paraplegia, shy character, and short stature. Am. J. Hum. Genet., 88, 788–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moreno-De-Luca A., Helmers S.L., Mao H., Burns T.G., Melton A.M., Schmidt K.R., Fernhoff P.M., Ledbetter D.H., Martin C.L. (2011) Adaptor protein complex-4 (AP-4) deficiency causes a novel autosomal recessive cerebral palsy syndrome with microcephaly and intellectual disability. J. Med. Genet., 48, 141–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kong X.F., Bousfiha A., Rouissi A., Itan Y., Abhyankar A., Bryant V., Okada S., Ailal F., Bustamante J., Casanova J.L., et al. (2013) A novel homozygous p.R1105X mutation of the AP4E1 gene in twins with hereditary spastic paraplegia and mycobacterial disease. PLoS ONE, 8, e58286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Novarino G., Fenstermaker A.G., Zaki M.S., Hofree M., Silhavy J.L., Heiberg A.D., Abdellateef M., Rosti B., Scott E., Mansour L., et al. (2014) Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science, 343, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Najmabadi H., Hu H., Garshasbi M., Zemojtel T., Abedini S.S., Chen W., Hosseini M., Behjati F., Haas S., Jamali P., et al. (2011) Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature, 478, 57–63. [DOI] [PubMed] [Google Scholar]
- 26.Verkerk A.J., Schot R., Dumee B., Schellekens K., Swagemakers S., Bertoli-Avella A.M., Lequin M.H., Dudink J., Govaert P., van Zwol A.L., et al. (2009) Mutation in the AP4M1 gene provides a model for neuroaxonal injury in cerebral palsy. Am. J. Hum. Genet., 85, 40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yap C.C., Murate M., Kishigami S., Muto Y., Kishida H., Hashikawa T., Yano R. (2003) Adaptor protein complex-4 (AP-4) is expressed in the central nervous system neurons and interacts with glutamate receptor delta2. Mol. Cell Neurosci., 24, 283–295. [DOI] [PubMed] [Google Scholar]
- 28.Hirst J., Bright N.A., Rous B., Robinson M.S. (1999) Characterization of a fourth adaptor-related protein complex. Mol. Biol. Cell, 10, 2787–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dell'Angelica E.C., Mullins C., Bonifacino J.S. (1999) AP-4, a novel protein complex related to clathrin adaptors. J. Biol. Chem., 274, 7278–7285. [DOI] [PubMed] [Google Scholar]
- 30.Matsuda S., Miura E., Matsuda K., Kakegawa W., Kohda K., Watanabe M., Yuzaki M. (2008) Accumulation of AMPA receptors in autophagosomes in neuronal axons lacking adaptor protein AP-4. Neuron, 57, 730–745. [DOI] [PubMed] [Google Scholar]
- 31.Burgos P.V., Mardones G.A., Rojas A.L., daSilva L.L., Prabhu Y., Hurley J.H., Bonifacino J.S. (2010) Sorting of the Alzheimer's disease amyloid precursor protein mediated by the AP-4 complex. Dev. Cell, 18, 425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lo G.T., Lombardi F., Santorelli F.M., Kawarai T., Orlacchio A. (2014) Hereditary spastic paraplegia: clinical-genetic characteristics and evolving molecular mechanisms. Exp. Neurol., 261, 518–539. [DOI] [PubMed] [Google Scholar]
- 33.EuroEPINOMICS-RES Consortium, Epilepsy Phenome/Genome Project, Epi4 K Consortium (2014) De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am. J. Hum. Genet., 95, 360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Unichenko P., Yang J.W., Luhmann H.J., Kirischuk S. (2014) Glutamatergic system controls synchronization of spontaneous neuronal activity in the murine neonatal entorhinal cortex. Pflugers Arch., doi 10.1007/s00424-014-1600-5. [DOI] [PubMed] [Google Scholar]
- 35.Lemke J.R., Hendrickx R., Geider K., Laube B., Schwake M., Harvey R.J., James V.M., Pepler A., Steiner I., Hortnagel K., et al. (2014) GRIN2B mutations in west syndrome and intellectual disability with focal epilepsy. Ann. Neurol., 75, 147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brusa R., Zimmermann F., Koh D.S., Feldmeyer D., Gass P., Seeburg P.H., Sprengel R. (1995) Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science, 270, 1677–1680. [DOI] [PubMed] [Google Scholar]
- 37.Beyer B., Deleuze C., Letts V.A., Mahaffey C.L., Boumil R.M., Lew T.A., Huguenard J.R., Frankel W.N. (2008) Absence seizures in C3H/HeJ and knockout mice caused by mutation of the AMPA receptor subunit Gria4. Hum. Mol. Genet., 17, 1738–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Menuz K., Nicoll R.A. (2008) Loss of inhibitory neuron AMPA receptors contributes to ataxia and epilepsy in stargazer mice. J. Neurosci., 28, 10599–10603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goytain A., Hines R.M., El-Husseini A., Quamme G.A. (2007) NIPA1(SPG6), the basis for autosomal dominant form of hereditary spastic paraplegia, encodes a functional Mg2+ transporter. J. Biol. Chem., 282, 8060–8068. [DOI] [PubMed] [Google Scholar]
- 40.Allison R., Lumb J.H., Fassier C., Connell J.W., Ten M.D., Seaman M.N., Hazan J., Reid E. (2013) An ESCRT-spastin interaction promotes fission of recycling tubules from the endosome. J. Cell Biol., 202, 527–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Renvoise B., Chang J., Singh R., Yonekawa S., FitzGibbon E.J., Mankodi A., Vanderver A., Schindler A., Toro C., Gahl W.A., et al. (2014) Lysosomal abnormalities in hereditary spastic paraplegia types SPG15 and SPG11. Ann. Clin. Transl. Neurol., 1, 379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Khundadze M., Kollmann K., Koch N., Biskup C., Nietzsche S., Zimmer G., Hennings J.C., Huebner A.K., Symmank J., Jahic A., et al. (2013) A hereditary spastic paraplegia mouse model supports a role of ZFYVE26/SPASTIZIN for the endolysosomal system. PLoS Genet., 9, e1003988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Drmanac R., Sparks A.B., Callow M.J., Halpern A.L., Burns N.L., Kermani B.G., Carnevali P., Nazarenko I., Nilsen G.B., Yeung G., et al. (2010) Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science, 327, 78–81. [DOI] [PubMed] [Google Scholar]
- 44.Reumers J., De R.P., Zhao H., Liekens A., Smeets D., Cleary J., Van L.P., Van Den Bossche M., Catthoor K., Sabbe B., et al. (2012) Optimized filtering reduces the error rate in detecting genomic variants by short-read sequencing. Nat. Biotechnol., 30, 61–68. [DOI] [PubMed] [Google Scholar]
- 45.Dibbens L.M., Tarpey P.S., Hynes K., Bayly M.A., Scheffer I.E., Smith R., Bomar J., Sutton E., Vandeleur L., Shoubridge C., et al. (2008) X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat. Genet., 40, 776–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schubert J., Siekierska A., Langlois M., May P., Huneau C., Becker F., Muhle H., Suls A., Lemke J.R., de Kovel C.G., et al. (2014) Mutations in STX1B, encoding a presynaptic protein, cause fever-associated epilepsy syndromes. Nat. Genet., 46, 1327–1332. [DOI] [PubMed] [Google Scholar]
- 47.Jameel M., Klar J., Tariq M., Moawia A., Altaf M.N., Seema W.S., Abdullah U., Naeem K.T., Raininko R., Baig S.M., Dahl N. (2014) A novel AP4M1 mutation in autosomal recessive cerebral palsy syndrome and clinical expansion of AP-4 deficiency. BMC Med. Genet., 15, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.