

Abstract
Osteoprogenitor cells combined with supportive biomaterials represent a promising approach to advance the standard of care for bone grafting procedures. However, this approach faces challenges, including inconsistent bone formation, cell survival in the implant, and appropriate biomaterial degradation. We have developed a collagen–hydroxyapatite (HA) scaffold that supports consistent osteogenesis by donor derived osteoprogenitors, and is more easily degraded than a pure ceramic scaffold. Herein, the material properties are characterized as well as cell attachment, viability, and progenitor distribution in vitro. Furthermore, we examined the biological performance in vivo in a critical-size mouse calvarial defect. To aid in the evaluation of the in-house collagen–HA scaffold, the in vivo performance was compared with a commercial collagen–HA scaffold (Healos®, Depuy). The in-house collagen–HA scaffold supported consistent bone formation by predominantly donor-derived osteoblasts, nearly completely filling a 3.5 mm calvarial defect with bone in all samples (n=5) after 3 weeks of implantation. In terms of bone formation and donor cell retention at 3 weeks postimplantation, no statistical difference was found between the in-house and commercial scaffold following quantitative histomorphometry. The collagen–HA scaffold presented here is an open and well-defined platform that supports robust bone formation and should facilitate the further development of collagen–hydroxyapatite biomaterials for bone tissue engineering.
Keywords: tissue engineering, stem cells, bone, collagen, bone marrow
Introduction
Cell-based tissue engineering of bone has shown promising clinical1 and preclinical2,3 results, however, this approach is not yet well understood and faces several challenges before becoming a transformative clinical procedure. One such challenge is the delivery of progenitor cells in a supportive biomaterial that provides the microenvironmental cues for cell survival, vascular invasion, and bone formation. Additionally, the biomaterial scaffold must be degradable, leaving only new tissue behind when the repair is complete. Thus far, an ideal scaffold for bone tissue engineering has not been identified.4
Bone is primarily composed of hydroxyapatite [Ca10(PO4)6OH2] (∼69% by weight) substituted with ions such as carbonate and magnesium.5,6 Type I collagen forms ∼90% of the organic fraction of bone, with the remaining ∼10% being proteins, such as osteonectin, fibronectin, and osteopontin, among many others.6 Following a biomimetic design paradigm, we have developed a type I collagen–hydroxyapatite scaffold (herein termed Col–HA).7 The type I collagen phase is easily degraded by cell-secreted collagenases, supports differentiation of progenitor cells into bone forming osteoblasts,7,8 and contains unique binding motifs for the attachment of other ECM molecules, such as fibro nectin.9 In addition, the degradation of collagen into gelatin may provide a means for cells to modulate the availability of RGD sites, normally hidden within the collagen triple helix.10 Xenogeneic collagen can elicit an immune response from human patients, however this is quite rare clinically and patients can be prescreened for collagen sensitivity prior to exposure to collagen-containing devices.11
Several collagen–calcium phosphate materials are currently FDA-approved for use with bone marrow extract as a bone void filler, such as Healos® (Depuy), Collagraft® (Zimmer), Ossimend® (Collagen Matrix), and Mozaik® (Integra LifeSciences). However, osteoprogenitors represent only a very small fraction of the bone marrow population,12 and it has been found previously that the number of progenitors strongly influences osteogenesis in vivo.13 One study has indicated that fresh bone marrow combined with a coral scaffold did not form bone when implanted in a ovine segmental defect, whereas culture-expanded bone marrow stro mal cells (BMSCs), seeded to the same material, did form new bone.2 Thus culture expansion, which greatly increases the number of osteoprogenitors, is a likely requirement for successful cell-based bone tissue engineering. Currently, there is no clinical precedent for the use of commercial collagen–HA scaffolds with culture-expanded osteoprogenitors.
Commercial scaffolds are useful for the development of cell-based therapies for bone tissue engineering, due to their sterility, use-history and consistency. However design modifications by the researcher are generally out of reach. Additionally, in order to understand how scaffold properties influence cell-based outcomes it would be of benefit to the researcher to have full control over scaffold properties. This would ultimately allow for a deeper understanding of cell-scaffold interactions, ultimately leading to optimal scaffold design and contributing to the long-term goal of safe and efficient cell-based therapies in the clinic.
The goals of this work were to (i) fabricate a sterile collagen–HA scaffold (ii) evaluate cell attachment, viability, and progenitor number in the scaffold just prior to implantation, (iii) achieve consistent osteogenesis in vivo with the Col–HA scaffold combined with culture-expanded BMSCs, and (iv) compare the level of early bone formation in the Col–HA scaffold to an established commercial collagen–HA biomate rial. The Col–HA scaffold presented here supports robust osteogenesis and is fully defined, open, and modifiable. This platform is ideal for basic research and facilitates the devel opment of collagen–hydroxyapatite biomaterials for bone tissue engineering.
Experimental
Scaffold fabrication by collagen–hydroxyapatite co-precipitation and freeze casting
The main component of the scaffold, type I collagen, was derived from rat tail tendons following Rajan et al.14 Collagen fibers and HA nanoparticles were formed simultaneously by precipitation in a modified simulated body fluid (mSBF) solution.15 Briefly, the collagen solution was adjusted to 2.5 mg mL−1 by a twofold dilution in sterile ultrapure water at 4°C. For a 200 mL solution of mSBF, the following salts were added in the order they appear: 1.08 g NaCl, 0.1428 g K2PO4 0.0622 g MgCl2, 2.4 g HEPES, 0.1758 g CaCl2, and 0.294 g NaHCO3. While kept cold, the pH of the solution was adjusted to 7.0 with sodium hydroxide solution and then transferred to a water bath at 40° C for 24 h to allow in situ coprecipitation. The gel precipitate was centrifuged at 11,000g and 4°C for 12 min. The supernatant was discarded and the pellet was freeze-dried (Labconco). The collagen–HA precipitate was reconstituted with water at a concentration of 100 mg mL−1, briefly homogenized to obtain a uniform slurry, and placed in a polystyrene culture dish. To impart a porous structure, the sample was freeze-dried starting from room temperature to −40°C at a cooling rate of −0.37°C min−1. The dried scaffold was immersed in a solution of 20 mg mL−1 EDC [1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride] for 24 h at 4°C to covalently crosslink the collagen fibers. The scaffold was then rinsed in a solution of 5% (w/w) glycine in sterile water overnight to block unreacted EDC, followed by three sequential rinses in sterile water for 15 min each at 4°C. Finally, rinsed scaffolds were freeze-dried again, cut to a thickness of ∼500 μm with a milling machine and punched to a diameter of 3.5 mm.
Healos® is composed of bovine type I collagen fibers mineralized with a thin coating of calcium phosphate, and formed into a high porosity sponge containing 4–200 μm pores. This material was received sterile. In-house scaffolds were terminally sterilized with a 24-h exposure to ethylene oxide gas using a bench-top sterilizer (Anprolene AN74i, Anderson Products), followed by a 24-h purge of excess gas in a vented hood. Before implantation, sterility of Col–HA scaffolds was assessed by incubating sterilized and unsterilized scaffolds, a negative control, in tryptic soy broth (Sigma) at 40°C for 2 weeks. The sterility of the scaffold was confirmed by the absence of bacterial growth in vials containing ethylene oxide sterilized scaffolds under these culture conditions.
Characterization of collagen–hydroxyapatite scaffold
The ratio of inorganic (HA) to organic (collagen) content was determined by thermogravimetric analysis (TGA Q-500, TA Instruments). The inorganic material was examined by X-ray diffraction (D2 Phaser, Bruker) performed on the resultant powder from a proteinase-K (Invitrogen) digestion of the scaffold. Diffraction peaks were acquired from a 2θ of 10° to 90° at a scan speed of 2.4° min−1.
Infrared absorbance spectra from 4000 to 400 cm−1 was acquired at a resolution of 4 cm−1 over 32 scans (Magna 560, Nicolet). Spectra were then analyzed with Know It All V9.5 software (BioRad).
Electron micrographs were acquired with a field emission scanning electron microscope (JSM-6335F, JEOL). Prior to imaging, Col–HA scaffolds were sputter coated (Polaron E5100) with a thin layer of gold-palladium.
To enable examination of scaffolds with X-ray tomography, scaffolds were stained with 1% iodine in ethanol over night to enhance radiopacity16 1500 images were acquired at an angle of −103° to 103° with an exposure time of 4 s, source power of 8 W, a voltage of 55 kV, and a 20× objective (MicroXCT-400, Xradia). Tomography images were reconstructed with XM Reconstructor (Xradia) and viewed with the 3D viewer plugin for the FIJI distribution of NIH ImageJ.17
Scaffold porosity, wall thickness, and anisotropy were calculated from tomography data from dry scaffolds with the BoneJ plugin for FIJI.18 Since very small pore interconnects may result in a near 100% interconnectivity, but have minimal contribution to mass transport, we excluded pores with a size <15 μm from the interconnectivity measurement. This was accomplished by performing a sphere-filling algorithm (thickness, BoneJ)18 to the void volume. A thresh old was then performed to select the fraction of the void volume with a caliber from 0-15 μm. This was added to the original solid structure to fill the small holes (≤15 μm) in the scaffold walls. Finally, the interconnectivity (connected void volume/total void volume) was calculated using the 3D objects counter19 applied to the void volume. The largest object was used as the interconnected volume.
Scaffold pore size was determined from histological sections of the scaffold using the mean linear intercept method.20 Briefly, transverse sections of scaffolds were obtained by cryosectioning (CM3050 S Cryostat, Leica) and imaged with darkfield optics (Axio Scan.Z1, Zeiss). Image processing was performed with ImageJ. Four sections were analyzed by drawing four lines regions of interest (ROIs) at different orientations for each section. The mean pore size was calculated as an average of the pore lengths falling on the line ROIs.
Scaffold permeability, k (m4 Ns−1), was measured with water and a custom flow cell using the following equation found in Ref. 21
where Δx = scaffold length (m), A = scaffold cross sectional area (m2), MB1 = mass flow rate without scaffold (g s−1), MB2 = mass flow rate with scaffold (g s−1), and r = radius of the outlet (m).
In vitro cell culture with primary mouse osteoprogenitors
Mouse BMSCs were isolated from the femur and tibia of CD1 wild type animals. Cells were added to 100 mm culture dishes in α-MEM (Gibco) containing 10% fetal bovine serum, and 1% penicillin–streptomycin. Following isolation, cells were allowed to attach to the culture dish in a humidified incubator at 5% CO2, ambient O2 and 37°C. To allow hematopoietic cells to contribute to early colony formation by osteoprogenitors, the culture medium was changed 4 days after seeding the bone marrow pellet. After 6 days of incubation, cells were trypsinized and seeded dropwise on to the top of either the Col–HA scaffold or a bovine collagen–hydroxyapatite scaffold (Healos®, Depuy) at a density of 1.0 × 106 cells in 15 μL of culture medium to immerse the scaffold. After seeding, cells were allowed to settle for 30 min before an additional 1 mL of culture medium was added to the culture well. Cell permeation and attachment were assessed at 1 minute, 1 hour and 12 h after seeding. Cell-seeded constructs were also incubated for a 1-week period to evaluate cell attachment, distribution, and to check for dramatic scaffold degradation associated with the sterilization method.
Following in vitro incubation, scaffolds were fixed in 10% formalin and stained for F-actin (Rhodamine-Phalloidin, Invitrogen) and nuclear DNA (Hoescht, Invitrogen). Cell viability and progenitor status was examined after 1 and 5 days of in vitro incubation. Viability was assessed using calcein and ethidium homodimer-1 provided as a kit (Live/Dead, Invitrogen). Osteoprogenitor cells were labeled using the α-SMAmCherry fluorescent reporter. This reporter marks a population of multipotent cells and consists of a transgene containing the mCherry fluorescent protein driven by the smooth muscle alpha actin (α-SMA) promoter.22
To enable viewing of the scaffold cross section, the scaffold was cut in half and glued to the bottom of a culture well, with the cross section facing up. The cross section of the scaffold was scanned in three dimensions to a depth of 100 μm with a 2-photon microscope (Ultima IV, Prairie technologies). Several tiles were stitched together to form a high-resolution (20 × objective) wide field view. Second harmonic images were acquired at an excitation wavelength of 900 nm, and data were collected at 450 nm, corresponding to the frequency doubling of the excitation wavelength as it interacts with the noncentrosymmetric structure of the collagen triple helix.
Image processing and analysis of in vitro data
Cell distribution from the outside edge was assessed by a distance analysis.24 The cell seeding efficiency (CSE), defined as the number of cells in the scaffold divided by the total number of cells seeded, was calculated using the 3D particle counter to identify and count cell nuclei in 3D stacks of seeded scaffolds.
In vivo bone formation in a mouse calvarial defect
Two groups (n = 5) of NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) immunodeficient mice were used as hosts. 1 day after scaffold seeding, host mice were anesthetized with a ketamine (135 mg kg−1)—xylazine (15 mg kg−1) blend and a 3.5-mm diameter critical size defect was created in the right parietal lobe using a bur trephine (RAL #229-030, Benco Dental). Extreme care was taken to prevent damage to the dura mater beneath the calvarium. Animals were given postoperative analgesic (bupronephrine, 0.08 mg kg−1). At 3 weeks postimplantation, animals were sacrificed for X-ray and histological analysis. To mark areas of active mineralization, calcein and alizarin complexone labels were injected intraperitoneally at 2 and 3 weeks, respectively.
In order to track cell fate after transplantation, donor cells were derived from bone marrow from CD1 animals carrying a Col3.6Cyan reporter gene. The reporter consists of the cyan fluorescent protein driven by a type I collagen promoter region that is activated in osteoblasts.25 The bone marrow was flushed with a 27-gauge needle from the femur and tibia of CD1 Col3.6Cyan animals and plated in α-MEM (Gibco) containing 10% FBS and 1% penicillin–streptomycin, incubated at 37°C, 5% CO2, and ambient O2. After 6 days, adherent cells were removed with 0.25% trypsin-EDTA (Gibco). One million cells in 15 lL of culture medium were seeded dropwise to each dry Col–HA and Healos® (Depuy) scaffold. After seeding, cells were allowed to settle for 30 min before one mL of culture medium was added. Cells were incubated with scaffolds overnight to ensure full cell attachment to the scaffold surface. All procedures in this study were approved by the UConn Health Center Institutional Animal Care and Use Committee (IACUC).
Histomorphometric analysis of in vivo data
To quantify the bone area fraction and donor cell area fraction for the test and control group, a region of interest (ROI) outlining the defect area was manually drawn on radiographs and histological sections. A threshold was then applied to the images to demarcate areas of bone.
To evaluate the edge integration of the constructs from radiographs, a line ROI was manually drawn along the perimeter of the implant, but inside the defect area. The line ROI contains the peripheral regions of the implant that were contacting host bone. If the implant was connected to the host bone, this would appear as a bone pixel in the line ROI placed between the construct and the edge of the host bone. To find the fraction of the host bone perimeter that was connected to the implant by bone, the number of bone pixels was divided by the total number of pixels in the line region.
Donor cell colocalization with the mineral label was determined using the distance analysis mentioned in the in vitro image analysis section. To exclude donor cells not associated with the label surface, a cutoff value of 14 μm was applied, corresponding to the mean distance from the edge of the label surface to the far edge of a donor cell on the label surface.
Statistical analysis
A t test was used to determine statistical significance (p < 0.05) between the Col–HA and Healos groups (n = 5).
Results
Characterization of the collagen–hydroxyapatite scaffold
Following freeze casting, the scaffold contained 15.36% water, 60.94% collagen, and 23.70% HA by thermogravimetric analysis (Supporting Information Figure S1). The identity of the inorganic phase was confirmed as HA by analysis of the XRD pattern of the resultant powder following proteinase K digestion of the collagen–HA composite [Figure 1(f)]. The XRD pattern was indicative of a poorly crystalline HA with broad peaks, namely at a 2Θ of 25.9° and 31.8° corresponding to the (002) plane, and overlap of the (211), (112), and (300) planes of HA, respectively.
Figure 1.
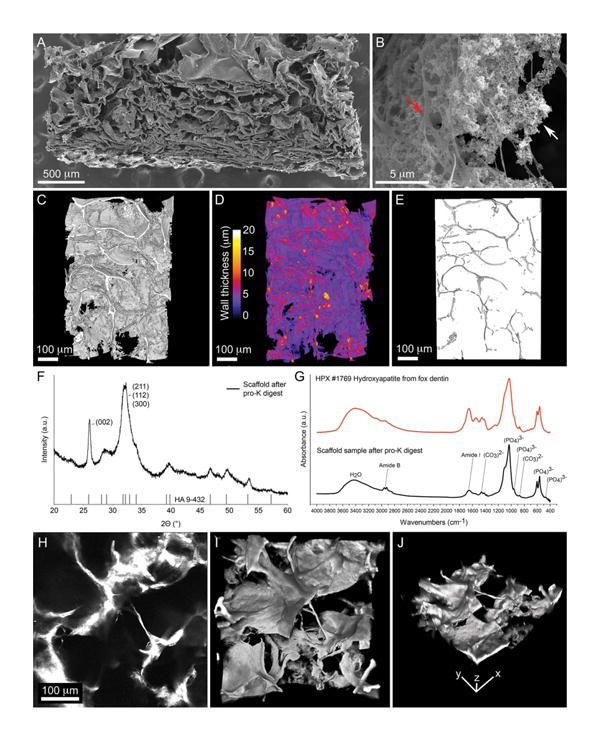
Characterization of Col–HA scaffold. (A) Electron micrograph of a Col–HA scaffold cut in cross section. (B) Magnification of hydroxy-apatite nanoparticles (white arrow) interspersed with collagen fibers (red arrow). (C) 3D reconstruction of the scaffold acquired by X-ray microtomography. (D) 3D reconstruction of the scaffold color-coded to indicate wall thickness. (E) Scaffold pore volume in 3D acquired by X-ray microtomography. (F) X-ray diffraction pattern of the inorganic phase of the scaffold when compared with a hydroxyapatite reference (vertical bars). (G) Infrared absorbance spectra of the inorganic phase of the scaffold (black line) compared with a hydroxyapatite reference spectra (red line). (H) A single slice of a 3D stack of images representing the collagen–HA scaffold imaged by its second harmonic signal. (I) Top and (J) iso-metric view of a 3D reconstruction of the collagen–HA scaffold viewed with second harmonic microscopy. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
FT-IR absorbance spectra of the digested scaffold sample further indicated that the inorganic phase is a carbonated HA, sharing similarities with the absorbance spectra of hydroxyapatite from dentin [Figure 1(g)]. Phosphate vibrational bands were observed at 961 (ν1), 475 (ν1), 1046 (ν3), 603 (ν4), and 567 (ν4) cm−1.26. Carbonate vibrational bands were observed at 873 (ν2) and 1400-1580 (ν3) cm−1.27 Amide I and Amide B bands from residual collagen in the sample were detected at 1653 and 2923 cm−1, respectively.27 Taken together, these results indicate the inorganic phase of the scaffold is a poorly crystalline carbonated hydroxyapatite similar to that found in native bone.
The collagenous phase of the scaffold produced a strong second harmonic signal when examined with 2-photon microscopy [Figure 1(h–j)]. Since the second harmonic signal from collagen is due to the repeating noncentrosymmetric structure of the triple helical protein,23 this indicates the scaffold contains collagen in a triple helical conformation rather than a denatured conformation (gelatin) which does not produce a second harmonic signal.28
The scaffold microstructure has a cellular organization with sheet-like walls [Figure 1(a,c,d)]. Near the nanoscale, the scaffold structure consists of agglomerated HA nanoparticles interspersed within collagen fibers [Figure 1(b)]. By X-ray microtomography the porosity of the scaffold was found to be 93% (Table I). The mean pore size was 101 ± 71 μm, with a scaffold wall thickness of 5.5 ± 2.4 μm [Figure 1(d)]. Pore alignment was largely isotropic with an anisotropy value of 0.3 (one being a perfectly aligned structure and zero being random). The scaffold permeability and degree of interconnectivity was 1.68 × 10−10 m4 Ns−1 and 99%, respectively (Table I). This scaffold architecture and composition should be supportive of cell infiltration, attachment, and osteogenesis.
Table I. Scaffold architectural properties.
| Sample | Porosity (%) | Pore Size (μm) | Wall Thickness (μm) | Interconnectivity (%) | Permeability (m4 Ns−1) | Anisotropy |
|---|---|---|---|---|---|---|
| Col–HA | 93 | 101±71 | 5.5±2.4 | 99 | 1.68 × 10−10 | 0.3 |
The interconnectivity is defined as the connected void volume divided by the total void volume. The value of the anisotropy parameter tends toward zero for randomly oriented structures and to one for parallel flat plates.
Cell seeding and attachment to collagen–hydroxyapatite scaffolds
To evaluate the time required for culture-expanded mouse BMSCs to attach to the scaffold, cells were seeded to Col–HA scaffolds and Healos®. Subsequently, scaffolds were harvested at 1 min, 1 and 12 h after seeding. It was found that cells were well-attached after 12 h of in vitro culture, but not within 1 h or, not surprisingly, within 1 min [Figure 2(a-f)]. The 1-min condition represents the initial conditions at the time of implantation when cells are seeded to a scaffold and immediately implanted.
Figure 2.
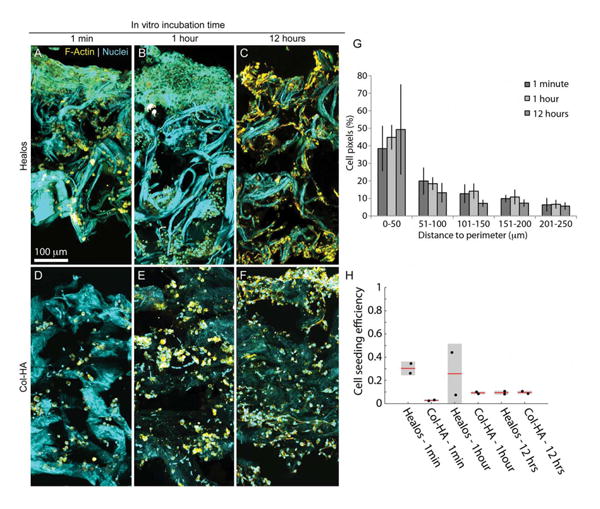
In vitro characterization with mouse BMSCs. Maximum intensity projections of scaffolds cut in half and imaged in 3D normal to the cross section. Healos scaffold after (A) 1 min, (B) 1 h, and (C) 12 h of incubation with mouse BMSCs. Col–HA scaffold after (D) 1 min, (E) 1 h, and (F) 12 h of incubation with mouse BMSCs. The collagen content generates a second harmonic signal (cyan). Cells were stained for F-actin (yellow) and nuclei (punctate cyan signal). (G) Quantification of cell distribution relative to the outside edge. (H) Cell seeding efficiency at 1 min, 1 and 12 h of incubation after seeding (initial seeding density was 1 million cells/scaffold, approximately 3.5 mm in diameter and 500 μm thick). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Cells were present throughout the thickness of both scaffolds at all time points, indicating cells were able to penetrate through interconnections in the void volume [Figure 2(a-f)]. However, cell density decreased with increasing distance from the outer surface of the scaffold [Figure 2(g)]. The cell seeding efficiency was found to be ∼10% when the initial seeding number was one million cells. This corresponds to about 100,000 cells seeded to a scaffold, prepared in the geometry of a 3.5 mm diameter mouse criticalsize calvarial defect. Furthermore, large clumps of cells on top of the scaffold were observed at 1 min and 1 h after seeding [Figure 2(a,b) and Supporting Information Figure S2). In a separate experiment, these clumps were not present after a few weeks of implantation,29 suggesting that these cells do not survive after implantation. Similarly, cell clumping on the loading side of the scaffold was not observed after 12 h of in vitro culture, indicating that unattached or poorly attached cells may not survive beyond a few hours. These results indicate that a 12-h incubation time is sufficient for culture-expanded mouse BMSCs to fully attach to the scaffold. To ensure cell attachment before implantation, a 12-h incubation period was used in all experiments described hereafter.
Sterilizing protein-based biomaterials is challenging due to the potential for material damage as a result of the sterilization process. Immersion in 70% ethanol is not a robust sterilization method, since hydrophilic virus and bacterial spores are resistant to this method.30 Previously, we observed rapid degradation of our scaffolds (<1 week) when sterilized with gamma irradiation and cultured with mouse BMSCs (data not shown). Herein, ethylene oxide gas was employed to sterilize Col–HA scaffolds. Sterility was confirmed by the absence of bacterial growth after 2 weeks in bacterial culture medium at 40°C.
To determine if the sterilization procedure had a negative effect on scaffold degradation, as observed with irradiation sterilization, scaffolds were cut into disks and seeded with culture-expanded mouse BMSCs and incubated for 1 week. In contrast to gamma irradiation (data not shown), ethylene oxide sterilized scaffolds did not exhibit rapid degradation when cultured with cells for 1 week [Figure 3(b)]. The Col–HA scaffolds maintained their original circularity, while the Healos® control group expanded anisotropically upon hydration [Figure 3(a–d)]. When stained for F-actin filaments after 1 week in vitro, cells were well attached to the scaffold surface and distributed throughout the thickness [Figure 3(e–g)]. These results suggest ethylene oxide is a suitable method for producing sterile collagen–hydroxyapatite scaffolds without resulting in rapid degradation (<1 week in vitro) and maintaining good cell attachment to the biomaterial.
Figure 3.
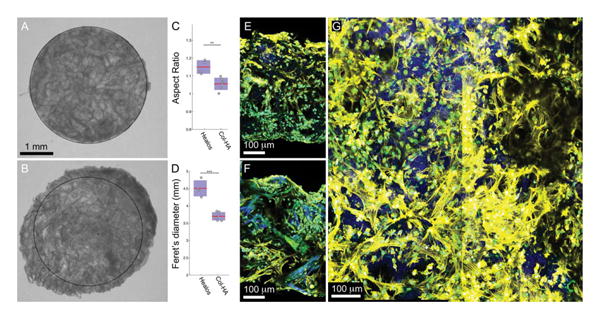
Sterilized scaffolds seeded with mouse BMSCs after 1 week in vitro. (A) Ethylene oxide gas sterilized Col–HA scaffold, and (B) Healos® after 1 week in vitro with mouse BMSCs. Quantification of the (C) aspect ratio (long axis/short axis) and (D) longest diameter (Feret's diameter) Col–HA and Healos® scaffolds after 1 week in vitro. Maximum intensity projection of (E) a Col–HA and (F) a Healos® scaffold seeded with mouse BMSCs cut in cross section after 1 week in vitro. (G) Maximum intensity projection of a 2-photon stack acquired from a top view of a Col–HA scaffold. Cells (E)–(G) are stained for F-actin (yellow), nuclei (cyan), and the scaffold produces a second harmonic signal (blue). [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Previous work has indicated that BMSCs from the donor population are predominantly responsible for bone formation in the calvarial model.3,8,29 This observation underscores the need for a viable progenitor population within the seeded scaffold to achieve consistent osteogenesis. Here, we examined the cell viability and the distribution of osteoprogenitor cells at 1 and 5 days after seeding Col–HA scaffolds. At day 1, the majority of cells were viable; however, cells were not uniformly distributed in the radial direction of the scaffold [Figure 4(a)]. In contrast, at day 5, cells were well distributed throughout the scaffold [Figure 4(b)], high lighting the ability of cells to migrate into available space. Upon closer inspection with 2-photon microscopy, a subset of smaller diameter nonviable cells was found within the scaffold walls [Figure 4(e)]. To examine cell viability throughout the thickness of the scaffold, samples were cut into half and viewed in cross-section. Cells appeared pre dominantly viable throughout the scaffold thickness 5 days after seeding [Figure 4(f)], suggesting that strictly diffusive transport in the relatively thin scaffold is sufficient to support cell viability over this period. Similarly, when cells containing a fluorescent marker for osteoprogenitor cells, α-SMAmCherry,22 were seeded to Col–HA scaffolds, they were not homogenously distributed by day 1 [Figure 4(c)], but were homogenously distributed by day 5 after seeding [Figure 4(d)]. Taken together, these results suggest that at 1 day after seeding, representing the initial conditions at the time of implantation used here, cells were well attached to the scaffold, viable, and the population contained large numbers of osteoprogenitors.
Figure 4.
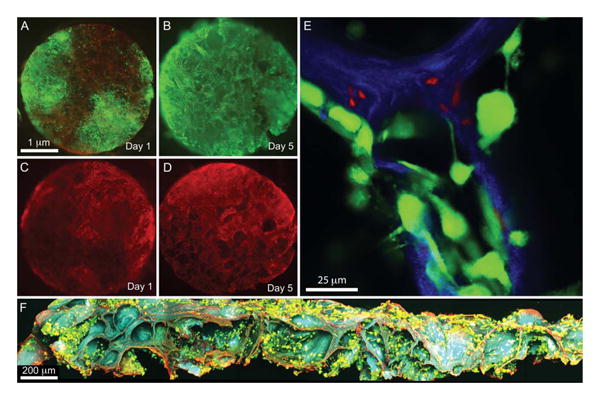
Cell viability and progenitor status. Col–HA scaffolds seeded with culture expanded mouse BMSCs and incubated for (A) 1 and (B) 5 days in vitro. Scaffolds were stained for live (green) and dead (red) cells. (A slight red background is visible, due to scaffold adsorbance of the dead stain). Col–HA scaffolds seeded with culture expanded mouse BMSCs carrying a SMAAmCherry (red) reporter for osteoprogenitor cells and incubated for (C) 1 and (D) 5 days in vitro. (E) Single image of a 2-photon micrograph showing live (green) overlying the scaffold (blue) and smaller dead (red) cells that have invaded the scaffold walls after 1 day in vitro. (F) Maximum intensity projection of a 3D stack of the scaffold containing cells stained for live (green) and dead (red) viewed in cross section after 5 days in vitro. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
In vivo calvarial defect model of bone repair
After 3 weeks of implantation, calvaria containing critical size defects filled with BMSCs combined with either the Col–HA scaffold or Healos® were harvested. Viewed with a stereomicroscope (Supporting Information Figure S3), the implant regions were more opaque than the surrounding host bone. All samples contained bone by radiographic dis crimination and several implants appeared well integrated to the host bone [Figure 5(a–d,g–i)]. Two samples were not well connected to the host bone [Figure 5(f,h), orange arrows] and two others exhibited significant gaps between the implant and host [Figure 5(e,j), orange arrows]. Some implants appeared to overlap the edge of the host bone rather than filling the defect [Figure 5(e,g,h,i), blue arrows]. Since all scaffolds were placed in the defect at the time of implantation, this would require the edge of the scaffold to pop out sometime after.
Figure 5.
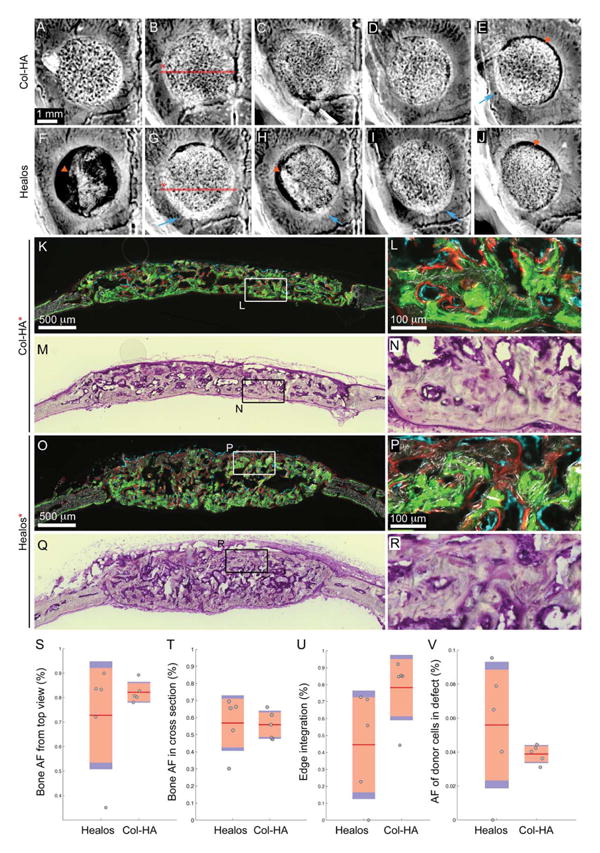
Evaluation of bone formation within mouse critical size calvarial defects after 3 weeks. Radiographs of calvarial defects filled with mouse BMSCs in (A)–(E) Col–HA and (F)–(J) Healos® scaffolds after 3 weeks. Blue arrows indicate areas where the scaffold popped out of the defect. Orange arrows indicate sites of scaffold degradation. (K) Fluorescence and (M) toluidine-blue stained sections from (K) to (N) Col–HA and (O) to (R) Healos® scaffolds. The samples, (B) and (G), from which the sections, (K) and (O), came are indicated on the radiographs by the red line. Fluorescence images contain donor cells (cyan), mineralization label delivered at 2 weeks (green) and one day before sacrifice at 3 weeks (red), overlayed on a darkfield image of the mineralized tissue. Quantification of (S) bone area fraction from radiographs, (T) bone area fraction from histological sections, (U) edge integration from radiographs, and (V) area fraction of donor cells from histological sections. (S)–(V) Red line indicates the mean, pink bars indicate 95% confidence intervals and blue bars indicate one standard deviation. Individual samples are represented by dots. [Color figure can be viewed in the online issue, which is available at wileyonlinelibrary.com.]
Mineralized tissue was observed between the red and green mineralization labels, given at 2 and 3 weeks, respectively [Figure 5(k,l,o,p)], indicating that new bone had formed as opposed to mistaking the calcium phosphate phase of the scaffold for new bone. Donor cells carrying the Col3.6Cyan25 osteoblast reporter gene were found lining the active mineralizing surface labeled with alizarin red [Figure 5(l,p)]. Furthermore, 75±0.10 and 67±0.05% of the mineral label was lined with donor cells for Healos® and Col–HA, respectively (Supporting Information Figure S5) suggesting that the donor source played a dominant role in early bone formation (<3 weeks). Taken together, these results indicate that all samples contained mineralized tis sue deposited primarily by donor cells and that some of the samples appeared to have degraded.
The mean bone area fraction (AF) from radiographs was slightly higher for the Col–HA group compared with the Healos group, however this difference was not significant [Figure 5(s)]. In cross-section, the mean bone AF was similar between the groups [Figure 5(t)]. The Col–HA scaffolds, when viewed from the top, were slightly more integrated with host than the Healos® group, however, this result (p=0.077) was not significant [Figure 5(u)]. When the mean AF of donor cells in the defects viewed in cross-section was compared, the Healos® group was slightly higher, although this difference was not significant [Figure 5(v)]. The in-group standard deviation for each of the four parameters investigated was smaller for the Col–HA group compared with the Healos® group [Figure 5(s–v)], blue bars indicate one standard deviation. Taken together, these results indicate the Col–HA group exhibited consistent bone formation and appeared well attached to the host in four out of five samples. When compared with Healos®, the Col–HA scaffold performed favorably, providing evidence for the efficacy of the Col–HA scaffold for cell-based bone repair.
Discussion
Biomaterials for cell delivery have enabled considerable progress towards consistent bone regeneration in defect sites.2,31,32 However, challenges remain, such as cell survival in the implant33 and realizing the ideal degradation rate of the biomaterial.34 We have developed a type I collagen–hydroxyapatite scaffold for cell-based bone tissue engineering that is relatively simple to fabricate, highly porous, and is easily remodeled by cells. Here we show that this biomaterial retains progenitor cells, supports robust new bone formation after only 3 weeks in vivo, and for the metrics and time point examined here, is comparable in performance to a clinical grade material (Healos®), used here as a benchmark. Unlike the commercial benchmark, the Col–HA material is an open scaffold produced in an academic lab, facilitating further modification and development of this class of biomaterials towards healing bone defects in a clinical setting with culture-expanded osteoprogenitors.
The microstructure of the Col–HA scaffold is an interspersed composite of collagen fibers and HA nanoparticles [Figure 1(b)], generated by simultaneous collagen fibrillogenesis and precipitation of HA nanoparticles. In contrast, Healos® is composed of collagen fibers with a thin coating of hydroxyapatite. The collagen component of the Col–HA scaffold is arranged in a triple-helical conformation, indicated by second harmonic imaging [Figure 1(h–k)]. Cell attachment to collagen has been reported to be strongly dependent on protein conformation.10,35,36 The availability of collagen motifs on the scaffold surface may facilitate binding of cells, ECM proteins, and provide signals to infiltrating cells as the collagen is remodeled.
Employed here to fabricate the Col–HA scaffold, freeze casting is a relatively gentle process that accommodates the incorporation of proteins and generates a highly porous and thin-walled architecture. Due to the compliant mechanical properties of the collagen–HA scaffold, it may be best suited for non-load-bearing indications such as craniofacial repair. However, many bone fractures require fixation in combination with bone grafting, thus a compliant collagen–HA scaffold could theoretically be applied in a load bearing application when combined with mechanical fixation. If new bone quickly fills a defect, it will supersede the poor mechanical properties of the original scaffold, just as the soft cartilaginous callus becomes rigid bone following nor mal fracture healing. In addition, a more radiolucent biomaterial, such as the Col–HA scaffold, enables better radiographic discrimination of the progression of new bone formation in a cell-seeded construct.
Marcacci et al. found that a pure hydroxyapatite scaffold was not fully degraded after 6 years of implantation with culture-expanded BMSCs in humans. In contrast, the collagen–HA scaffold described here could degrade faster (∼weeks) than pure hydroxyapatite scaffolds owing to MMP-mediated degradation of the collagen phase.
A few of the collagen–HA scaffolds tested herein exhib ited more rapid degradation (<3 weeks) at the peripheral edge. This type of rapid degradation impairs host integration and reduces the amount of bone formed. Rapid degradation can result from early MMP-mediated degradation of type I collagen by host cells of the innate immune system.37–39 In a separate experiment, it was found that repeated surgery to expose the implant for live-animal imaging resulted in a similar radial gap between the host and implant, whereas a control group that did not undergo additional surgery displayed much better integration, further pointing to an inflammatory mechanism behind peripheral scaffold degradation.29 Neutrophil-derived MMP-8 has been reported to act on type I collagen scaffolds implanted in mouse myocardium.39 Interestingly, when empty scaffolds were implanted in mouse calvarial defects, scaffolds persist without noticeable degradation (data not shown), suggesting a dependence on the presence of cells for peripheral scaffold degradation. BMSCs have been shown to play an immu nomodulatory role in vitro and in vivo, with an anti- or proinflammatory effect proving at times variable and strongly dependent on the microenvironment they experience.40,41 One possibility regarding the samples exhibiting degradation herein, is that the wound microenvironment experienced by the donor BMSCs did not drive them towards immunosuppression. Instead, primed by such a milieu, seeded BMSCs could have promoted neutrophil invasion and MMP-mediated degradation of the scaffold.40
Donor cells were predominantly responsible for bone for mation in both groups of scaffolds, echoing earlier reports.3,8,29 Here we observed Col3.6Cyan donor osteoblasts overlying a red mineralization label, providing a rigorous indicator that donor cells were responsible for de novo tissue formation. Since the scaffold is relatively thin (400-500 μm), diffusion of nutrient and waste removal appears sufficient to support cells through the early period following implantation when a functioning vasculature has not yet formed. Larger defects will likely require a more advanced vascular plexus prior to implantation to ensure donor cell survival. The limi tations of this study include the relatively early time frame examined, the non-load-bearing model, and the small sample size (n = 5). In the future, longer time points in a load-bearing defect combined with mechanical testing of the functional properties of new bone should be examined to more critically evaluate this approach.
Conclusion
We have developed a sterile collagen–HA scaffold for cell based bone tissue engineering and examined its material properties and biological performances in vitro and in vivo combined with culture-expanded mouse BMSCs. The scaffold is composed of triple-helical type I collagen and poorly crys talline carbonated apatite arranged in a porous (93%) cellular architecture. When seeded to the scaffold, cells percolate through the full thickness of the thin geometry (∼500 μm thick) and are fully attached to the biomaterial after 12 h in vitro. The population of seeded cells just before implantation contained predominantly viable cells and large numbers of osteoprogenitors. When the Col–HA and control scaffolds were seeded with culture-expanded mouse BMSCs and implanted into a critical size calvarial defect, robust bone formation was observed in both groups at 3 weeks post-implantation. For the number of scaffolds (n = 5) and early time point investigated here, quantitative histomorph ometry indicated no significant difference between the Col–HA and control scaffold in terms of bone formation and donor cell retention. The Col–HA scaffold is an open and well-defined platform that enables the researcher to have greater control of a cell-biomaterial model system, facilitating further development of collagen–hydroxyapatite biomaterials for cell delivery and bone defect repair.
Supplementary Material
Acknowledgments
The authors would like to thank Li Chen, Dr. Xi Jiang, Dr. Peter Maye, Dr. Mark Kronenberg, Dr. Yusuf Khan, Dr. Nat Dyment, Gary Lavigne, Laura Pinatti, Dr. Lichun Zhang, Dr. Sina Shah-bazmohamadi, Dr. Zengmin Xia, Dr. Erica Kramer, and Michael Zilm for their technical expertise and helpful discussions. The authors also wish to express their gratitude to Drew Clearfield for his careful reading of the manuscript.
Contract grant sponsor: GAANN pre-doctoral fellowship from the Department of Education; contract grant number: P200A09315
Contract grant sponsor: NIH; contract grant number: 1R21AR059962-01A1, 3 R01 AR064381-01S1
Contract grant sponsor: NSF; contract grant number: CBET-1133883
Footnotes
Additional Supporting Information may be found in the online version of this article.
References
- 1.Quarto R, Mastrogiacomo M, Cancedda R, Kutepov SM, Mukhachev V, Lavroukov A, Kon E, Marcacci M. Repair of large bone defects with the use of autologous bone marrow stroma cells. New Engl J Med. 2001;344:385–386. doi: 10.1056/NEJM200102013440516. [DOI] [PubMed] [Google Scholar]
- 2.Petite H, Viateau V, Bensaid W, Meunier A, de Pollak C, Bourguignon M, Oudina K, Sedel L, Guillemin G. Tissue-engineered bone regeneration. Nat Biotechnol. 2000;18:959–963. doi: 10.1038/79449. [DOI] [PubMed] [Google Scholar]
- 3.Cowan CM, Shi YY, Aalami OO, Chou YF, Mari C, Thomas R, Quarto N, Contag CH, Wu B, Longaker MT. Adipose-derived adult stromal cells heal critical-size mouse calvarial defects. Nat Bio technol. 2004;22:560–567. doi: 10.1038/nbt958. [DOI] [PubMed] [Google Scholar]
- 4.Szpalski C, Wetterau M, Barr J, Warren SM. Bone tissue engineering: Current strategies and techniques—Part I: Scaffolds. Tissue Eng Part B Rev. 2012;18:246–257. doi: 10.1089/ten.TEB.2011.0427. [DOI] [PubMed] [Google Scholar]
- 5.Pate FD. Bone chemistry and paleodiet. J Archaeol Method Theory. 1994;1:161–209. [Google Scholar]
- 6.Boskey AL, Robey PG. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism. Hoboken, NJ: Wiley; 2013. The composition of bone. [Google Scholar]
- 7.Yu X, Wang L, Peng F, Jiang X, Xia Z, Huang J, Rowe D, Wei M. The effect of fresh bone marrow cells on reconstruction of mouse calvarial defect combined with calvarial osteoprogenitor cells and collagen-apatite scaffold. J Tissue Eng Regen Med. 2013;7:974–983. doi: 10.1002/term.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu X, Xia Z, Wang L, Peng F, Jiang X, Huang J, Rowe D, Wei M. Controlling the structural organization of regenerated bone by taioring tissue engineering scaffold architecture. J Mater Chem. 2012;22:9721. [Google Scholar]
- 9.Di Lullo GA. Mapping the ligand-binding sites and disease associated mutations on the most abundant protein in the human, type I collagen. J Biol Chem. 2001;277:4223–4231. doi: 10.1074/jbc.M110709200. [DOI] [PubMed] [Google Scholar]
- 10.Davis GE, Bayless KJ, Davis MJ, Meininger GA. Regulation of tis sue injury responses by the exposure of matricryptic sites within extracellular matrix molecules. Am J Pathol. 2000;156:1489–1498. doi: 10.1016/S0002-9440(10)65020-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lynn AK, Yannas IV, Bonfield W. Antigenicity and immunogenic ity of collagen. J Biomed Mater Res. 2004;71B:343–354. doi: 10.1002/jbm.b.30096. [DOI] [PubMed] [Google Scholar]
- 12.Pittenger MF. Multilineage potential of adult human mesenchy mal stem cells. Science. 1999;284:143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- 13.Hernigou PH, Poignard A, Beaujean F, Rouard H. Percutaneous autologous bone-marrow grafting for nonunions: Influence of the number and concentration of progenitor cells. J Bone Joint Surg. 2005;87:1430–1437. doi: 10.2106/JBJS.D.02215. [DOI] [PubMed] [Google Scholar]
- 14.Rajan N, Habermehl J, Cote MF, Doillon CJ, Mantovani D. Prepa ration of ready-to-use, storable and reconstituted type I collagen from rat tail tendon for tissue engineering applications. Nat Pro toc. 2007;1:2753–2758. doi: 10.1038/nprot.2006.430. [DOI] [PubMed] [Google Scholar]
- 15.Qu H, Xia Z, Knecht D, Wei M. Synthesis of dense collagen/apatite composites using a biomimetic method. J Am Ceram Soc. 2008;91:3211–3215. [Google Scholar]
- 16.Metscher BD. MicroCT for developmental biology: A versatile tool for high-contrast 3D imaging at histological resolutions. Dev Dyn. 2009;238:632–640. doi: 10.1002/dvdy.21857. [DOI] [PubMed] [Google Scholar]
- 17.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: An open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doube M, Klosowski MM, Arganda-Carreras I, Cordelieres FP, Dougherty RP, Jackson JS, Schmid B, Hutchinson JR, Shefelbine SJ. BoneJ: Free and extensible bone image analysis in ImageJ. Bone. 2010;47:1076–1079. doi: 10.1016/j.bone.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bolte S, Cordelieres FP. A guided tour into subcellular colocaliza-tion analysis in light microscopy. J Microsc. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 20.O'Brien FJ, Harley BA, Yannas IV, Gibson L. Influence of freezing rate on pore structure in freeze-dried collagen-GAG scaffolds. Biomaterials. 2004;25:1077–1086. doi: 10.1016/s0142-9612(03)00630-6. [DOI] [PubMed] [Google Scholar]
- 21.Kemppainen JM, Hollister SJ. Differential effects of designed scaffold permeability on chondrogenesis by chondrocytes and bone marrow stromal cells. Biomaterials. 2010;31:279–287. doi: 10.1016/j.biomaterials.2009.09.041. [DOI] [PubMed] [Google Scholar]
- 22.Grcevic D, Pejda S, Matthews BG, Repic D, Wang L, Li H, Kronenberg MS, Jiang X, Maye P, Adams DJ, Rowe DW, Aguila HL, Kalajzic I. In vivo fate mapping identifies mesenchymal pro genitor cells. Stem Cells. 2012;30:187–196. doi: 10.1002/stem.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams RM, Zipfel WR, Webb WW. Interpreting second-harmonic generation images of collagen I fibrils. Biophys J. 2005;88:1377–1386. doi: 10.1529/biophysj.104.047308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lo Celso C, Fleming HE, Wu JW, Zhao CX, Miake-Lye S, Fujisaki J, Cote D, Rowe DW, Lin CP, Scadden DT. Live-animal tracking of individual haematopoietic stem/progenitor cells in their niche. Nature. 2009;457:92–96. doi: 10.1038/nature07434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalajzic I, Kalajzic Z, Kaliterna M, Gronowicz G, Clark SH, Lichtler AC, Rowe D. Use of type I collagen green fluorescent protein transgenes to identify subpopulations of cells at different stages of the osteoblast lineage. J Bone Miner Res. 2002;17:15–25. doi: 10.1359/jbmr.2002.17.1.15. [DOI] [PubMed] [Google Scholar]
- 26.Rehman I, Bonfield W. Characterization of hydroxyapatite and car bonated apatite by photo acoustic FTIR spectroscopy. J Mater Sci Mater Med. 1997;8:1–4. doi: 10.1023/a:1018570213546. [DOI] [PubMed] [Google Scholar]
- 27.Chang MC, Tanaka J. FT-IR study for hydroxyapatite/collagen nanocomposite cross-linked by glutaraldehyde. Biomaterials. 2002;23:4811–4818. doi: 10.1016/s0142-9612(02)00232-6. [DOI] [PubMed] [Google Scholar]
- 28.Zeugolis DI, Khew ST, Yew ESY, Ekaputra AK, Tong YW, Yung LYL, Hutmacher DW, Sheppard C, Raghunath M. Electro-spinning of pure collagen nanofibres—Just an expensive way to make gel atin? Biomaterials. 2008;29:2293–2305. doi: 10.1016/j.biomaterials.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 29.Villa MM, Wang L, Huang J, Rowe DW, Wei M. Visualizing osteogenesis in vivo within a cell-scaffold construct for bone tissue engineering using two-photon microscopy. Tissue Eng Part C Methods. 2013;19:839–849. doi: 10.1089/ten.tec.2012.0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holy CE, Cheng C, Davies JE, Shoichet MS. Optimizing the sterili zation of PLGA scaffolds for use in tissue engineering. Biomaterials. 2001;22:25–31. doi: 10.1016/s0142-9612(00)00136-8. [DOI] [PubMed] [Google Scholar]
- 31.Krebsbach PH, Mankani MH, Satomura K, Kuznetsov SA, Robey PG. Repair of craniotomy defects using bone marrow stromal cells. Transplantation. 1998;66:1272–1278. doi: 10.1097/00007890-199811270-00002. [DOI] [PubMed] [Google Scholar]
- 32.Bruder SP, Kraus KH, Goldberg VM, Kadiyala S. The effect of implants loaded with autologous mesenchymal stem cells on the healing of canine segmental bone defects. J Bone Joint Surg. 1998;80:985–996. doi: 10.2106/00004623-199807000-00007. [DOI] [PubMed] [Google Scholar]
- 33.Zeitouni S, Krause U, Clough BH, Halderman H, Falster A, Blalock DT, Chaput CD, Sampson HW, Gregory CA. Human mesenchymal stem cell-derived matrices for enhanced osteoregeneration. Sci Transl Med. 2012;4:132ra55, 132ra55. doi: 10.1126/scitranslmed.3003396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marcacci M, Kon E, Moukhachev V, Lavroukov A, Kutepov S, Quarto R, Mastrogiacomo M, Cancedda R. Stem cells associated with macroporous bioceramics for long bone repair: 6- to 7-year outcome of a pilot clinical study. Tissue Eng. 2007;13:947–955. doi: 10.1089/ten.2006.0271. [DOI] [PubMed] [Google Scholar]
- 35.Davis GE. Affinity of integrins for damaged extracellular matrix: αv β3 binds to denatured collagen type I through RGD sites. Bio chem Biophys Res Commun. 1992;182:1025–1031. doi: 10.1016/0006-291x(92)91834-d. [DOI] [PubMed] [Google Scholar]
- 36.Fields GB. The collagen triple-helix: Correlation of conformation with biological activities. Connective Tissue Res. 1995;31:235–243. doi: 10.3109/03008209509010815. [DOI] [PubMed] [Google Scholar]
- 37.Parks WC. Matrix metalloproteinases in repair. Wound Repair Regen. 1999;7:423–432. doi: 10.1046/j.1524-475x.1999.00423.x. [DOI] [PubMed] [Google Scholar]
- 38.Werb Z, Gordon Sa. Secretion of a specific collagenase by stimu lated macrophages. J Exp Med. 1975;142:346–360. doi: 10.1084/jem.142.2.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van Amerongen MJ, Harmsen MC, Petersen AH, Kors G, van Luyn MJA. The enzymatic degradation of scaffolds and their replacement by vascularized extracellular matrix in the murine myocardium. Biomaterials. 2006;27:2247–2257. doi: 10.1016/j.biomaterials.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 40.English K, Wood KJ. Mesenchymal stromal cells in transplantation rejection and tolerance. Cold Spring Harb Perspect Med. 2013;3(5):a015560. doi: 10.1101/cshperspect.a015560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Auletta JJ, Deans RJ, Bartholomew AM. Emerging roles for multi potent, bone marrow-derived stromal cells in host defense. Blood. 2012;119:1801–1809. doi: 10.1182/blood-2011-10-384354. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.